

Redox-responsive nanomedicine of miR-519c and gemcitabine effectively inhibits pancreatic tumor growth in mice.
Abstract
Desmoplastic and hypoxic pancreatic cancer microenvironment induces aberrant expression of miRNAs and hypoxia-inducible factor-1α (HIF-1α) responsible for gemcitabine (GEM) resistance. We demonstrated that miR-519c was down-regulated in pancreatic cancer and transfection of miR-519c in GEM-resistant pancreatic cancer cells inhibited HIF-1α level under hypoxia. We synthesized redox-sensitive mPEG-co-P(Asp)-g-DC-g-S-S-GEM polymer, with GEM payload of 14% (w/w) and 90% GEM release upon incubation with l-glutathione. We synthesized mPEG-co-P(Asp)-g-TEPA-g-DC for complex formation with miRNA. Chemical modification of miR-519c with 2′-O-methyl phosphorothioate (OMe-PS) at 3′ end enhanced its stability and activity without being immunogenic. Epidermal growth factor receptor targeting peptide GE11 decoration increased tumor accumulation of micelles after systemic administration and significantly inhibited orthotopic desmoplastic pancreatic cancer growth in NSG mice by down-regulating HIF-1α and genes responsible for glucose uptake and cancer cell metabolism. Our multifunctional nanomedicine of GEM and OMe-PS–miR-519c offers a novel therapeutic strategy to treat desmoplasia and hypoxia-induced chemoresistance in pancreatic cancer.
INTRODUCTION
Pancreatic cancer is one of the most chemotherapy-resistant cancers and a leading cause of cancer-related mortality with a dismal 5-year survival rate of 8 to 10% (https://seer.cancer.gov/statfacts/html/pancreas.html). Gemcitabine (GEM) is a first-line therapy for pancreatic cancer either alone or in combination with other drugs such as nanoparticle albumin-bound paclitaxel or nab-paclitaxel (1). However, the clinical effect of single-agent GEM is modest due to its rapid metabolism, inefficient delivery to the desmoplastic tumor, and development of chemoresistance (2, 3). The combination therapy of GEM and nab-paclitaxel versus GEM alone increases survival along with an increase in grade 3 neutropenia, fatigue, and neuropathy (4). The use of FOLFIRINOX (5-fluorouracil, oxaliplatin, irinotecan, and leucovorin combination), as compared to GEM, significantly prolongs survival but at the expense of higher grade 3 or 4 toxicities (5).
Most patients with pancreatic cancer are present with metastatic or inoperable disease, and therefore, systemic polychemotherapy remains the treatment of choice. The median survival of patients with metastatic pancreatic cancer is still less than a year with either FOLFIRINOX or GEM and nab-paclitaxel (6, 7). Therefore, safer and more effective combination of GEM and novel drugs further need to be developed particularly for older or less fit patients or those with significant comorbidities, who cannot tolerate intensive chemotherapy.
Chemotherapy resistance in pancreatic cancer involves multiple mechanisms. GEM-resistant cells mainly represent cancer stem cells and epithelial-to-mesenchymal transformation (EMT) phenotype (8, 9). Desmoplasia produces hypoxic microenvironment, stimulating hypoxia-inducible factor-1α (HIF-1α), which, in turn, modulates tumor metabolism, induces profibrotic and angiogenic responses, and mediates the overexpression of adenosine triphosphate–binding cassette superfamily G member 2 (ABCG2). As a member of the ABC transporter superfamily, ABCG2 functions as a drug efflux pump and has been reported to play a role in GEM resistance in pancreatic cancer (10, 11). Because of an increase in high-glucose metabolism in GEM-resistant cells, the flux of glycolytic intermediates is directed toward nonoxidative pentose phosphate pathway (PPP), leading to an increase in pyrimidine biosynthesis and pool of deoxycytidine triphosphate (dCTP). The structural similarity between dCTP and GEM, a nucleoside analog, results in a molecular competition to bind the replicating DNA, reducing the efficacy of GEM. Hypoxia is responsible for an increase in sonic hedgehog (SHH) signaling and smoothened (SMO) expression in pancreatic cancer and activation of the transcription factor glioma-associated oncogene (GLI) (12). These changes result in further generation of fibrosis and a decrease in blood flow, creating a desmoplasia-hypoxia vicious cycle.
MicroRNAs (miRNAs) regulate gene expression by the degradation of target mRNAs, repressing of mRNA translation, or up-regulating of target genes. Aberrant expressions of miRNAs correlate with altered expression of genes responsible for stemness, metastasis, tumor metabolism, hypoxia, and chemoresistance (13). We previously reported differential down-regulation of miR-205 and miR-let7b in human pancreatic cancer tissues and cells (14). Further, we demonstrated that the combination of GEM-conjugated polymeric micelles and miR-205 significantly inhibited pancreatic tumor growth in mice after systemic administration, as miR-205 sensitized resistant pancreatic cells to GEM (8, 15). miR-519c is of particular interest in pancreatic cancer, as it is down-regulated in pancreatic cancer, binds to HIF-1α mRNA, and can inhibit HIF-1α expression, leading to sensitization of pancreatic cancer cells (16, 17). We hypothesize that a combination of GEM and miR-519c can target HIF-1α and ABCG2, resensitize resistant pancreatic cancer cells to GEM, and result in decreased desmoplasia. Moreover, 2′-O-methyl phosphorothioate (2′-OMe-PS) modification of RNA based on our previous study (18) was applied to miR-519c to increase its stability, binding affinity, and functional potential and decrease immunostimulatory properties.
Nanoparticulate delivery systems have shown distinct advantages in increasing drug accumulation at the tumor site after systemic administration. GEM delivery is limited by its poor pharmacokinetic profiles and dense desmoplasia, which may be overcome by a GEM-conjugated delivery system. We recently developed methoxy poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate)-graft-gemcitabine-graft-dodecanol (mPEG-b-PCC-g-GEM-g-DC), which conjugates GEM via stable amide bonds and self-assembles into micelles, with 12% (w/w) GEM loading and 2.5-fold higher GEM accumulation in orthotopic pancreatic cancer compared to free GEM (2). Redox-sensitive polymeric micelles are very promising due to their controlled drug release inside the tumor cells in the presence of intracellular stimuli, including glutathione (GSH) (19). GSH concentration in tumor cells is several times higher than that in the extracellular microenvironment and normal tissues (20). Epidermal growth factor receptor (EGFR) has been overexpressed in 40 to 80% of pancreatic cancers. Cells that overexpress EGFR will accumulate the functionalized particles, resulting in greater cytotoxicity. We hypothesized that EGFR targeted GE11 peptide mixed micelles system based on tetraethylene pentamine (TEPA) for complex formation with miRNA and GSH-sensitive polypeptide for GEM conjugation via disulfide bond for targeted delivery of GEM and OMe-PS–miR-519c will result in a synergistic inhibition of pancreatic cancer without excess toxicity. Our in vitro and in vivo studies confirmed that mixed OMe-PS–miR-519c–complexed and GEM-conjugated mixed micelles effectively inhibit pancreatic tumor growth.
RESULTS
Overexpression of HIF-1α in patient pancreatic cancer tissues and cell lines
We first established a GEM-resistant pancreatic ductal adenocarcinoma (PDAC) cell model by incubating wild-type MIA PaCa-2 cells with increasing concentrations of GEM over 2 months. The resulting GEM-resistant MIA PaCa-2R cells were 25-fold more resistant compared to the wild-type MIA PaCa-2 cells, as reflected by the pronounced increase in the median inhibitory concentration value from 2 to 50 μM. Our immunohistochemical analysis demonstrated a high expression of HIF-1α protein in patient-derived pancreatic cancer tissues but a negligible expression in adjacent healthy patient tissues (fig. S1A). Western blot analysis of MIA PaCa-2R cells demonstrated that the expression level of HIF-1α was very low under normoxic conditions but significantly increased, reaching the highest level at 4 hours after incubation of MIA PaCa-2R cells under hypoxic condition (fig. S1B).
miR-519c inhibits HIF-1α, Hh signaling, and tumor cell invasion
miR-519c was significantly down-regulated in pancreatic cancer tissue compared to adjacent normal tissues, as determined by real-time reverse transcription polymerase chain reaction (RT-PCR) (Fig. 1A). Similarly, we observed a significantly low miR-519c expression level in pancreatic cancer cell lines such as HPAF-II, Capan-1, and MIA PaCa-2R, compared to nonmalignant pancreatic cells HPDE (human pancreatic duct epithelial) (Fig. 1B). With target prediction by miRTarBase, we confirmed that miR-519c directly targets HIF-1α and ABCG2, indicating its potential utility to reverse hypoxia-induced chemoresistance.
Fig. 1. miR-519c expression is down-regulated in pancreatic cancer, and its overexpression inhibits HIF-1α, Hh signaling, and cell migration under hypoxic condition.
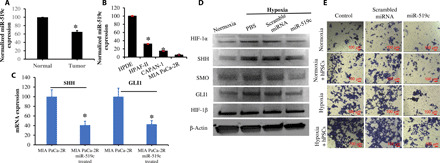
miR-519c was down-regulated in patient tissues (A) and pancreatic cancer cell lines (B), compared to paired normal tissues and normal pancreatic cells such as HPDE. Further, miR-519c inhibited the expression of HIF-1α and Hh ligands such as SHH, SMO, and GLI1 after the transfection of MIA PaCa-2R cells with Lipofectamine/miR-519c under hypoxic condition (C and D). (E) miR-519c also inhibited cell migration under hypoxic condition when MIA PaCa-2R cells were cultured on Transwell chamber with human pancreatic stellate cells (hPSCs). *P < 0.05, two-paired t test.
Expression of SHH, SMO, and GLI1 was significantly higher under hypoxic versus normoxic conditions, as determined by Western blot analysis. Transfection of pancreatic cancer cells with Lipofectamine/miR-519c complexes significantly inhibited the expression of HIF-1α and Hh ligands, such as SHH, GLI1, and SMO (Fig. 1, C and D). We observed that addition of human pancreatic stellate cells (hPSCs)–conditioned medium to MIA PaCa-2R cells induced tumor cell invasion, especially under hypoxic condition (Fig. 1E), indicating that the exposure of MIA PaCa-2R cells to desmoplastic microenvironment enhances their invasion capacity. Additional treatment of MIA PaCa-2R cells with miR-519c inhibited cell migration under hypoxic condition and desmoplasia (Fig. 1E).
miR-519c inhibits glucose influx and metabolism under hypoxia
Flow cytometry data demonstrated an up-regulation of glucose transporter 1 (GLUT1) expression in MIA PaCa-2R cells under hypoxic conditions. Transfection of these cells with miR-519c, but not scrambled miRNA, significantly inhibited GLUT1 expression under hypoxic conditions (Fig. 2A and fig. S2). As a glucose analog, the uptake of 2-deoxyglucose (2-DG) by MIA PaCa-2R cells was significantly increased under hypoxic conditions even when these cells were transfected with scrambled miRNA. This is expected, as GLUT1 is a well-known HIF-1α target. Transfection of these cells with miR-519c resulted in a significant decrease in 2-DG uptake under hypoxic conditions (Fig. 2B), suggesting that miR-519c negates hypoxia-induced elevation in glucose uptake, using 2-DG as a GLUT1 substrate. The concentration of dCTP was significantly higher under hypoxic conditions but declined following the transfection of MIA PaCa-2R cells with Lipofectamine/miR-519c complexes (Fig. 2C), indicating that the intracellular level of dCTP is severely suppressed by miR-519c. Hypoxia is known to increase glycolytic influx and pyrimidine biosynthesis in tumor cells. The mRNA expression levels of several known targets of HIF-1α were up-regulated under the hypoxic condition. These targets included NME/NM23 nucleoside diphosphate kinase (NME4) involved in PPP and nucleotide biosynthesis pathways, as well as lactate dehydrogenase (LDH), hexokinase 2 (HKII), and transketolase (TKT) involved in glucose metabolism. The expression level of these targets genes was significantly inhibited after the transfection of MIA PaCa-2R cells with miR-519c but not by scrambled miRNA (Fig. 2D). Together, these results provide strong evidence that miR-519c repletion attenuates glucose metabolism and reduces intracellular dCTP pool.
Fig. 2. miR-519c inhibits glucose metabolism to inhibit GEM resistance to pancreatic cancer cells under hypoxic condition.
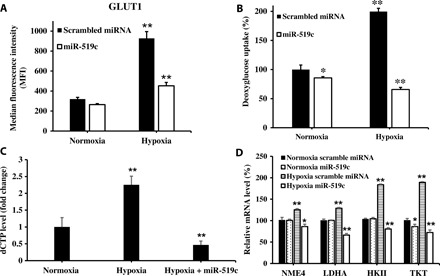
(A and B) miR-519c inhibited 2-DG uptake, as well as expression of GLUT1 and dCTP, but not scrambled miRNA under hypoxic condition after transfection of MIA PaCa-2R cells with Lipofectamine miR-519c. Cells transfected with Lipofectamine/scrambled miRNA complexes were used as the control. (C) miR-519c decreased dCTP level under hypoxic condition. (D) In addition, miR-519c but not scrambled miRNA inhibited NME4, HKII, LDHA, and TKT expressions under hypoxic condition. Results are presented as the mean ±SD (n = 3). *P < 0.05 and **P < 0.001, two-paired t test.
miR-519c sensitizes hypoxic GEM-resistant tumor cells to cell killing by GEM
Because of an increase in high-glucose metabolism in GEM-resistant cells, the flux of glycolytic intermediates is directed toward nonoxidative PPP, leading to an increase in pyrimidine biosynthesis and pool of dCTP. As GEM is a nucleoside analog, its efficacy is affected by both nucleoside synthesis and their cellular uptake. The presence of dC resulted in a dose-dependent decrease in cytotoxicity after treatment with GEM (Fig. 3A), suggesting that treatment with dCTP increased GEM resistance in MIA PaCa-2R cells. The combination of miR-519c and GEM demonstrated a dose-dependent synergistic effect on cell killing. In contrast, treatment with scrambled miRNA had little effect on cell killing (Fig. 3B). Apoptosis assay further confirmed these observations (fig. S3A). The effect of miR-519c on cell cycle of MIA PaCa-2R cells was determined by flow cytometry after the transfection of MIA PaCa-2R cells with miR-519c under normoxic and hypoxic conditions. Treatment with miR-519c compared to scrambled miRNA under hypoxic conditions resulted in a significant increase in the percentage of cells in the G2 phase (Fig. 3C and fig. S3A). These data suggest that miR-519c inhibited pancreatic cancer cell proliferation by inducing G2 phase cell cycle arrest.
Fig. 3. miR-519c and GEM synergistically inhibit pancreatic cancer cell proliferation and promote apoptosis.

(A) Coincubation of GEM with 100 μM dC-reduced drug potency. (B) Significant decrease in cell viability when MIA PaCa-2R cells were transfected with a combination of GEM and miR-519c under the hypoxic condition for 48 hours, but GEM combination with scrambled miRNA was less effective. (C) miR-519c inhibited viability by arresting cell cycle in the G2-M phase. (D and E) miR-519c inhibits ABCG2, c-Myc, and programmed death-ligand 1 (PD-L1) expressions, which are up-regulated under hypoxic condition, but scrambled miRNA had little effect. (F) Combination of miR-519c and GEM was more effective in apoptotic cell death of tumor spheroids generated using MIA PaCa-R cells than either of them alone. The confocal laser scanning microscopy images were captured at a depth of 15 μm. *P < 0.05 and **P < 0.001, two-paired t test.
The Western blot analysis demonstrated that drug resistance–related protein ABCG2 and transcription factor c-MYC (which promotes cancer cell proliferation) and programmed death-ligand 1 (PD-L1) were up-regulated under hypoxic condition. These data indicated that the hypoxic-dependent up-regulation of ABCG2- and c-MYC–magnified GEM resistance and PD-L1 can promote immune escape of tumor cells from cytotoxic T lymphocytes and contribute to the immunosuppression in tumors under hypoxia. Then, the apoptosis rate was investigated by the coculturing of MIA PaCa-2R and peripheral blood lymphocytes with or without miR-519c treatment under normoxic and hypoxic conditions (fig. S4). We observed a significant increase in apoptosis of MIA PaCa-2R cells in the coculture system with miR-519c transfection under hypoxia. Treatment of MIA PaCa-2R cells with miR-519c but not scrambled miRNA inhibited expression of ABCG2, c-MYC, and PD-L1 (Fig. 3, D and E). These data suggest that miR-519c has the potential to improve the cytotoxicity of activated T cells and immunotherapy.
Three-dimensional (3D) desmoplastic tumor spheroids were developed to mimic the hypoxic microenvironment in pancreatic cancer by mixing and culturing hPSCs and MIA PaCa-2R cells in hanging drop plates. Live and dead assay was carried out after treatment with miR-519c and GEM for 48 hours by confocal laser scanning microscopy. Fluorescein isothiocyanate–labeled calcein and propidium iodide binding to nucleic acids presented live and dead cells, respectively. Treatment with the combination of miR-519c and GEM for 9 days exhibited a significantly higher inhibition of tumor growth, leading to the collapse of tumor spheroids. In contrast, treatment with miR-519c or GEM alone failed to destroy tumor spheroids (Fig. 3F). These results suggest that miR-519c facilitates the resensitization of pancreatic cancer cells to GEM.
Chemical modification with OMe-PS increases the stability and targeting efficiency of miR-519c without eliciting proinflammatory response
Chemical modifications can protect miRNA against nuclease degradation and affect their nonspecific interaction with plasma proteins and cell membrane components. Therefore, modification of its 3′ end with OMe-PS stabilized miR-519c, which in good agreement with our previous study (18). We determined the effect of nucleases on miRNA stability by incubating unmodified miR-519c and OMe-PS–miR-519c in 50% fetal bovine serum (FBS). miR-519c was degraded within an hour in 50% FBS at 37°C. When three-terminal nucleotides at the 3′ end of miR-519c guide strand were modified, with 2′-OMe-PS, its stability was substantially increased, resulting in significant levels of intact miRNA remained even after 24 hours of incubation at 37°C (Fig. 4A). Further, there was no loss of activity in cell killing due to this chemical modification. Incubation of MIA PaCa-2R cells with the combination of 100 nM OMe-PS–miR-519c or miR-519c and increasing concentrations of GEM demonstrated a dose-dependent synergistic effect on cell killing, with OMe-PS–miR-519c being more potent than miR-519c (Fig. 4B).
Fig. 4. Chemically modified OMe-PS–miR-519c was more stable and effective but a little immunogenic compared to unmodified miR-519c.

(A) Synergistic effect of OMe-PS–miR-519c and GEM on cell viability after transfection of MIA PaCa-2R cells with 100 μM OMe-PS–miR-519c and increasing concentrations of GEM for 48 hours under hypoxic conditions. (B) Stability of OMe-PS–miR-519c and unmodified miR-519c in 50% FBS. (C and D) Effect of miR-519c and OMe-PS–miR-519c on proinflammatory cytokines interleukin-2 (IL-2) and IL-4, as analyzed by enzyme-linked immunosorbent assay. (E) Association of OMe-PS–miR-519c with endogenous AGO2 protein. (F) HIF-1α and ABCG2 expression levels after miR-519c and OMe-PS–miR-519c treatment for 48 hours under hypoxic conditions, as determined by Western blot analysis. *P < 0.05 and **P < 0.001, two-paired t test.
To examine whether the chemical modification of miR-519c induces proinflammatory cytokine production, we determined the levels of interleukin-2 (IL-2) and IL-4 after incubating OMe-PS–miR-519c and unmodified miR-519c with human peripheral blood mononuclear cells (hPBMCs) for 24 hours and measured cytokines released in hPBMC cultures using a cytokine multianalyte ELISArray kit. The antigen standard cocktail was added to the plate as the positive control. There was a little induction of cytokines due to miRNA treatment. Compared to miR-519c–treated groups, OMe-PS–miR-519c–treated hPBMCs showed the lower release of cytokines, including IL-2 and IL-4 (Fig. 4, C and D).
Argonaute 2 (AGO2) protein is a ribonuclease (RNase) and is an essential component of the RNA-induced silencing complex (RISC) where one strand of the mature miRNA duplex is first loaded onto AGO2 protein. AGO2-bound miRNA interacts with complementary regions of target mRNA and leads to either AGO2-mediated endonuclease cleavage of the mRNA or reduction in its translation efficiency. Therefore, we assessed the effect of chemical modification of miR-519c on its association with AGO2 compared to unmodified miR-519c and OMe-PS–miR-519c in the cytoplasm. As shown in Fig. 4E, the transfection of MIA PaCa-2R with miR-519c and OMe-PS–miR-519c was resulting in 3- and 16-fold higher RISC loading than that of the control group. Consequently, higher RISC loading resulted in improved inhibition of HIF-1α and ABCG2 (Fig. 4F). Notably, the AGO2 association did not differ between normoxic and hypoxic conditions.
Redox-sensitive nanocarriers achieve combined delivery of OMe-PS–miR-519c and GEM
On the basis of the results above, we selected OMe-PS–miR-519c over miR-519c for in vivo studies due to its higher stability, enhanced RISC loading, and affinity. We used our mPEG-co-P(Asp)-g-TEPA-g-DC cationic copolymer for complex formation with miRNA and used for preparing mixed micelles with mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC for codelivery of OMe-PS-miR-591c and GEM. 1H nuclear magnetic resonance (NMR) of mPEG-co-P(Asp)-g-TEPA-g-DC and mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC is shown in Fig. 5 (A and B). The percentage of GEM loading in mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC was calculated to be 14% on the basis of the integration of peak at 3.5 parts per million (ppm) for the CH2 of mPEG (f) and peaks at 7.8 to 8.22, 7.09, 5.29 to 6.16, 4.36, 4.27, 3.0, and 2.93 ppm (a, b, c, d, e, g, and h), respectively (protons on GEM-S-S-NH2). The rest of the carboxylic acid groups in the polymer were also reacted with dodecylamine (DC) to provide sufficient hydrophobicity for micelle formation, as indicated by the peaks at 0.85, 0.98, and 1.23 ppm (i, j, and k), respectively. In TEPA polymer, the broad peak at 2.98 ppm for TEPA and the characteristic peaks at 1.38, 1.24, and 0.85 ppm (e, f, and g) for DC chain suggest their successful conjugation (Fig. 5B). 1H NMR spectra of intermediate products, such as mPEG-PBLA (Poly-β-benzyl-L-aspartate) in dimethyl sulfoxide (DMSO)–d6, Boc-Ala-SS-OH in CDCl3, Diboc-GEM in DMSO-d6, Boc-Ala-SS-Diboc-GEM in CDCl3, Ala-SS-GEM in DMSO-d6, and GE11-PEG-co-P(Asp)-g-DC in DMSO-d6, are also shown in fig. S5.
Fig. 5. Conjugation of GEM to redox-sensitive polymer via disulfide bond demonstrates rapid GEM release in the presence of GSH but negligible GEM release in the absence of GSH at pH 7.4.
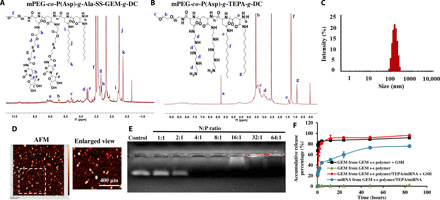
(A and B) 1H NMR spectra of mPEG-co-P(Asp)-g-TEPA-g-DC and mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC. (C) Particle size distribution of redox-sensitive mixed micelles, as determined by dynamic light scattering. (D) Surface morphology and particle size distribution of mixed micelles determined by atomic force microscopy. (E) mPEG-co-P(Asp)-g-TEPA-g-DC forms complexes with miRNA at the N:P (TEPA:miRNA) ratio of 4:1 and above, as determined by gel electrophoresis. (F) GEM and miRNA release profiles from micelles with or without miR-519c demonstrate GSH-responsive GEM release and controlled release of miRNA in the presence of GSH.
The particle size distribution of miR-519c–complexed and GEM-conjugated redox-sensitive mixed micelles was prepared using mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC and mPEG-b-PCC-g-DC-g-TEPA (Fig. 5C). As determined by atomic force microscopy, these mixed micelles were spherical in shape and narrow particle size distribution, with the mean particle size of 129.5 ± 5 nm (Fig. 5D). GEM loading was 14% in the polymer conjugate, as determined after alkaline hydrolysis and analysis with high-performance liquid chromatography–ultraviolet (HPLC-UV), which was consistent with the NMR calculation. Micelles prepared using mPEG-co-P(Asp)-g-TEPA-g-DC had the mean particle size of 77.18 nm and formed a complex with miRNA at a very low N:P ratio (4:1) (fig. S6). The particle size of mixed micelles based on the above two polymers was 160 nm. Drug-polymer conjugates and miRNA were analyzed for their drug release profiles at different conditions. In the absence of GSH, GEM conjugates were stable and released only up to 5% of GEM within 4 days. Disulfide conjugates released significant amounts of GEM within an hour in 10 mM GSH. These results confirmed that disulfide conjugates were stable at physiological conditions but rapidly released GEM in the reducing environment. The release profile demonstrated a controlled release of miRNA from GEM conjugates/TEPA micelles over 2 days (Fig. 5F).
EGFR-targeted nanocarriers effectively deliver GEM and OMe-PS–miR-519c to desmoplastic pancreatic tumor in an orthotopic mouse model
To optimize GE11 peptide concentration for enhanced EGFR receptor–meditated endocytosis, we determined the effect of GE11 peptide on the cellular uptake of micelles after incubation of MIA PaCa-2R cells with coumarin G6–loaded mixed micelles prepared at 10:90, 20:80, and 30:70 ratios of GE11 peptide–decorated micelles and nontargeted micelles. There was an increase in cellular uptake with increasing GE11 peptide concentration, but the level of cellular uptake for the mixed micelles at 20:80 and 30:70 ratios was similar (fig. S7A). To avoid the effect of high GE11 peptide concentration on micellar stability and systemic toxicity, we did not formulate targeted micelles beyond 30:70 ratio. Preincubation of cells with excess of free GE11 peptide significantly reduced the cellular uptake of the mixed micelles, indicating that the uptake was receptor mediated (fig. S7A). Then, we determined the biodistribution of GEM to the tumor at 6 and 24 hours after systemic administration of nontargeted and EGFR-targeted GE11-decorated GEM-conjugated OMe-PS–miR-519c mixed micelles into desmoplastic pancreatic tumor–bearing nonobese diabetic/severe combined immunodeficient–gamma null (NSG) mice at the doses of GEM (20 mg/kg) and miRNA (1 mg/kg). Desmoplastic orthotopic mouse model recapitulates hypoxic tumor microenvironment responsible for decreased drug delivery and chemoresistance in pancreatic cancer. At 6 and 24 hours after administration, GEM and miRNA were extracted from the tumor, and the liver and their concentrations were determined by liquid chromatography–tandem mass spectrometry (LC-MS/MS) (using 5-Aza-2′-dC as an internal standard) and real-time PCR, respectively. We observed significantly higher GEM and miR-519c concentrations in the tumor at 6 and 24 hours after administration of GE11 peptide–decorated combination mixed micelles compared to nontargeted combination mixed micelles (Fig. 6A). In contrast, GEM concentrations in the liver at 6 and 24 hours after administration of GE11 peptide–decorated combination mixed micelles were significantly lower than those of the mice injected with nontargeted combination mixed micelles (fig. S8A). The relative levels of miR-519c were higher in the tumor at 6 and 24 hours after administration of GE11 peptide–decorated combination mixed micelles compared to nontargeted combination mixed micelles (fig. S8C).
Fig. 6. Biodistribution and antitumor efficacy of GEM-conjugated OMe-PS–miR-519c–complexed redox-sensitive micelles after systemic administration into orthotopic desmoplastic pancreatic tumor–bearing mice.

(A) Biodistribution of GEM and OMe-PS–miR-519c at 6 and 24 hours after injection of nontargeted (NT) and GE11-targeted micelles following intravenous injection at GEM dose of 20 mg/kg and miR-519c dose of 1 mg/kg. GEM concentration was determined by LC-MS/MS using 5-Aza-2′-dC as an internal standard for GEM quantification. miR-519c concentration was determined by real-time PCR. (B) Bioluminescence images of tumors and isolated tumor images at the end of the experiment. (C) Tumor weight after treatment and (D) body weight. (E) Representative microscopic pictures of immunohistochemical staining for HIF-1α, α-SMA, Ki-67, and cleaved caspase-3. Scale bars (10×), 200 μm. Photo credit: Xiaofei Xin, University of Nebraska Medical Center. *P < 0.05 and *P < 0.001, two-paired t test.
Combination therapy of GEM and OMe-PS–miR-519c via EGFR-targeted nanocarriers exerts superior efficacy against orthotopic pancreatic tumor growth
Desmoplastic orthotopic mouse model recapitulates hypoxic tumor microenvironment responsible for decreased drug delivery and chemoresistance in pancreatic cancer. Micelles were injected intravenously three times a week for 2 weeks, and tumor growth was monitored by in vivo imaging system (IVIS) bioluminescence imaging. The rationale of doses and schedules of individual drugs were designed on the basis of previous studies (21). Notably, the combination therapy had lower doses of individual drug and miRNA. GEM micelles and OMe-PS–miR-519c micelles separately controlled tumor growth significantly more than saline or free GEM. Nontargeted combination micelles demonstrated synergy and had greater tumor growth inhibition than either GEM-conjugated micelles or OMe-PS–miR-519c–complexed micelles, reflecting the ability of OMe-PS–miR-519c to down-regulate HIF-1α and resensitize pancreatic cancer to GEM. EGFR-targeted GE11 peptide–decorated mixed micelles were even more effective than nontargeted mixed micelles in controlling tumor growth (Fig. 6B). The tumor weight was the lowest for GE11 peptide–decorated combination micelles–treated group compared to all treated groups (Fig. 6C). We did not observe a decrease in body weight or morbidity in mice (Fig. 6D).
Hematoxylin and eosin (H&E) staining of tumor tissues confirmed the extensive apoptotic and necrotic cells throughout the tumor in the control group and the inhibition of proliferation of tumor cells in the treated groups. Compared to the control and free GEM group, the tumor samples from GEM conjugated redox-sensitive micelles–treated group exhibited with limited metastatic cells (Fig. 6E). Coinjection of hPSCs with MIA PaCa-2R cells stimulated tumor growth by inducing fibrosis, as evidenced by the overexpression of α–smooth muscle actin (α-SMA) in the tumor of the saline-treated group (Fig. 6E). Treatment of the mice with GEM-conjugated redox-sensitive micelles or miR-519c–complexed micelles showed a significantly less α-SMA–positive fibrotic area (Fig. 6E). However, the mice treated with the combination therapy showed the least α-SMA–positive staining in the tumor.
Ki-67 protein is an excellent marker for determining the cell proliferation. Ki-67 staining of tumor tissues confirmed the extensive cell proliferation in the nontreated control group compared to the free GEM-treated group. Tumor tissues treated with GEM-conjugated redox-sensitive micelles–treated group showed a significantly lower number of cells Ki-67–positive cells (Fig. 6E). Furthermore, cleaved caspase-3 staining of tumor tissues indicated the induction of significant apoptosis by treatment with GEM redox-sensitive micelles and miR-519c micelles–treated groups, compared to free GEM- and saline-treated group. GE11 peptide–decorated combination micelles–treated group showed the lowest Ki-67 and highest cleaved caspase-3 expression compared to all other treatment groups (Fig. 6E). In addition, we determined the systemic toxicity of GE11-decorated combination micelles–treated groups and saline-treated control group by histological analysis of the major organs. There were no noticeable histological changes observed in the livers, spleens, kidneys, and hearts from the treatment groups (fig. S9), suggesting that the mice tolerated GE11 peptide–decorated combination micellar formulation of GEM and miR-519c treatment well.
These findings indicated that EGFR-targeted micelles were able to deliver miR-519c and GEM to the tumor sites and release the antitumor agents via redox responsiveness, thus resulting in maximum synergy against pancreatic cancer without additional toxicities.
DISCUSSION
PDAC is a recalcitrant disease characterized by highly aggressive cancer cells, extensive desmoplastic reaction, and hypovascularization. These unique features endow PDAC tumors with an array of resistance mechanisms against standard-of-care chemotherapy treatments (22). It is, therefore, urgent to identify novel targets that can sensitize tumor cells. Desmoplastic and hypoxic pancreatic tumor microenvironments play multifaceted roles in inducing chemotherapy resistance, promoting distant metastasis, and serving as a barrier to drug delivery (23). Hypoxic microenvironment activates quiescent PSCs in extracellular matrix (ECM) by up-regulation of Hh signaling ligands (Fig. 1D). Strong desmoplasia severely affects vascular function, resulting in hypovascularization of the tumor, which up-regulates HIF-1α expression (fig. S1). Therefore, a cycle of hypoxia and desmoplasia is amplifying each other. This cycle could be blocked by HIF-1α inhibition, which was found sufficient enough to impede Hh signaling (24).
Aberrant expression of miRNA correlates with altered expression of genes responsible for stemness, metastasis, cancer metabolism, hypoxia, and chemoresistance (25). The advantage of miRNAs lies in their ability to modulate multiple cellular pathways simultaneously, which are difficult to target by small molecules and therapeutic proteins. miR-519c is down-regulated in pancreatic cancer patient tissues and different cell lines (Fig. 1, A and B) but can target HIF-1α and Hh ligands such as SHH and GLI, as determined by Western blotting (Fig. 1, C and D). Therefore, miR-519c could be a promising strategy in the inhibition of desmoplasia of pancreatic cancer and other hypoxia-related genes. The presence of miR-519c prevented migration of MIA PaCa-2R cells in the presence of hPSC-conditioned medium (Fig. 1E), indicative of miR-519c–mediated inhibition of tumor cell–stromal metabolic interactions. The tumor microenvironment promotes the interactions between cancer cells and their surrounding cancer-associated fibroblasts (CAFs; PSCs in this case). This feedback is reciprocal, and CAFs can both promote and impair cancer progression, while cancer cells promote PSC activation, proliferation, migration, and ECM remodeling capability via the Hh pathway. Activated PSCs secrete numerous growth factors including platelet-derived growth factor, transforming growth factor–β, and inflammatory cytokines in the tumor (26). Many reports suggest that HIF-1α induces migration in pancreatic cancer cells by regulating genes such as EMT regulators Snail, chemokine (C-X3-C motif) receptor 1 (CX3CR1), and glucose metabolism (27, 28). We have shown that inhibiting HIF-1α decreased several metabolism-related genes such as GLUT1, NME4, LDHA, HKII, and TKT (Fig. 2, A and D), thus providing the rationale for reduced migration of MIA PaCa-2R cells in the presence of miR-519c.
Resistance to chemotherapeutic drugs in pancreatic cancer could have many causes, such as intrinsic resistance mechanisms and desmoplastic microenvironments that promotes cancer cell resistance by providing an environment that hampers drug delivery. HIF-1α–mediated metabolic alterations that facilitate GEM resistance in pancreatic cancer cells have been recently reported (29). GEM-resistant pancreatic cancer cells maintain a higher dC pool. Increased glucose metabolism fuels GEM resistance in pancreatic cancer cells, supporting de novo synthesis of dC through nonoxidative PPPs. Here, we show that GEM resistance in hypoxic pancreatic cancer cells can be reversed by miR-519c repletion. The coordinated transcription induction of genes encoding glycolytic enzymes and pyrimidine synthesis in response to hypoxia is mediated by HIF-1α. Consistent with HIF-1α down-regulation by miR-519c, we found that miR-519c may counteract hypoxia-induced increase in glycolytic influx and intracellular dCTP level (Figs. 2 and 3).
Hypoxia up-regulates glucose metabolism, angiogenesis, and drug-resistant genes (30). HIF-1 signaling transactivates a multitude of target genes that enhance glucose uptake, as determined by an increase in 2-DG (Fig. 2B), glycolysis, and oxidative phosphorylation, thus facilitating cancer cell survival under oxygen- and glucose-deprived microenvironments. HIF-1α also results in an increase in pancreatic cancer, leading to increased dCTP pool and GEM resistance (Figs. 2D and 3A). Hypoxia and overexpression of HIF-1α ultimately promote induction of an invasive and treatment-resistant phenotype of pancreatic cancer.
We have reported previously that under in vitro conditions, GEM is highly effective in mediating toxicity against human pancreatic cancer cells. In contrast, orthotopic tumors in the pancreas were resistant to systemic therapy of GEM. Many factors affect GEM efficacy in vivo, including higher-rate metabolism, dose-limiting toxicity, competition with cellular dCTP pool, various efflux mechanisms, and poor penetration into the bulk of the tumor.
ABCG2 is an efflux transporter that is often observed in drug-resistant cancer cells and contributes to a multidrug resistance phenotype. ABCG2 may help provide a survival advantage during conditions of hypoxia and allow cells to escape the toxic effects of chemotherapeutic drugs. HIF-1α binds to the hypoxia response element, and binding of latter to the ABCG2 promoter increases its transcription in pancreatic cancer cells (31). Our data suggest that miR-519c, by inhibiting HIF-1α, decreases ABCG2-mediated GEM resistance. Over the decades, researchers faced the technical hurdles associated with bringing a therapeutic oligonucleotide product to market. A key hurdle includes getting small interfering RNA/miRNA molecules into the right cells in vivo. Delivery of miRNAs is difficult because they are relatively large, chemically unstable, immunogenic, and negatively charged molecules that do not cross the cell membrane (32). Previously, we have shown that rationally designed chemically modified miRNA could provide an excellent solution by increasing their stability and decreasing their immunogenicity (18). Here, we chemically modified miR-519c with OMe-PS at its 3′ end to enhance stability, in vivo half-life, and loading affinity into the RISC (Fig. 4).
The therapeutic efficacy of GEM depends on its delivery to the tumor site, activation by dC kinase to GEM monophosphate, and evading deactivation by cytidine deaminase. As discussed above in detail, desmoplasia makes it difficult for GEM to achieve adequate accumulation inside the tumor after systemic administration. Previously, we have shown that PEGylation prolongs the circulation of GEM-conjugated micelles and improves its overall therapeutic effect (14).
EGFR overexpression is observed in pancreatic cancer cells (33). EGFR-binding monoclonal antibody cetuximab (C225) blocks the binding of EGFR ligands to the receptor. Although cetuximab can exert an anticancer effect in patients expressing wild-type KRAS, pancreatic cancer that expresses mutated KRAS (Kirsten rat sarcoma) in 95% of cases is resistant to cetuximab therapy. EGFR expression is also high in distant metastatic pancreatic cancer (34), hence provides an ideal candidate for targeted drug delivery. Previously, we demonstrated higher tumor accumulation when cetuximab-conjugated micelles were administered into orthotopic PC (pancreatic cancer)–bearing NSG mice, which resulted in low tumor burden compared to immunoglobulin G–targeted and nontargeted micelles (21). To avoid high mitogenic potential of full-length EGFR antibody cetuximab, we developed GE11 peptide (CYHWYGYTPQNVI)–decorated micelles, reported equal target binding capacity as of cetuximab, and showed higher drug accumulation and better efficacy (35). Further, small-sized PEGylated nanoparticles and micelles are preferentially accurate at the tumor and inflammatory sites via passive targeting (2).
To further improve our delivery system, here, we developed a new redox-sensitive therapeutic strategy to enhance the chemotherapeutic efficacy in pancreatic cancer, while reducing systemic side effects at the same time. GEM conjugated with the polymer via an amide bond shows very slow release from the micelles, thus proved to be a bottleneck for effective drug concentration. A conjugate with disulfide bonds, which can be easily cleaved by reducing GSH into sulfhydryl groups, causes the degradation of micelles and facilitates the release of the drug. The disulfide bond in mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC can be readily broken by GSH, the concentration of which is significantly higher in tumor than the normal tissues. That can facilitate the precise release of GEM in the tumor site and limit its toxicity to normal organs.
For codelivery, we synthesized mPEG-co-P(Asp)-g-DC-g-S-S-GEM, mPEG-P(Asp)-g-TEPA-g-DC, and GE11-PEG-P(Asp)-g-DC to load GEM and miR-519c in the hydrophobic core (Fig. 5). Because of the complexity of different components, we formulated mixed micelles using different ratios of these polymers. Our novel formulations offer distinct advantages such as targeted delivery of GEM and miR-519c, prevention of rapid deamination of GEM, and the intratumoral release of GEM based on intracellular stimuli such as GSH (Fig. 5F), thus reducing toxicity to normal tissues, and enhanced stability of miRNA. Moreover, PEG shell can prevent aggregation and impart a high degree of stability and stealth effect, leading to the enhanced mean residence time of the drug and miRNA.
EGFR expression plays an important role in metastasis, especially liver metastasis, and recurrence of human pancreatic cancer (34). EGFR facilitates metastasis in pancreatic cancer through activation of Akt and extracellular signal–regulated kinase pathway. Metastatic nodules have higher EFGR expression level than the primary tumor, but we did not observe those nodules in the liver, as evidenced by H&E staining (Fig. 6E). The total mass balance of the injected dose might be the reason for lower accumulation of GE11 combination micelles in the liver than the nontarget micelles (fig. S8, A and B).
The rationale of combination therapy that combines two or more therapeutic agents is to lower the doses of individual drug and miRNA to achieve better therapeutic outcome and minimize dose-related side effects (36). Since miR-519c resensitizes pancreatic cancer cells to GEM, we decreased the dose of GEM from 40 to 20 mg/kg and the dose of OMe-PS–miR-519c from 2 to 1 mg/kg when used in combination to determine whether there is synergism in their therapeutic efficacy. GEM-conjugated OMe-PS–miR-519c–complexed mixed micelles exhibited the highest synergy in tumor regression after systemic administration in orthotopic pancreatic tumor–bearing mice (Fig. 6, B and C), with no weight loss (Fig. 6D) or any sign of toxicity in the vital organs of mice from any group during or after the treatment. We observed a significant decrease in HIF-1α, α-SMA, and Ki-67 expression and a decrease in the cleaved caspase-3 expressions in the combination treatment group (Fig. 6E). This might be attributed to GE11 peptide–decorated micelles achieving higher concentrations of GEM and OMe-PS–miR-519c in the tumor. High α-SMA in pancreatic cancer is associated with dense stroma reaction and worse patient outcome. α-SMA is expressed by activated PSCs, in response to hypoxia and Hh signaling.
Particle size distribution, shape, and surface morphology of nanoparticles affect their biodistribution and accumulation at the tumor site (37, 38). The particle size distribution of mPEG-co-P(Asp)-g-TEPA-g-DC/miR-519c–complexed micelles, GEM-conjugated mPEG-co-P(Asp)-g-Ala-S-S-GEM-g-DC micelles, and their combination micelles was 100 ± 20 nm (Fig. 5, C and D). On the basis of our previous studies (2, 39), small particle size and PEGylated surface prolong the blood circulation of these micelles after systemic administration. However, stroma acts as a barrier for nanoparticle delivery to the pancreatic tumor. Since miR-519c targets hypoxia and reduces the desmoplastic barrier, we observed a decrease in α-SMA expression in the tumor (Fig. 6E). Therefore, there was significant accumulation of miR-519c and GEM in the tumor after systemic administration of combination micelles (Fig. 6A and fig. S8C).
The combination of GEM and miR-519c showed a synergistic effect in the nonmetastatic orthotopic desmoplastic pancreatic cancer mouse model (Fig. 6), which suggests that this treatment could be useful for patients with stage 0 to III pancreatic cancer. Although we did not observe liver metastasis during the therapeutic study, EGFR expression is expected to be higher in hepatic metastatic nodules than in the primary tumor. Hence, GE11-based nanoparticle delivery platform has the potential to target the metastatic nodules in the liver or other distant metastasis in stage IV (34). More data related to the therapeutic effect study in the orthotopic pancreatic tumor mouse model of liver metastasis will be needed to testify this in the future.
In conclusion, the key hurdles of the clinical translation of nanomedicine are biological barriers, large-scale manufacturing, biocompatibility, and safety (40, 41). The characterization of our combination micelles shows sufficient drug loading of GEM and miRNA, redox-responsive release of GEM, and controlled release of miRNA. The GE11-targeting mixed micelle system proposed in this study leads to active EGFR targeting, significant accumulation in desmoplastic pancreatic tumor, precise release of GEM and miR-519c at tumor sites with the disulfide bond, and efficient synergistic therapeutic effect in vivo. In addition, the PEG corona prolonged the circulation time of micelles and has no observed toxicity in major organs, which enhance the biocompatibility and safety of this platform. Further, scale-up of this delivery system is feasible and can support a first-in-human phase 1 clinical trial.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM) high-glucose medium, 0.25% trypsin, and Dulbecco’s phosphate-buffered saline (DPBS) were purchased from HyClone (Logan, UT). Keratinocyte-SFM (serum-free medium) medium, bovine pituitary extract, and human recombinant EGF were purchased from Gibco (Chevy Chase, MD). Heat-inactivated FBS, antibiotic-antimycotic for cell culture, radioimmunoprecipitation assay (RIPA) buffer for cell lysis and phosphatase inhibitor cocktail (100×), live and dead assay kit, miR-519c-3p mimics, Pierce BCA (bicinchoninic acid) protein assay kit, RediPlate 96 RiboGreen RNA quantitation kit, and FxCycle PI/RNase staining solution (F10797) were purchased from Thermo Fisher Scientific (Waltham, MA). Hepes buffer was purchased from Millipore Sigma (St. Louis, MO). Human c-Myc primary antibody was purchased from Proteintech (Manchester, UK). GLUT1, GLI1, SHH, β-actin, and SMO primary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Laemmli buffer (4×), 10× tris/glycine/SDS protein electrophoresis running buffer (pH 8.3), and 10× tris-buffered saline buffer were purchased from Bio-Rad (Hercules, CA). HIF-1α and ABCG2 primary antibodies were purchased from Abcam (Cambridge, MA). HIF-1β primary antibody was purchased from Novus Biologicals LLC (Centennial, CO). AGO2 primary antibody was purchased from Boster Bio (Pleasanton, CA). Glucose uptake cell-based assay kit (item no. 600470) was purchased from Cayman Chemicals (Ann Arbor, MI). RNeasy mini kit, miScipt II RT kit, miScript SYBR Green kit, Hs_miR-519c-3p miScript primer assay, and Hs_RnU6 miScript primer assay were purchased from QIAGEN (Germantown, MD).
Cell culture
MIA PaCa-2 cell line was purchased from the American Type Culture Collection (Manassas, VA), and MIA PaCa-2–resistant (MIA PaCa-2R) cells were generated from MIA PaCa-2 by incubating with GEM in a high-glucose DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C, 5% CO2, and 100% humidity and were split when confluent. HPDE cells were cultured in Keratinocyte-SFM–supplemented bovine pituitary extract and human recombinant EGF in an identical atmosphere. hPSCs were cultured in stellate cell basal medium supplemented with 10% FBS, stellate cell growth supplement, and 1% penicillin/streptomycin. A hypoxic chamber filled with 94% nitrogen, 5% CO2, 1% O2, and 100% humidity was used to generate hypoxic conditions as needed.
hPSC-conditioned medium was obtained using the following steps: Subconfluent PSCs were washed with PBS and incubated with stellate cell basal medium under above conditions for 48 hours. Then, the medium was collected and centrifuged for further use.
Cell migration assay
MIA PaCa-2R cells of 5 × 105 per well were seeded in six-well plate, and, after 24 hours, cells were treated with PBS, scrambled miRNA, and miR-519c with or without PSC-conditioned medium for 6 hours under normoxia and hypoxia, respectively. Then, 1 × 105 cells were collected from each well, resuspended with 200 μl of serum-free DMEM, and added to Transwell upper chambers. One milliliter of DMEM containing 20% FBS that served as the chemoattractant was in the lower chamber, and the cells were incubated for 24 hours under normoxic and hypoxic conditions. A cotton-tipped swab was used to remove the cells that did not migrate through the pores. The chambers were fixed in 4% paraformaldehyde and stained with crystal violet.
Glucose uptake assay and GLUT1 expression
MIA PaCa-2R cells of 5 × 104 per well were seeded in a 96-well plate. After 24 hours, cells were treated with miR-519c and scrambled miRNA in 100 μl of glucose-free culture media for another 24 hours under normoxic and hypoxic conditions. Glucose uptake cell-based assay kit was applied to evaluate glucose uptake in MIA PaCa-2R cells as following: 2-NBDG (2-deoxy-2-((7-nitro-2,1,3-benzoxadiazol-4-yl)amino)-glucose) (150 μg/ml) was added in glucose-free medium for 1.5 hours under normoxic and hypoxic conditions. The plates were centrifuged for 5 min at 400g at room temperature. The supernatants were discarded, and 200 μl of the cell-based assay buffer was added to each well for washing cells twice. At the end, 100 μl of cell-based assay buffer was added, and the plates were analyzed by a fluorescent reader with an excitation/emission of 485/535 nm to quantify 2-NBDG taken up by cells. Samples were normalized by the fluorescence intensity in scrambled miRNA under normoxia. The concentration of dCTP was quantified by LC-MS/MS.
For GLUT1 expression, MIA PaCa-2R cells at a dose of 2 × 105 per well were seeded in a six-well plate. After 24 hours, cells were incubated under normoxic and hypoxic conditions. Then, cells were harvested, washed with cell staining buffer three times, incubated with GLUT1 primary antibody in cell staining buffer for 1 hour, followed by Cy5-conjugated secondary antibody for another 30 min, and analyzed by flow cytometer.
RT–quantitative PCR and Western blot
MIA PaCa-2R cells at a dose of 2 × 105 per well were plated in a six-well plate overnight and transfected by Lipofectamine/miR-519c under normoxic and hypoxic conditions for 24 hours. Then, all samples were subjected to total RNA isolation by QIAGEN RNeasy mini kit. Reverse transcription PCR was carried out using QIAGEN miScript II reverse transcription kit. MiR-519c and mRNA levels of SHH, GLI1, NME4, LDHA, HKII and TKT were quantitatively assayed in Roche Light Cycler 480 using miScript SYBR Green PCR kit. MiR-519c and mRNA were normalized to U6 small nuclear RNA and glyceraldehyde-3-phosphate dehydrogenase levels, respectively. Primers were designed as follows: HKII-1, 5′-GAGCCACCACTCACCCTACT-3′ (forward) and 5′-ACCCAAAGCACACACGGAAGTT-3′ (reverse); TKT-1, 5′-TCCACACCATGCGCTACAAG-3′ (forward) and 5′-CAAGTCGGAGCTGATCTTCCT-3′ (reverse); NME4-1, 5′-AGGGTACAATGTCGTCCGC-3′ (forward) and 5′-GACGCTGAAGTCACCCCTTAT-3′ (reverse).
For Western blot assay, all samples were isolated with RIPA buffer on ice within 5 min to prevent HIF-1α degradation when reexposed to O2, and the protein concentration was determined with Pierce BCA protein assay kit. After that, cell lysates were mixed with Laemmli loading buffer, boiled at 100°C for 5 min, loaded in the wells of 4 to 15% SDS–polyacrylamide gel electrophoresis gel, transferred by electroporation to polyvinylidene difluoride membrane, and incubated with blocking buffer for 1 hour at room temperature first and then with anti–HIF-1α, anti–c-Myc, anti-ABCG2, anti–HIF-1β, and anti–β-actin primary antibody overnight at 4°C. IR-680 fluorescent dye–labeled secondary antibodies were added, followed by imaging in iBright FL1000. HIF-1β and β-actin were both used as the loading control.
Cell cycle analysis
MIA PaCa-2R cells at a dose of 2 × 105 per well were plated in a six-well plate and treated with scrambled miRNA and miR-519c under normoxic and hypoxic conditions for 24 hours. Then, cells were harvested, fixed with 70% ethanol in 4°C for 1.5 hours, and washed three times with PBS to remove ethanol. Subsequently, cells were centrifuged, a pellet of cells was collected in tubes, and 0.5 ml of FxCycle PI/RNase staining solution was added to each flow cytometry sample for 30 min at room temperature, avoiding any exposure to light. Flow cytometry was used to test samples at an excitation/emission of 532/585 nm.
Cytotoxicity, apoptosis, and efficacy in desmoplastic tumor spheroid model
Cytotoxicity of GEM, miR-519, and their combination was determined by MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) assay. MIA PaCa-2R cells were plated at a density of 5000 cells per well in a 96-well plate overnight. Then, GEM, miR-519c-3p, and the combination of GEM and miR-519c were added to 96-well plates with a GEM concentration ranging from 0.1 to 50 μM and miR-519c concentration from 5 to 100 nM under hypoxic condition. After 48 hours of incubation, MTT solution at a concentration of 5 mg/ml was added, and cells were cultured for another 3 hours. The supernatant was discarded, and 200 μl of DMSO was added to each well. Cell viability was determined using a spectrophotometer.
hPSC and MIA PaCa-2R cells (1:2; a total of 600 cells in 40 μl per well) in DMEM with 10% FBS were seeded onto Perfecta3D 96-Well Hanging Drop Plates (3D Biomatrix Inc., Ann Arbor MI) and incubated with 5% CO2 at 37°C. On the fourth day, the spheroids were treated with GEM, miR-519c, and their combination. Cells treated with PBS and scrambled miRNA were used as controls. Morphologies of tumor spheroids were observed under a Zeiss microscope on days 6, 8, 10, and 12 to determine cytotoxicity. Live and dead assay kit was applied following the manufacturer’s protocol to visualize the apoptosis in desmoplastic tumor spheroids by confocal laser scanning microscopy.
AGO2 association
Targeting gene silencing by RNA interference requires RISC. AGO protein is a core protein binding to miRNA. We loaded AGO2 with miRNAs by the action of a specialized assembly RISC-loading complex. To assess the difference of miRNA loading between unmodified miR-519c and OMe-PS–miR-519c, MIA PaCa-2R cells at a dose of 2 × 105 per well were seeded in a six-well plate and then transfected with unmodified miR-519c and OMe-PS–miR-519c using Lipofectamine for 24 hours under normoxic and hypoxic conditions. Cells were washed three times with PBS and lysed using Pierce IP lysis buffer. A Pierce BCA assay kit was applied to quantify protein concentration, and all samples were diluted to 1000 μg/ml by cell lysis buffer. Ten micrograms of AGO2 primary antibody and 500 μl of cell lysis buffer were mixed and incubated overnight at 4°C. Then, the antigen sample/antibody mixture was added to 1.5 ml of microcentrifuge tubes containing prewashed magnetic beads and incubated for 1 hour at room temperature. After that, beads were collected with a magnetic stand and washed twice with tris-buffered saline and ultrapure water, respectively. One hundred microliters of elution buffer was incubated with samples for 10 min at room temperature. The supernatant of miRNA-AGO2 complexes was harvested after separating from beads with a magnetic stand, following which 15 μl of neutralization buffer was added. Synthetic miR-39 from Caenorhabditis elegans (Cel-miR-39) was selected as a spike-in control for miRNA quantification by RT–quantitative PCR. For this purpose, first, all samples were isolated by QIAGEN RNeasy mini kit, then reverse transcribed by miScript II RT kit, and amplified with miScript SYBR Green PCR kit using Roche LightCycler 480.
Synthesis of redox-responsive GEM-conjugated copolymer, TEPA-containing copolymer, and GE11 peptide–conjugated copolymer
Redox-responsive GEM-conjugated copolymer, TEPA-containing copolymer for complex formation with miRNA, and EGFR-targeting GE11 peptide–conjugated copolymer were synthesized, as illustrated in Fig. 7 as described below:
Fig. 7. Synthesis schemes of redox-sensitive GEM-conjugated copolymer, TEPA-containing polymer for complex formation with miRNA, and EGFR-targeting GE11 peptide–conjugated polymer.
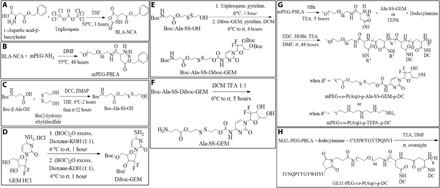
GE11 peptide, CYHWYGYTPQNVI. (A) Synthesis of BLA-NCA. (B) Synthesis of mPEG-PBLA. (C-F) Synthesis of Ala-SS-GEM. (G) Synthesis of mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC and mPEG-P(Asp)-g-TEPA-g-DC. (H) Synthesis of GE11-PEG-co-P(Asp)-g-DC. rt: room temperature, EDC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, HoBt: Hydroxybenzotriazole, TEA: triethylamine.
Step 1: Synthesis of BLA-NCA: We synthesized β-benzyl l-aspartate N-carboxy anhydride (BLA-NCA) by reacting l-aspartic acid β-benzyl ester with triphosgene in tetrahydrofuran (THF) under N2 atmosphere. (Fig. 7A).
Step 2: Synthesis of amphiphilic polyamino copolymer mPEG-P(Asp): We synthesized mPEG-PBLA by ring-opening polymerization of BLA-NCA using mPEG-NH2 (weight-average molecular weight, 5000) as a macroinitiator in dry N,N-dimethylformamide at 55°C under the N2 atmosphere for 48 hours (Fig. 7B).
Step 3: Synthesis of Ala-SS-GEM: We synthesized Ala-SS-GEM using the following four steps: First, Boc-β-Ala-OH was reacted with bis(2-hydroxyethyl) disulfide using dicyclohexylcarbodiimide (DCC) coupling reaction in the presence of 4-dimethylaminopyridine in THF. The crude product was purified by flash column chromatography to obtain the intermediate Boc-Ala-SS-OH as a colorless liquid (Fig. 7C). Second, Diboc-GEM was synthesized by reacting GEM HCl, and Boc-anhydride was dissolved in a mixture of dioxane and 1.0 M KOH at room temperature. After completion of the reaction, the reaction mixture was extracted with EtOAc (Ethyl acetate) and washed; dried organic layer was concentrated and, again, treated with Boc-anhydride in 1.0 M KOH solution. After completion of the reaction, the mixture was washed and dried, and the combined organic layer was evaporated and purified by column chromatography to give diboc-GEM a white solid product (Fig. 7D). Third, the intermediate Boc-Ala-SS-OH and diboc-GEM were reacted with triphosgene in the presence of pyridine in anhydrous dichloromethane (DCM) for 1 hour at 0°C. The reaction mixture was stirred for additional 4 hours at room temperature. The crude product was then purified using column chromatography to afford Boc-Ala-SS-Diboc-GEM as a colorless oil (Fig. 7E). Last, Ala-SS-GEM was synthesized by removing the Boc groups in a mixture of DCM:trifluoroacetic acid (TFA) (1:1) (Fig. 7F).
Step 4: Ala-SS-GEM and DC (DC-NH2) were conjugated to mPEG-PBLA polymer by aminolysis reaction in dry dimethylformamide at room temperature for 48 hours to afford mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC (Fig. 7G). The structure and molecular weight of mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC was determined by 1H NMR and gel permeation chromatography, respectively. Similarly, mPEG-P(Asp)-g-TEPA-g-DC was synthesized by replacing Ala-SS-GEM with TEPA.
Step 5: GE11 peptide (CYHWYGYTPQNVI) and DC were conjugated to MAL-PEG-PBLA polymer in dry dimethylformamide at room temperature overnight to get GE11-PEG-co-P(Asp)-g-DC (GE11-targeting peptide; Fig. 7H).
Preparation and characterization of micelles and drug release profile
The mixture of mPEG-co-P(Asp)-g-TEPA-g-DC and mPEG-co-P(Asp)-g-Ala-SS-GEM-g-DC copolymers dissolved in acetone/methanol (1:1, v/v) was added drop by drop to aqueous solution containing miR-519c. To remove residual acetone and methanol, samples were evaporated in a rotary evaporator. The mixture was filtered through polycarbonate syringe filters of 200-nm pore size. Particle size was measured using Malvern Zetasizer. In vitro GEM release from redox-responsive micelles was performed under pH 7.4 with or without 10 mM GSH. Dialysis bags (molecular weight cutoff, 3500 Da) were loaded with 1 ml of redox-responsive micelles, immersed into 30 ml of PBS buffer solutions, and shaken at a speed of 100 rpm at 37°C (n = 3). GEM concentration was determined by HPLC-UV under the following conditions: C18 column (5 μm, 250 × 4.6 mm), flow rate (1 ml/min), acetonitirle and water (90:10, v/v) as mobile phase, an injection volume of 20 μl, and a wavelength of 267 nm. In vitro miRNA release from TEPA micelles was carried out by suspending the formulation to PBS buffer solutions under pH 7.4 at each time point and shaking at a speed of 100 rpm at 37°C (n = 3). The samples were centrifuged, and the supernatants were collected at each time point. The concentration of released miRNA was tested by RiboGreen RNA quantification kit.
Biodistribution and therapeutic studies
All the animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center, Omaha, NE and carried out as per the National Institutes of Health guidelines. The orthotopic human pancreatic cancer mouse model was developed using 6- to 8-week-old male NSG mice (20 to 25 g). Luciferase stably expressing MIA PaCa-2R cells (0.5 × 106) and hPSCs (0.5 × 106) were mixed with Matrigel (2:1) using ice-cold instruments and syringes. Then, mice were anesthetized using isoflurane in an induction chamber, and 30 μl of Matrigel cell suspension was orthotopically injected into the pancreas tail. Tumor growth was monitored using the Spectrum IVIS (PerkinElmer, Hopkinton, MA) after intraperitoneal administration of luciferin (20 mg/ml) (150 μl per mouse). Three weeks after tumor injection, free GEM and OMe-PS–miR-519c, combination micelles, and GE11 combination micelles were injected to mice via the tail vein as a single administration at a dose (or dose equivalent) of GEM (20 mg/kg) and miR-519c (1 mg/kg). Mice were euthanized either at 6 or 24 hours, and major organs such as the tumor, heart, liver, spleen, lung, and kidney were collected for LC-MS/MS analysis (4000 QTRAP, AB, Sciex Inc.). Briefly, 50 mg of tissue samples were homogenized in 1 ml of PBS, spiked with 5-aza-dC as the internal standard and transferred on ice immediately. Subsequently, 3.0 ml of cold acetonitrile was added, followed by vortexing and high-speed centrifugation in 4°C. The supernatant was evaporated to dryness, and the residues were reconstituted with 200 μl of acetonitrile:water (10:90, v/v). LC-MS/MS data acquisition will be performed using the Analyst software on a QTRAP 4000 mass spectrometer. The mass spectrometer will be operated in the positively selected reaction monitoring for GEM [mass/charge ratio (m/z), 264.0/112.0) and internal standard (m/z, 229.0/113.0). Total RNA from tissue samples were isolated by QIAGEN RNeasy mini kit, and RT-PCR assay was applied for miR-519c quantification.
For therapeutic study, mice were randomly divided into six groups (n = 5 per group) when the bioluminescence reached 1 × 106. GEM at a dose of 40 mg/kg for free GEM and GEM micelles, miR-519c at the dose of 2 mg/kg for miR-519c micelles, and GEM at a dose of 20 mg/kg and miR-519c at 1 mg/kg for combination micelles were administered via tail vein every 3 days in a total of five injections. Body weights were measured before each dose administration. Three days after the fifth administration, the animals were euthanized to harvest major organs such as the tumor, heart, liver, spleen, lung, and kidney. Examination included H&E staining, immunohistochemical, and Western blot analyses.
Statistical analysis
Results are presented as the means ± SD. One-way analysis of variance (ANOVA) was used to assess the statistical significance of differences between groups. A cutoff of P < 0.05 was used to indicate a significant difference.
Supplementary Material
Acknowledgments
Funding: The NIH (1R01GM113166) and the Faculty Start-up fund of the University of Nebraska Medical Center to R.I.M. are duly acknowledged for providing financial support for this work. V.R.B. is supported by the National Institute of General Medical Sciences, 1 U54 GM115458. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Author contributions: X.X., Virender Kumar, and R.I.M. designed the study. X.X., F.L., Vinod Kumar, and R.B. performed the experiments and collected data. X.X., Virender Kumar, C.T., and R.I.M. analyzed the data. All authors interpreted the study results. X.X. and R.I.M. wrote the manuscript. All authors provided critical feedback and gave their final approval. Competing interests: All the authors except V.R.B. have declared that they have no competing interests. V.R.B. reports receiving consulting fees from Takeda, Omeros, Agios, Abbvie, Partner Therapeutics, and Incyte, research funding (institutional) from Jazz, Incyte, Tolero Pharmaceuticals, and National Marrow Donor Program, and drug support for a trial from Oncoceutics. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabd6764/DC1
REFERENCES AND NOTES
- 1.Philip P. A., Lacy J., Portales F., Sobrero A., Pazo-Cid R., Manzano Mozo J. L., Kim E. J., Dowden S., Zakari A., Borg C., Terrebonne E., Rivera F., Sastre J., Bathini V., López-Trabada D., Asselah J., Saif M. W., Shiansong Li J., Ong T. J., Nydam T., Hammel P., Nab-paclitaxel plus gemcitabine in patients with locally advanced pancreatic cancer (LAPACT): A multicentre, open-label phase 2 study. Lancet Gastroenterol. Hepatol. 5, 285–294 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Kattel K., Mondal G., Lin F., Kumar V., Mahato R. I., Biodistribution of self-assembling polymer–gemcitabine conjugate after systemic administration into orthotopic pancreatic tumor bearing mice. Mol. Pharm. 14, 1365–1372 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liang C., Shi S., Meng Q., Liang D., Ji S., Zhang B., Qin Y., Xu J., Ni Q., Yu X., Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 49, e406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saltz L. B., Bach P. B., Albumin-bound paclitaxel plus gemcitabine in pancreatic cancer. N. Engl. J. Med. 370, 478 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Conroy T., Hammel P., Hebbar M., Abdelghani M. B., Wei A. C., Raoul J.-L., Choné L., Francois E., Artru P., Biagi J. J., Lecomte T., Assenat E., Faroux R., Ychou M., Volet J., Sauvanet A., Breysacher G., Fiore F. D., Cripps C., Kavan P., Texereau P., Bouhier-Leporrier K., Khemissa-Akouz F., Legoux J.-L., Juzyna B., Gourgou S., O’Callaghan C. J., Jouffroy-Zeller C., Rat P., Malka D., Castan F., Bachet J.-B.; Canadian Cancer Trials Group and the Unicancer-GI–PRODIGE Group , FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 379, 2395–2406 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Von Hoff D. D., Ervin T., Arena F. P., Chiorean E. G., Infante J., Moore M., Seay T., Tjulandin S. A., Ma W. W., Saleh M. N., Harris M., Reni M., Dowden S., Laheru D., Bahary N., Ramanathan R. K., Tabernero J., Hidalgo M., Goldstein D., Van Cutsem E., Wei X., Iglesias J., Renschler M. F., Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conroy T., Desseigne F., Ychou M., Bouché O., Guimbaud R., Bécouarn Y., Adenis A., Raoul J.-L., Gourgou-Bourgade S., de la Fouchardière C., Bennouna J., Bachet J.-B., Khemissa-Akouz F., Péré-Vergé D., Delbaldo C., Assenat E., Chauffert B., Michel P., Montoto-Grillot C., Ducreux M.; Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup , FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Chaudhary A. K., Mondal G., Kumar V., Kattel K., Mahato R. I., Chemosensitization and inhibition of pancreatic cancer stem cell proliferation by overexpression of microRNA-205. Cancer Lett. 402, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh S., Chitkara D., Kumar V., Behrman S. W., Mahato R. I., miRNA profiling in pancreatic cancer and restoration of chemosensitivity. Cancer Lett. 334, 211–220 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Mimeault M., Batra S. K., Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell. Mol. Med. 17, 30–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang R., Cheng L., Xia J., Wang Z., Wu Q., Wang Z., Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1α in pancreatic cancer cells. Curr. Cancer Drug Targets 14, 407–417 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Katagiri T., Kobayashi M., Yoshimura M., Morinibu A., Itasaka S., Hiraoka M., Harada H., HIF-1 maintains a functional relationship between pancreatic cancer cells and stromal fibroblasts by upregulating expression and secretion of sonic hedgehog. Oncotarget 9, 10525–10535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chitkara D., Mittal A., Mahato R. I., miRNAs in pancreatic cancer: Therapeutic potential, delivery challenges and strategies. Adv. Drug Deliv. Rev. 81, 34–52 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Mittal A., Chitkara D., Behrman S. W., Mahato R. I., Efficacy of gemcitabine conjugated and miRNA-205 complexed micelles for treatment of advanced pancreatic cancer. Biomaterials 35, 7077–7087 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Kumar V., Mondal G., Slavik P., Rachagani S., Batra S. K., Mahato R. I., Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol. Pharm. 12, 1289–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nong K., Zhang D., Chen C., Yang Y., Yang Y., Liu S., Cai H., MicroRNA-519 inhibits hypoxia-induced tumorigenesis of pancreatic cancer by regulating immune checkpoint PD-L1. Oncol. Lett. 19, 1427–1433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.To K. K. W., Robey R. W., Knutsen T., Zhan Z., Ried T., Bates S. E., Escape from hsa-miR-519c enables drug-resistant cells to maintain high expression of ABCG2. Mol. Cancer Ther. 8, 2959–2968 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar V., Kumar V., Luo J., Mahato R. I., Therapeutic potential of OMe-PS-miR-29b1 for treating liver fibrosis. Mol. Ther. 26, 2798–2811 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu L., Torchilin V. P., Stimulus-responsive nanopreparations for tumor targeting. Integr. Biol. 5, 96–107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bansal A., Simon M. C., Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 217, 2291–2298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mondal G., Almawash S., Chaudhary A. K., Mahato R. I., EGFR-targeted cationic polymeric mixed micelles for codelivery of gemcitabine and miR-205 for treating advanced pancreatic cancer. Mol. Pharm. 14, 3121–3133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nevala-Plagemann C., Hidalgo M., Garrido-Laguna I., From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 17, 108–123 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Kuninty P. R., Bansal R., De Geus S. W. L., Mardhian D. F., Schnittert J., van Baarlen J., Storm G., Bijlsma M. F., van Laarhoven H. W., Metselaar J. M., Kuppen P. J. K., Vahrmeijer A. L., Östman A., Sier C. F. M., Prakash J., ITGA5 inhibition in pancreatic stellate cells attenuates desmoplasia and potentiates efficacy of chemotherapy in pancreatic cancer. Sci. Adv. 5, eaax2770 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spivak-Kroizman T. R., Hostetter G., Posner R., Aziz M., Hu C., Demeure M. J., Von Hoff D., Hingorani S. R., Palculict T. B., Izzo J., Kiriakova G. M., Abdelmelek M., Bartholomeusz G., James B. P., Powis G., Hypoxia triggers hedgehog-mediated tumor–stromal interactions in pancreatic cancer. Cancer Res. 73, 3235–3247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilles M.-E., Hao L., Huang L., Rupaimoole R., Lopez-Casas P. P., Pulver E., Jeong J. C., Muthuswamy S. K., Hidalgo M., Bhatia S. N., Slack F. J., Personalized RNA medicine for pancreatic cancer. Clin. Cancer Res. 24, 1734–1747 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Marzoq A. J., Mustafa S. A., Heidrich L., Hoheisel J. D., Alhamdani M. S. S., Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep. 9, 5303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai Y.-P., Wu K.-J., Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 19, 102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z., Jia X., Duan Y., Xiao H., Sundqvist K. G., Permert J., Wang F., Excess glucose induces hypoxia-inducible factor-1α in pancreatic cancer cells and stimulates glucose metabolism and cell migration. Cancer Biol. Ther. 14, 428–435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shukla S. K., Purohit V., Mehla K., Gunda V., Chaika N. V., Vernucci E., King R. J., Abrego J., Goode G. D., Dasgupta A., Illies A. L., Gebregiworgis T., Dai B., Augustine J. J., Murthy D., Attri K. S., Mashadova O., Grandgenett P. M., Powers R., Ly Q. P., Lazenby A. J., Grem J. L., Yu F., Matés J. M., Asara J. M., Kim J.-w., Hankins J. H., Weekes C., Hollingsworth M. A., Serkova N. J., Sasson A. R., Fleming J. B., Oliveto J. M., Lyssiotis C. A., Cantley L. C., Berim L., Singh P. K., MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 32, 71–87.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai X., Kaluz S., Jiang Y., Shi L., Mckinley D., Wang Y., Wang B., Van Meir E. G., Tan C., A novel small-molecule arylsulfonamide causes energetic stress and suppresses breast and lung tumor growth and metastasis. Oncotarget 8, 99245–99260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He X., Wang J., Wei W., Shi M., Xin B., Zhang T., Shen X., Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1α and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 17, 188–198 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F., Mahato R. I., miRNAs as targets for cancer treatment: Therapeutics design and delivery. Adv. Drug Deliv. Rev. 81, v–vi (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yokoi K., Fidler I. J., Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 10, 2299–2306 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Tobita K., Kijima H., Dowaki S., Kashiwagi H., Ohtani Y., Oida Y., Yamazaki H., Nakamura M., Ueyama Y., Tanaka M., Inokuchi S., Makuuchi H., Epidermal growth factor receptor expression in human pancreatic cancer: Significance for liver metastasis. Int. J. Mol. Med. 11, 305–309 (2003). [PubMed] [Google Scholar]
- 35.Mondal G., Kumar V., Shukla S. K., Singh P. K., Mahato R. I., EGFR-targeted polymeric mixed micelles carrying gemcitabine for treating pancreatic cancer. Biomacromolecules 17, 301–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayat Mokhtari R., Homayouni T. S., Baluch N., Morgatskaya E., Kumar S., Das B., Yeger H., Combination therapy in combating cancer. Oncotarget 8, 38022–38043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J., Mao W., Lock L. L., Tang J., Sui M., Sun W., Cui H., Xu D., Shen Y., The role of micelle size in tumor accumulation, penetration, and treatment. ACS Nano 9, 7195–7206 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Toy R., Peiris P. M., Ghaghada K. B., Karathanasis E., Shaping cancer nanomedicine: The effect of particle shape on the in vivo journey of nanoparticles. Nanomedicine 9, 121–134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar V., Mundra V., Peng Y., Wang Y., Tan C., Mahato R. I., Pharmacokinetics and biodistribution of polymeric micelles containing miRNA and small-molecule drug in orthotopic pancreatic tumor-bearing mice. Theranostics 8, 4033–4049 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shreffler J. W., Pullan J. E., Dailey K. M., Mallik S., Brooks A. E., Overcoming hurdles in nanoparticle clinical translation: The influence of experimental design and surface modification. Int. J. Mol. Sci. 20, 6056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hua S., de Matos M. B. C., Metselaar J. M., Storm G., Current trends and challenges in the clinical translation of nanoparticulate nanomedicines: Pathways for translational development and commercialization. Front. Pharmacol. 9, 790 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabd6764/DC1