

The Parkinson disease kinase LRRK2 translocates to the lysosomal membrane triggering lysosomal sorting through JIP4.
Abstract
Genetic variation around the LRRK2 gene affects risk of both familial and sporadic Parkinson’s disease (PD). However, the biological functions of LRRK2 remain incompletely understood. Here, we report that LRRK2 is recruited to lysosomes after exposure of cells to the lysosome membrane–rupturing agent LLOME. Using an unbiased proteomic screen, we identified the motor adaptor protein JIP4 as an LRRK2 partner at the lysosomal membrane. LRRK2 can recruit JIP4 to lysosomes in a kinase-dependent manner via the phosphorylation of RAB35 and RAB10. Using super-resolution live-cell imaging microscopy and FIB-SEM, we demonstrate that JIP4 promotes the formation of LAMP1-negative tubules that release membranous content from lysosomes. Thus, we describe a new process orchestrated by LRRK2, which we name LYTL (LYsosomal Tubulation/sorting driven by LRRK2), by which lysosomal tubulation is used to release vesicles from lysosomes. Given the central role of the lysosome in PD, LYTL is likely to be disease relevant.
INTRODUCTION
Mutations in LRRK2 are a relatively common cause of familial late-onset Parkinson’s disease (PD) (1, 2), and variations at the LRRK2 locus have also been linked to the more numerous sporadic PD (3, 4). LRRK2 encodes leucine-rich repeat kinase 2, a large protein with extensive protein scaffolding sequences as well as two enzymatic activities. Most of the proven pathogenic mutations are located in the ROC (Ras of complex proteins)–COR (C terminus of ROC) bidomain or adjacent kinase domain that control guanosine triphosphate (GTP) hydrolysis and kinase activity, respectively. The majority of evidence suggests that mutations lead to a toxic function of the protein (5). At the cellular level, evidence suggests that LRRK2 can regulate membrane trafficking events via phosphorylation of a subset of RAB GTPases (6–8), although the precise relationship(s) between LRRK2 mutations, RAB phosphorylation, and neurodegeneration remains uncertain.
Recent data have pointed to the lysosome as a crucial organelle in PD. Genetically, mutations in genes encoding for lysosomal proteins have been identified in familial cases of PD (9) and have been nominated as risk factors for sporadic PD (3), leading to the suggestion that PD should be considered a lysosomal disease (10). An accumulation of lysosomal damage with age in kidneys, which normally express high levels of LRRK2, has been documented in knockout (KO) mice (11). Pathogenic LRRK2 mutations affect lysosomal structure and function in cultured astrocytes (12) and other cell types (13, 14). However, the mechanistic basis by which LRRK2 affects lysosome function is unclear. In addition, because LRRK2 can be localized to a wide range of other membrane-bound structures in cells (7, 15, 16), whether LRRK2-mediated lysosomal defects are primary events or secondary effects driven by toxicity to other cellular components is uncertain.
Here, we describe that LRRK2 translocates to the lysosomal surface in response to lysosomal membrane permeabilization, leading to the phosphorylation and recruitment of Ras-related in brain 35 (RAB35) and RAB10. As a consequence, both RAB proteins promote the translocation of the motor adaptor protein c-Jun N-terminal kinase (JNK)–interacting protein 4 (JIP4). JIP4 is present in, and helps to form, lysosomal associated membrane protein 1 (LAMP1)–negative tubular structures stemming from lysosomes. Live-cell super-resolution microscopy reveals that these tubules bud, extend, and release small vesicular structures, suggesting a scenario where lysosomes (undergoing membrane permeabilization) sort membranous content that can then interact with other lysosomes. We call this newly described process LYsosomal Tubulation/sorting driven by LRRK2 (LYTL).
RESULTS
LRRK2, along with the motor adaptor protein JIP4, gets recruited to the membrane of a subset of lysosomes
To understand how LRRK2 might affect lysosomal function, we first examined the localization of LRRK2 in cells. For these experiments, we used mouse primary astrocytes as follows: (i) primary astrocytes express LRRK2 endogenously (fig. S1A), (ii) their flattened morphology allows us to monitor membrane trafficking events, and (iii) they have been proposed to play a role in the pathobiology of PD through non–cell-autonomous effects on microglia and neurons (17).
Exogenously expressed LRRK2 has been widely reported in the literature to have a diffuse cytosolic distribution (7, 18, 19). However, when expressed in mouse primary astrocytes, LRRK2 colocalized with a subset of structures positive for the late-endosomal/lysosome (LE/LYS) membrane marker LAMP1 in about half of the cells examined. Specifically, LRRK2 is present at a subset of LE/LYS (2.72 ± 0.41 LRRK2-positive LE/LYS structures per cell) (Fig. 1A) that is also positive for the LE/LYS marker RAB7 and the lysosomal marker LAMP2. These structures only occasionally contain the lysosomal enzyme cathepsin B (CTSB) and were negative for the early endosomal marker EEA1 (Fig. 1B). The combination of the presence of multiple LE/LYS markers with variable levels of CTSB suggests that LRRK2 is recruited to LYS with a low degradative capacity (20).
Fig. 1. LRRK2 and JIP4 are localized at the membrane of a subset of lysosomes in primary astrocytes.
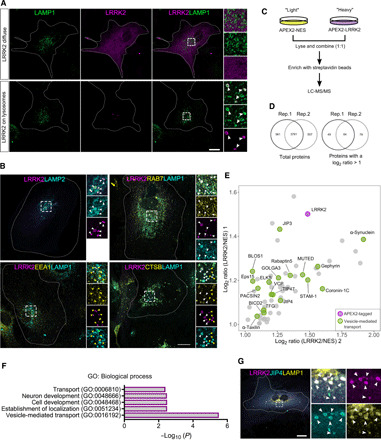
(A) Representative confocal images of 3xflag-LRRK2 and LAMP1 expression in mouse primary astrocytes. (B) Representative confocal images of astrocytes expressing 3xflag-LRRK2 costained with LAMP2, RAB7, EEA1, and CTSB. (C) Outline of the APEX2 proteomic approach to detect LRRK2-membrane interactors. (D) Venn diagrams showing the number of common proteins detected in both replicates (left) and the number of proteins selected as candidates due to having a twofold enrichment in LRRK2 versus negative control in both replicates (right). (E) Scatter plot depicting the 64 LRRK2-interacting candidates from the APEX2 screening. LRRK2 is marked in red, and proteins involved in vesicle-mediated transport are marked in green. (F) Gene Ontology (GO) search of the top 5 enriched terms for biological process of the 64 LRRK2 potential interacting partners, with P values adjusted using a Bonferroni correction indicated on the horizontal axis. (G) Representative confocal image of an astrocyte expressing 3xflag-LRRK2 and GFP-JIP4 and stained for LAMP1. White arrowheads show colocalization, and yellow arrowheads show structures without localization. Scale bar, 20 μm.
We next asked whether LRRK2 enzymatic activity could play a role in its recruitment to LYS by expressing the hyperactive and pathogenic mutations G2019S (kinase domain) and R1441C (GTPase domain), along with the artificial inactive mutations K1906M (kinase dead) and T1348N (GTP-binding null) (fig. S1B). An increase in the recruitment of LRRK2 to LYS was observed in both hyperactive mutations compared to the wild-type (WT) form, whereas a decrease was seen with either inactive mutations (fig. S1, C to E). Domain-specific LRRK2 constructs (ΔHEAT, ΔWD40, HEAT, ROC-COR-Kinase, and ROC-COR) (fig. S1F) showed much less recruitment to LYS compared to the full-length construct (fig. S1, G to I). However, comparing the domains to each other, we detected a higher amount of cells with lysosomal LRRK2 for the HEAT and ΔWD40 domains compared to the other three domains (fig. S1, G and J), suggesting that although every domain seems to be important to maintain LRRK2 at the lysosomal membrane, recruitment likely happens through its N-terminal region.
These results suggest that while not all LRRK2 is lysosomal, it is likely to play a role in a small subset of these organelles. To identify lysosome-specific functional interactors of LRRK2, we used the unbiased proximity-based biotinylation protein-protein interaction method ascorbate peroxidase 2 (APEX2) (21), followed by quantitative proteomics in human embryonic kidney (HEK) 293FT cells with stable isotope–labeled amino acids. As a cytosolic control to exclude nonspecific interactions, APEX2 was tagged to a nuclear export signal (NES) (Fig. 1C). Both vectors (APEX2-3xflag-LRRK2 and APEX2-3xflag-NES) were successfully validated using immunostaining and Western blot (fig. S2, A and B). From the mass spectrometry hits found in two independent replicates with the LRRK2 vector but not APEX-NES (table S1), 64 proteins were selected as possible candidates for LRRK2 interaction (Fig. 1, D and E; fig. S2C; and table S2). Among these candidates, six have already been linked to LRRK2 in previous studies (α-synuclein, JIP3, JIP4, coronin-1C, gephyrin, and PACSIN2) (22–24), suggesting that this method can detect authentic LRRK2 interactors. Fifteen of the candidate proteins have a role in vesicle-mediated transport (Fig. 1F), and six (BLOS1, Muted, JIP3, JIP4, BICD2, and STAM1) have been linked to lysosome biogenesis or dynamics in previous studies. To further prioritize the lysosomal-related hits, we tested whether they were recruited to LRRK2-positive LYS in astrocytes. JIP4 was the only candidate that colocalized with LRRK2-positive LYS (Fig. 1G and fig. S2D). We were able to validate that there was a physical interaction between these two proteins by overexpressing LRRK2 and blotting for endogenous JIP4 in cells (fig. S2E). We therefore considered JIP4, previously nominated by several independent laboratories using different techniques (8, 23), to be a reliable LRRK2 interacting protein and a candidate for mediating any functional effects of LRRK2 on the lysosome.
LRRK2 recruitment to lysosomes occurs as a response to lysosomal membrane rupture, independent of lysophagy
JIP4 is a cytosolic scaffolding protein, associated with multiple aspects of vesicle-mediated transport by acting as an adaptor for both dynein and kinesin motor proteins (25). JIP4 has also been linked to stress response (26), leading us to speculate that the LRRK2:JIP4 complex might respond to lysosomal damage. Such a role would be consistent with the data above showing that LRRK2-positive LYS has low levels of CTSB (Fig. 1B), which is seen when the lysosomal membrane is ruptured and the organellar contents leak into the cytosol (27). To test the hypothesis that lysosomal membrane damage might trigger LRRK2:JIP4 recruitment to LYS, we treated primary astrocytes with the lysosomotropic reagent l-leucyl-l-leucine methyl ester (LLOME). LLOME enters the cell via endocytosis and is transported to the lysosomes where it undergoes condensation by cathepsin C, leading to lysosomal membrane rupture (28). Exposure of cells to 1 mM LLOME triggered a notable and time-dependent increase in LRRK2 recruitment to the lysosomal membrane (Fig. 2A and fig. S3A). We confirmed that LRRK2 is recruited to inactive putative lysosomes by showing that these structures are negative for both LysoTracker and Magic Red CTSB (Fig. 2, B and C), fluorescent probes that measure lysosomal pH and activity, respectively.
Fig. 2. Lysosomal membrane permeabilization enhances LRRK2 recruitment to the lysosomal membrane.
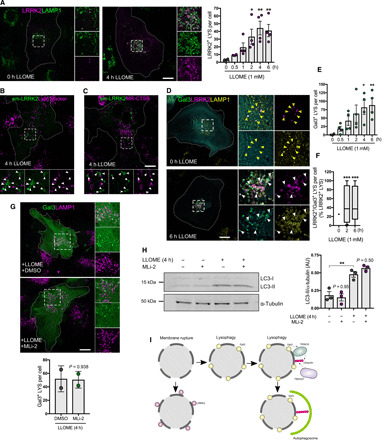
(A) Confocal images of astrocytes untreated or treated with LLOME expressing 3xflag-LRRK2 and LAMP1. The histogram shows the number of LRRK2-positive lysosomes per cell. Data are mean ± SEM (n = 20 cells per N, N = 5). One-way ANOVA with Dunnett’s. (B and C) Live-cell confocal images of astrocytes expressing Emerald-GFP-LRRK2 exposed to LysoTracker Red DND-99 (B) or Magic Red CTSB (C) and treated with LLOME. White arrowheads show absence of colocalization between LRRK2 and the two dyes. (D to F) Confocal images of astrocytes expressing 3xflag-LRRK2, EGFP-Gal3, and LAMP1, untreated or treated with LLOME (D). Yellow arrowheads indicate absence of colocalization, while white arrowheads show colocalization. (E) Histogram shows the number of Gal3-positive lysosomes per cell in cotransfected cells. Data are means ± SEM. One-way ANOVA with Dunnett’s (n = 10 to 20 cells per N, N = 4). (F) Colocalization analysis using n = 20 cells from a single experiment. The percentage of LRRK2-positive/Gal-positive lysosomes normalized by the total number of LRRK2-positive lysosomes was measured in each cell. Box plot shows the median, and the whiskers show the 10th to 90th percentile. One-way ANOVA with Dunnett’s. (G) Confocal images of astrocytes expressing EGFP-Gal3 and LAMP1, pretreated with DMSO or MLi-2 and incubated with LLOME (4 hours). Data are means ± SD. Unpaired t test (n = 20 to 39 cells, N = 2). (H) Western blot of astrocytes pretreated with MLi-2 before adding LLOME. Histogram shows normalized LC3-II levels using two-way ANOVA with Tukey’s. Data are means ± SEM from n = 3. AU, arbitrary units. (I) Working model suggesting that the function of LRRK2 at ruptured lysosomes is independent of lysophagy. Scale bar, 20 μm. *P < 0.05; **P < 0.01, ***P < 0.001.
One widely reported effect of lysosome membrane disruption is the induction of lysophagy, a mechanism to clear ruptured lysosomes from cells via selective autophagy. Galectin-3 (Gal3) is diffusely distributed in the cell under normal conditions but is recruited to the ruptured membrane and initiates lysophagy after a high degree of lysosomal membrane damage (29). In our experimental paradigm, Gal3 was recruited to a subset of lysosomes after LLOME treatment (Fig. 2, D and E). However, LRRK2-positive LYS was partially positive for Gal3 (Fig. 2, D and F), suggesting that LRRK2:JIP4 are recruited to a different lysosomal pool than those that will be degraded by lysophagy.
To further determine if LRRK2 modifies lysophagy, we pretreated cells with the potent LRRK2 kinase inhibitor MLi-2 before adding LLOME. MLi-2 was able to completely block endogenous LRRK2 kinase activity in mouse primary astrocytes as documented by inhibition of both the LRRK2 autophosphorylation site pS1292 and pT73 on RAB10, a known substrate of LRRK2 (fig. S2F). In contrast, LRRK2 kinase inhibition did not affect Gal3 recruitment to the LYS in the presence of LLOME (Fig. 2G) and did not modify the autophagic response triggered by LLOME, as measured by LC3 lipidation (Fig. 2H). This was further confirmed in stable HEK293T cells inducibly expressing LRRK2 (fig. S3, F and G). Together, our data suggest that LRRK2 plays a lysophagy-independent role in response to lysosomal membrane permeabilization (Fig. 2I). We therefore considered whether LRRK2 recruitment occurs before the lysosomal membrane is ruptured enough to recruit Gal3 and trigger lysophagy and whether JIP4 might be important in mediating effects of LRRK2 on lysosomal function.
LRRK2 recruits JIP4 to ruptured lysosomes in a kinase-dependent manner
As expected, exogenously expressed JIP4 translocates to the membrane of LRRK2-positive LYS in a time-dependent manner (Fig. 3A and fig. S4D). However, we noted the presence of several LRRK2-positive/JIP4-negative LYS, even after 6 hours of LLOME exposure (Fig. 3A). This result raised the possibility that exogenous tagged JIP4 may be competing with endogenous JIP4 in mouse astrocytes. We were unable to directly address this question in mouse cells as the JIP4 antibody did not produce signal above background in primary astrocytes. However, the same antibody did produce staining in human cells, and we therefore instead used inducible HEK293T lines. Endogenous JIP4 was observed on LRRK2-positive LYS after LLOME treatment in HEK293T cells (fig. S4, B and C), at relatively more rapid time points than with exogenous JIP4, being visible at 2 hours of treatment. We also noted that the addition of LLOME did not alter protein levels of endogenous JIP4 (fig. S4A). MLi-2 treatment numerically decreased the lysosomal localization of LRRK2, but this effect was not statistically significant (P = 0.09; fig. S3, C to E), whereas this treatment completely blocked the recruitment of JIP4 to LRRK2-positive LYS (Fig. 3B), demonstrating that LRRK2 kinase activity is required for JIP4 translocation to the lysosomal membrane. JIP4 translocation was also seen in experiments where we did not transfect cells with LRRK2 (Fig. 3C and fig. S4D), thus showing that LRRK2 is able to recruit JIP4 while expressed at endogenous levels. Pharmacological kinase inhibition of endogenous LRRK2 was also able to arrest JIP4 lysosomal membrane localization (Fig. 3C). With endogenous LRRK2, the recruitment of JIP4 to the lysosomal membrane occurs 10 hours after LLOME addition (fig. S4D). These results demonstrate that increasing expression of LRRK2 accelerates JIP4 recruitment to the LYS surface in a kinase-dependent manner.
Fig. 3. LRRK2 recruits JIP4 through its kinase activity.
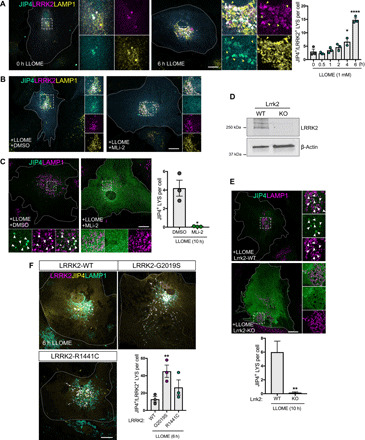
(A) Representative confocal images of astrocytes expressing 3xflag-LRRK2, GFP-JIP4, and LAMP1 untreated or treated with LLOME. Histogram depicts the number of JIP4-positive lysosomes per cell after LLOME (n = 20 cells per N, N = 3). Data are means ± SEM. One-way ANOVA with Dunnett’s post hoc test. (B) Representative confocal images of astrocytes expressing 3xflag-LRRK2, GFP-JIP4, and LAMP1. Cells were pretreated with DMSO or MLi-2 and incubated with LLOME (6 hours). (C) Astrocytes expressing GFP-JIP4 and LAMP1 were pretreated with DMSO or MLi-2 before adding LLOME (10 hours). Histogram shows the number of JIP4-positive lysosomes per cell (n = 20 to 30 cells per N, from N = 3). Data are means ± SEM using unpaired t test with Welch’s correction. (D) Western blot confirming lack of LRRK2 expression in KO astrocytes compared to WT. (E) Images of Lrrk2-WT or Lrrk2-KO astrocytes transfected with GFP-JIP4 and treated with LLOME (10 hours). Statistical analysis used an unpaired t test with Welch’s correction (n = 13 cells per condition in a single experiment). (F) Astrocytes expressing GFP-JIP4 were cotransfected with 3xflag-LRRK2-WT, 3xflag-LRRK2-G2019S, or 3xflag-LRRK2-R1441C, stained for LAMP1, and treated with LLOME (6 hours). Histogram shows the number of JIP4-positive lysosomes per cell. Data are means ± SEM (n = 20 cells per N, from N = 3). One-way ANOVA with Dunnett’s. Yellow arrowheads indicate LRRK2-positive/JIP4-negative lysosomes. White arrowheads show colocalization. Scale bar, 20 μm. *P < 0.05; **P < 0.01,****P < 0.0001.
To confirm that these observations were not due to off-target effects of MLi-2, we compared the amount of lysosomal JIP4 in Lrrk2-WT and Lrrk2-KO astrocytes in the presence of LLOME. Consistent with previous data, cells lacking endogenous LRRK2 did not recruit JIP4 to the LYS membrane (Fig. 3, D and E). Conversely, cells expressing the G2019S mutation (fig. S3H) show a nearly threefold increase in the number of JIP4-positive LYS per cell compared to cells only expressing the WT form of the protein (Fig. 3F). These results further confirm that endogenous LRRK2 recruits JIP4 to damaged lysosomes in a kinase-dependent manner.
LRRK2 phosphorylates RAB10 and RAB35 at the lysosomal membrane
Because JIP4 recruitment to the lysosomal membrane is slower than LRRK2, we speculated that LRRK2 might require intermediate partners to recruit JIP4 after lysosomal membrane damage. LLOME addition was able to trigger LRRK2-lysosomal localization in HEK293FT cells tagged with APEX2 (fig. S5A), so we repeated the APEX2 proteomic analysis in these cells in the presence or absence of LLOME (Fig. 4A). From this screen, we found several endolysosomal markers, including LAMP2, LAMTOR2, PSAP, RAB25, and GABARAPL1/2, confirming by proteomics the enrichment of LRRK2 in the endolysosomal system after LLOME treatment (Fig. 4B and table S3). Two known substrates of LRRK2, RAB35 and RAB10, were also enriched by LLOME treatment (Fig. 4B). By staining for endogenous RAB35 in primary astrocytes, we observed a LLOME-driven recruitment of RAB35 to LRRK2-positive LYS (Fig. 4C), with similar results for a GFP-tagged version of RAB10 (Fig. 4D). We therefore asked whether lysosomal membrane permeabilization triggers an LRRK2-dependent phosphorylation of both RAB proteins. LLOME addition induces a strong increase in phospho-RAB10 (pT73) that is not seen in cells expressing the kinase-dead (K1906M) mutant LRRK2 or in cells treated with MLi-2 (Fig. 4E). Because there are no commercially available antibodies for phospho-RAB35, we used Phos-tag gels that allow the recognition of phosphorylated forms of proteins due to altered motility in acrylamide gels. Cells treated with LLOME show a nearly threefold increase in phospho-RAB35 levels (Fig. 4F), which was blocked by coincubation with MLi-2. The LRRK2 autophosphorylation site pS1292 was not sensitive to the addition of LLOME. To ensure that LRRK2-mediated RAB phosphorylation occurs at the lysosomal membrane, we exogenously expressed LRRK2 and RAB10 in the presence of LLOME and stained for phospho-RAB10 using the RAB10-pT73 antibody. As expected, the RAB10-pT73 signal colocalizes with LRRK2 in the lysosomal surface (Fig. 4G and fig. S5B). Collectively, these results show that lysosomal membrane damage triggers increased kinase activity toward RAB substrates.
Fig. 4. LRRK2 phosphorylates RAB35 and RAB10 in the membrane of ruptured lysosomes.
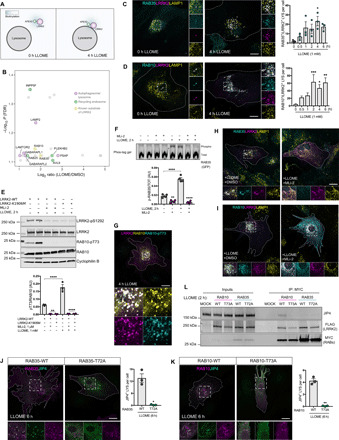
(A) Cartoon of the APEX2 screening in HEK293FT cells in the presence or absence of LLOME. (B) Volcano plot showing the subset of proteins with fold change > 2 and false discovery rate (FDR)–corrected P < 0.08 (n = 3) in proximity with LRRK2 in LLOME-treated cells. (C and D) Astrocytes expressing 3xflag-LRRK2, RAB35 (C), GFP-RAB10 (D), and LAMP1 treated or not with LLOME. The number of RAB35-positive (C) (n = 20 cells per N, N = 3) or RAB10-positive (D) (n = 20 cells in a single experiment) lysosomes per cell was quantified. Data are means ± SEM. One-way ANOVA with Dunnett’s. (E) Western blot images of HEK293FT cells transfected with 3xflag-LRRK2 (WT or K1906M) and pretreated with DMSO or MLi-2 before LLOME was added. Data are means ± SEM (n = 4). Two-way ANOVA with Tukey’s. (F) Phos-tag gel image of HEK293FT cells expressing 3xflag-LRRK2 and GFP-RAB35, pretreated with DMSO or MLi-2 and incubated with LLOME. Normalized phospho-RAB35 levels were measured (n = 4). Data are means ± SEM. Two-way ANOVA with Tukey’s. (G) Astrocyte expressing 3xflag-LRRK2, 2xmyc-RAB10, and RAB10-T73. (H and I) Astrocytes expressing 3xflag-LRRK2, RAB35 (H), GFP-RAB10 (I), and LAMP1 pretreated with DMSO or MLi-2 and incubated with LLOME. (J and K) Astrocytes were transfected with GFP-JIP4 and 2xmyc-RAB35-(WT/T72A) (J) or 2xmyc-RAB10-(WT/T73A) (K) and treated with LLOME. JIP4-positive lysosomes per cell were quantified (n = 20 cells per N, N = 3). Unpaired t test with Welch’s. Data are means ± SEM. (L) HEK293FT cells transfected with 3xflag-LRRK2 along with 2xmyc-RAB10-(WT/T73A) or 2xmyc-RAB35-(WT/T72A) were treated with LLOME, and lysates were subjected to immunoprecipitation with anti-myc antibodies. White arrowheads indicate colocalization. Scale bar, 20 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
LRRK2 recruitment of JIP4 occurs through RAB35 and RAB10 phosphorylation
We next asked whether LRRK2 could also recruit RAB35 and RAB10 to the lysosomal membrane in a kinase-dependent manner, similar to JIP4. LRRK2 pharmacological kinase inhibition completely prevented RAB35 and RAB10 recruitment to LRRK2-positive LYS (Fig. 4, H and I). In astrocytes expressing LRRK2 at endogenous levels, both RAB35 and RAB10 were recruited to the lysosomal membrane after LLOME treatment (fig. S5, C and D), and this was also blocked by MLi-2 (fig. S5, E and F). These results show that endogenous LRRK2 is able to relocalize both RAB proteins to LYS in a kinase-dependent manner. As both RAB35 and RAB10 are recruited to the LYS in a similar fashion to LRRK2, we wanted to determine if RAB35 or RAB10 was important to maintain LRRK2 at the lysosomal membrane. However, depletion of RAB35 and RAB10 expression (fig. S5, G and H) did not alter the ability of LRRK2 to translocate to lysosomes (fig. S5I). Together, our data indicate that LRRK2 precedes and mediates the recruitment of RAB35 and RAB10 to LYS in a kinase-dependent fashion.
In astrocytes transiently transfected with tagged versions of LRRK2, JIP4, and RAB35 and stained for endogenous RAB10, all four proteins are present in the same structures (fig. S6A), suggesting a possible link between the two RAB proteins and JIP4. JIP4 has been previously shown to require several RAB GTPases to ensure its presence in the recycling endosomal membrane (30), so we hypothesized that LRRK2 is able to recruit JIP4 to ruptured LYS via phosphorylation of RAB35 and RAB10. First, we examined the response of RAB35/RAB10 mutants that cannot be phosphorylated by LRRK2 (T72/73A) to LLOME compared to their WT counterparts. Both RAB phospho-null mutants had a significantly lower lysosomal localization after LLOME treatment and were instead found diffusely distributed in the cytosol (fig. S6, B and C). Next, we asked whether the phospho-null version of RAB GTPases affects the recruitment of JIP4 to LYS. For both RAB35 and RAB10, cells expressing the phospho-null mutation were unable to recruit JIP4 to the lysosomal membrane, even in those cells where RAB35-T72A and RAB10-T73A had a lysosomal localization (Fig. 4, J and K). Furthermore, cells knocked down for RAB10 showed significantly lower JIP4 presence on lysosomes (fig. S6D) after LLOME addition, while the effect of RAB35 depletion did not reach statistical significance (P = 0.09). Using coimmunoprecipitation in cells treated with LLOME, we were able to see endogenous JIP4 physically interacting with RAB10-WT but not RAB10-T73A (Fig. 4L). Under the same conditions, we failed to detect a physical interaction between JIP4 and RAB35. Together, our data show that LRRK2 recruits JIP4 through the phosphorylated form of RAB10, and JIP4 is a RAB downstream interactor in the context of lysosomal membrane damage.
JIP4 enhances the formation of tubular structures emanating from lysosomes
To further investigate the JIP4 in the lysosomal membrane, we imaged primary astrocytes transfected with LRRK2 and JIP4 using an Airyscan detector. The improved resolution of this approach allowed us to observe the presence of JIP4-positive/LRRK2-negative/LAMP1-negative tubular structures that stem from LYS in LLOME-treated astrocytes (Fig. 5, A and B, and movie S1). Consistent with the observation that the G2019S mutation leads to higher recruitment of JIP4 to the LYS, this mutation was also associated with a higher number of tubules in the cell (Fig. 5C). JIP4-positive lysosomal tubules were also negative for LAMP1 when imaging living cells (fig. S6E). Furthermore, these tubules were negative for LIMP2, another typical lysosomal membrane marker (fig. S6F), and for the lysosomal luminal marker Dextran-555 (fig. S6G).
Fig. 5. JIP4 promotes the formation of LAMP1-negative lysosomal tubular structures.
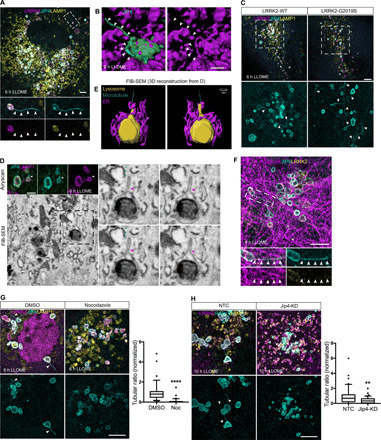
(A) Representative Airyscan image of a LLOME-treated astrocyte (6 hours) expressing GFP-JIP4, 3xflag-LRRK2, and LAMP1. (B) Three-dimensional (3D) surface reconstruction was done in astrocytes treated with LLOME (6 hours) and previously transfected with 3xflag-LRRK2, mNeonGreen-JIP4, and LAMP1-HaloTag. (C) Super-resolution images comparing the number of JIP4-positives tubules in cells expressing LRRK2-WT and G2019S after LLOME treatment (6 hours). (D) FIB-SEM image of a tubule stemming from a lysosome in a 3xflag-LRRK2-G2019S–transfected astrocyte. Top panel shows the Airyscan image of two LAMP1-HaloTag/mNeonGreen-JIP4–labeled lysosomes. Bottom panel shows the correlated EM image, and right panel shows a lysosome with a tubule in different z planes. (E) 3D surface reconstruction of (D) showing a microtubule and the endoplasmic reticulum (ER) in contact with the lysosome. (F) Super-resolution image of an astrocyte expressing 3xflag-LRRK2-G2019S, GFP-JIP4, and α-tubulin. (G) Representative super-resolution images of astrocytes expressing 3xflag-LRRK2-G2019S, GFP-JIP4, and LAMP1 and treated with LLOME (6 hours). Cells were treated with DMSO or nocodazole (Noc). JIP4 tubulation index was measured using an unpaired t test with Welch’s correction (n = 36 to 38 cells pooled from three experiments). Box plot shows the median, and the whiskers show the 10th to 90th percentile. (H) Confocal super-resolution images of astrocytes expressing 3xflag-LRRK2-G2019S, RAB10, and LAMP1 and incubated with LLOME (10 hours). RAB10 tubulation index was measured using an unpaired t test with Welch’s correction (n = 41 cells pooled from two experiments). Box plot shows the median, and the whiskers show the 10th to 90th percentile. White and red (FIB-SEM picture) arrowheads indicate lysosomal tubular structures, and blue arrowheads indicate microtubules. Scale bar, 5 μm or 2 μm (B and D). *P < 0.05, **P < 0.01, ****P < 0.0001.
Next, we analyzed the tubular structures using focused ion beam scanning electron microscopy (FIB-SEM) on astrocytes incubated with LLOME. We first observed that the JIP4/LAMP1-positive compartments contained electron-dense structures (Fig. 5D, fig. S7A, and movie S2), consistent with their lysosomal nature. Typically, the parent lysosomes contained what appear to be partially degraded content, confirming our previous assumption that these are enzymatically inactive lysosomes. By analyzing the JIP4-positive tubules at the structural level, we were able to confirm that the tubular membrane originates from the lysosomal membrane (red arrowheads in Fig. 5D and fig. S7, A and B). We also noted that JIP4-positive lysosomal tubules display variable morphologies, particularly in relation to overall length and thickness (Fig. 5D; fig. S7, A and B; and movie S2), suggesting a dynamic process of their generation and resolution.
We detected microtubules connecting with JIP4-positive tubules (Fig. 5D, blue arrowheads). It has been shown that JIP4, through its interaction with motor proteins and microtubules, is required for the formation of endosomal sorting tubules (31). Consistent with the FIB-SEM observations, we saw JIP4-positive tubules that colocalized with α-tubulin (Fig. 5E) and disruption of microtubules with nocodazole was associated with a prevention of tubule formation (Fig. 5F), indicating that these structures are dependent on microtubules. Endogenous RAB10, but not RAB35, was present in a subset of JIP4-positive tubules (fig. S7, C and D), consistent with the coimmunoprecipitation results obtained in Fig. 4L. This observation allowed us to use RAB10 as a marker protein to determine whether JIP4 was necessary for tubule formation. Overexpression of JIP4 leads to an increase in the number of RAB10-labeled tubules (fig. S7F), and cells knocked down for JIP4 (fig. S7E) showed fewer RAB10-positive tubules (Fig. 5G) in LLOME-exposed astrocytes. Therefore, JIP4 is required for the extension of lysosomal membranes into tubules, likely via an interaction with microtubules.
JIP4-positive tubules release vesicular structures that interact with other lysosomes
We next used super-resolution imaging to observe tubular dynamics in living cells, without fixation after LLOME treatment. JIP4-positive tubules bud, extend, and release from lysosomes to form vesicular structures that were released into the cytosol (Fig. 6, A and E, and movies S3 and S4). JIP4-positive vesicles were observed to have several different behaviors, in that the scission can occur at the base of the tubule or from the tip (Fig. 6A and movie S3). Alternatively, tubules can retract into a vesicle that is ejected to the cytosol (Fig. 6, A to E, and movies S3 and S4). The absence of SNX1, SNX3, and SNX27 at the tubule suggests a different sorting mechanism than that previously described (32) to recycle cargo such as CI-M6PR from endosomes to the TGN (fig. S7, G to I). Although JIP4-positive vesicles are motile in the cytosol, we often detected stationary vesicles in contact with other lysosomes (Fig. 6B). We could also identify moving vesicles that stop to interact with a lysosome transiently that are then released to move elsewhere (movie S5). Furthermore, using FIB-SEM, we were able to find a JIP4-positive vesicle contacting a lysosome (red arrowhead in Fig. 6C). The recipient lysosomes appear to be active, as the interactions occur at Magic Red CTSB–positive lysosomes for period of time up to 4 min (white arrowhead in Fig. 6D and movie S6). Last, we occasionally observed a resolved tubule forming a vesicle that is then able to interact with other lysosomes (Fig. 6E and movie S4). Together, our data identify that LLOME-treated LYS forms JIP4-positive tubules to release vesicular structures that can then contact other lysosomes.
Fig. 6. JIP4-positive tubules are resolved in vesicular structures that contact with other lysosomes.
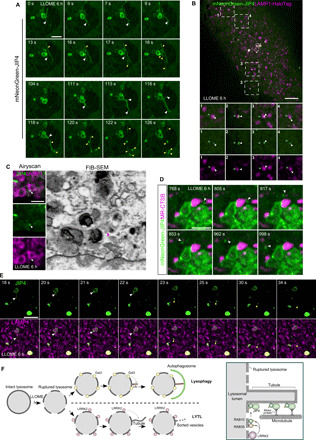
(A) Time-lapse fast Airyscan confocal images of a LLOME-treated (6 hours) astrocyte expressing 3xflag-LRRK2-G2019S and mNeonGreen-JIP4 showing a group of JIP4-positive lysosomes for over 2 min, where the different tubular dynamics were under display (15 slices per frame were taken at 1 frame per second). (B and C) Astrocytes expressing 3xflag-LRRK2-G2019S, mNeonGreen-JIP4, and LAMP1-HaloTag and treated with LLOME (6 hours) were analyzed with a SoRa spinning disk super-resolution microscope. (B) A single frame of an astrocyte, where several JIP4-positive vesicles (white arrowhead) are in close proximity to other lysosomes (blue arrowhead). (C) Time-lapse confocal images of a resolved tubule that, after ejection to the cytosol, contacts other lysosomes (15 slices per frame, 1 frame per second). (D) FIB-SEM image of a JIP4-positive vesicle associated with a lysosome in a 3xflag-LRRK2-G2019S–transfected astrocyte after LLOME treatment (6 hours). Top panel shows the Airyscan image of a group of lysosomes (LAMP1) and a vesicle labeled with JIP4 (white arrow). Bottom panel shows the correlated EM image, with a red arrowhead pointing to the vesicle. (E) LLOME-treated astrocytes (6 hours) expressing 3xflag-LRRK2-G2019S and mNeonGreen-JIP4 and stained with Magic Red CTSB were analyzed with a fast Airyscan confocal microscope for almost 4 min, at 6.05 s per frame. (F) Schematic representation of our working model. White arrowhead marks a JIP4-positive vesicle associated with an active lysosome (red). White arrowheads indicate JIP4-positive lysosomal tubules, and yellow arrowheads show resolved tubules (vesicular structures and scissioned tubules) (A to C). Scale bars, 2 μm (D), 2.5 μm (A, C, and E), and 5 μm (B).
Our overall model is that LRRK2 is recruited to LYS that is not sufficiently damaged to trigger lysophagy. At the lysosomal membrane, LRRK2 is able to phosphorylate RAB35 and RAB10, leading to their retention in the membrane. This event is required to bring the motor adaptor protein JIP4 to the lysosomal membrane where it helps to form tubular structures along microtubules that can secondarily generate vesicles that can interact with other lysosomes (Fig. 6F). We call this newly described process LYsosomal Tubulation/sorting driven by LRRK2, or LYTL.
DISCUSSION
Although a genetic link between LRRK2 and PD was first reported in 2004 (1, 33), the role of this kinase in the cell remains uncertain. The localization of LRRK2 to intracellular membranes (34–36) and the observation that RAB GTPases are kinase substrates for LRRK2 (6, 7, 37) suggest that LRRK2 might be involved in vesicle-mediated transport (38). However, the major phenotype seen in LRRK2 KO animals or in animals treated with LRRK2 kinase inhibitors is accumulation of lysosomal damage (11, 22, 39). This suggests that a major role for endogenous LRRK2 is related to lysosomal function, but a specific mechanism that might explain this observation has not been identified to date. Here, we propose that mechanism is LYTL and that LRRK2 controls dynamic generation of vesicles from damaged lysosomes.
We speculate that LRRK2 may be recruited to lysosomes at a stage of membrane damage that is temporally before the presentation of Gal3 on the surface of the organelles but have not formally proven this conjecture at the present time. A deeper understanding of why LRRK2 is recruited to lysosomes, and what differentiates LRRK2 from Gal3 recruitment, will need to be addressed in the future. A notable feature of LYTL is the activation of LRRK2 kinase activity. This is reminiscent of activation at other membranes, including at the trans-Golgi network (TGN) (7, 40). However, activation of the TGN requires strong overexpression of RAB29, whereas LYTL requires only modest lysosomal membrane permeabilization, at levels less than those required to trigger lysophagy. Consequentially to activation, LRRK2 phosphorylates RAB35 and RAB10, leading to their retention in the lysosomal membrane, likely, in turn, due to diminished binding of p-RABs to GDI1/2 (15). At the same time, p-RABs are able to recruit JIP4 to the lysosomal surface, consistent with previous data showing enhanced binding of p-RABs to this motor adaptor protein (8). Previous literature suggests that phosphorylation-deficient RABs, including RAB8A (6) and RAB10 (16), retain their ability to interact with their respective guanine nucleotide exchange factors (GEFs), and we infer that is also likely to be true for RAB35. We therefore favor the hypothesis that the lack of ability of T72/73A versions of RAB10/RAB35 to be enriched on lysosomes is driven by diminished LRRK2-dependent phosphorylation rather than a lack of GEF-dependent activation. Our data suggest that RAB10 is the primary driver in the recruitment of JIP4; it is therefore possible that RAB35 plays a secondary role in bringing JIP4 to the lysosomal membrane and/or plays a different role once recruited by LRRK2. It is also plausible that the link between the RAB proteins and JIP4 requires other steps or proteins beyond the phosphorylation by LRRK2. Thus, JIP4 acts as a RAB downstream interactor in the lysosomal membrane, similar to the role previously described for JIP4 in recycling endosomes (30).
Recently, Eguchi et al. (16) observed LRRK2 relocalization to LE/LYS in cells treated with chloroquine, a different lysosomotropic reagent that increases pH in lysosomes and late endosomes. Eguchi et al. proposed a scenario where LRRK2 promotes lysosomal exocytosis to release nondegraded cargo to the extracellular space. Our model is distinct in that JIP4-positive lysosomes failed to show a net movement toward the plasma membrane using live-cell imaging. The difference in lysosomal behavior may be related to the use of different cellular models (mouse primary astrocytes versus cancer cells) or the compound used to damage the lysosome (LLOME versus chloroquine). Exploration of the differences between different lysosomal damaging agents in different cell types may be mechanistically informative in future studies.
Sorting at other cellular compartments such as endosomes occurs through the formation of tubular structures that bud and extend from organelles. Tubular structures are then severed to produce vesicles that travel to the plasma membrane or the TGN (41). Our data show that JIP4 promotes the formation of tubular structures at the lysosome, which are to our knowledge described here for the first time, but may be similar to structures at other organelles including endosomes (31). It is likely that JIP4 mediates tubule dynamics by recruiting motor proteins (25), because we have demonstrated that tubule extension requires microtubules. JIP4-positive tubules undergo scission to generate smaller vesicular structures that travel through the cytosol and interact with other lysosomes and possibly other cellular organelles. Membrane contact sites have been shown to have different functions, such as the interchange of lipids, calcium, or other metabolites from one compartment to another (42). Neither the tubules nor the resolved material is positive for lysosomal membrane markers (LAMP1 or LIMP2), and the JIP4-positive tubules were resistant to paraformaldehyde (PFA) fixation. These observations argue against LYTL being involved in proto-lysosome formation through lysosomal reformation (LR), because LR tubules are amenable to PFA fixation (43) and positive for LAMP1 (44). LRRK2 has been associated (along with RAB2A) with the sorting of lysozyme from dense core vesicles (DCVs) in the gut (45). As astrocytes are considered a highly secretory cell in the central nervous system (46), LYTL could be used as an alternative sorting process beyond exosomes, DCVs, or secretory lysosomes, among others. Thus, the function of LYTL, including why lysosomes release membranous content and the overall effect of this sorting process in the cell, will require additional future investigations.
We view the activation of LRRK2 by lysosomal membrane permeabilization as an analogous process to the activation of another PD-associated kinase, PINK1, by mitochondrial damage (47). Specifically, one of the proposed functions of the PINK1/Parkin system during mitochondrial stress is the release of mitochondria-derived vesicles to lysosomes (48). These considerations may not directly suggest functional convergence of mitochondrial and lysosomal pathways but do indicate that there are multiple kinase-dependent events that can control function of organelles relevant to PD. However, indirectly, the ability of LRRK2 mutants to cause retention of RAB10 at the lysosomal membrane could prevent RAB10 from being recruited to depolarized mitochondria and, as a consequence, limit mitophagy (49).
Overall, we have identified a newly described cellular process that promotes lysosomal sorting material from lysosomes after induction of lysosomal membrane permeabilization. LYTL is controlled by LRRK2 kinase activity because the recruitment of all the downstream components is completely blocked by MLi-2. Conversely, the G2019S mutation in LRRK2 that is pathogenic for PD increases both the lysosomal localization of LRRK2 and JIP4 recruitment and tubulation. Considering the proposed centrality of lysosome biology in PD pathogenesis, it is possible that LYTL is involved in disease mechanisms. As increased lysosomal membrane damage has been associated with aging (50, 51), and aggregates or fibrils taken up by endocytosis can trigger lysosomal membrane permeabilization (52), it is possible that LYTL could be more active during aging and further enhanced in patients carrying LRRK2 pathogenic mutations, potentially contributing to neurodegeneration.
METHODS
Cell culture
All procedures with animals followed the guidelines approved by the Institutional Animal Care and Use Committee of the National Institute on Aging. Astrocyte cultures were prepared from C57BL/6J newborn (postnatal day 0) pups. Dissected mouse cortices were incubated in 1 ml/cortex Basal Medium Eagle (BME) (Sigma-Aldrich), containing 5 U of papain/(Worthington) for 30 min at 37°C. Five micrograms of deoxyribonuclease I was added to each cortex preparation, and brain tissue was dissociated into a cellular suspension that was washed twice with 10 volumes of BME and counted. Astrocyte cultures were plated in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (FBS) (Lonza) into 75-cm2 tissue culture flasks. For the preparation of purified astrocyte cultures, 7- to 10-day primary cultures were vigorously shaken to detach microglia and oligodendrocytes. Culture purity was assessed with GFAP for astrocytes, and OLIG2 and IBA1 to exclude oligodendrocytes and microglia. Cultures had >90% of astrocytes in all experiments. Astrocytes were used from passage 2 to passage 3.
HEK293FT cells were maintained in DMEM containing glucose (4.5 g/liter), 2 mM l-glutamine, and 10% FBS at 37°C in 5% CO2. The HEK293T-inducible GFP-LRRK2-WT cell line was obtained from D. Alessi (University of Dundee), and expression was induced by addition of doxycycline (53). HEK293FT cells, HEK293T GFP-LRRK2 doxycycline-induced cells, and primary astrocytes were seeded on 12-mm coverslips precoated with Matrigel.
Reagents
LLOME (Sigma-Aldrich, L7393) was diluted in dimethyl sulfoxide (DMSO) and added at 1 mM for the indicated times. Nocodazole (Sigma-Aldrich, M1404) was diluted in DMSO and added at 10 μM 2 hours before fixation. Fixable Dextran–Alexa Fluor 555, 10 kDa (Thermo Fisher Scientific, D34679) was incubated for 6 hours (2.5 mg/ml). Cells were then washed three times with phosphate-buffered saline (PBS), and fresh medium was added to chase Dextran overnight (18 to 24 hours) before treating astrocytes with LLOME. MLi-2 (Tocris, 5756) was used at 1 μM, 90 min before LLOME addition. Magic Red CTSB was obtained from ImmunoChemistry Technologies (938) and incubated with cells at a dilution of 1:250 in medium for 30 min. Then, cells were washed three times with PBS before imaging. LysoTracker Red DND-99 (Thermo Fisher Scientific, L7528) was added at 1 μM for 30 min before cells were washed three times with PBS and analyzed them using confocal microscopy. LAMP1-HaloTag–transfected cells were incubated with the JF646 peptide (Promega, GA1121) at 200 nM for 15 min, and cells were then washed three times and incubated with fresh medium before treated with LLOME.
Antibodies
The following primary antibodies were used: mouse anti-FLAG M2 [Sigma-Aldrich, F3165; 1:500 for immunocytochemistry (ICC) and 1:10,000 for western-blot (WB)], rabbit anti-JIP4 (Cell Signaling Technology; 5519, 1:1000 for WB and 1:100 for ICC), rat anti-LAMP1 [Developmental Studies Hybridoma Bank (DSHB), 1D4B; 1:100 for ICC], rat anti-LAMP2 (DSHB, 1:100 for ICC), mouse anti-LAMP2 (DSHB, H4B4; 1:100 for ICC), rat anti-FLAG (BioLegend, 637302; 1:100 for ICC), rabbit anti-RAB7A (Abcam, ab137029; 1:200 for ICC), rat anti-myc (Chromotek, 9e1-100; 1:500 for ICC), chicken anti-GFP (Aves Lab, GFP-1020; 1:500 to 1:1000 for ICC), mouse anti-GFP (Roche, 11814460001; 1:10,000 for WB), goat anti-CTSB (R&D Systems, AF965; 1:500 for ICC), mouse anti–α-tubulin (Cell Signaling Technology, 3873; 1:350 for ICC and 1:15,000 for WB), rabbit anti-RAB35 (Proteintech, 11329-2-AP; 1:100 for ICC and 1:1000 for WB), rabbit anti-RAB10 (Abcam, ab237703; 1:100 for ICC and 1:1000 for WB), rabbit anti-RAB10 (phospho-T73) (Abcam, ab241060; 1:100 for ICC and 1:1000 for WB), rabbit anti-LRRK2 (Abcam, ab133474; 1:1000 for WB), rabbit anti-LRRK2 (phospho-S1292) (Abcam, ab203181; 1:1000 for WB), rabbit anti-EEA1 (Cell Signaling Technology, 3288; 1:100 for ICC), mouse anti–β-actin (Sigma-Aldrich, A5441; 1:15,000 for WB), rabbit anti-LC3B (Cell Signaling Technology, 2775; 1:1000 for WB), and rabbit anti–cyclophilin B (Abcam, ab16045; 1:15,000 for WB).
For ICC, unless otherwise stated, the secondary antibodies were purchased from Thermo Fisher Scientific. The following secondary antibodies were used: donkey anti-mouse Alexa Fluor 568 (A10037, 1:500), donkey anti-rabbit Alexa Fluor 488 (A-21206, 1:500), donkey anti-mouse Alexa Fluor 568 (A-21202, 1:500), donkey anti-rat Alexa Fluor 488 (A-21208, 1:500), donkey anti-goat Alexa Fluor 488 (A-11055, 1:500), donkey anti-rabbit Alexa Fluor 568 (A10042, 1:500), donkey anti-mouse Alexa Fluor 647 (A-31571, 1:500), and goat anti-rat Alexa Fluor 647 (A-21247, 1:250 to 1:500). Donkey anti-chicken Alexa Fluor 488 (703-545-155, 1:500) and donkey anti-rat Alexa Fluor 405 (712-475-153, 1:100) were obtained from Jackson ImmunoResearch.
Cloning
Constructs for 3xflag-tagged LRRK2 WT/full length and domains, and GUS have been described previously (7, 54). JIP3, JIP4, and BICD2 Gateway PLUS shuttle clones were obtained from GeneCopoeia. BLOS1, MUTED, and STAM1 complementary DNA (cDNA) were obtained from Dharmacon. RAB35, RAB10, and APEX-NES cDNA were acquired from Addgene (Addgene, #49552, #49472, and #49386) (55–57). cDNAs were amplified with polymerase chain reaction (PCR) and cloned into pCR8/GW/TOPO vector (Thermo Fisher Scientific). Each was then subcloned into pDEST vectors using Gateway technology (Thermo Fisher Scientific). Full-length LRRK2 was subcloned into the pDEST-Emerald GFP and p3xflag-APEX2-DEST vectors; JIP3, STAM1, BICD2, BLOS1, and MUTED were subcloned into pDEST-53; JIP4 was also subcloned into pDEST-NeonGreen; RAB35 was subcloned into pDEST-53 and the pCMV-2xmyc-DEST vectors; and RAB10 was subcloned into pCMV-2xmyc-DEST plasmid. 2xmyc-RAB10-T73A and 2xmyc-RAB35-T72A were generated by directed mutagenesis from the WT vectors using the QuikChange Lightning Kit (Agilent). EGFP-RAB10 (Addgene, #49472) (56), EGFP-Gal3 (Addgene, #73080) (29), and LAMP1-RFP (Addgene, #1817) (58) were purchased from Addgene. The LAMP1-HaloTag construct was provided by the Bonifacino laboratory, and the LIMP2-myc vector was a gift from M. Schwake (59). All primers used for cloning are in table S5, and the expression constructs used in this study are summarized in table S4.
Transfection
Transient transfections of HEK293FT cells and astrocytes were performed using Lipofectamine 2000 and Stem reagents, respectively (Thermo Fisher Scientific). HEK293FT cells were transfected for 24 hours, and mouse primary astrocytes were transfected for 48 hours. For small interfering RNAs (siRNAs), cells were transfected with the SMARTpool ON-TARGET (Dharmacon) plus scramble or Rab35, Rab10, or Jip4 siRNAs using Lipofectamine RNAiMAX (Thermo Fisher Scientific) transfection reagent for astrocytes. Astrocytes were incubated with siRNA for a total of 4 days before fixation or lysis.
APEX2 reaction
HEK293FT cells were plated in 150-cm2 flasks previously coated with Matrigel and transfected with the appropriate vectors. Twenty-four hours later, 500 μM biotin-phenol was preincubated with cells for 30 min at 37°C. H2O2 (1 mM) was added for 1 min while gently mixing, and the cells were subsequently washed twice with quenching buffer [tris-buffered saline (TBS) supplemented with 1 mM CaCl2, 10 mM sodium ascorbate, 1 mM sodium azide, and 1 mM Trolox], once with PBS, and twice with quenching buffer for 1 min per wash. Cells were collected in 10 ml of quenching buffer and centrifuged at 3000g for 10 min. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Pierce) supplemented with 10 mM sodium ascorbate, 1 mM sodium azide, 1 mM Trolox, 1 mM dithiothreitol, and protease inhibitors (Roche Complete). Samples were briefly sonicated and pelleted at 10,000g for 10 min, and then equal amounts of proteins were applied to streptavidin magnetic beads (Thermo Fisher Scientific) and incubated for 1 hour at room temperature (RT) on a rotator. Cells were then washed sequentially with KCl (1 M), Na2CO3 (0.1 M), and urea (2 M) in 0.1 M tris buffer followed by two final washes with RIPA buffer. Beads were eluted by boiling in 45 μl of 2× protein loading buffer supplemented with 2 mM biotin and β-mercaptoethanol for 10 min. Protein lysates were loaded on an acrylamide gel and run for 1 hour at 150 V. The gel was then stained with GelCode Blue stain (Thermo Fisher Scientific) for 30 min and washed with dH2O overnight.
Mass spectrometry analysis
Protein was quantified, and the lysates were mixed in a 1:1 ratio (light:heavy), with a final number of two to three replicates. Lysates were separated on a 4% to 20% polyacrylamide gel (Bio-Rad). Eight bands were cut from each replicate. Gel bands were reduced with tris(2-carboxyethyl)phosphine, alkylated with N-ethylmaleimide (NEM), and digested with trypsin. The system used for data acquisition is an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) coupled with an UltiMate 3000 high-pressure liquid chromatography instrument (Thermo Fisher Scientific). Extracted peptides were desalted and then separated on an ES803 column (Thermo Fisher Scientific) using a gradient with mobile phase B (MP B) increasing from 5 to 27% over 60 min. The composition of MP A and MP B is 0.1% formic acid in 100% high-performance liquid chromatography (HPLC) acetonitrile. The liquid chromatography–tandem mass spectrometry (LC-MS/MS) data were acquired in data-dependent mode. The survey scan was performed in Orbitrap with the following parameters: mass range, 350 to 1500 mass/charge ratio (m/z); resolution, 120,000 at m/z 400; automatic gain control (AGC) target, 4 × 105 ions. The product scan was performed in ion trap with the following parameters: collision-induced dissociation on as many precursor ions as allowed in 3 s; isolation window, 1.6 Da. Database search and ratio calculation were performed using Proteome Discoverer 2.2 software. Conditions used in the database are listed below. Database: Sprot Human database; modifications: 13C(6) (R), 13C(6) (K), oxidation (M), NEM(C). Heavy/Light (H/L) ratios are calculated for each sample with (Fig. 4) or without (Fig. 1) normalization.
Phos-tag gels
HEK293FT cells were transfected with 3xflag-LRRK2 and GFP-RAB35 plasmids and, 24 hours later, treated with LLOME or DMSO for 2 hours. Cells were then lysed in 10× cell lysis buffer (Cell Signaling Technology) supplemented with EDTA-free protease inhibitor (Roche) and 1× Halt phosphatase inhibitor. Lysates were cleared by centrifugation at 20,000g. Phos-tag SDS–polyacrylamide gel electrophoresis (PAGE) was performed following the vendor’s instructions (Wako). Briefly, gels for Phos-tag SDS-PAGE consisted of a stacking gel [4.5% acrylamide, 350 mM bis-tris/HCl, tetramethylethylenediamine (TEMED), and 0.05% ammonium persulfate (APS)] and a separating gel (12% acrylamide, 350 mM bis-tris/HCl, 50 μM Phos-tag solution, 100 μM ZnCl2, TEMED, and 0.05% APS). Lysates were electrophoresed at 80 V for the stacking gel and at 150 V for the separating gel. After SDS-PAGE, the gels were washed twice (20 min each) with transfer buffer containing 10 mM EDTA. Gels were transferred to membranes by a semidry Trans-Blot Turbo transfer system (Bio-Rad).
Confocal microscopy
Confocal images were taken using a Zeiss LSM 880 microscope equipped with a 63× 1.4 numerical aperture (NA) objective. Super-resolution imaging was performed using the Airyscan mode. Raw data were processed using Airyscan processing in “autostrength” mode with Zen Black software version 2.3. Live-cell super-resolution imaging was performed in Fast Airyscan mode on an inverted Zeiss LSM 880 Fast Airyscan microscope equipped with a 63× 1.4 NA objective, and environmental chamber to maintain cells at 37°C with humidified 5% CO2 gas during imaging. Immersion oil for 37°C was used during imaging.
For spinning disk super-resolution microscopy, we used a W1-SoRa super-resolution spinning disk microscope (Nikon) with a 60× 1.49 NA oil immersion objective and a ×2.8 intermediate magnification (×168 combined), with an offset micro-lensed SoRa disk and environmental chamber to maintain cells at 37°C with humidified 5% CO2 gas during imaging. For deconvolution, we used 10 iterations of the Landweber algorithm. Images from two channels were acquired simultaneously using a Cairn twin-cam emission splitter and two Photometrics prime 95b sCMOS cameras, a 565LP DM, and appropriate emission cleanup filters. Unless otherwise stated, pictures are maximum intensity projection from Z stacks.
Tubular ratio measurements
Astrocytes were transfected with the 3xflag-LRRK2-G2019S plasmid and cotransfected or not with the GFP-JIP4 vector. After fixation and staining, cells were imaged with a confocal microscope using the Airyscan module to enhance resolution. The tubular ratio in each cell was measured as follows: Ratio = #JIP4 + tubules/#JIP4 + lysosomes. A similar approach was used for endogenous RAB10. For each independent replicate, the ratio was normalized by the average value of the control group (i.e., DMSO, NTC, and MOCK). Data were generated from cells pooled across at least two independent experiments. Only cells with 10 or more JIP4 or RAB10-positive lysosomes per cell were imaged.
Coimmunoprecipitation
HEK293FT cells transfected with 3xflag-LRRK2, 2xmyc-RAB10, 2xmycRAB10-T73A, 2xmyc-RAB35, and 2xmyc-RAB35-T72A plasmids were lysed in IP buffer [20 mM tris-HCl (pH 7.5), 300 mM NaCl, 1 mM EDTA, 0.3% Triton X-100, 10% glycerol, 1× Halt phosphatase inhibitor cocktail (Thermo Fisher Scientific), and protease inhibitor cocktail (Roche)] for 30 min on ice. Lysates were centrifuged at 4°C for 10 min at 20,000g, and the supernatant was further cleared by incubation with EZview™ protein G agarose beads (Sigma-Aldrich) for 30 min at 4°C (only for FLAG IP). After agarose bead removal by centrifugation, lysates were incubated with anti-flag M2 agarose beads (Sigma-Aldrich) or myc-Trap agarose beads (Chromotek) for 1 hour at 4°C on a rotator. Beads were washed six times with IP wash buffer [20 mM tris-HCl (pH 7.5), 300 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 10% glycerol] and eluted in 1× kinase buffer (Cell Signaling Technology) containing 150 mM NaCl, 0.02% Triton X-100, and 3xflag peptide (150 ng/μl) (Sigma-Aldrich) by shaking for 30 min at 4°C (for FLAG IP) or by boiling samples in 2× loading buffer with β-mercaptoethanol for 5 min (for MYC IP).
Immunostaining
Primary cultures of astrocytes or HEK293FT cells were fixed with 4% PFA for 10 min, permeabilized with PBS/0.1% Triton X-100 for 10 min, and blocked with 5% donkey serum for 1 hour at RT. Primary antibodies were diluted in blocking buffer (1% donkey serum) and incubated overnight at 4°C. After three 5-min washes with PBS/0.1% Triton X-100, secondary fluorescently labeled antibodies were diluted in blocking buffer (1% donkey serum) and incubated for 1 hour at RT. Coverslips were washed twice with 1× PBS and an additional two times with dH2O and mounted with ProLong Gold antifade reagent (Thermo Fisher Scientific).
SDS-PAGE and Western blotting
Proteins were resolved on 4 to 20% Criterion TGX precast gels (Bio-Rad) and transferred to membranes by a semi-dry Trans-Blot Turbo transfer system (Bio-Rad). The membranes were blocked with Odyssey Blocking Buffer (LI-COR, catalog no. 927-40000) and then incubated for 1 hour at RT or overnight at 4°C with the indicated primary antibody. The membranes were washed in TBS-Triton (TBS-T) (3 × 5 min) followed by incubation for 1 hour at RT with fluorescently conjugated goat anti-mouse, rat, or rabbit IRDye 680 or 800 antibodies (LI-COR). The blots were washed in TBST (3 × 5 min) and scanned on an Odyssey CLx (LI-COR). Quantitation of Western blots was performed using Image Studio (LI-COR).
Sample preparation for FIB-SEM
Following Airyscan microscopy, cells were fixed and processed largely as previously described (60) with small modifications. After dehydration, the MatTek glass coverslip was removed from the plastic using propylene oxide. The removed glass coverslip was then rinsed in 100% ethanol followed by immersion in mixtures of Durcupan ACM and ethanol with the following ratios: 25:75 for 1.5 hours, 50:50 for 1.5 hours, 75:25 overnight. The sample was then immersed in 100% Durcupan ACM for 4 to 5 hours with replacement of fresh Durcupan every hour. The glass coverslip was then removed, and excess Durcupan was removed using filter paper. The coverslip was then placed in an oven at 60°C for 10 min, after which the sample was placed vertically in a 50-ml Falcon tube in folded filter papers and centrifuged for 15 min at 37°C and 750 relative centrifugal force (RCF). The glass coverslip was then placed in an oven at 60°C under vacuum and left to polymerize over 2 days. The sample was then sputter-coated with 50-nm gold and painted with silver paint, followed by drying under vacuum. The samples were imaged inside a Zeiss Crossbeam 540 FIB-SEM microscope. Platinum and carbon were deposited over the region of interest, and the run was set up and controlled by Atlas software (Fibics) SEM settings: 1.5 kV; 2.0 nA; milling probe: 300 pA. The slice thickness and the voxel size were set to 6 nm. The total volume acquired per tissue sample (XYZ) was 44.21 m × 4.22 m × 27.38 m.
Statistical analysis
Analyses based on cell counts were performed by an investigator blinded to treatment/transfection status. Statistical analysis for experiments with two treatment groups used Student’s t tests with Welsh’s correction for unequal variance. For more than two groups, we used one-way analysis of variance (ANOVA) or two-way ANOVA where there were two factors in the model. Tukey’s post hoc test was used to determine statistical significance for individual comparisons in those cases where the underlying ANOVA was statistically significant and where all groups were compared; Dunnett’s multiple comparison test was used where all groups were compared back to a single control group. Individual points in histograms represent the mean of each independent replicate (N). Unless otherwise stated, graphed data are presented as means ± SEM. Comparisons considered statistically significant are indicated: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Supplementary Material
Acknowledgments
Funding: This research was supported, in part, by the Intramural Research Program of the NIH, National Institute on Aging (MRC) and the NIA Postdoctoral Funding Opportunity (L.B.-P.). We thank D. C. Gershlick (Cambridge University), J. S. Bonifacino (NIH), and D. Roosen (FMP Berlin) for critical feedback and also M. Schwake (Universitat Bielefeld) for providing the LIMP2-myc plasmid. We also thank E. Balzer (Nikon) for assistance using the SoRa spinning disk super-resolution microscope. Author contributions: Conceptualization: L.B.-P. and M.R.C.; methodology: L.B.-P., A.B., C.D.W., E.L., C.K.E.B., S.S.-A., A.M., N.L., and R.K.; formal analysis: L.B.-P. and M.R.C.; investigation: L.B.-P., A.B., E.L., Y.L., C.D.W., and J.H.K.; writing: L.B.-P. and M.R.C.; supervision: M.R.C.; funding: M.R.C., Y.L., and L.B.-P. Competing interests: The authors have no competing interests related to this work. R.K. is currently an employee of Abcam. Data and materials availability: All the data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabb2454/DC1
REFERENCES AND NOTES
- 1.Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Martí Carrera I., Pena A. S., de Silva R., Lees A., Martí-Massó J. F., Pérez-Tur J., Wood N. W., Singleton A. B., Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Lesage S., Leutenegger A.-L., Ibanez P., Janin S., Lohmann E., Dürr A., Brice A.; French Parkinson’s Disease Genetics Study Group , LRRK2 haplotype analyses in European and North African families with Parkinson disease: A common founder for the G2019S mutation dating from the 13th century. Am. J. Hum. Genet. 77, 330–332 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang D., Nalls M. A., Hallgrímsdóttir I. B., Hunkapiller J., van der Brug M., Cai F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team, Kerchner G. A., Ayalon G., Bingol B., Sheng M., Hinds D., Behrens T. W., Singleton A. B., Bhangale T. R., Graham R. R., A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nalls M. A., Blauwendraat C., Vallerga C. L., Heilbron K., Bandres-Ciga S., Chang D., Tan M., Kia D. A., Noyce A. J., Xue A., Bras J., Young E., von Coelln R., Simón-Sánchez J., Schulte C., Sharma M., Krohn L., Pihlstrøm L., Siitonen A., Iwaki H., Leonard H., Faghri F., Gibbs J. R., Hernandez D. G., Scholz S. W., Botia J. A., Martinez M., Corvol J.-C., Lesage S., Jankovic J., Shulman L. M., Sutherland M., Tienari P., Majamaa K., Toft M., Andreassen O. A., Bangale T., Brice A., Yang J., Gan-Or Z., Gasser T., Heutink P., Shulman J. M., Wood N. W., Hinds D. A., Hardy J. A., Morris H. R., Gratten J., Visscher P. M., Graham R. R., Singleton A. B.; 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium , Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M. P., Beilina A., Blackinton J., Thomas K. J., Ahmad R., Miller D. W., Kesavapany S., Singleton A., Lees A., Harvey R. J., Harvey K., Cookson M. R., Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Steger M., Tonelli F., Ito G., Davies P., Trost M., Vetter M., Wachter S., Lorentzen E., Duddy G., Wilson S., Baptista M. A. S., Fiske B. K., Fell M. J., Morrow J. A., Reith A. D., Alessi D. R., Mann M., Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 5, e12813 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beilina A., Rudenko I. N., Kaganovich A., Civiero L., Chau H., Kalia S. K., Kalia L. V., Lobbestael E., Chia R., Ndukwe K., Ding J., Nalls M. A.; International Parkinson’s Disease Genomics Consortium; North American Brain Expression Consortium, Olszewski M., Hauser D. N., Kumaran R., Lozano A. M., Baekelandt V., Greene L. E., Taymans J.-M., Greggio E., Cookson M. R., Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 111, 2626–2631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steger M., Diez F., Dhekne H. S., Lis P., Nirujogi R. S., Karayel O., Tonelli F., Martinez T. N., Lorentzen E., Pfeffer S. R., Alessi D. R., Mann M., Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 6, e31012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramirez A., Heimbach A., Gründemann J., Stiller B., Hampshire D., Cid L. P., Goebel I., Mubaidin A. F., Wriekat A.-L., Roeper J., Al-Din A., Hillmer A. M., Karsak M., Liss B., Woods C. G., Behrens M. I., Kubisch C., Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Klein A. D., Mazzulli J. R., Is Parkinson’s disease a lysosomal disorder? Brain 141, 2255–2262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R. J. III, Shen J., Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9879–9884 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henry A. G., Aghamohammadzadeh S., Samaroo H., Chen Y., Mou K., Needle E., Hirst W. D., Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 24, 6013–6028 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Hockey L. N., Kilpatrick B. S., Eden E. R., Lin-Moshier Y., Brailoiu G. C., Brailoiu E., Futter C. E., Schapira A. H., Marchant J. S., Patel S., Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 128, 232–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orenstein S. J., Kuo S.-H., Tasset I., Arias E., Koga H., Fernandez-Carasa I., Cortes E., Honig L. S., Dauer W., Consiglio A., Raya A., Sulzer D., Cuervo A. M., Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez R. C., Wawro P., Lis P., Alessi D. R., Pfeffer S. R., Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J. Cell Biol. 218, 4157–4170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eguchi T., Kuwahara T., Sakurai M., Komori T., Fujimoto T., Ito G., Yoshimura S.-I., Harada A., Fukuda M., Koike M., Iwatsubo T., LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. U.S.A. 115, E9115–E9124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.di Domenico A., Carola G., Calatayud C., Pons-Espinal M., Muñoz J. P., Richaud-Patin Y., Fernandez-Carasa I., Gut M., Faella A., Parameswaran J., Soriano J., Ferrer I., Tolosa E., Zorzano A., Cuervo A. M., Raya A., Consiglio A., Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 12, 213–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stafa K., Trancikova A., Webber P. J., Glauser L., West A. B., Moore D. J., GTPase activity and neuronal toxicity of Parkinson’s disease–Associated LRRK2 is regulated by ArfGAP1. PLOS Genet. 8, e1002526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mamais A., Chia R., Beilina A., Hauser D. N., Hall C., Lewis P. A., Cookson M. R., Bandopadhyay R., Arsenite stress down-regulates phosphorylation and 14-3-3 binding of leucine-rich repeat kinase 2 (LRRK2), promoting self-association and cellular redistribution. J. Biol. Chem. 289, 21386–21400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng X.-T., Xie Y.-X., Zhou B., Huang N., Farfel-Becker T., Sheng Z.-H., Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J. Cell Biol. 217, 3127–3139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung V., Udeshi N. D., Lam S. S., Loh K. H., Cox K. J., Pedram K., Carr S. A., Ting A. Y., Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 11, 456–475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pellegrini L., Hauser D. N., Li Y., Mamais A., Beilina A., Kumaran R., Wetzel A., Nixon-Abell J., Heaton G., Rudenko I., Alkaslasi M., Ivanina N., Melrose H. L., Cookson M. R., Harvey K., Proteomic analysis reveals co-ordinated alterations in protein synthesis and degradation pathways in LRRK2 knockout mice. Hum. Mol. Genet. 27, 3257–3271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu C. H., Chan D., Wolozin B., LRRK2 and the stress response: Interaction with MKKs and JNK-interacting proteins. Neurodegener Dis. 7, 68–75 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bae E.-J., Kim D.-K., Kim C., Mante M., Adame A., Rockenstein E., Ulusoy A., Klinkenberg M., Jeong G. R., Bae J. R., Lee C., Lee H.-J., Lee B.-D., Di Monte D. A., Masliah E., Lee S.-J., LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 9, 3465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montagnac G., Sibarita J.-B., Loubéry S., Daviet L., Romao M., Raposo G., Chavrier P., ARF6 interacts with JIP4 to control a motor switch mechanism regulating endosome traffic in cytokinesis. Curr. Biol. 19, 184–195 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Tanaka T., Iino M., Goto K., Knockdown of Sec8 enhances the binding affinity of c-Jun N-terminal kinase (JNK)-interacting protein 4 for mitogen-activated protein kinase kinase 4 (MKK4) and suppresses the phosphorylation of MKK4, p38, and JNK, thereby inhibiting apoptosis. FEBS J. 281, 5237–5250 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Aits S., Kricker J., Liu B., Ellegaard A.-M., Hämälistö S., Tvingsholm S., Corcelle-Termeau E., Høgh S., Farkas T., Holm Jonassen A., Gromova I., Mortensen M., Jäättelä M., Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 11, 1408–1424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thiele D. L., Lipsky P. E., Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: Dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc. Natl. Acad. Sci. U.S.A. 87, 83–87 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maejima I., Takahashi A., Omori H., Kimura T., Takabatake Y., Saitoh T., Yamamoto A., Hamasaki M., Noda T., Isaka Y., Yoshimori T., Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 32, 2336–2347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi H., Etoh K., Ohbayashi N., Fukuda M., Rab35 promotes the recruitment of Rab8, Rab13 and Rab36 to recycling endosomes through MICAL-L1 during neurite outgrowth. Biol. Open 3, 803–814 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchesin V., Castro-Castro A., Lodillinsky C., Castagnino A., Cyrta J., Bonsang-Kitzis H., Fuhrmann L., Irondelle M., Infante E., Montagnac G., Reyal F., Vincent-Salomon A., Chavrier P., ARF6-JIP3/4 regulate endosomal tubules for MT1-MMP exocytosis in cancer invasion. J. Cell Biol. 211, 339–358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cullen P. J., Steinberg F., To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 19, 679–696 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., Asmus F., Müller-Myhsok B., Dickson D. W., Meitinger T., Strom T. M., Wszolek Z. K., Gasser T., Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Alegre-Abarrategui J., Christian H., Lufino M. M. P., Mutihac R., Venda L. L., Ansorge O., Wade-Martins R., LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 18, 4022–4034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biskup S., Moore D. J., Celsi F., Higashi S., West A. B., Andrabi S. A., Kurkinen K., Yu S.-W., Savitt J. M., Waldvogel H. J., Faull R. L. M., Emson P. C., Torp R., Ottersen O. P., Dawson T. M., Dawson V. L., Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 60, 557–569 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Berger Z., Smith K. A., LaVoie M. J., Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511–5523 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacLeod D. A., Rhinn H., Kuwahara T., Zolin A., Di Paolo G., McCabe B. D., Marder K. S., Honig L. S., Clark L. N., Small S. A., Abeliovich A., RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77, 425–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonet-Ponce L., Cookson M. R., The role of Rab GTPases in the pathobiology of Parkinson’ disease. Curr. Opin. Cell Biol. 59, 73–80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tong Y., Giaime E., Yamaguchi H., Ichimura T., Liu Y., Si H., Cai H., Bonventre J. V., Shen J., Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 7, 2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purlyte E., Dhekne H. S., Sarhan A. R., Gomez R., Lis P., Wightman M., Martinez T. N., Tonelli F., Pfeffer S. R., Alessi D. R., Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 38, e101237 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNally K. E., Cullen P. J., Endosomal retrieval of cargo: Retromer is not alone. Trends Cell Biol. 28, 807–822 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Prinz W. A., Toulmay A., Balla T., The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 21, 7–24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sridhar S., Patel B., Aphkhazava D., Macian F., Santambrogio L., Shields D., Cuervo A. M., The lipid kinase PI4KIIIβ preserves lysosomal identity. EMBO J. 32, 324–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu L., McPhee C. K., Zheng L., Mardones G. A., Rong Y., Peng J., Mi N., Zhao Y., Liu Z., Wan F., Hailey D. W., Oorschot V., Klumperman J., Baehrecke E. H., Lenardo M. J., Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q., Pan Y., Yan R., Zeng B., Wang H., Zhang X., Li W., Wei H., Liu Z., Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 16, 918–926 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Verkhratsky A., Matteoli M., Parpura V., Mothet J.-P., Zorec R., Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 35, 239–257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pickles S., Vigié P., Youle R. J., Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McLelland G.-L., Soubannier V., Chen C. X., McBride H. M., Fon E. A., Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wauters F., Cornelissen T., Imberechts D., Martin S., Koentjoro B., Sue C., Vangheluwe P., Vandenberghe W., LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16, 203–222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirkegaard T., Roth A. G., Petersen N. H. T., Mahalka A. K., Olsen O. D., Moilanen I., Zylicz A., Knudsen J., Sandhoff K., Arenz C., Kinnunen P. K. J., Nylandsted J., Jäättelä M., Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature 463, 549–553 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Gabandé-Rodríguez E., Pérez-Cañamás A., Soto-Huelin B., Mitroi D. N., Sánchez-Redondo S., Martínez-Sáez E., Venero C., Peinado H., Ledesma M. D., Lipid-induced lysosomal damage after demyelination corrupts microglia protective function in lysosomal storage disorders. EMBO J. 38, e99553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papadopoulos C., Meyer H., Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr. Biol. 27, R1330–R1341 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Nichols R. J., Dzamko N., Morrice N. A., Campbell D. G., Deak M., Ordureau A., Macartney T., Tong Y., Shen J., Prescott A. R., Alessi D. R., 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 430, 393–404 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greggio E., Zambrano I., Kaganovich A., Beilina A., Taymans J.-M., Daniëls V., Lewis P., Jain S., Ding J., Syed A., Thomas K. J., Baekelandt V., Cookson M. R., The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 283, 16906–16914 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang B., Hubber A., McDonough J. A., Roy C. R., Scidmore M. A., Carlyon J. A., The Anaplasma phagocytophilum-occupied vacuole selectively recruits Rab-GTPases that are predominantly associated with recycling endosomes. Cell. Microbiol. 12, 1292–1307 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rzomp K. A., Scholtes L. D., Briggs B. J., Whittaker G. R., Scidmore M. A., Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect. Immun. 71, 5855–5870 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lam S. S., Martell J. D., Kamer K. J., Deerinck T. J., Ellisman M. H., Mootha V. K., Ting A. Y., Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sherer N. M., Lehmann M. J., Jimenez-Soto L. F., Ingmundson A., Horner S. M., Cicchetti G., Allen P. G., Pypaert M., Cunningham J. M., Mothes W., Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4, 785–801 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Conrad K. S., Cheng T.-W., Ysselstein D., Heybrock S., Hoth L. R., Chrunyk B. A., am Ende C. W., Krainc D., Schwake M., Saftig P., Liu S., Qiu X., Ehlers M. D., Lysosomal integral membrane protein-2 as a phospholipid receptor revealed by biophysical and cellular studies. Nat. Commun. 8, 1908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bleck C. K. E., Kim Y., Willingham T. B., Glancy B., Subcellular connectomic analyses of energy networks in striated muscle. Nat. Commun. 9, 5111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabb2454/DC1