

Cell populations can switch strategies between bet-hedging and adaptive tracking using only posttranslational processes.
Abstract
In the face of uncertainty, cell populations tend to diversify to enhance survival and growth. Previous studies established that cells can optimize such bet hedging upon environmental change by modulating gene expression to adapt both the average and diversity of phenotypes. Here, we demonstrate that cells can tune phenotypic diversity also using posttranslational modifications. In the chemotaxis network of Escherichia coli, we find, for both major chemoreceptors Tar and Tsr, that cell-to-cell variation in response sensitivity is dynamically modulated depending on the presence or absence of their cognate chemoeffector ligands in the environment. Combining experiments with mathematical modeling, we show that this diversity tuning requires only the environment-dependent covalent modification of chemoreceptors and a standing cell-to-cell variation in their allosteric coupling. Thus, when environmental cues are unavailable, phenotypic diversity enhances the population’s readiness for many signals. However, once a signal is perceived, the population focuses on tracking that signal.
INTRODUCTION
A central question in cell biology is how a population of cells deals with an ever-changing environment. A canonical paradigm for cellular responses to environmental challenges is the genetic switch, perhaps best exemplified by the lac operon (1), where cells sense changes in environmental factors and respond by changing gene expression. The response time scale of this strategy is limited by that of transcription and translation (~1 hour), leaving cells vulnerable to rapid fluctuations in the environment. A contrasting strategy that allows cells to cope with uncertain or rapidly changing environments is bet hedging, where cell populations diversify their phenotypes even within stable environments, by exploiting inherent stochasticity in cellular processes (2–6). Bet hedging allows subpopulations of cells to be prepared in advance, by maintaining a heterogeneous distribution of phenotypes matched to the repertoire of environments they might encounter in the future. Although genetic switching and bet hedging provide contrasting survival strategies with distinct advantages, they are not mechanistically exclusive. Bacteria can control the degree of phenotypic diversity in an environment-dependent manner by dynamically modulating gene expression noise (7–9). Here, we demonstrate that cells can modulate their phenotypic diversity even in the absence of gene expression changes through posttranslational processes, thus implementing fast control of phenotypic diversity.
The chemotaxis signaling pathway of Escherichia coli detects and responds to temporal changes in the extracellular concentrations of chemoeffector molecules through receptor-kinase complexes consisting of thousands of interacting two-state chemoreceptor proteins and kinases (6, 10). An adaptation mechanism, mediated by methylation and demethylation of the chemoreceptors, modulates the sensitivity of the system. Like in many other sensory systems in biology (11–13), bacteria respond to relative changes in the signal (14), thus following the Weber-Fechner’s law of psychophysics (15–17) and fold-change detection (FCD) (18, 19). The wealth of quantitative data that has been collected for this system through population-averaged measurements (14, 20–23) provides a powerful foundation for examining how individual cells differ from the average in their sensory response and whether and how such diversity is modified upon adaptation.
To quantify the sensitivity of individual cells in the presence of different background-stimulus levels, we combined single-cell fluorescence resonance energy transfer (FRET) measurements of the chemotaxis signaling activity (24, 25) with a microfluidic system for fast stimulus modulation. We found that in the absence of any background signal, the individual cells’ sensitivities are distributed over about one decade of concentration, but upon adaptation to a background signal, the distribution of sensitivities narrows down to about one tenth of a decade, thus focusing the population’s sensitivities to the relevant signal range. Combining experiments and mathematical analyses, we show how the population of cells exploits a standing variation in the degree of allosteric receptor coupling and the environment-dependent covalent modification of the receptors to tune the diversity in signaling sensitivities, which emerges in a class of allosteric models of two-state receptor activity. Crucially, this modulation of sensory diversity does not require any changes in the expression level of proteins, and hence, this mechanism can operate rapidly and in the absence of growth. Rather, it is a network-level property that arises through an adaptive change in the nonlinear mapping between molecular states and sensory phenotype.
RESULTS
High-throughput measurements of individual cell responses reveal substantial phenotypic and temporal variability
To quantify the sensitivity of the chemotaxis system in individual E. coli cells, we stimulated them with short pulses of α-methylaspartate (MeAsp)—a nonmetabolizable analog of the chemoattractant aspartate (26)—while monitoring the output of the signaling pathway using an in vivo single-cell FRET measurement of the activity of the kinase CheA (Fig. 1) (24, 25). To measure “instantaneous” responses of cells without confounding effects due to adaptation, we developed a polydimethylsiloxane (PDMS)–based microfluidic device capable of fast (~0.1 s) switching between stimulus levels (see Materials and Methods, Fig. 1A, and fig. S1), nearly 100-fold faster than the cells’ adaptation time scale upon a small stimulus, which is on the order of 10 s (20). Cells grown to mid-exponential phase [optical density (OD) = 0.46 ± 0.01] were washed in motility buffer and gently loaded in the device where they attached to the surface of the coverslip. We then subjected the cells to a sequence of eight identical subsaturating step stimuli presented over zero background while measuring the FRET level in each individual cell (~100 cells per experiment) (Fig. 1B). To avoid sampling highly correlated responses from a single cell due to the relatively slow temporal fluctuation in the kinase activity [correlation time ~12 s; (25)], measurements were conducted over >100 s with 15-s intervals between consecutive stimuli. A saturating step of MeAsp (>0.5 mM) was used to determine the FRET level corresponding to zero kinase activity at the beginning of each measurement (Fig. 1B). The FRET level after subtracting this zero-kinase level, hereafter called the FRET signal, is proportional to the kinase activity [see Materials and Methods; (21)].
Fig. 1. Statistics of signaling variability from single-cell FRET with fast stimulus modulation.
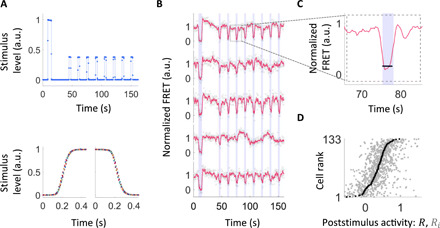
(A) Fast and precise control of input stimuli within our bespoke microfluidic device. Top: Temporal profile of the ligand stimulus within the device, measured using a fluorescent dye. Our stimulus protocol involves one large stimulus followed by eight subsaturating step stimuli. Bottom: Superimposing multiple stimulus time series (each in a different color) demonstrates fast and highly reproducible relaxation for both steps up (left) and steps down (right). For a drawing and more details of the device, see fig. S1. (B) Responses are highly variable both across isogenic cells from the same growth culture and over time within the same cell. Response time series (FRET signal normalized by its steady-state level) for 5 representative cells out of the 133 measured in a single experiment are shown. Blue shading indicates times at which MeAsp step stimuli were applied (4 μM, except the first stimulus, which was 0.5 mM). Gray circles indicate FRET response, and red lines indicate its moving average with a 1.5-s window. (C) Poststimulus activity is defined as the median FRET signal level (black line) during the 3-s step stimulus (blue shading) relative to the steady-state FRET level. (D) Summary of response variability upon 4 μM MeAsp steps for all 133 cells measured in the experiment of (B). All responses (poststimulus activities) Ri (light gray) upon repeated application of identical steps are shown for every measured cell, sorted by rank of their median response R (dark gray). Note that Ri and R can take negative values due to measurement noise (fig. S2). The cumulative distribution of median response (traced out by the R point series) is broad, indicating extensive diversity across cells. a.u., arbitrary units.
The instantaneous response of a cell to a step stimulus was quantified by the poststimulus activity defined as the FRET signal relative to the steady state: Ri = Fi/Fss, where Fi is the median of the FRET signal over the 3 s during the ith step stimulus and Fss is the steady-state FRET signal, defined as the average over the entire time series except the time points during and right after the step stimuli (see Materials and Methods; Fig. 1C). The mean level of measurement noise for each individual response was 17% of the steady-state FRET signal (fig. S2), and the response distributions were stationary during the measurements (fig. S3). Sorting cells by their median poststimulus activity reveals substantial cell-to-cell variability (Fig. 1D), consistent with previous reports of phenotypic variability in this system (6, 25, 27–30). Within each cell, we also observe large variations in the responses to identical stimuli (Fig. 1D, light gray dots), consistent with previous reports of temporal (behavioral) variability in individual cells adapted to a constant environment (6, 24, 25, 31). We ruled out cell cycle phase as a source of the cell-to-cell variation in kinase responses, as the latter demonstrated no correlation with cell length (fig. S4).
Determination of the distribution of sensitivities
The standard method for determining the sensitivity of a signaling pathway is to fit the sigmoidal to dose-response measurements and determine 1/K1/2 as the sensitivity of the cell. This approach has been used to quantify the dose response of populations of E. coli cells using FRET-based methods (21, 23, 32) and non-adapting single cells (25). However, this approach becomes impractical for measuring the response of single cells in the presence of adaptation because of the limited photon budget in single-cell FRET (25). Therefore, we devised an alternative strategy for determining the distribution of K1/2 within a cell population without the need to measure dose-response curves from individuals (Fig. 2).
Fig. 2. Determining the distribution of K1/2 across cells without dose-response measurements.
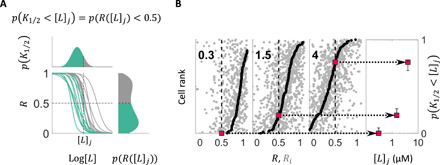
(A) Principle of extracting the K1/2 distribution, p(K1/2), without dose-response measurements. K1/2, defined as the stimulus level that yields half-maximal poststimulus activity (R = 0.5), is typically determined by measuring dose-response curves (middle), which can vary from cell to cell. Here, we instead measure the distribution of R upon a stimulus of magnitude [L]j, p(R([L]j)), because the fraction of cells with K1/2 smaller than a given stimulus magnitude [L]j (p(K1/2 < [L]j); colored at the top) is equal to the fraction of cells whose poststimulus activity R([L]j) is less than one-half (p(R([L]j) < 0.5); colored at the bottom). (B) By repeating experiments of the type depicted in Fig. 1 at different stimulus step sizes [L]j, we build up the cumulative distribution of K1/2, p(K1/2 < [L]j). Each of the three panels on the left shows the summary of responses (as in Fig. 1D) for an experiment with a different [L]j (added MeAsp concentration, given in μM by bold-faced numbers within panels), where sorting cells by their median poststimulus activity R (dark gray dots) provides the cumulative distribution of R, p(R([L]j) < r), corresponding to the fraction of cells whose response to [L]j is smaller than r (0 ≤ r ≤ 1). Using the identity illustrated in (A), the cumulative distribution of K1/2 (p(K1/2 < [L]j); rightmost) can be constructed by reading off values for p(R([L]j) < r) at r = 0.5 for each applied stimulus level [L]j. Error bars in the right show 95% bootstrap CIs.
To determine the distribution of K1/2, we exploited a simple identity relating the distribution of K1/2 to that of R, the (median) poststimulus activity of individual cells, which states that the fraction of cells with K1/2 smaller than a given stimulus magnitude [L] is equal to the fraction of cells whose (median) poststimulus activity R([L]) is less than one-half (Fig. 2A)
| (1) |
Thus, from the distributions of the within-cell median poststimulus activities, one can construct the cumulative distribution function (CDF) of K1/2 of the population by determining, for each step stimulus intensity [L], the relative rank of the cell whose median poststimulus activity is 0.5 (Fig. 2B). Equation 1 is valid for any monotonic dependence of R on [L] and does not assume any specific steepness of a cell’s response curve or variation of it across cells.
Diversity in response sensitivity is modulated by the environment without changes in gene expression
Following this approach, we first determined the median of the poststimulus activity of individual cells adapted to a uniform environment with no MeAsp in the background, by stimulating cells with step stimuli that ranged from 0 to 30 μM MeAsp (Fig. 3A). From these data, we extracted the distribution of K1/2 (inverse sensitivity), which was well approximated by a log-normal distribution (see Materials and Methods; Fig. 3, B and C). We found that in zero background, the sensitivity of individual cells to MeAsp was distributed over a wide range covering about one decade (~1 μM < K1/2 < ~10 μM).
Fig. 3. K1/2 distributions at different background concentrations reveal diversity tuning of response sensitivity.
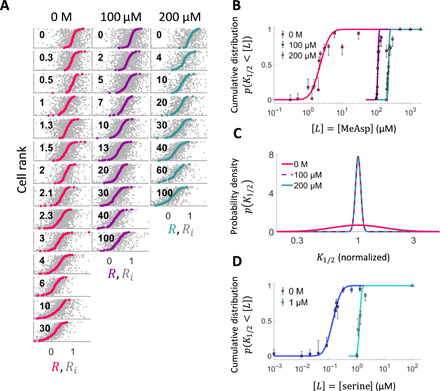
(A) Summary of responses to step stimulation by MeAsp (gray dots: response to individual steps Ri, colored dots: median response of each cell R). Background concentration ([L]0) and step size (Δ[L]) are shown in μM at the top and within each panel, respectively. Cells are sorted by their median response. (B) Cumulative distribution of K1/2, p(K1/2 < [L]), of responses to MeAsp in cells adapted to three different background concentrations of MeAsp, [L]0 = {0,100,200} μM, constructed from the data in (A) through the procedure outlined in Fig. 2. Curves represent fits by log-normal distributions. Error bars are 95% bootstrap CIs. The concentrations of stimuli used to define saturating responses are indicated by the triangles. (C) The distributions of K1/2 computed from the fits in (B) reveal that diversity in K1/2 is strongly attenuated upon adaptation to both 100 and 200 μM MeAsp. Note that in this panel, the distribution at each background concentration is centered by normalizing K1/2 by the mode of the distribution to facilitate visual comparison. (D) Cumulative distribution of K1/2, p(K1/2 < [L]), of responses to serine in cells adapted to different background concentrations of serine, [L]0 = {0,1} μM.
Given the well-characterized adaptation to ambient chemoattractant concentration at the population level, we wondered whether and how the single-cell distribution of sensitivities could be affected by adaptation to a constant nonzero background of MeAsp. Consistent with the population-level FRET measurements (14, 21), the average of the K1/2 distribution shifted with the background stimulus level due to sensory adaptation when cells were adapted to 100 μM MeAsp before step stimulation (Fig. 3B). Unexpectedly, the diversity in response sensitivity across cells also changed drastically, with the K1/2 distribution becoming much narrower upon adaptation (Fig. 3C). A similar collapse in the K1/2 distribution width was found to occur for cells adapted to a higher (200 μM) background of MeAsp. We further determined that this sensory diversity tuning is not specific to the MeAsp receptor Tar, as the distribution for serine, the cognate ligand of the other major chemoreceptor Tsr, demonstrated a similar collapse in width upon adaptation (Fig. 3D and fig. S5). Thus, the environment-dependent tuning of response diversity is not specific to a single receptor species and appears to be a general property of the bacterial chemotaxis network.
Recent studies have shown that cell populations can control the level of phenotypic diversity in an environment-dependent manner by modulating the variance of the protein abundance distributions (7–9). Here, by contrast, experiments were carried out under conditions in which neither the cognate receptors nor any other protein can be synthesized (due to auxotrophic limitation; see Materials and Methods). The observed tuning of sensory diversity must therefore be attributable to a mechanism that involves posttranslational processes rather than changes in gene expression.
Response diversity arises from, but is not tuned by, variation in receptor coupling
To understand the molecular mechanism underlying this adaptive tuning of diversity in cell response sensitivities (Fig. 3), we turned to modeling. The receptor-kinase complexes of the chemotaxis system in E. coli and other species are arranged in hexagonal arrays of trimers of dimers that respond cooperatively to signals (33, 34). The activity of such clusters can be modeled using an extension of the Monod-Wyman-Changeux (MWC) model of allostery (35) and has been shown to agree with a large body of experimental data (20, 23, 36–42). In this model (Fig. 4A and Supplementary Text), allosteric interactions between n coupled receptors form “signaling teams” within which all n receptors (and associated kinase molecules) share the same activity states (active or inactive). The free energy difference between the two receptor states is determined not only by the ligand concentration [L] (analogous to the oxygen concentration in the classical MWC model for hemoglobin) but also by the average methylation level m of receptors. Because of feedback from downstream adaptation enzymes, the value of m at steady state, in turn, depends on the background stimulus level [L]0, i.e., m = m([L]0). Kinase activity upon a step change in input from a given background [L]0 to another stimulus level [L] depends on two parameters n and m*, where m* is the receptor methylation level in the absence of ligand stimulus. Values of the parameters n and m* for E. coli chemoreceptors have been constrained by a large body of population FRET data (20, 21, 36, 37, 39, 42) and have been shown to vary as a function of expression level ratios between key chemotaxis signaling proteins (23, 25). Given that these ratios are affected by stochastic gene expression, the values of n and m* can vary across individual cells of the population (25), whereas values of other biochemical parameters (e.g., the dissociation constants of the receptors) are intrinsic to the structure of relevant proteins, which can be assumed invariant across isogenic populations of cells (see Supplementary Text).
Fig. 4. Adaptive MWC model for chemoreceptors reveals origin of sensory diversity and its tuning mechanism.
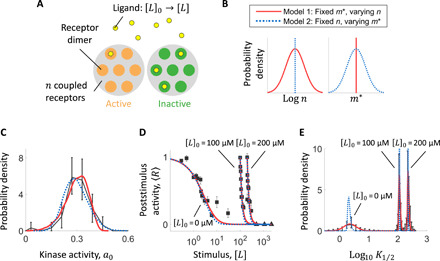
(A) Schematic for allosteric MWC model of the receptor kinase complex. The effective number of coupled receptor dimers n affects the response of kinase activity a upon a step change in ligand concentration from [L]0 to [L], through the expression a = (1 + exp (f(n, m*, [L]0, [L])))−1, where m* is the methylation level of the receptors in the absence of ligand. Both n and m* can vary across cells due to differences in gene expression. (B) Two limiting cases of cell-to-cell variation in the model parameters. Model 1 (red solid lines): m* is fixed, but n varies across cells. Model 2 (blue dotted lines): n is fixed, but m* varies across cells. (C to E) Fits of models 1 and 2 to the distribution of steady-state kinase activity a0 (C), population-averaged dose-response curves (D), and distribution of logK1/2 (E). Black corresponds [in (C) and (D)] to measured data and [in (E)] to probability density computed from model fits to cumulative distributions (see fig. S7). Error bars represent 95% bootstrap CIs.
The observed diversity in K1/2 values might thus reflect cell-to-cell differences in the value of n, m*, or both. To discriminate between these possibilities, we first considered two models that represent limiting cases (Fig. 4B and fig. S6). In model 1, the value of m* is fixed across cells, but the value of n varies across the population. In model 2, n is fixed and m* varies. Both models could fit the distribution of steady-state kinase activity (Fig. 4C) previously measured in isogenic populations (25), as well as the population-averaged dose-response data (Fig. 4D). However, the two models yield contrasting predictions for the underlying diversity in single-cell sensitivity. Whereas model 1 with variability only in the size of receptor coupling n demonstrated a tuning of K1/2 diversity upon adaptation to MeAsp in close agreement with the experimental data, model 2 with variability only in m* demonstrated little or no diversity tuning, with the width of the K1/2 distribution remaining approximately constant, with or without adaptation to MeAsp (Fig. 4E). A more general model in which both n and m* vary across cells also yielded consistent results: Fitting with this model yielded a broad distribution for n (CV(n) = 0.41) but a very narrow one for m* (CV(m*) = 0.02) (fig. S8). In a similar manner, the observed diversity tuning of response sensitivity to serine stimuli could also be explained by the variation in the number of coupled Tsr receptors while keeping m* fixed (fig. S5).
Thus, MWC modeling implicates as the predominant source of response diversity a single parameter, the degree of allosteric coupling n for the receptor cognate to the applied ligand stimulus. The model yields excellent fits to the changes in the shape of the K1/2 distribution p(K1/2) upon adaptation without assuming any changes in the underlying parameter distribution p(n). Consistently, further model-based analysis of the dose-response data (fig. S9 and Supplementary Text) did not detect significant changes in the distribution p(n) over the different backgrounds [L]0 across which diversity tuning (i.e., a change in the width of p(K1/2)) is observed. These modeling results thus suggest that while variation in n is the key ingredient for response diversity, the posttranslational mechanism that accounts for adaptive tuning of that diversity does not require a change in the degree of variation in n across cells.
Posttranslational receptor modification implements an environment-dependent “diversity switch”
To pinpoint the mechanism responsible for diversity tuning with the MWC model, we focused on the simplest variant (model 1) that reproduced the observed diversity tuning assuming cell-to-cell variation in only a single parameter, n. We first investigated how diversity in K1/2 (as quantified by its coefficient of variation, CV(K1/2)) depends on the adapted state background [L]0 in this model while holding fixed the distribution p(n). CV(K1/2) demonstrated two plateaus: At low [L]0, K1/2 is highly variable with CV(K1/2) approaching 0.5, whereas at high [L]0, diversity is strongly suppressed with CV(K1/2) attenuated by nearly an order of magnitude (Fig. 5A, gray curve). Thus, diversity in response sensitivity demonstrates two regimes: high diversity at low [L]0 and low diversity at high [L]0.
Fig. 5. Prediction and experimental corroboration of a diversity switch.
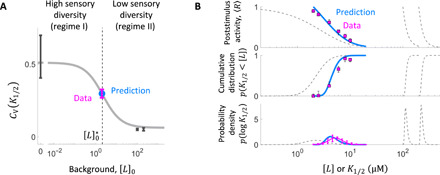
(A) The adaptive MWC model predicts a switch from high to low sensory diversity as the background stimulus level [L]0 is increased from zero. The coefficient of variation of K1/2 (CV(K1/2)) at [L]0 = 0 μM and [L]0 = {100,200} μM MeAsp (black points) falls within the high- and low-diversity regimes, respectively, predicted by model 1 (gray curve). To test the predicted transition regime, we measured the K1/2 distribution at the crossover point (dotted line). The measured CV (magenta) for cells adapted to is in excellent agreement with the model prediction (blue point). All CV values were computed from parameters of the log-normal distributions fitted to the CDF of K1/2 (fig. S12). Error bars were computed by propagating the SE of the parameters. (B) The model accurately predicts the full distribution of K1/2 diversity and the population-level response at . Model prediction (blue) and experimental results (magenta) for the population dose-response curve (top), CDF (middle), and probability density function (PDF, bottom) of K1/2 at [L]0 = 2 μM MeAsp. Model parameters were constrained only by the data at [L]0 = {0,100,200} μM data (Fig. 4), with no additional fit parameters for the [L]0 = 2 μM data. Model behavior at [L]0 = {0,100,200} μM backgrounds is shown for reference (gray dashed curves). Error bars represent 95% bootstrap CIs.
A key quantity that determines how the diversity in n affects diversity in K1/2 is the susceptibility of K1/2 with respect to n, defined by the absolute partial derivative ∣∂n log (K1/2)∣. In broad terms, when this susceptibility is high, variation in n contributes strongly to diversity in K1/2; when it is low, the effects of variation in n can be suppressed. We found that the susceptibility ∣∂n log (K1/2)∣ computed using the MWC model (and evaluated at the population mean, n = 〈n〉) also exhibits a decreasing profile as a function of [L]0 with two plateaus (fig. S10), closely mirroring the CV(K1/2) profile (Fig. 5A, gray curve).
The existence of two regimes with contrasting susceptibilities ∣∂n log (K1/2)∣ has been predicted theoretically for chemoreceptor MWC models [fig. S11; (37)]. In this class of models, the methylation-dependent free energy difference between the active and inactive states of ligand-unbound receptors follows a linear relationship fm = − α(m([L]0) − m0), where α corresponds to the free energy per methyl group, and m0 is an offset methylation level at which fm = 0. Because of nonlinearities arising in the allosteric mechanism, the dependence of K1/2 on n changes qualitatively as the methylation level crosses m0 [fig. S11; (37)]. When the methylation level is low (i.e., m([L]0) < m0), K1/2 becomes inversely proportional to n (specifically, K1/2 ∝ KI/n, where the dissociation constant of the inactive receptor KI sets the concentration scale), and therefore, the degree of receptor coupling n strongly affects sensitivity. In this regime, the susceptibility ∣∂n log (K1/2)∣ is thus high, and we can expect cell-to-cell variation in n to cause substantial K1/2 diversity across cells. Conversely, when the methylation level is high (i.e., m([L]0) > m0), K1/2 becomes independent of n (37). In this regime, the susceptibility ∣∂n log (K1/2)∣ is thus low, and we can expect K1/2 diversity to be suppressed. The crossover between the two regimes is set by the offset methylation level m0 at which the free-energy contribution from covalent modification feedback vanishes (i.e., fm = − α(m([L]0) − m0) = 0). Thus, the drastic difference in diversity we found at zero and high (100 and 200 μM) background concentrations (Fig. 5A, black points) could be explained by the switch from high to low susceptibility ∣∂n log (K1/2)∣ as the adapted-state covalent modification level m([L]0) increases beyond m0.
The success of the adaptive MWC model in explaining the observed response diversity motivated us to further test its predictive power: Given the model calibrated by data obtained so far in the high- and low-diversity regimes, how accurately could we predict K1/2 diversity at an as-yet unmeasured background concentration? Using experimentally determined values for the parameters KI, α, m0 (14, 20), and m*, the crossover background concentration at which the adapted state modification level reaches m0 (i.e., ) is readily computed from the model (see the Supplementary Materials) as μM MeAsp (Fig. 5A, vertical dashed line), at which the model predicts an intermediate level of K1/2 diversity (Fig. 5A, blue point). We thus opted to measure the distribution of K1/2 at a background of 2 μM (Fig. 5 and fig. S12). The results are in excellent quantitative agreement with model predictions not only at the level of CV(K1/2) (Fig. 5A, magenta point) but also for the entire shape of the distribution (Fig. 5B, middle and bottom) and population-level response (Fig. 5B, top).
Thus, the adaptive MWC model of chemoreceptors provides not only a mechanistic explanation for but also predictive power over the observed diversity tuning in the bacterial chemotaxis system, in which posttranslational receptor modification mediates the transition between two regimes of sensory diversity: When the background stimulus level is low (regime I), receptor modification falls below m0 and diversity is augmented; when the background stimulus is high (regime II), modification exceeds m0 and response diversity is attenuated.
DISCUSSION
By combining a novel microfluidic device with a single-cell FRET assay, we characterized the diversity of chemoeffector responses and its dependence on background stimulus conditions within isogenic populations of E. coli. We found that the width of the sensitivity distribution is strongly modulated in an environment-dependent manner under experimental conditions (auxotrophic limitation) that do not permit gene expression changes. Mathematical modeling provided remarkably accurate predictions and a mechanistic explanation for this diversity tuning that requires only a change in the posttranslational modification of signaling proteins. Below, we discuss the molecular requirements and functional implications of this novel mechanism for diversity tuning, as well as the significance of its implementation without changes in gene expression.
The molecular source of response diversity and its implications for sensory preference
It has long been known that the intracellular variable modulating bacterial chemotactic sensitivity upon sensory adaptation is the covalent modification level m of chemoreceptors (43, 44). Naively, therefore, one might expect the diversity in sensitivity we observed across cells to be the result of cell-to-cell differences in this key internal variable. Our MWC model analysis revealed, however, that the main contribution to response diversity comes not from m but instead from n, the degree of chemoreceptor coupling. While n is a coarse-grained parameter that can be affected by both the size and composition of receptor clusters, the likely dominant contribution to its variation is the expression-level ratio between the two major receptor species Tar and Tsr, which has been shown to vary strongly across cells (45). A recent study in adaptation-deficient cells found that the diversity in dose-response parameters (K1/2 and the Hill coefficient, H) across cells could be largely explained by variation in this ratio (25). Varying the Tar/Tsr ratio determines the direction of chemotactic cell migrations when subjected to two conflicting chemoeffector gradients (46)—whereas cells with high Tar/Tsr ratios migrate preferentially toward MeAsp (the cognate ligand for Tar), cells with low Tar/Tsr ratios do so toward serine (the cognate ligand for Tsr). Thus, the diversity in response sensitivity we observed in our FRET experiments can be interpreted to reflect diversity in sensory preference, which could, in turn, significantly affect population-level chemotactic performance in environments that present multiple stimuli.
Diversity tuning enables transitions between bet-hedging and tracking response strategies
Optimal strategies for biological adaptation depend on accessible information about the environment (47, 48). When environmental cues provide sufficiently accurate information, “tracking strategies” that accordingly adjust phenotypes can provide an advantage. When environmental cues do not carry sufficient information, bet-hedging strategies can provide “readiness” of different individuals to different environments.
For sensory adaptation in bacterial chemotaxis, the zero-background condition is singular in that there is no information about the nature of future environmental signals. E. coli cells express five types of chemoreceptors (Tar, Tsr, Tap, Trg, and Aer) that sense a variety of stimuli (10). Given that the relative expression levels of these receptors are highly variable across cells (45) and that different receptor species are mixed within clusters (49), the combinations of the effective degree of coupling for each receptor type realized in a cell population are numerous.
The switch-like transition in K1/2 diversity we observed (Fig. 5) enables cells to diversify their response sensitivities (and hence sensory preference) for different ligands when all ligand concentrations are near zero and uncertainty is at a maximum (Fig. 6A). This could be beneficial in improving the readiness of the isogenic population for many future signals—a sensory bet-hedging strategy. By contrast, once a relevant signal is detected, such as a gradient of aspartate, cell-to-cell variability in sensitivity can lead to detrimental desensitization (when sensitivity is too low) or sensory saturation (when sensitivity is too high) that precludes effective tracking of the signal as cells climb the gradient by chemotaxis. Our experiments revealed that the width of the distribution of sensitivities is markedly reduced upon adaptation to higher ligand concentrations, therefore focusing the population on tracking that signal. In summary, this novel mechanism of sensory diversity tuning could enable an isogenic population to be ready for any signal when the environment is uncertain but switch to tracking a specific signal once it is detected.
Fig. 6. Posttranslational modulation of sensory diversity allows cell populations to switch rapidly between bet-hedging and tracking strategies.
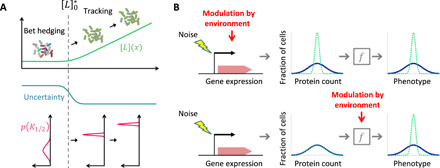
(A) Diversity tuning in chemotactic response sensitivity. In environments with low background signals below the crossover level (), uncertainty about future signals is high, and the population diversifies its sensory preference. In environments with high background signals above the crossover level (), the population can attenuate its sensory diversity and switch to tracking the perceived signal. (B) Phenotypic diversity can be tuned by environmental modulation of either gene expression or posttranslational processes. Top: Gene expression–dependent diversity tuning involves modulation of stochastic gene expression in response to environment changes (7–9), leading to different distributions of expressed protein counts across cells in different environments. This mechanism can tune phenotypic diversity without environmental modulation of posttranslational expression-phenotype mappings (represented here by the box labeled by f). Bottom: By contrast, the posttranslational diversity tuning mechanism we found in this study involves environmental modulation of the expression-phenotype mapping (f, implemented in bacterial chemotaxis by covalent modification of allosteric chemoreceptors). This mechanism requires no environmental modulation of gene expression and hence can achieve rapid tuning of phenotypic diversity.
Sensory diversity breaks the trade-off between response gain and response range
Another challenge unique to the zero-background signal condition is that there is no information about the magnitude of the future signal. In dealing with the uncertainty in the signal strength, two key performance measures of a cell population as a sensory system are the width of the range of input signals over which it can respond (response range; fig. S13A) and the degree to which the input signal is amplified at the output level (gain; fig. S13A). For a homogeneous cell population with a sigmoidal stimulus-response curve, it is known that there is an inherent trade-off between these two performance measures (50, 51). A large response range requires a shallow response curve, but this inevitably reduces the response gain, and vice versa (fig. S13B). To understand whether and how the diversified sensitivity contributes to the performance of a cell population, we computed the performance measures of gain and response range in the zero-stimulus background condition using the MWC model and compared a cell population with diversity in the number of coupled receptors n to a hypothetical homogenous population (fig. S13B). As expected from the diversity in the sensitivity due to the variation in n, the diverse population exhibits a broader response range than the homogeneous population (fig. S13B). On the other hand, each individual cell in the diverse population maintains a high response gain (fig. S13B), reflecting the insensitivity of the gain to the variation in n in the low-background signal regime of the coupled two-state receptors (37). Thus, a population with diverse sensitivity can outperform homogeneous populations in dealing with the unpredictability associated with the zero background.
Pleiotropic functionality of allosteric signaling regimes
As noted above, the high- and low-diversity regimes we found here were identified in an earlier theoretical study as regimes I and II, respectively, of cooperative chemosensing (37). In that study, it was found that cooperativity (i.e., n > 1) extends the dynamic range of sensing to lower concentrations due to the 1/n scaling of K1/2 in regime I, whereas it increases signal gain by increasing the steepness of response (i.e., the Hill coefficient, H > 1) in regime II. Subsequent studies found that when E. coli cells are adapted to higher concentrations in regime II ([L]0 ≫ KI), responses to step changes (39) and time-varying signals (18, 19) depend only on relative changes of chemoeffector concentrations (14). This property of FCD provides a robust sensory strategy in many natural contexts where absolute signal intensities tend to carry less information than relative contrast (19). By contrast, in regime I of cooperative sensing (), the sensory response becomes proportional to the absolute change in chemoeffector input. Although this connection between the cooperative sensing regimes and FCD is intriguing, we note that the diversity tuning we found here is not causally related to FCD. One can construct a network model that demonstrates the linear-response/FCD transition but does not demonstrate diversity tuning (see the Supplementary Materials and fig. S14). Evidently, cooperativity in E. coli chemoreceptors provides multiple benefits in sensory performance: increased dynamic range/signal gain, FCD, and diversity tuning of response sensitivity. The molecular parameters that define cooperativity and the resulting signaling regimes are thus likely under pleiotropic selection (52) and would provide fertile ground for future studies of trade-offs and optimality (53) in the design of allosteric signaling systems.
Modulation of expression-to-phenotype maps as a general mechanism for adapting phenotypic diversity
Recent pioneering studies have provided a handful of examples of how bacteria can modulate phenotypic diversity in an environment-dependent manner, by changing the distribution of protein abundance across cells [Fig. 6B, top; (7–9)]. By contrast, the diversity-tuning mechanism we found here is implemented by posttranslational processes (Fig. 6B, bottom). The mechanism hinges on a nonlinear relationship (represented by the box with label f in Fig. 6B, bottom) between the phenotype of interest (here, the response sensitivity or its inverse, K1/2), a gene expression–dependent parameter (here, the degree of receptor coupling, n), and a posttranslational variable that varies in response to the environment (here, the covalent modification state of chemoreceptors, m).
An important difference between gene expression–dependent and posttranslational mechanisms of diversity tuning lies in the achievable speed for environment-dependent modulation of diversity. Whereas the former is limited by the time scale of gene expression (typically measured in minutes), the latter can be implemented by much faster biochemical processes (the fastest covalent modifications occur on subsecond time scales). Another significant difference is in biochemical costs and requirements: Gene expression–dependent diversity tuning requires synthesis of new proteins and hence may be rendered useless under nutrient-limited conditions, whereas the posttranslational mechanism studied here could be operational in any environment that supports the required type of covalent modification (here, methylation). Thus, posttranslational diversity tuning could be advantageous when cell populations need to adapt to fast-switching environments such as the gut (54, 55) and/or under poor nutrient conditions such as marine environments (56). Given the ubiquity of nonlinear functions throughout cellular biochemistry, we expect that posttranslational diversity tuning could play a role in the survival of a broad range of cell types in a variety of biological contexts.
MATERIALS AND METHODS
Strains and plasmids
The strain used is a derivative of E. coli K-12 strain RP437 (HCB33). The FRET acceptor-donor pair (CheY-mRFP and CheZ-mYFP) is expressed in tandem from plasmid pSJAB106 (25) under an isopropyl-β-d-thiogalactopyranoside (IPTG)–inducible promoter with induction level of 50 μM IPTG. The glass-adhesive mutant of FliC (FliC*) was expressed from a sodium salicylate (NaSal)–inducible pZR1 plasmid (25) with induction level of 3 μM NaSal. We transformed the plasmids in VS115, a cheY cheZ fliC mutant of RP437 [a gift of V. Sourjik; (25)], referred to as “wild-type” strain in the main text.
Microfluidic device design, fabrication, and deployment
Microfluidic devices were constructed from PDMS on a 24 mm × 60 mm cover glass (#1.5) following standard soft lithography protocols (57). Briefly, the master molds for the device were created with a positive photoresist (AZ 9260, MicroChemicals) on a silicon wafer using a standard photolithography technique (57). Approximately 20-μm-high master molds were created. To fabricate the device, the master molds were coated with a 5-mm-thick layer of degassed 10:1 PDMS-to-curing agent ratio (Sylgard 184, Dow Corning). The PDMS layer was cured at 80°C for 1 hour and then cut and separated from the wafer, and holes were punched for the inlets and outlet. The PDMS device was then bonded to a cover glass. The PDMS was cleaned with transparent adhesive tape (Magic Tape, Scotch) followed by rinsing with (in order) isopropanol, methanol, and Millipore-filtered water. The glass was rinsed with acetone, isopropanol, methanol, and Millipore-filtered water. The PDMS device was tape-cleaned an additional time before the surfaces of the device and coverslip were treated with a plasma generated by a corona treater. Then, the PDMS device was laminated to the coverslip and then baked at 80°C overnight.
Sample preparation in the microfluidic device was conducted as follows: Of the five inlets of the device (fig. S1A), four inlets are connected to reservoirs (liquid chromatography columns, C3669; Sigma-Aldrich) filled with motility media containing various concentrations of MeAsp through polyethylene tubing (Fine Bore Polythene Tubing, 0.58 mm inside diameter, 0.96 mm outer diameter, Smiths Medical). Another inlet (located at the extremity) is connected to a reservoir filled with motility media containing fluorescein, which enables us to observe the flow of the solution and allows us to calibrate the pressure applied to the reservoirs before each experiment. The tubing was connected to the PDMS device through stainless steel pins that were directly plugged into the inlets or outlet of the device. Cells washed and suspended in motility media were loaded in the device from the outlet of the device and attached to the glass surface in the microfluidic device by reducing the flow speed inside the chamber. The pressure inside the reservoirs connected to the inlets was controlled by computer-controlled solenoid valves (MH1, Festo) that promptly switches between atmospheric pressure and higher pressure introduced from a source of pressurized air. The pressure applied to the reservoirs was adjusted before each experiment by observing the flow of the fluorescent solution under the microscope so that all stimulus solutions are delivered to imaging areas. The FRET measurements were conducted at three different positions in a microfluidic device, and an identical stimulus protocol was repeated at every position.
In vivo single-cell FRET microscopy
Single-cell FRET microscopy and cell culture were carried out essentially as described previously (25). In brief, cells from a saturated overnight culture were grown to OD 0.45 to 0.47 in 10 ml of tryptone broth (1% bacto-tryptone and 0.5% NaCl) in the presence of ampicillin (100 μg/ml), chloramphenicol (34 μg/ml), 50 μM IPTG, and 3 μM NaSal. Cells were collected by centrifugation (5 min at 5000 rpm) and washed twice with motility media [10 mM KPO4, 0.1 mM EDTA, 1 μM methionine, and 10 mM lactic acid (pH 7)] and then resuspended in 2 ml of motility media.
FRET imaging in the microfluidic device was performed using an inverted microscope (Eclipse Ti-E, Nikon) equipped with an oil immersion objective lens (CFI Apo TIRF 60× Oil, Nikon). Yellow fluorescent protein (YFP) was illuminated with a light-emitting diode illumination system (pE-4000, CoolLED, for experiments with MeAsp stimuli, and SOLA SE, Lumencor, for experiments with serine stimuli) through an excitation bandpass filter (FF01-500/24-25, Semrock) and a dichroic mirror (F01-542/27-25F, Semrock), and the fluorescence emission was led into an emission image splitter (OptoSplit II, Cairn) and further split into donor and acceptor channels with a second dichroic mirror (FF580-FDi01-25x36, Semrock) and collected through emission bandpass filters (FF520-Di02-25x36 and FF593-Di03-25x36, Semrock) with a sCMOS (scientific CMOS) camera (ORCA-Flash4.0 V2, Hamamatsu). Red fluorescent protein (RFP) was illuminated in the same way as YFP except that an excitation bandpass filter (FF01-562/40-25 for experiments with MeAsp stimuli and FF01-575/05-25 for experiments with serine; both from Semrock) and a dichroic mirror (FF593-Di03-25x36, Semrock) were used. Additional excitation filter (59026x, Chroma) was used for experiments with serine stimuli. Before each time-lapse measurement, an acceptor image (RFP excitation and RFP emission) and a donor image (YFP excitation and YFP emission) were taken to estimate the RFP expression level and cell volume of each cell used for data analysis. In time-lapse imaging, images were acquired every 0.3 to 0.5 s.
FRET analysis
The FRET level of each cell was calculated essentially in the same way as described previously (21, 25, 32). After flat-field correction of the fluorescent images, fluorescent signals, i.e., donor signal (obtained from donor channel: YFP excitation and YFP emission) and FRET-acceptor signal (obtained from FRET-acceptor channel: YFP excitation and RFP emission), were extracted from the images for each individual cell using an image segmentation technique. The extracted raw fluorescent time series were corrected for bleaching by fitting both donor and FRET-acceptor signals with a biexponential function and dividing out the decay to yield donor signal D(t) and FRET-acceptor signal A(t).
We define the FRET index as the decrease in the donor signal D(t), ΔD (≥0), due to FRET between the donor (mYFP) and acceptor (mRFP1) molecule, normalized by the intensity of donor illumination reaching a cell through the donor excitation filter, νD, and cell volume, Vcell
where νD was extracted from the flat-field image, and Vcell was estimated from the no-binning YFP image. The FRET index was chosen because ΔD/(νDVcell) is proportional to the concentration of active CheA. To show this, we consider the following. First, ΔD can be decomposed as
where εD, EFRET, QD, LD, SD, tDD, and [DA] are respectively the absorption coefficient of donor, the FRET efficiency of the complex, the quantum yield of donor, the throughput of the donor emission light-path, the quantum sensitivity of the camera for donor emission, the exposure time for the donor image, and the concentration of the donor-acceptor complex. Because εD, EFRET, QD, LD, SD, and tDD are all constants once the experimental system is fixed, by introducing C = εDEFRETQDLDSDtDD, we write ΔD as
So, the FRET index FRET(t) is proportional to [DA] or the concentration of CheYp-CheZ complex [CheYp · CheZ]. The concentration of the complex reaches a quasi–steady state on the time scale larger than the time scale of hydrolysis of phosphorylated CheY, CheYp, catalyzed by CheZ [~0.5 s; (22)] due to the balance between phosphorylation and dephosphorylation of CheY. Thus, the following holds
where kA and kZ are respectively the rate constants for autophosphorylation of CheA and that for hydrolysis of CheYp by CheZ, a (0 < a < 1) is the fraction of active CheA, and [CheA]T is the total concentration of CheA (25). Given the conservation equation [CheA]T = [CheA] + [CheAp], the last step of the above equations holds when [CheAp] ≪ [CheA]T. This is achieved when sufficient amount of CheY-RFP and CheZ-YFP is present in the cell as verified previously (25), and therefore, we exclude cells from analysis whose concentrations of CheY-mRFP1 and CheZ-mYFP are low.
Together, the FRET index is
where C′ = CkA/kZ, which is invariant between cells. To increase the signal-to-noise ratio, ΔD can be computed, rather than directly extracting from D(t), as
where r(t) ≡ A(t)/D(t) = r0 + Δr(t); r0 and D0 are the ratio r and donor signal D, respectively, in the absence of FRET, which is obtained by applying saturating stimulus to cells (21); and α = ∣ΔA/ΔD∣ is the absolute value of the ratio of changes in the fluorescent signals due to FRET, which is a constant dependent on a measurement system (21, 25, 32). Fss was defined as the average FRET index excluding time points during and right after stimuli (15 s for a saturating stimuli and 6 s for subsaturating step stimuli). Fi was defined as the median FRET index during a step stimulus (10 time points). Response to each step stimulus Ri was defined as Ri = Fi/Fss.
Measurement noise evaluation
A measured FRET time series FRET(t) can be conceptually decomposed into the “true” signal FRETtrue(t), which is proportional to the concentration of active CheA, and the measurement noise arising from the finite number of photons ξ(t)
Because the true signal is also fluctuating, it is not trivial to estimate the magnitude of the measurement noise in general. However, we can exploit the fact that, when a saturating stimulus is presented, the true signal, and therefore also its variance, goes to zero (21) and hence
Thus, the variance of the FRET time series during a saturating stimulus can be equated with the measurement noise
When evaluating a FRET level upon a stimulus, we used the median value of n (=10) consecutive measurement points to mitigate the contribution of measurement noise. Because the measurement noise is delta correlated, the contribution of measurement noise is . We estimated this quantity by computing the SE of the mean from n consecutive data points during saturating stimuli from each cell and then computed the ensemble average of the value (fig. S2).
Estimation of the distribution of the response constant K1/2
Using the identity p(K1/2 < [L]) = p(R < 0.5) (Fig. 2), the response distributions were converted to the CDF of the response constant K1/2 as schematically shown in Fig. 2A. The error bar of the estimated p(K1/2 < [L]) was obtained by bootstrapping over single responses (95% CI). The data were fitted by the CDF of the log-normal distribution by the weighted least square fitting method. The weights were given by the inverse of the width of the 95% CI except the data points of Δ[L] = 0 (no step stimulus) and Δ[L] = Δ[L]sat (saturating stimulus), which were weighted with an arbitrary high value. Extracted parameters and their SE were (μ, σ) = (0.822 ± 0.069,0.51 ± 0.12) for [L]0 = 0 M, (μ, σ) = (1.659 ± 0.039,0.291 ± 0.027) for [L]0 = 2 μM, (μ, σ) = (4.715 ± 0.012,0.051 ± 0.006) for [L]0 = 100 μM, and (μ, σ) = (5.442 ± 0.014,0.052 ± 0.010) for [L]0 = 200 μM.
Extraction of MWC model parameters
For MeAsp responses, we considered the following three different types of the MWC model, each of which has different variations in the parameter values of m* and n. Model 1: m* is fixed, but n varies. Model 2: m* varies, but n is fixed. Model 3: both m* and n vary. When the parameters are allowed to vary, we assumed the normal distribution for m*, , and the log-normal distribution for n, . We determined the parameters (m*, μn, σn) for model 1 by the least square method, using the distribution of kinase activity a0, the dose-response curves, and the cumulative distribution of K1/2 (Fig. 4, C to E). The obtained values were (m*, μn, σn) = (0.445,2.018,0.387). For model 2, both the mean values of m* and n were fixed to the same values as the means of those of model 1, 〈m*〉 = 0.445 and 〈n〉 = 8.1. The SD of m*, σm, was chosen to minimize the mean squared error in fitting to the distribution of kinase activity a0, which gave σm = 0.02. For model 3, all the parameter values were determined in the same way as model 1. The obtained parameter values were (μm, σm, μn, σn) = (0.446,0.010,2.035,0.385), and the correlation coefficient between m* and log(n) was 0.144. For serine responses, we considered only variation in n (model 1). The best-fit parameter values were (m*, μn, σn) = (0.4838,2.322,0.4174). In fig. S9 (A to D), model 1 was fitted to the data, allowing the log-normal distribution of n to depend on different background conditions, i.e., the parameters m*, μn,0, σn,0, μn,100, σn,100, μn,200, and σn,200 were estimated simultaneously, where the numbers in the subscript indicate the corresponding background MeAsp concentrations. The estimated parameters were m* = 0.455 (0.447 to 0.464), μn,0 = 2.030 (1.950 to 2.148), σn,0 = 0.409 (0.254 to 0.557), μn,100 = 2.206 (2.048 to 2.323), σn,100 = 0.426 (0.264 to 0.670), μn,200 = 2.057 (1.896 to 2.171), and σn,200 = 0.603 (0.449 to 0.872), where the maximum likelihood values and 95% CIs (shown in the parentheses) were obtained from the likelihood function estimated by the Metropolis-Hastings sampling method. The log likelihood function was defined as , where Na, Nd, and NK are the numbers of data points of the kinase activity distribution (Fig. 4C), the dose-response curves (Fig. 4D), and the CDF of K1/2 (Fig. 3B), respectively; x and σ are the data point and its uncertainty (SD estimated as 68% CI); is model prediction; and C is the normalization constant of the likelihood function (which need not be specified for Metropolis-Hastings sampling). The weights in front of the summations were chosen such that it computes the average value of the residuals in each experiment, which gives more equal weight across experiments that might have different statistical power, and that the two datasets (i.e., one that gives the kinase activity distribution and one that gives both the dose-response curves and the CDF of K1/2) have an equal weight.
Supplementary Material
Acknowledgments
We thank S. Parkinson and V. Sourjik for strains; N. Frankel, A. Waite, and Y. Dufour for help with the microfluidics; S. Boskamp and Z. Rychanavska for cell culture and technical assistance; M. Kamp for microscopy assistance; E. Clay, B. Ait Said, and M. Konijnenburg for software and electronic support; F. Avgidis and S. Grannetia for help with experiments; and H. Mattingly and J. van Zon for discussions and critical reading of the manuscript. Funding: This study was supported by the Allen Distinguished Investigator Program (grant 11562) through the Paul G. Allen Frontiers Group, NIH award R01GM106189, NWO VIDI award 680-47-515, and NWO/FOM Projectruimte grant 11PR2958. Author contributions: K.K., T.E., and T.S.S. designed the research. T.E. and T.S.S. supervised the project and secured funding. K.K., J.M.K., T.E., and T.S.S. conceived the method. K.K. performed the experiments. K.K., T.E., and T.S.S. performed the data analysis and mathematical modeling. K.K. and J.L. performed theoretical analyses in the Supplementary Materials. K.K., J.M.K., T.E., and T.S.S. wrote the manuscript with input from J.L. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc1087/DC1
REFERENCES AND NOTES
- 1.B. Müller-Hill, The lac Operon (De Gruyter, 1996).
- 2.Kussell E., Leibler S., Phenotypic diversity, population growth, and information in fluctuating environments. Science 309, 2075–2078 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Acar M., Mettetal J. T., van Oudenaarden A., Stochastic switching as a survival strategy in fluctuating environments. Nat. Genet. 40, 471–475 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Frankel N. W., Pontius W., Dufour Y. S., Long J., Hernandez-Nunez L., Emonet T., Adaptability of non-genetic diversity in bacterial chemotaxis. eLife 3, e03526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ackermann M., A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol. 13, 497–508 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Waite A. J., Frankel N. W., Emonet T., Behavioral variability and phenotypic diversity in bacterial chemotaxis. Annu. Rev. Biophys. 47, 595–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roggiani M., Goulian M., Oxygen-dependent cell-to-cell variability in the output of the escherichia coli tor phosphorelay. J. Bacteriol. 197, 1976–1987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schreiber F., Littmann S., Lavik G., Escrig S., Meibom A., Kuypers M. M. M., Ackermann M., Phenotypic heterogeneity driven by nutrient limitation promotes growth in fluctuating environments. Nat. Microbiol. 1, 16055 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Carey J. N., Mettert E. L., Roggiani M., Myers K. S., Kiley P. J., Goulian M., Regulated stochasticity in a bacterial signaling network permits tolerance to a rapid environmental change. Cell 175, 1989–1990 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkinson J. S., Hazelbauer G. L., Falke J. J., Signaling and sensory adaptation in Escherichia coli chemoreceptors: 2015 update. Trends Microbiol. 23, 257–266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorur-Shandilya S., Demir M., Long J., Clark D. A., Emonet T., Olfactory receptor neurons use gain control and complementary kinetics to encode intermittent odorant stimuli. eLife 6, e27670 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamino K., Kondo Y., Rescaling of spatio-temporal sensing in eukaryotic chemotaxis. PLOS ONE 11, e0164674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamino K., Kondo Y., Nakajima A., Honda-Kitahara M., Kaneko K., Sawai S., Fold-change detection and scale invariance of cell-cell signaling in social amoeba. Proc. Natl. Acad. Sci. U.S.A. 114, E4149–E4157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazova M. D., Ahmed T., Bellomo D., Stocker R., Shimizu T. S., Response rescaling in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 108, 13870–13875 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.E. H. Weber, De Pulsu, Resorptione, Auditu et Tactu: Annotationes Anatomicae et Physiologicae, Auctore (prostat apud CF Koehler, 1834).
- 16.G. Fechner, Elements of Psychophysics (Holt, Rinehart and Winston.[DL], 1860).
- 17.Stevens S. S., On the psychophysical law. Psychol. Rev. 64, 153–181 (1957). [DOI] [PubMed] [Google Scholar]
- 18.Shoval O., Goentoro L., Hart Y., Mayo A., Sontag E., Alon U., Fold-change detection and scalar symmetry of sensory input fields. Proc. Natl. Acad. Sci. U.S.A. 107, 15995–16000 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adler M., Alon U., Fold-change detection in biological systems. Curr. Opin. Syst. Biol. 8, 81–89 (2018). [Google Scholar]
- 20.Shimizu T. S., Tu Y., Berg H. C., A modular gradient-sensing network for chemotaxis in Escherichia coli revealed by responses to time-varying stimuli. Mol. Syst. Biol. 6, 382 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sourjik V., Berg H. C., Receptor sensitivity in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 99, 123–127 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sourjik V., Berg H. C., Binding of the Escherichia coli response regulator CheY to its target measured in vivo by fluorescence resonance energy transfer. Proc. Natl. Acad. Sci U.S.A. 99, 12669–12674 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sourjik V., Berg H. C., Functional interactions between receptors in bacterial chemotaxis. Nature 428, 437–441 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Colin R., Rosazza C., Vaknin A., Sourjik V., Multiple sources of slow activity fluctuations in a bacterial chemosensory network. eLife 6, e26796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keegstra J. M., Kamino K., Anquez F., Lazova M. D., Emonet T., Shimizu T. S., Phenotypic diversity and temporal variability in a bacterial signaling network revealed by single-cell FRET. eLife 6, e27455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mesibov R., Adler J., Chemotaxis toward amino acids in Escherichia coli. J. Bacteriol. 112, 315–326 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dufour Y. S., Gillet S., Frankel N. W., Weibel D. B., Emonet T., Direct correlation between motile behavior and protein abundance in single cells. PLOS Comput. Biol. 12, e1005041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kollmann M., Løvdok L., Bartholomé K., Timmer J., Sourjik V., Design principles of a bacterial signalling network. Nature 438, 504–507 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Spudich J. L., Koshland D. E., Non-genetic individuality: Chance in the single cell. Nature 262, 467–471 (1976). [DOI] [PubMed] [Google Scholar]
- 30.Waite A. J., Frankel N. W., Dufour Y. S., Johnston J. F., Long J., Emonet T., Non-genetic diversity modulates population performance. Mol. Syst. Biol. 14, e8149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korobkova E., Emonet T., Vilar J. M., Shimizu T. S., Cluzel P., From molecular noise to behavioural variability in a single bacterium. Nature 428, 574–578 (2004). [DOI] [PubMed] [Google Scholar]
- 32.V. Sourjik, A. Vaknin, T. S. Shimizu, H. C. Berg, Methods in Enzymology (Elsevier, 2007), vol. 423, pp. 365–391. [DOI] [PubMed] [Google Scholar]
- 33.Briegel A., Li X., Bilwes A. M., Hughes K. T., Jensen G. J., Crane B. R., Bacterial chemoreceptor arrays are hexagonally packed trimers of receptor dimers networked by rings of kinase and coupling proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 3766–3771 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piñas G. E., Frank V., Vaknin A., Parkinson J. S., The source of high signal cooperativity in bacterial chemosensory arrays. Proc. Natl. Acad. Sci. U.S.A. 113, 3335–3340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monod J., Wyman J., Changeux J.-P., On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12, 88–118 (1965). [DOI] [PubMed] [Google Scholar]
- 36.Mello B. A., Tu Y., An allosteric model for heterogeneous receptor complexes: Understanding bacterial chemotaxis responses to multiple stimuli. Proc. Natl. Acad. Sci. U.S.A. 102, 17354–17359 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keymer J. E., Endres R. G., Skoge M., Meir Y., Wingreen N. S., Chemosensing in Escherichia coli: Two regimes of two-state receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 1786–1791 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skoge M. L., Endres R. G., Wingreen N. S., Receptor-receptor coupling in bacterial chemotaxis: Evidence for strongly coupled clusters. Biophys. J. 90, 4317–4326 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu Y., Shimizu T. S., Berg H. C., Modeling the chemotactic response of Escherichia coli to time-varying stimuli. Proc. Natl. Acad. Sci. U.S.A. 105, 14855–14860 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Endres R. G., Oleksiuk O., Hansen C. H., Meir Y., Sourjik V., Wingreen N. S., Variable sizes of Escherichia coli chemoreceptor signaling teams. Mol. Syst. Biol. 4, 211 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clausznitzer D., Oleksiuk O., Løvdok L., Sourjik V., Endres R. G., Chemotactic response and adaptation dynamics in Escherichia coli. PLOS Comput. Biol. 6, e1000784 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu Y., Quantitative modeling of bacterial chemotaxis: Signal amplification and accurate adaptation. Annu. Rev. Biophys. 42, 337–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunten P., Koshland D., Tuning the responsiveness of a sensory receptor via covalent modification. J. Biol. Chem. 266, 1491–1496 (1991). [PubMed] [Google Scholar]
- 44.Borkovich K. A., Alex L. A., Simon M. I., Attenuation of sensory receptor signaling by covalent modification. Proc. Natl. Acad. Sci. U.S.A. 89, 6756–6760 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoney A., Salman H., Precision and variability in bacterial temperature sensing. Biophys. J. 108, 2427–2436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalinin Y., Neumann S., Sourjik V., Wu M., Responses of Escherichia coli bacteria to two opposing chemoattractant gradients depend on the chemoreceptor ratio. J. Bacteriol. 192, 1796–1800 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gremer J. R., Kimball S., Venable D. L., Within-and among-year germination in Sonoran Desert winter annuals: Bet hedging and predictive germination in a variable environment. Ecol. Lett. 19, 1209–1218 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Xue B., Sartori P., Leibler S., Environment-to-phenotype mapping and adaptation strategies in varying environments. Proc. Natl. Acad. Sci. U.S.A. 116, 13847–13855 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ames P., Studdert C. A., Reiser R. H., Parkinson J. S., Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc. Natl. Acad. Sci. 99, 7060–7065 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldbeter A., Koshland D. E., An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. U.S.A. 78, 6840–6844 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madar D., Dekel E., Bren A., Alon U., Negative auto-regulation increases the input dynamic-range of the arabinose system of Escherichia coli. BMC Syst. Biol. 5, 111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paaby A. B., Rockman M. V., The many faces of pleiotropy. Trends Genet. 29, 66–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shoval O., Sheftel H., Shinar G., Hart Y., Ramote O., Mayo A., Dekel E., Kavanagh K., Alon U., Evolutionary trade-offs, Pareto optimality, and the geometry of phenotype space. Science 336, 1157–1160 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Arnoldini M., Cremer J., Hwa T., Bacterial growth, flow, and mixing shape human gut microbiota density and composition. Gut Microbes 9, 559–566 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.T. J. Wiles, B. H. Scholmann, E. S. Wall, R. Betancourt, R. Parthasarathy, K. Guillemin, Swimming motility and chemotaxis control the spatial organization, persistence, and inflammatory activity of a model intestinal pathobiont. bioRxiv 779983 [Preprint]. 24 September 2019. 10.1101/779983. [DOI]
- 56.Stocker R., Marine microbes see a sea of gradients. Science 338, 628–633 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Qin D., Xia Y., Whitesides G. M., Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 5, 491–502 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Hansen C. H., Sourjik V., Wingreen N. S., A dynamic-signaling-team model for chemotaxis receptors in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 107, 17170–17175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goentoro L., Shoval O., Kirschner M. W., Alon U., The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol. Cell 36, 894–899 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc1087/DC1