

A conserved SUMO-ubiquitin-proteasome pathway protects cells from DNA damage induced by topoisomerases.
Abstract
Topoisomerases form transient covalent DNA cleavage complexes to perform their reactions. Topoisomerase I cleavage complexes (TOP1ccs) are trapped by camptothecin and TOP2ccs by etoposide. Proteolysis of the trapped topoisomerase DNA-protein cross-links (TOP-DPCs) is a key step for some pathways to repair these lesions. We describe a pathway that features a prominent role of the small ubiquitin-like modifier (SUMO) modification for both TOP1- and TOP2-DPC repair. Both undergo rapid and sequential SUMO-2/3 and SUMO-1 modifications in human cells. The SUMO ligase PIAS4 is required for these modifications. RNF4, a SUMO-targeted ubiquitin ligase (STUbL), then ubiquitylates the TOP-DPCs for their subsequent degradation by the proteasome. This pathway is conserved in yeast with Siz1 and Slx5-Slx8, the orthologs of human PIAS4 and RNF4.
INTRODUCTION
DNA lesions range from abasic sites, chemical adducts, single- and double-strand breaks (DSBs), and DNA-protein cross-links (DPCs) (1). Defects in DNA damage repair compromises genome integrity, leading to mutagenesis, carcinogenesis, and neurodegeneration (2). DPC formation frequently occurs through the action of topoisomerases, which cleave DNA by the formation of a protein-DNA phosphotyrosyl intermediate, termed topoisomerase cleavage complex (TOPcc) (3, 4). While TOPccs are typically transient reaction intermediates, a variety of conditions can inhibit topoisomerase-mediated DNA rejoining, resulting in persistent and abortive TOPccs (which we refer to as TOP-DPCs) that may interfere with DNA metabolisms if they persist (4). Although TOP1 and TOP2 are distinct in terms of their structures and catalytic mechanisms, DPCs arising from either enzyme can be generated by enzymatic malfunctions (5), preexisting DNA alterations (4), or small-molecule inhibitors (6). The generation of DPCs is the basis of antitumor activity for anticancer agents such as camptothecin (CPT) derivatives that target TOP1 and etoposide (ETP) or anthracyclines that target TOP2 (6).
A variety of pathways have been shown to repair TOP1- and TOP2-DPCs (7–10). Since the lesions include a protein component that conceals the DNA breaks, proteolysis plays a critical role in the repair of these lesions (11–15). Early studies suggested that cells process TOP-DPCs by using the ubiquitin (Ub)–proteasome pathway (UPP) to digest the protein component of the DPC (11, 12). Although the proteasome likely plays a key role in proteolytic steps needed for repairing TOP-DPCs, other proteases have recently been shown to remove the protein component of TOP-DPCs. This is the case of the metalloprotease SPRTN/WSS1, a component of the replisome, which targets TOP-DPCs for proteolysis (16, 17). Very recently, other nonproteasomal protease pathways for the repair of TOP-DPCs have been described (18–20). While the proteasome might accomplish removal of the protein component of the DPCs, the presence of a phosphotyrosyl DNA adduct at the end of the broken DNA requires additional processing that cannot be accomplished by proteolysis alone.
A second set of nucleolytic enzymes are required to generate unmodified DNA ends that can be used by DSB repair. Two specialized enzymes, tyrosine-DNA phosphodiesterase-1 (TDP1) and TDP2, were identified that efficiently hydrolyze the tyrosyl-DNA bond (8, 9). Both enzymes appear to require proteolysis of the protein component of the DPC before cleaving the phosphotyrosyl bond (21, 22). In addition to these specialized nucleases, other nucleolytic enzymes such as MRE11, CtIP, XPF, and XPG can also process DPCs (4, 7), although a requirement for prior proteolytic digestion for these enzymes is unclear.
A key issue is how cells distinguish between a TOP-DPC and a TOPcc. Signaling pathways may recognize topoisomerases when aberrantly bound to DNA, e.g., when the enzymes fail to dissociate appropriately and trigger posttranslational modification(s) of abortive TOPccs. This signaling could be related to the UPP and would result in polyubiquitylation of the enzymes. In addition to Ub, small Ub-like modifiers (SUMO) including SUMO-1 and SUMO-2/3 have also been shown to modify topoisomerases upon exposure to their inhibitors (23–26); however, the consequences of this modification and the fates of modified trapped enzymes remain largely unknown. A recent study suggests a specific role of SUMO-2/3 modification in the repair of TOP2-DPCs by allowing TDP2 to process the DPC without evoking proteasomal degradation (26). In some cases, polymeric SUMO chains serve as a recognition signal for SUMO-targeted Ub ligases (STUbLs), which attach Ub moieties on the SUMOylated substrates to induce their proteasomal degradation (27, 28). This SUMO-Ub pathway appears to orchestrate the turnover of DNA damage response (DDR) and repair enzymes including MDC-1, RPA, and 53BP1 (29–32).
The importance of SUMO modification in regulating consequences of TOP-DPCs has been studied in model systems. Schizosaccharomyces pombe cells require the SUMO ligase Nse2 in concert with Tdp1 to protect cells from TOP1-DPCs even in the absence of exogenous trapping conditions (33). Nse2 and the STUbL Slx8 repair TOP1-mediated DNA damage by facilitating Rad16-Swi10–mediated homologous recombination. Slx8 and Pli1, another SUMO ligase in S. pombe, were found to cooperate and prevent TOP1-induced chromosomal recombination (34). Similarly, Pli1 and Slx8 are implicated as a SUMO ligase and a STUbL for TOP2-induced DNA damage in S. pombe (35). Yet, the relevant SUMO-dependent pathways for TOP-DPC have remained unexplored in higher eukaryotes.
Here, we describe a SUMO modification pathway that targets TOP1- and TOP2-DPCs for proteasomal degradation. We show that SUMO-2/3 acts as an early response to TOP-DPC formation, which is followed by SUMO-1 modification. These SUMO conjugations elicit the K48 polyubiquitylation of TOP-DPCs, which is then targeted by the proteasome regulatory subunit including PSMD14 (26S proteasome non-ATPase regulatory subunit 14) for deubiquitylation and the proteasome core subunit including PSMB5 (proteasome subunit beta type-5) for proteolytic destruction. We identify human PIAS4 (protein inhibitor of activated STAT protein 4), a Siz/PIAS family protein, as a shared SUMO ligase for TOP1-, TOP2α-, and TOP2β-DPCs by mass spectroscopy and biochemical analyses. We also identify RNF4 (RING finger protein 4) as the STUbL for TOP-DPCs, driving their proteasomal degradation via PIAS4-mediated SUMOylation. This SUMO-Ub pathway is conserved in Saccharomyces cerevisiae, with the SUMO ligase Siz1 responsible for the SUMOylation and the STUbL Slx5-Slx8 responsible for the subsequent ubiquitylation of both TOP1- and TOP2-DPCs. This SUMO-Ub pathway targets TOP-DPCs independently of replication, transcription and DDRs. Our results provide important molecular underpinnings for a major pathway repairing TOP-DPCs and provide a framework for further dissecting how different pathways can function in the repair of these important lesions.
RESULTS
Inhibiting SUMOylation prevents the repair of TOP1- and TOP2-DPCs in human cells
Although SUMO-2/3 modification of TOP2-DPCs has recently been shown to enable TDP2 to resolve the DPCs (26), whether direct inhibition of SUMOylation has an impact on the removal of both TOP1- and TOP2-DPCs had not been tested. Using ML-792, a potent inhibitor of the SUMO-activating enzyme (SAE) (36), we found by Western blotting (WB) that ML-792 alleviated both the CPT- and ETP-induced loss of cellular topoisomerases in human embryonic kidney–293 (HEK293) cells (Fig. 1, A and B, lanes 7). As expected, CPT- and ETP-induced degradation of TOP-DPCs was also prevented by the proteasome inhibitor MG132 and the Ub-activating enzyme inhibitor TAK-243 (Fig. 1, A and B, lanes 5 and 6). Using the in vivo complex of enzyme (ICE) assay to isolate and analyze TOP-DPCs (37), we observed that ML-792, TAK-243, or MG132 also increased the levels of TOP1-, TOP2α-, and TOP2β-DPCs in HEK293 cells (Fig. 1, C to H; fig. S1A), indicating that not only ubiquitylation and the proteasome but also SUMOylation are involved in the removal of the TOP-DPCs in human cells.
Fig. 1. Inhibiting SUMOylation prevents the repair of human TOP-DPCs.
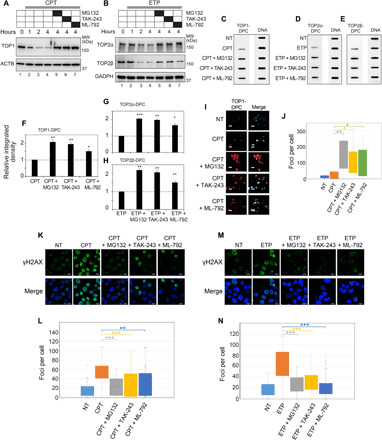
(A and B) Following treatment with MG132 (10 μM, 1 hour), TAK-243 (10 μM, 2 hours), or ML-792 (10 μM, 1 hour), HEK293 cells were treated with CPT (20 μM) or ETP (200 μM) for WB as indicated. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MW, molecular weight. (C to E) HEK293 cells were treated with CPT (1 μM) or ETP (10 μM) for 2 hours in the absence or presence of indicated inhibitors for ICE assay. (F to H) Quantitation of TOP-DPCs from triplicate experiments including blots in (C) to (E). Density of TOP-DPC/density of DNA of each group was normalized to that of cells treated with CPT or ETP alone. *P < 0.05 and **P < 0.01. (I) U2OS cells were treated with CPT (10 μM, 1 hour) in the absence or presence of indicated inhibitors for IF staining using anti–TOP1-DPC antibody. Scale bar, 20 μm. (J) Quantitation of TOP1-DPC foci of each treatment from (I). (K) U2OS cells were treated with CPT (1 μM, 1 hour) in the absence and presence of indicated inhibitors for IF staining using anti-γH2AX antibody. Scale bar, 20 μm. (L) Quantitation of γH2AX foci of each treatment from (K). ***P < 0.001. (M) Same as (K) except that cells were treated with ETP (5 μM, 1 hour). (N) Quantitation of γH2AX foci of each treatment group from (M).
Using an antibody that selectively recognizes TOP1-DPCs (38), immunofluorescence (IF) microscopy in human osteosarcoma U2OS cells showed that inhibition of SUMOylation by ML-792, inhibition of ubiquitylation by TAK-243, and inhibition of the proteasome by MG132 increased TOP1-DPCs (Fig. 1, I and J). While it would have been desirable to carry out a similar analysis for TOP2-DPCs, no antibody that preferentially recognizes TOP2-DPCs has yet been described.
As the induction of the DSB biomarker histone γH2AX by topoisomerase inhibitors requires proteolysis to expose the DSB otherwise concealed inside the TOP-DPC (15), we tested whether inhibiting SUMOylation and ubiquitylation also blocked the induction of γH2AX. Pretreatment with ML-792 or TAK-243, like MG132, diminished CPT- and ETP-induced γH2AX foci in U2OS cells (Fig. 1, K to N), implying that SUMOylation and ubiquitylation are required to activate the DDR downstream from TOP-DPCs. Next, we assessed whether ML-792 or TAK-243 could also specifically enhance the sensitivity of human cancer cells to topoisomerases inhibitors. Pretreatment with ML-792 or TAK-243 increased the sensitivity to CPT and ETP in HCT116 human colon carcinoma cells, although the effect of TAK-243 on etoposide sensitivity was modest (fig. S1, B and C). Together, these results demonstrate the importance of SUMO for the repair of TOP-DPCs in human cells.
Rapid and sequential SUMOylation and ubiquitylation of TOP1- and TOP2-DPCs in human cells
To directly characterize how SUMO-1, SUMO-2/3, and Ub modify topoisomerases, we performed pull-down experiments after transfection of constructs that express epitope-tagged topoisomerases in HEK293 cells. His-tagged TOP1 was found conjugated with SUMO-2/3 and Ub under unperturbed conditions, and CPT enhanced the SUMO-1, SUMO-2/3, and Ub modifications within 30 min (Fig. 2A). These modifications were further increased by MG132. Similarly, immunoprecipitation (IP) of FLAG-tagged TOP2α and TOP2β showed that TOP2 isozymes were SUMO-2/3–, SUMO-1–, and Ub-conjugated under unperturbed conditions and that treatment with ETP for 30 min increased the levels of these modifications, with a further increase upon cotreatment with MG132 (Fig. 2B). These results demonstrate that TOP1, TOP2α, and TOP2β are rapidly SUMOylated and ubiquitylated in response to TOP-DPCs.
Fig. 2. SUMOylation and ubiquitylation of human TOP-DPCs.
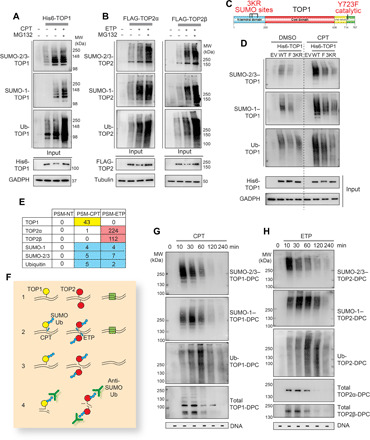
(A) 6× His-TOP1 expressing HEK293 cells were treated with DMSO or CPT (20 μM) ± MG132 for 30 min, followed by denaturing His pull-down and WB as indicated. (B) FLAG-TOP2α or FLAG-TOP2β expressing HEK293 cells were treated with DMSO or ETP (200 μM) ± MG132 for 30 min, followed by denaturing FLAG-IP and WB as indicated. (C) Domain schematic of human TOP1. (D) His-TOP1 WT, Y723F (F), or CPT3KR (3KR) expressing HEK293 cells were treated with DMSO or CPT for 30 min, followed by denaturing His pull-down and WB as indicated. (E) HEK293 cells were exposed to DMSO (NT), 20 μM CPT, or 200 μM ETP for 30 min, followed by ICE assay and high-performance liquid chromatography and tandem mass spectrometry (HPLC-MS/MS) analysis (PSM: peptide spectrum match). (F) Scheme of the DUST assay: (1) Cells are seeded and form reversible TOP1ccs (yellow) and TOP2ccs (red). (2) Drug-induced TOP-DPCs are modified by SUMO and Ub (blue hexagons). Other chromatin-bound proteins are shown as green square. (3) Cells are lysed using DNAzol and ethanol precipitated to isolate the DPCs. (4) Each sample is either digested with micrococcal nuclease for WB using antibodies targeting SUMOs, Ub, and topoisomerases or subjected to slot-blot using anti-double-stranded DNA (dsDNA) antibody. (G and H) HEK293 cells were exposed to 20 μM CPT or 200 μM ETP and collected at indicated time points for DUST assay to detect SUMO-2/3, SUMO-1 Ub, and total TOP-DPCs.
To clarify the relationship between SUMOylation and ubiquitylation of topoisomerases, we next used the non-SUMOylatable TOP1-3KR construct containing three mutations that prevent SUMOylation of TOP1 (K117R, K153R, and K103R; Fig. 2C) (25). These mutations not only blocked SUMOylation but also ubiquitylation of TOP1 (Fig. 2D; densitometric analyses in fig. S2A), indicating that SUMOylation is required for TOP1 ubiquitylation. A TOP1 catalytically dead (Y723F) mutant (Fig. 2C) also underwent SUMO modifications after CPT treatment, albeit to a lesser extent than TOP1 wild type (WT; Fig. 2D). This unexpected result suggests that covalent modification of topoisomerases is not solely due to the repair of trapped topoisomerases, as the active site tyrosine mutant cannot form a covalent complex with DNA.
To demonstrate that TOP1- and TOP2-DPCs are directly conjugated with SUMO-1, SUMO-2/3, and Ub, we performed high-performance liquid chromatography–tandem mass spectrometry (HPLC-MS/MS) of TOP-DPCs induced by CPT or ETP from HEK293 cells using the ICE assay. These analyses confirmed the presence of SUMO-1, SUMO-2, and Ub in the CPT- and ETP-treated, but not in the vehicle [dimethyl sulfoxide (DMSO)]–treated samples (Fig. 2E).
We next developed an assay to directly detect and follow the SUMOylation and ubiquitylation and potentially other types of posttranslational modifications (PTMs) of TOP-DPCs. For short, we refer to this assay as Detection of Ubiquitylated and SUMOylated TOP-DPCs (DUST; Fig. 2F). DUST is an extension of the RADAR assay, which was designed for quantitating TOP-DPCs and other DPCs (39). The RADAR (rapid approach to DNA adduct recovery) assay, similar to the ICE assay applied above, relies on generating extracts using strong denaturants to disrupt noncovalent protein/protein and protein/DNA interactions followed by nucleic acid purification. We applied the logic of the RADAR assay and used it to detect PTMs of the drug-induced TOP-DPCs (Fig. 2F). The DUST assay uses the same purification of TOP-DPCs and detects SUMOylated and ubiquitylated TOP-DPCs with antibodies targeting SUMO-2/3, SUMO-1, and Ub by WB. As with the RADAR protocol, topoisomerases that are not covalently bound to DNA do not copurify with nucleic acids, and therefore the assay specifically reports PTMs of TOP-DPCs.
Using antibodies targeting TOP1, TOP2α, and TOP2β, we observed by the DUST assay that TOP-DPCs appeared quickly (within 10 min) after addition of the topoisomerase inhibitors [Fig. 2, G and H, bottom panels; densitometric analyses in fig. S2 (B and C)], reached a peak at 30 min and gradually decreased to become undetectable after 4 hours in HEK293 cells. DUST assays also showed that TOP-DPCs were induced by topoisomerase inhibitors in a dose-dependent manner (fig. S2, D and E). With antibodies targeting SUMO-2/3, SUMO-1, and Ub, the DUST assay shows that SUMO-2/3, SUMO-1, and Ub modifications of TOP-DPCs peaked sequentially [at 10, 30 min, and 1 hour, respectively; Fig. 2, G and H; densitometric analyses in fig. S2 (B and C)], suggesting that these consecutive PTMs are coordinated with the clearance of the DPCs. Inhibition of proteasomal degradation with MG132 elevated the levels of total TOP-DPCs and ubiquitylated and SUMOylated TOP-DPC species at 30 min and 2 hours after treatments with the topoisomerase inhibitors (fig. S2, F and G).
As both TOP2α and TOP2β are trapped by ETP, the Ub- and SUMO-TOP2-DPCs presumably represent a mixed population comprising both TOP2α- and TOP2β-DPCs. To examine the TOP2 isozymes separately, we performed DUST assays in both WT and TOP2B CRISPR knockout (KO) HeLa cells with and without TOP2A small interfering RNA (siRNA) knockdown. These experiments showed that both TOP2α- and TOP2β-DPCs were SUMOylated and ubiquitinylated in response to ETP and that SUMOylation appeared most intense for the TOP2α-DPCs in the rapidly proliferating HeLa cells (fig. S2H). TOP2A down-regulation in TOP2B KO cells was found to deplete the SUMO and Ub signals (fig. S2H), further confirming that the SUMO and Ub signals detected by the DUST assay are specific to TOP-DPCs.
SUMO and Ub linkages of TOP-DPCs in human cells
Ub can be conjugated through one of its seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) to form homotypic poly-Ub chains (40). To determine the poly-Ub linkages of TOP-DPCs, we transfected hemagglutinin (HA)–tagged lysine-to-arginine mutants for each ubiquitylation residue and performed DUST assays in HEK293 cells. K11R, K48R, and K63R mutations reduced both TOP1- and TOP2-DPC ubiquitylation (fig. S3, A and B). The K48 and the K11 linkages are consistent with their known signaling role for proteasomal degradation (40). The K63 ubiquitylation of TOP-DPCs may serve to recruit other DDR proteins (40).
Given the established role of K11 of SUMO-2/3 in forming polymeric SUMO-2/3 chain (41), we transfected HA-SUMO-2 WT and K11R to HEK293 cells to determine the TOP-DPC SUMO-2/3 linkages. As expected, SUMO-2 K11R-transfected cells showed decreased SUMO-2 modification of both TOP1- and TOP2-DPCs (fig. S3, C and D, lanes 5 and 6). We also tested the linkages of the TOP-DPC SUMO-1 chain. Because biochemical studies have shown that SUMO-1 polymers can be assembled through their K7 residue (42), we performed DUST assays in cells transfected with HA-SUMO-1 WT or SUMO-1-K7R. Disruption of the K7 residue suppressed the SUMO-1 signal for both TOP1- and TOP2-DPCs (fig. S3, C and D, lanes 1, 2).These data indicate that the SUMO-1 polymers of TOP-DPCs are formed mainly through the K7 linkage, while the SUMO-2/3 polymers are formed through the K11 linkage.
To explore the interplay between SUMO-1 and SUMO-2/3 in TOP-DPC modifications, we used siRNA against SUMO-2/3 in the SUMO-1–transfected cells and observed shorter forms of SUMO-1–modified TOP-DPCs species (fig. S3, C and D, lane 3). Conversely, silencing of SUMO-1 in SUMO-2–transfected cells led to longer forms of SUMO-2–modified TOP-DPCs (fig. S3, C and D, lane 7). These results are in agreement with the SUMO literature that SUMO-2/3 moieties conjugation with SUMO-1 terminates SUMO-2/3 chain elongation (43). This conclusion is also consistent with our kinetics experiments showing the prior appearance and faster electrophoretic migration of the SUMO-2/3 conjugates on the TOP-DPCs (see Fig. 2, G and H). Our experiments suggest that SUMO-2/3 first forms polymeric chains on TOP-DPCs, which are subsequently capped by SUMO-1.
PIAS4 is a major SUMO ligase for both TOP1- and TOP2-DPCs in human cells
SUMOylation is executed through a cascade of reactions involving the SAEs or E1, the SUMO-conjugating enzyme UBC9 (ubiquitin carrier protein 9) or E2, and SUMO E3 ligases specific for each target protein. To identify the SUMO E3 ligase(s) catalyzing the SUMOylation of TOP1-DPCs, we expressed His-tagged TOP1 in HEK293 cells treated with CPT (or DMSO as solvent control) and purified TOP1 protein complexes for LC-MS/MS (Fig. 3A, left). Likewise, we performed IP of FLAG-tagged TOP2α and TOP2β in cells treated with ETP and analyzed TOP2-containing protein complexes by MS (Fig. 3A, right). All samples except for the empty vector control (pTrex) were consistently enriched with PIAS4 (Fig. 3A), a member of the Siz/PIAS SUMO ligase family implicated in the recruitment of TOP2α to centromeres for decatenation during mitosis (44) and in the recruitment of DDR proteins to DSB sites (29). To validate this result, we reciprocally pulled down PIAS4-containing protein complexes by expressing FLAG-PIAS4. TOP1, TOP2α, and TOP2β were retrieved by MS (Fig. 3B). We also performed IP–immunoblotting (IB) and proximity ligation assay (PLA), which confirmed the PIAS4 interaction with both TOP1 and TOP2 in the absence and presence of topoisomerase inhibitors (fig. S4, A to E). These results demonstrate the close association of the human topoisomerases with the SUMO E3 ligase PIAS4.
Fig. 3. Identification of PIAS4 as SUMO ligase and RNF4 as STUbL for human TOP-DPCs.
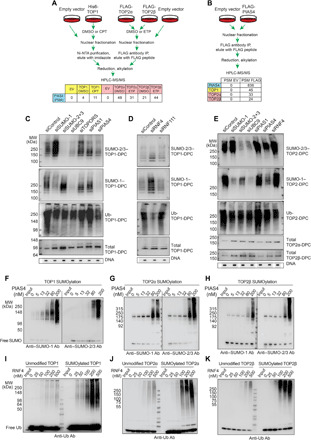
(A) Schemes of TOP1 His pull-down, TOP2α, and β FLAG-IP for LC-MS/MS. (B) Scheme of PIAS4 FLAG-IP for LC-MS/MS. (C) HEK293 cells were transfected with indicated siRNAs and treated with CPT (20 μM, 30 min) for DUST assay as indicated. (D) HEK293 cells were transfected with indicated siRNAs and treated with CPT (20 μM, 30 min) for DUST assay as indicated. (E) HEK293 cells were transfected with indicated siRNAs and treated with ETP (200 μM, 30 min) for DUST assay as indicated. (F) Left: SUMO conjugation assay with recombinant TOP1, SUMO E1, SUMO E2, SUMO-1, and increasing concentrations of PIAS4, followed by WB using anti–SUMO-1 antibody (Ab). Right: Same as left panel except that SUMO-2 and anti–SUMO-2/3 antibody were used. (G) Same as (F) except that recombinant TOP2α was used. (H) Same as (F) except that recombinant TOP2β was used. (I) Unmodified TOP1 (left) and SUMOylated TOP1 (right) were subjected to Ub conjugation assay in the presence of Ub E1, Ub E2, and RNF4 at indicated concentrations, followed by WB using anti-Ub antibody. (J) Same as (I) except that TOP2α was used. (K) Same as (I) except that TOP2β was used.
To demonstrate the functional role of PIAS4, we down-regulated PIAS4 in parallel with other SUMO ligases [PIAS1 and TOPORS (topoisomerase I-binding RING finger protein)] and main components of the SUMO system (SUMO-1, SUMO-2, SUMO-3, and UBC9) by siRNA (the efficacy of the knockdowns is shown in fig. S4F) and tested TOP-DPC SUMOylation and ubiquitylation by DUST assays in HEK293 cells. SUMO-1– and SUMO-2/3–conjugated TOP1-, TOP2α-, and TOP2β-DPCs were decreased by down-regulating PIAS4, while silencing PIAS1 or TOPORs did not have such an effect (Fig. 3, C to E). Ubiquitylation of the TOP-DPC was also reduced by silencing PIAS4. These experiments demonstrate that PIAS4 is the SUMO ligase largely responsible for the observed SUMOylation of TOP-DPCs and that it also regulates the subsequent ubiquitylation of TOP-DPCs. Perturbation of SUMOylation by down-regulation of SUMO expression, by preventing SUMO activation by UBC9 knockdown or by PIAS4 knockdown, all resulted in higher levels of TOP-DPCs (Fig. 3, C and E).
To confirm that PIAS4 can directly SUMOylate TOP-DPCs, we performed SUMOylation biochemical assays by incubating recombinant topoisomerases with PIAS4 in the presence of the SAE1/2 and the SUMO-conjugating enzyme UBC9. PIAS4 catalyzed TOP1 SUMOylation with preference for SUMO-1 over SUMO-2 conjugation (Fig. 3F). PIAS4 also catalyzed SUMO-1 and SUMO-2 conjugation of TOP2α and TOP2β (Fig. 3, G and H; fig. S4G shows PIAS4 auto-SUMOylation experiment). Together, our biochemical and cellular data demonstrate that PIAS4 is a SUMO ligase for TOP1-, TOP2α-, and TOP2β-DPCs.
RNF4 functions as a STUbL for both TOP1- and TOP2-DPCs in human cells
The finding that suppressing PIAS4, SUMO-2/3, SUMO-1, and UBC9 reduced TOP1- and TOP2-DPC ubiquitylation suggests that SUMOylation of the TOP-DPCs triggers their ubiquitylation. We hypothesized that RNF4, an STUbL implicated in DDR (30, 31), might ubiquitylate the TOP-DPCs following SUMOylation. Silencing RNF4 (the efficacy of the knockdown is shown in fig. S4F) reduced the ubiquitylation and enhanced the SUMOylation of both TOP1- and TOP2-DPCs (Fig. 3, D and E), whereas silencing of RNF111, another reported mammalian STUbL (28), did not affect the ubiquitylation of TOP1-DPCs (Fig. 3D).
In vitro ubiquitylation assays with recombinant TOP1 and TOP2 and the Ub system also showed that RNF4 readily catalyzed the ubiquitylation of topoisomerases (Fig. 3, I to K; fig. S4H shows RNF4 auto-ubiquitylation experiment). We also coupled the SUMOylation and ubiquitylation assays and found that RNF4 displayed stronger ubiquitylating activity toward SUMO-modified than unmodified topoisomerases (Fig. 3, I to K). These results indicate that RNF4 functions as a STUbL for both TOP1- and TOP2-DPCs.
Coordination and recruitment of PIAS4 and RNF4 for SUMOylation and Ubiquitylation of TOP-DPCs
To elucidate the roles of PIAS4 and RNF4 in the repair of TOP-DPCs, we performed time-course DUST assays in PIAS4 CRISPR KO HCT116 cells (confirmation of the KO by WB is shown in fig. S5A). TOP-DPC ubiquitylation and SUMOylation were reduced, and total TOP-DPCs were increased in the PIAS4 KO cells at 30 min and 2 hours after topoisomerase inhibitor treatments [Fig. 4, A and B; densitometric analyses in fig. S5 (B and C)]. These results are consistent with PIAS4 mediating the rapid TOP-DPC SUMOylation response and engaging the UPP pathway for TOP-DPC removal. After 6 hours, total TOP-DPCs in the KO cells diminished to undetectable levels, as they did in the WT cells, indicating that additional repair pathways act in response to persistent TOP-DPCs.
Fig. 4. Coordination of PIAS4 and RNF4 for SUMOylation and ubiquitylation of TOP-DPCs.
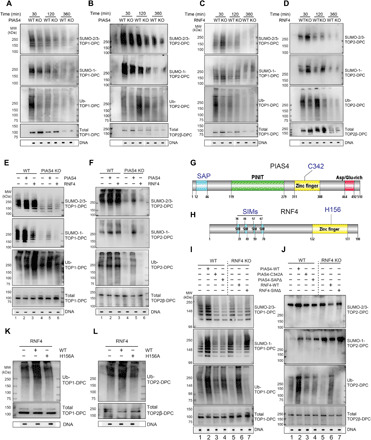
(A) HCT116 WT and PIAS4 KO cells were treated with CPT (20 μM), collected at indicated time points and analyzed by DUST assay as indicated. (B) Same as (A) except that ETP (200 μM) was used to induce TOP2-DPCs. (C) MCF7 WT and RNF4 KO cells were treated with CPT (20 μM), collected at indicated time points, and analyzed by DUST assay as indicated. (D) Same as (C) except that ETP (200 μM) was used. (E) HCT116 WT and PIAS4 KO cells were transfected with indicated plasmids, treated with CPT (20 μM, 1 hour), and analyzed by DUST assay as indicated. (F) Same as (E) except that ETP (200 μM) was used. (G) Domain schematic of human PIAS4. (H) Domain schematic of human RNF4. (I) MCF7 WT and RNF4 KO cells were transfected with indicated plasmids, treated with CPT (20 μM, 1 hour), and analyzed by DUST assay as indicated. (J) Same as (I) except that ETP (200 μM) was used. (K) MCF7 RNF4 KO cells were transfected with indicated plasmids, treated with CPT (20 μM, 1 hour), and analyzed by DUST assay as indicated. (L) Same as (K) except that ETP (200 μM) was used.
Using RNF4 CRISPR KO MCF7 cells (confirmation of the KO by WB is shown in fig. S5D), we also found delayed clearance of total and SUMOylated TOP-DPCs (Fig. 4, C and D; densitometric analyses in fig. S5 E and F), while ubiquitylation of TOP-DPCs was reduced. This result is consistent with the role of RNF4 in mediating rapid ubiquitylation and degradation of the SUMOylated TOP-DPCs. Yet, RNF4 inactivation did not fully block TOP-DPC removal, further confirming that other repair mechanisms are used to remove TOP-DPCs beyond the PIAS4-RNF4 pathway.
To further explore PIAS4 and RNF4 coordination, we performed DUST assays in PIAS4- and RNF4-overexpressing cells. PIAS4 overexpression stimulated both TOP-DPC SUMOylation and ubiquitylation, whereas RNF4 transfection only enhanced TOP-DPC ubiquitylation (Fig. 4, E and F, lanes 2 and 3). Expression of PIAS4 in the PIAS4 KO cells partially restored SUMOylation and ubiquitylation, whereas ectopic expression of RNF4 in PIAS4 KO cells failed to restore TOP-DPC ubiquitylation (Fig. 4, E and F, lanes 5 and 6). We conclude that RNF4 ubiquitylates TOP-DPCs in a PIAS4-dependent manner.
PIAS4 has been shown to localize to DSBs by binding DNA via its N-terminal SAP (SAF-A/B, Acinus and PIAS) domain (Fig. 4G) (29). Transfection of MCF7 cells with PIAS4 deleted for this domain (PIAS4 SAPΔ) and the PIAS4 catalytic-dead (C342A) mutant showed defective TOP-DPC SUMOylation and ubiquitylation (Fig. 4, I and J, lanes 3 and 4; fig. S5G for transfection efficiency controls), indicating that SUMOylation of TOP-DPCs is dependent on the recruitment of PIAS4 to DNA by its SAP domain.
To confirm that RNF4 is recruited through its SUMO-interacting motifs (SIMs) (45) (Fig. 4H), we transfected RNF4 WT and SIMΔ plasmids in RNF4 KO MCF7 cells (fig. S5H for transfection efficiency controls). The SIM mutant failed to induce the ubiquitylation of TOP-DPC in RNF4 KO cells while, as expected, not affecting TOP-DPC SUMOylation (Fig. 4, I and J, lanes 6 and 7). We also tested a putative RNF4 catalytic-dead mutant (H156A) (46) and found that it was unable to induce TOP-DPC ubiquitylation (Fig. 4, K and L; fig. S5H for transfection efficiency controls). PLA assays also showed that PIAS4 SAPΔ and RNF4 SIMΔ failed to colocalize with TOP1 and TOP2α (fig. S5, I and J). Together, our results lead to the conclusion that PIAS4 is recruited to TOP-DPCs through its SAP DNA binding domain and that TOP-DPC SUMOylation by PIAS4, in turn, recruits RNF4 through the RNF4 SIM motifs, leading to the ubiquitylation of both TOP1- and TOP2-DPCs.
SUMOylation and SUMO-targeted ubiquitylation of TOP-DPCs are induced independently of DNA replication, transcription, and DDR in human cells
Protein complexes that translocate along the DNA such as RNA polymerase II complexes are known to induce the ubiquitylation of cellular topoisomerases in response to topoisomerase inhibitors (13–15). To determine whether TOP-DPC SUMOylation and ubiquitylation are induced by transcription, replication, and/or DDR, we carried out DUST assays in HEK293 cells pretreated with the transcription inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), the DNA replication inhibitor aphidicolin (APH), the DNA-PKcs (DNA-dependent protein kinase, catalytic subunit) inhibitor VX984, the ATM (ataxia telangiectasia mutated) inhibitor KU55399, or the ATR (ataxia telangiectasia and Rad3-related protein) inhibitor AZD6738. None of these inhibitors showed an effect on the SUMOylation (fig. S6, A and B), indicating that the SUMOylation of TOP-DPCs is independent of collisions between TOP-DPCs, and it does not require activation of DDR kinases.
Both APH and DRB reduced TOP-DPC ubiquitylation (fig. S6, A and B), indicating that both TOP1- and TOP2-DPCs are ubiquitylated in response to transcription and replication independently of the PIAS4-RNF4 pathway. To further examine this point, we performed DUST assays in HEK293 cells transfected with empty vector and RNF4 overexpression plasmids. As expected from our conclusion that RNF4 is a TOP-DPC STUbL, the SUMO inhibitor ML-792 significantly reduced TOP-DPC ubiquitylation in cells transfected with RNF4 (fig. S6, C and D). Consistent with the results described above, both APH and DRB reduced endogenous TOP-DPC ubiquitylation without affecting TOP-DPC ubiquitylation in the cells transfected with RNF4 overexpression plasmid (fig. S6, C and D). These observations suggest that the PIAS4/RNF4 pathway modifies TOP-DPCs and evokes their RNF4-mediated ubiquitylation independently from perturbations of ongoing transcription, replication, or DDR activation.
RNF4 induces the proteasomal degradation of TOP-DPCs in human cells
To establish whether RNF4-mediated ubiquitylation of TOP-DPCs leads to their proteasomal degradation, we carried out ICE assays to measure the levels of TOP-DPCs in RNF4-transfected MCF7 WT cells. RNF4 overexpression reduced both TOP1- and TOP2-DPCs [Fig. 5, A to C; densitometric quantitation in fig. S7 (A to C)], and this reduction was abolished by MG132, indicating that RNF4 induces the proteasomal degradation of TOP-DPCs. Consistent with this result, MG132 failed to further increase TOP-DPCs in RNF4 KO cells, suggesting the epistasis between RNF4 and the proteasome in TOP-DPC clearance [Fig. 5, A to C; densitometric quantitation in fig. S7 (A to C)].
Fig. 5. RNF4 induces proteasomal degradation of human TOP-DPCs.
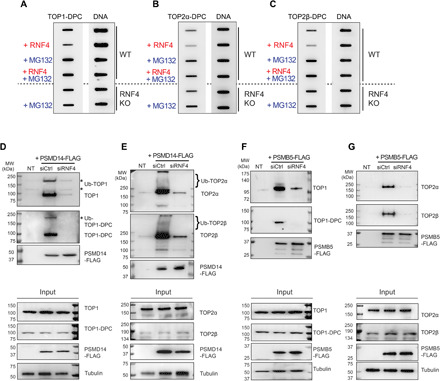
(A) MCF7 WT cells were transfected with or without RNF4 expression plasmid and pretreated with or without MG132, followed by CPT (1 μM, 2 hours) treatment. RNF4 KO cells were pretreated with or without MG132, followed by CPT treatment. Cells were subjected to ICE assay as indicated. (B) Same as (A) except that ETP (10 μM) was used to detect TOP2α-DPC. (C) Same as (A) except that ETP (10 μM) was used to detect TOP2β-DPC. (D) HEK293 cells transfected with control siRNA (siCtrl) or RNF4 siRNA and PSMD14-FLAG expression plasmid were pretreated with MG132 then cotreated with CPT (20 μM, 1 hour). Cells were subjected to FLAG-IP and WB with anti-TOP1, anti-TOP1-DPC, and anti-FLAG antibodies. NT, no transfection. (E) Same as (D) except that ETP (200 μM) was used to detect TOP2-PSMD14 interaction using anti-TOP2α and anti-TOP2β antibodies. (F) HEK293 cells transfected with control siRNA or RNF4 siRNA and PSMB5-FLAG expression plasmid were pretreated with MG132 then cotreated with CPT (20 μM, 1 hour). Cells were subjected to FLAG-IP and WB with anti-TOP1, anti–TOP1-DPC, and anti-FLAG antibodies. (G) Same as (F) except that ETP (200 μM) was used to detect TOP2-PSMB5 interaction using anti-TOP2α and anti-TOP2β antibodies.
As K48 ubiquitylation is the established modification for the UPP, we determined the Ub linkage(s) of the TOP-DPC mediated by RNF4 using DUST assays. Transfection of HEK293 cells with the single lysine Ub construct HA-Ub K48 or HA-Ub K63 showed that RNF4 overexpression induced K48 ubiquitylation of both TOP1- and TOP2-DPCs (fig. S7, D and E). This result is consistent with RNF4-mediated ubiquitylation of TOP-DPCs as a signal for their proteasomal degradation. The significance of the RNF4-mediated K63 ubiquitylation of the TOP-DPC warrants further investigations.
As proteasome inhibition precludes the induction γH2AX elicited by topoisomerase inhibitors (15, 47), we examined the contribution of PIAS4 and RNF4 for γH2AX induction after CPT or ETP treatment. IF microscopy in U2OS cells treated with CPT or ETP showed that knocking down PIAS4 and RNF4 reduced the γH2AX signal (fig. S7, F to I), in agreement with PIAS4-RNF4–mediated proteasomal degradation of TOP-DPCs. As described above, cells decreased TOP-DPCs to undetectable levels after 6 hours of topoisomerase inhibitor treatments regardless of PIAS4 or RNF4 status (Fig. 4, A to D), indicating that other pathways are acting to disjoin TOP-DPCs. In agreement with these observations, we showed by WB that γH2AX signal in PIAS4- and RNF4-deficient HEK293 cells elevated to the same levels as that in control cells after 8 hours of topoisomerase inhibitor treatments (fig. S7, J and K).
We next examined the interaction of two key proteasome components with the TOP-DPCs in RNF4-deficient cells. First, we assessed whether topoisomerases interact with PSMD14, the proteasome non–adenosine triphosphatase regulatory subunit 14 that deubiquitylates substrates before degradation (48). Following treatment with CPT or ETP in the presence of MG132, we performed FLAG-IP in PSMD14-FLAG expressing HEK293 cells and observed bands of higher molecular weights than the free TOP1 using anti-TOP1 and anti–TOP1-DPC antibodies (Fig. 5D, top and middle panels). Similarly, bands above the predicted size of free TOP2α and TOP2β were detected using anti-TOP2α and TOP2β antibodies (Fig. 5E, top and middle panels). We infer that the upper bands are poly-Ub-topoisomerase species. Down-regulation of RNF4 diminished the interaction of topoisomerases with PMSD14 (Fig. 5, D and E, right lane), suggesting that PSMD14 targets RNF4-ubiquitylated TOP-DPCs for deubiquitylation. In addition, we observed that unmodified topoisomerases are also coimmunoprecipitated by PSMD14-FLAG-IP (Fig. 5, D and E), raising the possibility that topoisomerases remain associated with PSMD14 after deubiquitylation and before translocation to other subunits of the proteasome.
We next transfected cells with a plasmid expressing FLAG-tagged PSMB5, a chymotrypsin-like proteolytic subunit of the 20S core particle, followed by treatment with CPT or ETP in the presence of MG132. FLAG-IP showed that PSMB5 interacted with unmodified TOP1, TOP2α, and TOP2β (Fig. 5, F, and G, top and middle panels). This finding is consistent with the translocation of the topoisomerase polypeptides into the proteasome catalytic core following their deubiquitylation. Down-regulation of RNF4 was found to reduce the TOP1, TOP2α, and TOP2β signals (Fig. 5, F and G, right lane). Together, these results demonstrate that the proteasome targets RNF4-ubiquitylated TOP1-, TOP2α-, and TOP2β-DPCs for degradation.
The SUMO-Ub-proteasome pathway for repairing TOP-DPCs functions in yeast
Previous work in yeast has lent support to the hypothesis that SUMOylation and SUMO-dependent ubiquitylation are a critical pathway for repairing TOP-DPCs in yeast (34, 35). We wished to examine the overall contribution of the orthologous SUMO-dependent ubiquitylation pathway in light of some of the other pathways that have been described in yeast. We decided to carefully revisit the importance of this pathway in S. cerevisiae. We examined HA-tagged TOP1 or TOP2 levels following exposure to CPT and ETP and found that these agents induced reduction in cellular TOP1 and TOP2, respectively (fig. S8, A and B), as previously observed in mammalian cells. MG132 blocked the drug-induced depletion of the tagged proteins (fig. S8, A and B), confirming that proteasome-mediated degradation of topoisomerases occurs upon TOP-DPC induction in yeast. Proteasome inhibition enhanced yeast cell sensitivity to both CPT and ETP (fig. S8, C and D). This result indicates that proteasomal degradation of TOP-DPCs likely functions as a repair pathway for these lesions.
We also examined whether topoisomerases are modified by SUMO and Ub upon DPC formation in yeast. HA-antibody IP showed that both TOP1 and TOP2 were conjugated with polymeric Smt3 (yeast SUMO-1) and Ub and that addition of CPT or ETP enhanced Smt3- and Ub-topoisomerase conjugates (Fig. 6, A and B). To assess levels of SUMO and Ub modifications of TOP-DPCs, we adapted the ICE assay for use in yeast (37). Low levels of TOP1- and TOP2-DPCs, as well as their ubiquitylation and SUMOylation, were detected in the absence of topoisomerase inhibitors (Fig. 6, C and D). This low level of DPCs may reflect either spontaneous levels of DPCs or a small amount of background carryover due to the challenges of rapid nucleic acid purification in yeast. Treatment with topoisomerase inhibitors clearly induced detectable TOP-DPCs and MG132 enhanced the TOP-DPCs, as well as their SUMOylation and ubiquitylation.
Fig. 6. The SUMO-Ub-proteasome pathway is conserved in yeast.
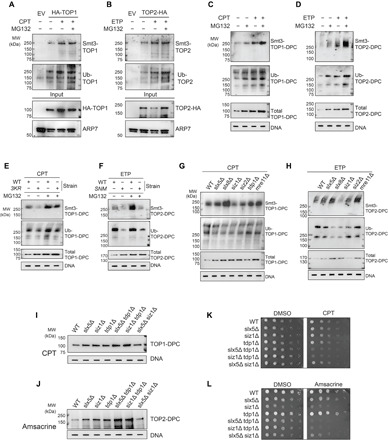
(A) HA-TOP1 expressing YMM10 cells were treated with CPT (20 μg/ml) ± MG132 for 30 min, followed by HA-IP-WB. (B) TOP2-HA expressing YMM10 cells were treated with ETP (200 μg/ml) ± MG132 for 30 min, followed by HA-IP-WB. (C) ICE assay in YMM10 cells expressing HA-TOP1 after 1-hour treatment with DMSO, MG132, or CPT (20 μg/ml) ± MG132. (D) ICE assay in YMM10 cells expressing TOP2-HA after 1-hour treatment with DMSO, MG132, or ETP (200 μg/ml) ± MG132. (E) ICE assay in YMM10 cells expressing HA-TOP1 WT or HA-top1 K65,91,92R after 1-hour treatment with CPT (20 μg/ml) ± MG132. (F) ICE assay in YMM10 cells expressing TOP2-HA WT or top2 SNM-HA after 1-hour treatment with ETP (200 μg/ml) ± MG132.(G) ICE assay in HA-TOP1 expressing BY4741 strains treated with CPT (20 μg/ml, 1 hour). (H) ICE assay in TOP2-HA expressing BY4741 strains treated with ETP (200 μg/ml, 1 hour). (I) ICE assay in HA-TOP1 expressing BY4741 strains treated with CPT (20 μg/ml, 1 hour). (J) ICE assay in TOP2-HA expressing BY4741 strains treated with amsacrine (200 μg/ml, 1 hour). (K) HA-TOP1 expressing BY4741 cells were plated on DMSO or CPT (0.5 μg/ml) containing plates and photographed after 2 days. (L) TOP2-HA expressing BY4741 cells were plated on DMSO or amsacrine (20 μg/ml) containing plates and photographed after 2 days.
To investigate whether SUMOylation of TOP-DPCs affects their ubiquitylation, we transformed the YMM10 strain (49) with the top1 K65R, K91R, K92R (top1 3KR) (50) or top2 Sumo No More (top2-SNM) (51) construct, both of which comprise mutations disrupting their respective SUMO consensus sites. The top1 3KR and top2 SNM showed decrease of SUMO-TOP-DPCs compared with the strains harboring the WT plasmids [Fig. 6, E and F; confirmation of the expressions by WB is shown in fig. S8 (E and F)]. Deficiency in SUMOylation reduced TOP-DPC ubiquitylation, suggesting that, like in human cells, SUMOylation induces TOP-DPC ubiquitylation in yeast.
To identify the SUMO ligase(s) and STUbL(s) for TOP-DPC, we performed ICE assays in yeast strains lacking the Slx5-Slx8 complex, the orthologous RNF4 proteins. Depletion of either Slx5 or Slx8 reduced the Ub-TOP-DPCs [Fig. 6, G and H; fig. S8 (G and H) shows cellular topoisomerase levels in each strain] and elevated the SUMO-TOP-DPCs, consistent with the interpretation that the Slx5-Slx8 complex removes SUMOylated TOP-DPCs. Gene deletion for Siz1, but not Siz2, the Siz/PIAS family proteins in S. cerevisiae, reduced the SUMOylation of TOP-DPCs and their ubiquitylation (Fig. 6, G and H), supporting the conclusion that SUMOylation of the TOP-DPC recruits the STUbL Slx5-Slx8 for their ubiquitylation.
An important pathway for repairing both TOP1- and TOP2-DPCs is TDP1 (8, 52), which hydrolyzes the phosphotyrosyl bond to excise the DPCs. We assessed genetic connections between SIZ1, SLX5, SLX8, and TDP1. slx5∆ tdp1∆ and siz1∆ tdp1∆ double mutants exhibited higher TOP-DPCs than did the respective single mutants [Fig. 6, I and J; fig. S8 (I and J) show cellular topoisomerase levels in each strain; fig. S8 (K and L) show densitometric analyses]. slx5∆ siz∆ double mutants and the respective single mutants exhibited similar levels of TOP-DPCs (Fig. 6, I and J), suggesting the epistasis between SIZ1 and SLX5 in TOP-DPC repair. Comparison of the sensitivity of the mutants to topoisomerase inhibitors by spotting cultures onto plates containing CPT or the TOP2 inhibitor amsacrine showed reduced growth of the slx5∆ tdp1∆ and siz1∆ tdp1∆ double-mutant strains (Fig. 6, K and L). These results demonstrate the functional role of the Siz1-Slx5 pathway that appears separate from the pathway defined by TDP1 for both TOP1 and TOP2-DPCs. Future experiments will be needed to delineate these two pathways with other repair functions such as those defined by WSS1 (17) and DDI1 (20).
DISCUSSION
We have demonstrated that an evolutionally conserved SUMO-Ub-proteasome pathway directly participates in the repair of both TOP1 and TOP2-DPCs in yeast and human cells. The relevant SUMO ligases are human PIAS4 and yeast Siz1 and the STUbLs human RNF4 and yeast Slx5-Slx8 (Fig. 7). For these studies, we designed the DUST assay to detect Ub- and SUMO-TOP-DPCs, which we used to show that SUMO-2/3, SUMO-1, and Ub are conjugated to TOP-DPCs consecutively. SUMO-2/3 conjugation occurs rapidly (in less than 10 min), while SUMO-1 and Ub conjugations reach maximal levels at slightly later times (at 30 and 60 min, respectively). Our finding that SUMO-2/3 down-regulation prevents both SUMO-1 and Ub modifications of TOP-DPCs (Fig. 4, C and E) suggests a regulatory role for SUMO-2/3 modification in the signaling and repair of TOP-DPCs. Note that a recent study showed that SUMO-2/3, but not SUMO-1, modification alters the conformation of TOP2-DPCs, thereby enables TDP2 to excise the tyrosyl-DNA bond (26). This suggests that SUMO-2/3 modification may play broad roles in regulating pathway choice in the repair of TOP2-DPCs and potentially TOP1-DPCs.
Fig. 7. Model for the SUMO-Ub-proteasome pathway in the repair of TOP-DPCs.
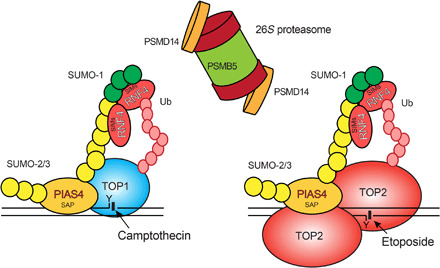
TOP1- and TOP2-DPCs are generated upon exposure to the topoisomerase inhibitors CPT and ETP. Both TOP1- and TOP2-DPCs are rapidly conjugated with SUMO-2/3, which is catalyzed by PIAS4 through its DNA binding SAP domain. PIAS4 subsequently deposits SUMO-1 moieties onto the SUMO-2/3 polymer to terminate its elongation. RNF4 recognizes and attaches K48-linked poly-Ub chains to the SUMOylated TOP-DPCs through its SUMO-interacting motif (SIM) domains. The 26S proteasome targets the ubiquitylated TOP-DPCs using the PSMD14 subunit for deubiquitylation and the PSMB5 subunit for proteolysis. The yeast orthologs of PIAS4 and RNF4 are Siz1 and Slx5-Slx8, respectively.
SUMO-1 modification of free TOP1 has been previously demonstrated in mammalian cells (53). Similarly, SUMOylation of TOP2 regulates the normal function of the enzyme in both yeast and mammalian cells (44, 54). We identified PIAS4 as a critical SUMO ligase for TOP1-, TOP2α-, and TOP2β-DPCs. PIAS4 was previously reported to catalyze SUMO-2 modification of TOP2α for its localization to centromeres to complete decatenation in Xenopus laevis (44). In agreement with previous work (44), we found that PIAS4 and topoisomerases interact in the absence of topoisomerase inhibitors (fig. S4, A to E). It is possible that PIAS4 SUMOylates topoisomerases and colocalizes with them to chromatin where the enzymes can continue to undergo additional SUMOylation. This finding provides additional support for the model that topoisomerases associate with factors that may provide for their removal if DNA rejoining is inhibited and that association between removal factors and the topoisomerases may occur even in the absence of formation of trapped TOP-DPCs (26). The regulatory role of PIAS4-mediated SUMOylation in DDR has been previously demonstrated (32). It is unclear at present whether TOP-DPC SUMOylation by PIAS4 is a subset of a more extensive SUMO response at DNA damage sites affecting other DDR and repair complexes including TDP1 (55), 53BP1, BRCA1, and Rad52 (29, 32, 56). Alternately, the initial SUMOylation of topoisomerases may be part of ordinary chromatin recruitment, and their continued presence on DNA can lead to the elevated levels of SUMOylation observed when the enzymes are trapped on DNA. By this latter model, the residence time of the enzyme would be the determinant of SUMOylation levels, and trapping of TOPccs would promote long residence times, elevate levels of SUMOylation, and initiate the repair mechanisms described here and potentially promote other repair pathways as well.
Our study connects SUMOylation of TOP-DPCs to their ubiquitylation through the STUbL RNF4 in humans and Slx5-Slx8 in yeast. Suppressing SUMOylation reduced TOP-DPC ubiquitylation. RNF4/Slx5-Slx8 are pivotal components, as demonstrated by our finding that depletion of RNF4 in human cells and Slx5-Slx8 in yeast reduce the levels of Ub-TOP-DPCs and elevates SUMO-TOP-DPCs. As a well-studied STUbL, RNF4 is activated upon formation of poly-SUMOylated substrates and recruited through its SIM domains and DNA binding motif within its RING domain (45). In yeast, the epistasis between SIZ1 and SLX5/SLX8 that we found for both levels of modifications and cell survival establishes the importance of the SIZ1-SLX5/SLX8 pathway for protecting from TOP-DPCs.
Our experiments show that PIAS4-mediated SUMOylation is activated independently of replication, transcription, and DDR and that RNF4-catalyzed TOP-DPC ubiquitylation is dependent on SUMOylation (fig. S6, C and D). These data signify a role of SUMOylation as an important protective factor against stalled TOP-DPCs, in part, by priming them for RNF4-mediated proteasomal degradation. While our work demonstrates that SUMOylation of human TOP1- and TOP2-DPCs induces proteasomal degradation, which is replication/transcription-independent; there are other proteasome pathways that respond specifically to replication and transcription. The original demonstration of proteolysis of trapped TOP1 and TOP2β was shown to be dependent on transcriptional elongation (13, 14) and replication fork progression (15). These transcription- and replication-mediated degradation pathways are likely modulated by the Cullin-RING Ub ligases (15) rather than STUbLs and may be SUMO-independent.
The metalloprotease SPRTN also engages in the proteolytic degradation of TOP-DPCs and general DPCs (3, 16). SPRTN activity does not require ubiquitylation of DPCs, whereas the proteasome primarily targets ubiquitylated DPCs (16, 57). In addition, SPRTN is predominantly associated with DNA synthesis as a component of the replisome and acts in a replication-dependent manner (16). Our work suggests that the proteasome can also be triggered by TOP-DPC SUMOylation independently of replication, transcription, and general DDR. Given that DPCs form independently of DNA transactions and cell cycle, it is conceivable that the proteasome plays a cardinal role in DPC repair throughout cell cycle to maintain genome stability.
Since TOP1 and TOP2 have distinct biochemical functions, the actions of drugs targeting these enzymes are also often regarded separately. Our work provides more direct connections between the repair of DNA damage induced by the enzymes individually and the relevant repair pathways, which at least in part are shared regardless of whether the damage is induced by TOP1 or TOP2 (Fig. 7). Given this commonality of the repair pathways, further studies are warranted to establish whether the SUMO-Ub-proteasome pathway described here extends to other enzymatic DPCs including those generated by DNA methyltransferases and those induced chemically by agents such as formaldehyde and chemotherapeutic cross-linking agents such as platinum derivatives (58).
MATERIALS AND METHODS
Chemicals
A full list of chemicals used in this study is available in table S1.
Human cell culture
A full list of human cell lines used in this study is available in table S2.
Generation of gene KO cells using CRISPR-Cas9
To stably knock out the genes encoding TOP2B, PIAS4, or RNF4, the CRISPR-Cas9 genome editing method was used. To delete TOP2B in HeLa cells, two 25–base pair [bp; minus the protospacer adjacent motifs (PAMs)] guide RNAs targeting TOP2B exon 7 were designed using the CHOPCHOP tool and cloned into the Bbs1 site of the Cas9 expressing guide RNA vectors pX458 and pX459, respectively. The plasmids were transfected using Lipofectamine 3000 (Thermo Fisher Scientific) in HeLa cells. Transfected cells were enriched by selection in puromycin (0.5 μg/ml) containing media for 3 days before isolation of single clones and screening for loss of TOP2β by WB. To delete PIAS4 in HCT116 cells, two guide RNAs targeting PIAS4 exon 2 were designed and cloned into pX458 and pX459, respectively. The plasmids were transfected in HCT116 cells using Lipofectamine 3000, followed by selection in media containing puromycin (1 μg/ml) for 3 days before isolation of single clones and screening for loss of PIAS4 by WB. To delete RNF4 gene in MCF7 cells, two guide RNAs targeting RNF4 exon 3 were designed using the CHOPCHOP tool and cloned into the Bbs1 site of the Cas9 expressing guide RNA vectors pX458 and pX459, respectively. The RNF4 guide constructs were transfected into MCF7 cells, followed by puromycin (0.5 μg/ml) selection and single clone isolation. Deletion of RNF4 gene was confirmed by Sanger sequencing and loss of RNF4 expression by WB. Sequences of the oligonucleotides encoding the guide RNAs are found in table S3.
Expression plasmids and siRNAs in human cells
For human TOP1 expression in HEK293 cells, N-terminally 6× His tagged TOP1 complementary DNA (cDNA) was amplified by polymerase chain reaction (PCR) using pGALhTOP1 yeast plasmid as template. The PCR product was inserted into Psp XI/Not I sites of a pT-REx-DEST Gateway vector (Invitrogen). For human TOP2α expression in HEK293 cells, N-terminally FLAG-tagged TOP2α cDNA was amplified by PCR using pMJ1hTOP2α yeast plasmid as a template and then inserted into Psp XI/Not I sites of the pT-REx-DEST Gateway vector. N-terminally FLAG-tagged TOP2β cDNA (derived from pHT500hTOP2β yeast plasmid) was PCR-amplified and inserted into Psp XI/Not I sites of the pT-REx-DEST Gateway vector. Sequences of the oligonucleotide primers are found in table S3. A full list of human expression plasmids used in this study is available in table S4.
siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. All siRNAs were used at a final concentration of 50 nM. A full list of siRNAs used in this study is available in table S5.
Site-directed mutagenesis in mammalian expression vectors
pTrex-6 × His-TOP1 Y723F and pTrex-6 × His-TOP1 K117, K153, and K103R (CPT3KR) were generated by the QuickChange II XL site-directed mutagenesis (SDM) Kit (Agilent, catalog no. 200521). HA-Ub K6R, K11R, K27R, K29R, K33R, K48R, K63R, HA-SUMO-1 K7R, HA-SUMO-2 K11R, FLAG-PIAS4 C342A, FLAG-PIAS4 SAPΔ, RNF4 SIM2, 3, 4 Δ-FLAG, and RNF4 H156A-FLAG were generated by the Q5 SDM Kit (NEB, catalog no. E0554S). Sequences of the oligonucleotide primers are found in table S3.
Antibodies
A full list of antibodies used in this study is available in table S6.
WB in human cells
CPT-induced TOP1 degradation and ETP-induced TOP2 degradation were monitored by WB of the alkaline lysates prepared from drug-treated HEK293 cells. Following treatment, cells were washed with Dulbecco’s modified Eagle medium and incubated at 37°C in a CO2 incubator for 30 min then lysed with 100 μl of an alkaline lysis buffer (200 mM NaOH and 2 mM EDTA). Alkaline lysates were neutralized by the addition of 100 μl of 1 M Hepes buffer (pH 7.3), followed by mixing with 10 μl 100 mM CaCl2, 1 μl 2 M dithiothreitol (DTT), and 2 μl 100× protease inhibitor cocktail (Thermo Fisher Scientific) and 200 units of micrococcal nuclease (Thermo Fisher Scientific; 100 units/μl). The resulting mixtures were incubated on ice for 1 hour after which 70 μl of 4× Laemmli buffer was added to each sample. The lysates were boiled for 10 min, analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotted with various antibodies as indicated. Other proteins were detected by lysing cells with radio-IP assay (RIPA) buffer [150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris (pH 7.5), 1 mM DTT, and protease inhibitor cocktail].
Viability assay in human cells
To measure the sensitivity of cells to drugs, mammalian cell lines were continuously exposed to various concentrations of the drugs and assessed for viability. Cells were trypsinized, and 10,000 cells were seeded in 96-well white plates (PerkinElmer Life Sciences, 6007680) in triplicate in 100 μl of medium per well overnight. The next day, cells were exposed to the drugs and incubated for 72 hours in the presence of the drugs. Cellular viability was determined using the ATPlite 1-step kits (PerkinElmer). Briefly, 50 μl of ATPlite solution was added in 96-well plates per well, respectively. After 5 min, luminescence was measured with an EnVision 2104 Multilabel Reader (PerkinElmer). The adenosine 5′-triphosphate (ATP) level in untreated cells was defined as 100%. Viability (%) of treated cells was defined as ATP-treated cells/ATP-untreated cells × 100.
ICE assay in human cells
TOP-DPCs were isolated and detected using ICE assay as previously described (37). Briefly, cells were lysed in sarkosyl solution [1% (w/v)] after treatment. Cell lysates were sheared through a 25-gauge 5/8 needle (10 strokes) to reduce the viscosity of DNA and layered onto CsCl solution [150% (w/v)], followed by centrifugation in NVT 65.2 rotor (Beckman coulter) at 42,000 rpm for 20 hours at 25°C. The resulting pellet containing nucleic acids and TOP-DPCs was obtained and dissolved in TE (Tris-EDTA) buffer. The samples were quantitated and subjected to slot-blot for immunoblotting with various antibodies as indicated. DNA (2 μg) was applied per sample. For mass spectrometric analysis, ICE samples were treated with ribonuclease A (RNase A) to eliminate RNA contamination. Experiments were performed in triplicate and TOP-DPCs were quantified by densitometric analysis using ImageJ.
DUST assay in human cells
The DUST assay is an extension of the RADAR assay (39). After topoisomerase inhibitor treatments, 1 × 106 human cells in 35-mm dish per sample were washed with 1× phosphate-buffered saline (PBS) and lysed with 600 μl of DNAzol (Invitrogen), followed by precipitation with 300 μl of 200 proof ethanol. The nucleic acids were collected, washed with 75% ethanol, resuspended in 200 μl of TE buffer, and then heated at 65°C for 15 min, followed by shearing with sonication (40% output for 10-s pulse and 10-s rest for four times). The samples were centrifuged at 15,000 rpm for 5 min at 4°C, and the supernatant were collected and treated with RNase A (100 μg/ml) for 1 hour at 4°C, followed by addition of 1:10 volume of 3 M sodium acetate sodium acetate and 2.5 volume of 200 proof ethanol. After 20-min full-speed centrifugation, the DNA pellet was recovered and resuspended in 100 μl of TE buffer. The sample (1 μl) was removed for spectrophotometric measurement of absorbance at 260 nm to quantitate DNA content (NanoDrop). DNA (10 μg) from each sample was digested with 50 units of micrococcal nuclease (100 units/μl; Thermo Fisher Scientific) in the presence of 5 mM CaCl2, followed by gel electrophoresis on 4 to 15% precast polyacrylamide gel (Bio-Rad) for immunodetection of total TOP-DPCs, SUMOylated TOP-DPCs, and ubiquitylated TOP-DPCs using specific antibodies. Because of the extremely low abundance of SUMOylated and ubiquitylated TOP-DPCs, samples were run in parallel gels to detect total, SUMOylated, and ubiquitylated TOP-DPCs separately instead of stripping and reprobing the same membrane for their detection. In addition, 2 μg of each sample was subjected to slot-blot for immunoblotting with anti–double-stranded DNA (dsDNA) antibody as a loading control to verify that amounts of DNA were digested with micrococcal nuclease.
His pull-down assay in denaturing and native conditions
One million human cells were washed with 1× PBS and incubated with 220 μl of IP lysis buffer [5 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.2% Triton X-100, 5% glycerol, 1 mM DTT, and 20 mM N-ethylmaleimide (Sigma Aldrich) and protease inhibitor cocktail] on a shaker for 15 min at 4°C, followed by sonication and centrifugation. The supernatant was collected and treated with 1 μl of benzonase (250 units/μl; EMD Millipore) for 1 hour. An aliquot (20 μl) of the lysate of each treatment group was saved as input. The rest of the lysates was divided in two groups: native pull-down and denaturing pull-down. For native pull-down, lysates were resuspended in 900 μl of IP lysis buffer containing 10 mM imidazole and 100 μl of equilibrated Ni–nitrilotriacetic acid (NTA)–agarose and rotated overnight at 4°C. The resin was spun down and washed with TI buffer two times [25 mM Tris-HCL and 20 mM imidazole (pH 6.8)], followed by resuspension in 2× Laemmli buffer for SDS-PAGE and immunoblotting with various antibodies as indicated. For denaturing pull-down, lysates were resuspended in 900 μl of buffer A [6 M guanidine-HCL, 0.1 M Na2HPO4/NaH2PO4 (pH 6.5), and 10 mM imidazole (pH 8.0)] containing 100 μl of equilibrated Ni-NTA–agarose and rotated overnight at 4°C. The samples were washed, processed, and immunoblotted as described for the native IP.
FLAG-IP in denaturing and native conditions
Human cells were washed with 1× PBS and processed exactly as described for the His pull-down assay. The lysates were again divided in two groups: native IP and denaturing IP. For native IP, lysates were resuspended in 900 μl IP lysis buffer containing 2.5 μg of anti-FLAG M2 antibody and rotated overnight at 4°C. Protein A/G PLUS-agarose slurry (50 μl) was added and incubated with the lysates for another 4 hours. After centrifugation, immunoprecipitates were washed with RIPA buffer two times and then resuspended in 2× Laemmli buffer for SDS-PAGE and immunoblotting with various antibodies as indicated. For denaturing pull-down, lysates were resuspended in 900 μl of RIPA buffer containing 2.5 μl of anti-FLAG M2 antibody and rotated overnight at 4°C. Protein A/G PLUS-agarose slurry (50 μl) was added and incubated with the lysates for another 4 hours. After centrifugation, immunoprecipitates were washed with RIPA buffer two times and then resuspended in 2× Laemmli buffer for SDS-PAGE and immunoblotting with various antibodies as indicated.
Mass spectrometry
Samples were either separated by SDS-PAGE for in-gel trypsin digestion or in-solution digested with trypsin following the filter-aided sample preparation protocol. Dried peptides were solubilized in 2% acetonitrile, 0.5% acetic acid, and 97.5% water for mass spectrometry analysis. They were trapped on a trapping column and separated on a 75 μm by 15 cm, 2-μm Acclaim PepMap reverse phase column (Thermo Fisher Scientific) using an UltiMate 3000 RSLCnano HPLC (Thermo Fisher Scientific). Peptides were separated at a flow rate of 300 nl/min followed by online analysis by tandem mass spectrometry using a Thermo Orbitrap Fusion mass spectrometer. Peptides were eluted into the mass spectrometer using a linear gradient from 96% mobile phase A (0.1% formic acid in water) to 55% mobile phase B (0.1% formic acid in acetonitrile). Parent full-scan mass spectra were collected in the Orbitrap mass analyzer set to acquire data at 120,000 full width at half maximum resolution; ions were then isolated in the quadrupole mass filter, fragmented within the HCD (higher-energy collisional dissociation) cell (HCD normalized energy of 32%; stepped, ±3%), and the product ions were analyzed in the ion trap. Proteome Discoverer 2.2 (Thermo Fisher Scientific) was used to search the data against human proteins from the UniProt database using SequestHT. The search was limited to tryptic peptides, with maximally two missed cleavages allowed. Cysteine carbamidomethylation was set as a fixed modification, and methionine oxidation was set as a variable modification. Diglycine modification to lysine was set as a variable modification for experiments to identify sites of enzymatic PTMs. The precursor mass tolerance was 10 parts per million, and the fragment mass tolerance was 0.6 Da. The Percolator node was used to score and rank peptide matches using a 1% false discovery rate.
Recombinant proteins
Human TOP1 was purified from baculovirus as previously described (59). Human TOP2α and TOP2β were purified from yeast strains JEL1 top1Δ transformed with 12-URA-B 6 × His-hTOP2α and JEL1 top1Δ transformed with 12-URA-C 6 × His-hTOP2β. TOP2 expression was induced by galactose. Yeast cells were lysed in equilibration buffer [300 mM KCl, 10 mM imidazole, 20 mM Tris-HCl (pH 7.7), 10% glycerol, and protease inhibitors] by glass bead homogenization. Lysates were incubated with Ni-NTA resin (Thermo Fisher Scientific) and washed using wash buffer #1 [300 mM KCl, 30 mM imidazole, 20 mM Tris-HCl (pH 7.7), 10% glycerol, and protease inhibitors] and then wash buffer #2 [150 mM KCl, 30 mM imidazole, 20 mM Tris-HCl (pH 7.7), 10% glycerol, and protease inhibitors]. hTOP2α and hTOP2β were eluted on a Poly-Prep chromatography column (Bio-Rad) with elution buffer [150 mM KCl, 300 mM imidazole, 20 mM Tris-HCl (pH 7.7), 10% glycerol, and protease inhibitors]. The peak protein fractions were dialyzed in dialysis buffer [750 mM KCl, 50 mM Tris-HCl (pH 7.7), 20% glycerol, 0.1 mM EDTA, and 0.5 mM DTT], and His tag was removed using TEV protease (Sigma-Aldrich). Recombinant human PIAS4 was purified from HEK293 cells overexpressing FLAG-PIAS4 using the FLAG IP Kit (Millipore, catalog no. FLAGIPT1). A full list of recombinant proteins used in this study is available in table S7.
In vitro SUMO conjugation assay
The 10 μl in vitro SUMOylation assay reactions in were prepared using 1× SUMO conjugation reaction buffer (R&D Systems, catalog no. B-60) containing 10 mM Mg2+-ATP solution (pH 7.0) (R&D Systems, catalog no. B-20), protease inhibitor cocktail (Thermo Fisher Scientific), 100 nM TOP1, TOP2α or TOP2β, 10 μM SUMO-1 or SUMO-2, 50 nM SUMO E1, and 0.1 μM UBC9 and PIAS4 of indicated concentrations. Reactions were incubated at 37°C for 30 min, followed by SDS-PAGE and immunoblotting with anti–SUMO-1 or anti–SUMO-2/3 antibodies.
In vitro Ub conjugation assay
The 10 μl in vitro ubiquitylation assay reactions were set up in 1× Ub conjugation reaction buffer (R&D systems, catalog no. B-70) containing 10 mM Mg2+-ATP solution (pH 7.0; R&D systems, catalog no. B-20), protease inhibitor cocktail, 100 nM TOP1, TOP2α, or TOP2β, 10 μM Ub, 50 nM Ub E1, 0.1 μM UbcH5a, and RNF4 of indicated concentrations. Reactions were incubated at 37°C for 30 min, followed by SDS-PAGE and immunoblotting with anti-Ub antibody.
In vitro SUMOylation-ubiquitylation coupled assay
In vitro SUMOylation assay reactions were conducted in 1× SUMO conjugation reaction buffer containing 350 nM TOP1, TOP2α, or TOP2β, 10 μM SUMO-1 or SUMO-2, and 50 nM SUMO E1, in the absence or presence of 0.1 μM UBC9 and 0.5 μM PIAS4 at 37°C for 30 min. The reaction was then aliquoted and diluted in 1× Ub conjugation reaction buffer containing 10 mM Mg2+-ATP solution (pH 7.0), protease inhibitor cocktail, 10 μM Ub, 50 nM Ub E1, 0.1 μM UbcH5a, and RNF4 of indicated concentrations. Reactions were incubated at 37°C for 30 min, followed by SDS-PAGE and immunoblotting with anti-Ub antibody.
TOP1-DPC IF
TOP1-DPC IF was performed using previously described protocol (38) with slight modification. U2OS cells were grown on chamber slides and treated with CPT in the absence or presence of indicated inhibitors. After treatments, cells were washed with PBS and fixed for 15 min at 4°C in 4% paraformaldehyde in 1× PBS and permeabilized with 0.25% Triton X-100 in PBS for 15 min at 4°C. The samples were incubated in 2% SDS at room temperature for 10 min, washed, and blocked with PBS containing 0.01% Triton X-100, 0.05% Tween 20, and 1% bovine serum albumin (PBSTT–1% BSA). After incubating overnight with TOP1-DPC antibody (Millipore Sigma) in PBSTT-BSA at 1:250 dilution at 4°C, cells were rinsed with PBSTT and incubated with Alexa Fluor 568–conjugated secondary antibody (Invitrogen) at 1:1000 in PBSTT-BSA for 1 hour in subdued light, washed, and mounted using mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; VECTASHIELD). Images were captured on an instant structured illumination microscope, processed using ImageJ, and analyzed using Imaris.
γH2AX IF
U2OS cells were seeded on chamber slides. After inhibitor treatment, cells were washed with PBS and fixed for 15 min at 4°C in 4% paraformaldehyde in PBS and permeabilized with 0.25% Triton X-100 in PBS for 15 min at 4°C. The samples were blocked with PBSTT–1% BSA, followed by overnight incubation with γH2AX antibody (Millipore Sigma) in PBSTT-BSA at 4°C, cells were rinsed with PBSTT, incubated with Alexa Fluor 4888–conjugated secondary antibody (Invitrogen) at 1:1000 in PBSTT-BSA for 1 hour in subdued light, washed, and mounted using mounting medium with DAPI (Vectashield). Images were captured on Zeiss LSM 880/Airyscan confocal microscope, processed using ImageJ, and analyzed using Imaris.
Proximity ligation assay
Duolink PLA fluorescence assay (Sigma-Aldrich , catalog no. DUO92101) was performed following manufacturer’s instruction. Briefly, U2OS cells were seeded on coverslips and treated with CPT or ETP. After inhibitor treatment, cells were washed with 1× PBS and fixed for 15 min at 4°C in 4% paraformaldehyde in PBS and permeabilized with 0.25% Triton X-100 in PBS for 15 min at 4°C. The coverslips were blocked with Duolink-blocking solution and incubated with indicated antibodies in the Duolink antibody diluent overnight, followed by incubation with PLUS and MINUS PLA probes, ligation, and amplification. Coverslips were then washed and mounted with using mounting medium with DAPI. Images were captured on wide field microscope, processed using ImageJ, and analyzed using Imaris.
Yeast strains and plasmids
BY4741 and YMM10 are two haploid strains of Saccharomyces cerevisiae, which were used as parental strains in this study. A full list of yeast strains used in this study in available is table S8. To generate disruption of TDP1 in BY4741 slx5Δ and siz1Δ strains, a PCR product containing the TDP1 ORF disrupted by the LEU2 marker was used in transformation of BY4741 slx5Δ and siz1Δ strains. To generate BY4741 slx5Δsiz1Δ double-mutant, BY4742 siz1Δ::KANMX4 and BY4741 slx5Δ::LEU2 were mated, and diploid cells were sporulated and microdissected. Mating type was determined using H317 MATa and H318 MATα ura2 tester strains. In the YMM10 strain, genes encoding nine different types of transmembrane drug-efflux pump proteins are deleted. For immunodetection of yeast TOP1, N-terminally HA-tagged TOP1 cDNA was amplified by PCR using primers and inserted in Nco I/Xho I sites of pYX112 vector. Strains were transformed with pYX112 as an empty vector or pYX112 HA-TOP1 in which TOP1 expression is driven from the TPI (triose phosphate isomerase) promoter. For immunodetection of yeast TOP2, strains were transformed with yCP50 as an empty vector or a yeast TOP2 to TOP3 × HA overexpression plasmid pDED1 TOP2 in which TOP2 expression is driven from the DED1 promoter. To enhance accumulation of CPT and ETP in yeast cells, Xho I–excised construct of DNA binding domain of PDR1 gene fused in frame to transcription repressor gene CYC8 from pBlueScript backbone was transformed to all BY4741 single-mutant derivatives to repress Pdr1-regulated genes (60). Sequences of the oligonucleotide primers are found in table S3.
Site-directed mutagenesis in yeast vectors
SUMOylation-deficient yeast TOP1 allele, top1-K65, 91, 92R, was constructed in pYX112 HA-TOP1 vector. SUMOylation-deficient yeast TOP2 allele, top2-SNM, was constructed in pDED1 TOP2 to TOP3 × HA vector. The mutagenesis reaction was conducted using the QuikChange II XL Mutagenesis Kit (Agilent, catalog no. 200521). Sequences of the oligonucleotide primers are found in table S3.
WB in yeast
Yeast containing deletions or plasmids were grown into log phase. After treatments, 2 × 108 cells were pelleted then washed with alkaline lysis buffer (200 mM NaOH and 2 mM EDTA). Cells were then resuspended in 700 μl of alkaline lysis buffer and lysed by homogenization using acid-washed glass beads (Millipore Sigma) at 4°C. After four cycles of homogenization (50 s each cycle, 5-min rest in between), lysates were centrifuged for 10 min at 14,000 rpm at 4°C, and 400 μl of supernatants was retrieved, followed by neutralization by the addition of 80 μl of 1 M HCl, 600 mM Tris (pH 8.0), 54 μl of 10× micrococcal nuclease buffer [50 mM CaCl2 and 500 mM Tris (pH 7.9)], and 1 μl of micrococcal nuclease (100 units/μl). The resulting mixtures were incubated on ice for 1 hour for releasing topoisomerases from the DNA by digestion. Protein concentration were determined by using the Bradford assay (Bio-Rad). The lysates were mixed with 4× Laemmli buffer and subjected to SDS-PAGE electrophoresis, followed by immunoblotting with indicated antibodies.
Clonogenic assays in yeast
Drug sensitivity assay in yeast cells was carried out in triplicate. Briefly, cells were grown on synthetic complete media lacking uracil (SC-URA) overnight then diluted to 2 × 106 cells/ml. After addition of CPT or amsacrine, cells were incubated, diluted, and plated at various time points as indicated onto SC-URA plates. Plates were incubated at 30°C, and the numbers of colonies were counted. Results were expressed relative to the number of viable colonies at the time of drug addition.
HA-IP in yeast
Yeast lysates were prepared by the alkaline lysis procedure and then neutralized as described for the yeast WB. The lysates were incubated with anti-HA antibody in yeast IP lysis buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1% NP-40, 0.2% Triton X-100, 5% glycerol, 1 mM DTT, 20 mM N-ethylmaleimide, and yeast protease inhibitor cocktail (Sigma Aldrich, P8215) at 4°C overnight. Protein A/G PLUS-agarose slurry (50 μl; Santa Cruz Biotechnology) was added and incubated with the lysates for another 4 hours. After centrifugation, immunoprecipitates were washed with yeast IP lysis buffer two times and then resuspended in 2× Laemmli buffer for SDS-PAGE and immunoblotting with indicated antibodies.
ICE assay in yeast
After treatments, yeast cells were pelleted and washed in 700 μl lysis buffer containing 6 M guanidinium thiocyanate, 1% sarkosyl, 4% Triton X 100, 10 mM Tris-HCl (pH 7.0), 20 mM EDTA, yeast protease inhibitor cocktail, 20 mM N-ethylmaleimide, and 1 mM DTT. Lysates were prepared as previously described (37) and ultracentrifuged for 18 hours at 42000 rpm in an NVT 65.2 rotor (Beckman coulter) at 25°C. Nucleic acids pellets containing TOP-DPCs were retrieved in ddH2O and digested with RNase A. Purified DNA samples were quantitated, and 10 μg of each sample was digested with micrococcal nuclease and subjected to SDS-PAGE for immunodetection of total TOP-DPCs, ubiquitylated TOP-DPCs, and SUMOylated TOP-DPCs. Each sample (2 μg) was subjected to slot-blot for immunoblotting with anti-dsDNA antibody to confirm equal amounts of DNA loading on the slot-blot.
Yeast growth in the presence of drug
For yeast growth on media containing drug, 3 μl of 1:5 serial dilution of growing cells was applied to SC-URA plates containing the indicated concentrations of drugs. Plates were incubated at 30°C for 2 days and photographed [photo credit: Yilun Sun, National Cancer Institute (NCI)/National Institutes of Health (NIH)].
Statistical analyses
Error bars on bar graphs represent standard deviation (SD), and P value was calculated using paired Student’s t test for independent samples.
Supplementary Material
Acknowledgments
We thank the Advanced Imaging and Microscopy (AIM) Resource of NIH and J. Chen, the co-operating director of AIM, for supporting some of our microscopic analyses; M. Lichten, Laboratory of Biochemistry and Molecular Biology, and NCI, for providing H317 MATa and H318 MATα ura2 tester strains; and J. Berger, Department of Department of Biophysics and Biophysical Chemistry, and John Hopkins University for providing 12-URA-B 6× His-hTOP2α and 12-URA-C 6× His-hTOP2β constructs. Funding: This study was supported by the Center for Cancer Research, the Intramural Program of the NCI (Z01 BC 006150) to Y.P., and by NIH grant R03 CA216010 (to J.L.N). Author contributions: Y.S., J.L.N., and Y.P. conceived the study. Y.S. designed the experiments, generated the reagents, and developed the methodology. Y.S. performed all the experiments except the mass spectrum analyses and the microscopic analyses in Fig. 1I and figs. S4 (D and E) and S5I, which were performed by L.M.M.J. and Y.P.S., respectively. K.C.N. assisted in certain experiments. Y.S., J.L.N., and Y.P. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and are deposited in Mendeley Data. Please use the following link to access the deposited dataset: https://data.mendeley.com/datasets/8tfbp8wkyh/draft?a=e55c6709-2dbc-4a72-b53a-f07a3eafbb22. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eaba6290/DC1
REFERENCES AND NOTES
- 1.E. C. Friedberg, S. J. Elledge, A. Lehmann, T. Lindahl, M. Muzi-Falconi, DNA Repair, Mutagenesis, and Other Responses to DNA Damage: A Subject Collection from Cold Spring Harbor Perspectives in Biology (Cold Spring Harbor perspectives in biology, Cold Spring Harbor Laboratory Press, 2014), pp. VII. [Google Scholar]
- 2.Jackson S. P., Bartek J., The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stingele J., Bellelli R., Boulton S. J., Mechanisms of DNA–protein crosslink repair. Nat. Rev. Mol. Cell Biol. 18, 563–573 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y., Sun Y., Huang S. N., Nitiss J. L., Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 17, 703–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fertala J., Vance J. R., Pourquier P., Pommier Y., Bjornsti M.-A., Substitutions of Asn-726 in the active site of yeast DNA topoisomerase I define novel mechanisms of stabilizing the covalent enzyme-DNA intermediate. J. Biol. Chem. 275, 15246–15253 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y., Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 8, 82–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Y., Saha S., Wang W., Saha L. K., Huang S. N., Pommier Y., Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair 89, 102837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pouliot J. J., Yao K. C., Robertson C. A., Nash H. A., Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 286, 552–555 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Cortes Ledesma F., El Khamisy S. F., Zuma M. C., Osborn K., Caldecott K. W., A human 5’-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461, 674–678 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Hartsuiker E., Neale M. J., Carr A. M., Distinct requirements for the Rad32Mre11 nuclease and Ctp1CtIP in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell 33, 117–123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao Y., Desai S. D., Ting C.-Y., Hwang J., Liu L. F., 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 276, 40652–40658 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Desai S. D., Liu L. F., Vazquez-Abad D., D’Arpa P., Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J. Biol. Chem. 272, 24159–24164 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Desai S. D., Zhang H., Rodriguez-Bauman A., Yang J.-M., Wu X., Gounder M. K., Rubin E. H., Liu L. F., Transcription-dependent degradation of topoisomerase I-DNA covalent complexes. Mol. Cell. Biol. 23, 2341–2350 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao H., Mao Y., Desai S. D., Zhou N., Ting C.-Y., Hwang J., Liu L. F., The topoisomerase IIβ circular clamp arrests transcription and signals a 26S proteasome pathway. Proc. Natl. Acad. Sci. U.S.A. 100, 3239–3244 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin C.-P., Ban Y., Lyu Y. L., Liu L. F., Proteasome-dependent processing of topoisomerase I-DNA adducts into DNA double strand breaks at arrested replication forks. J. Biol. Chem. 284, 28084–28092 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaz B., Popovic M., Newman J. A., Fielden J., Aitkenhead H., Halder S., Singh A. N., Vendrell I., Fischer R., Torrecilla I., Drobnitzky N., Freire R., Amor D. J., Lockhart P. J., Kessler B. M., McKenna G. W., Gileadi O., Ramadan K., Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Mol. Cell 64, 704–719 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stingele J., Schwarz M. S., Bloemeke N., Wolf P. G., Jentsch S., A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158, 327–338 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Borgermann N., Ackermann L., Schwertman P., Hendriks I. A., Thijssen K., Liu J. C., Lans H., Nielsen M. L., Mailand N., SUMOylation promotes protective responses to DNA-protein crosslinks. EMBO J. 38, e101496 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kojima Y., Machida Y., Palani S., Caulfield T. R., Radisky E. S., Kaufmann S. H., Machida Y. J., FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 11, 1318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serbyn N., Noireterre A., Bagdiul I., Plank M., Michel A. H., Loewith R., Kornmann B., Stutz F., The aspartic protease Ddi1 contributes to DNA-protein crosslink repair in yeast. Mol. Cell 77, 1066–1079.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Debéthune L., Kohlhagen G., Grandas A., Pommier Y., Processing of nucleopeptides mimicking the topoisomerase I–DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 30, 1198–1204 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao R., Schellenberg M. J., Huang S.-Y. N., Abdelmalak M., Marchand C., Nitiss K. C., Nitiss J. L., Williams R. S., Pommier Y., Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2·DNA and Top2·RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J. Biol. Chem. 289, 17960–17969 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao Y., Sun M., Desai S. D., Liu L. F., SUMO-1 conjugation to topoisomerase I: A possible repair response to topoisomerase-mediated DNA damage. Proc. Natl. Acad. Sci. U.S.A. 97, 4046–4051 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao Y., Desai S. D., Liu L. F., SUMO-1 conjugation to human DNA topoisomerase II isozymes. J. Biol. Chem. 275, 26066–26073 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Horie K., Tomida A., Sugimoto Y., Yasugi T., Yoshikawa H., Taketani Y., Tsuruo T., SUMO-1 conjugation to intact DNA topoisomerase I amplifies cleavable complex formation induced by camptothecin. Oncogene 21, 7913–7922 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Schellenberg M. J., Lieberman J. A., Herrero-Ruiz A., Butler L. R., Williams J. G., Muñoz-Cabello A. M., Mueller G. A., London R. E., Cortés-Ledesma F., Williams R. S., ZATT (ZNF451)–mediated resolution of topoisomerase 2 DNA-protein cross-links. Science 357, 1412–1416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatham M. H., Geoffroy M.-C., Shen L., Plechanovova A., Hattersley N., Jaffray E. G., Palvimo J. J., Hay R. T., RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 10, 538–546 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Poulsen S. L., Hansen R. K., Wagner S. A., van Cuijk L., van Belle G. J., Streicher W., Wikstrom M., Choudhary C., Houtsmuller A. B., Marteijn J. A., Bekker-Jensen S., Mailand N., RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. J. Cell Biol. 201, 797–807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galanty Y., Belotserkovskaya R., Coates J., Polo S., Miller K. M., Jackson S. P., Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462, 935–939 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin Y., Seifert A., Chua J. S., Maure J.-F., Golebiowski F., Hay R. T., SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 26, 1196–1208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galanty Y., Belotserkovskaya R., Coates J., Jackson S. P., RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 26, 1179–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson S. P., Durocher D., Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Heideker J., Prudden J., Perry J. J., Tainer J. A., Boddy M. N., SUMO-targeted ubiquitin ligase, Rad60, and Nse2 SUMO ligase suppress spontaneous Top1–mediated DNA damage and genome instability. PLOS Genet. 7, e1001320 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinacher R., Osman F., Lorenz A., Bryer C., Whitby M. C., Slx8 removes Pli1-dependent protein-SUMO conjugates including SUMOylated topoisomerase I to promote genome stability. PLOS ONE 8, e71960 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei Y., Diao L. X., Lu S., Wang H.-T., Suo F., Dong M.-Q., Du L.-L., SUMO-targeted DNA translocase Rrp2 protects the genome from Top2-Induced DNA Damage. Mol. Cell 66, 581–596.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 36.He X., Riceberg J., Soucy T., Koenig E., Minissale J., Gallery M., Bernard H., Yang X., Liao H., Rabino C., Shah P., Xega K., Yan Z. H., Sintchak M., Bradley J., Xu H., Duffey M., England D., Mizutani H., Hu Z., Guo J., Chau R., Dick L. R., Brownell J. E., Newcomb J., Langston S., Lightcap E. S., Bence N., Pulukuri S. M., Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 13, 1164–1171 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Anand J., Sun Y., Zhao Y., Nitiss K. C., Nitiss J. L., Detection of topoisomerase covalent complexes in eukaryotic cells. Methods Mol. Biol. 1703, 283–299 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Patel A. G., Flatten K. S., Peterson K. L., Beito T. G., Schneider P. A., Perkins A. L., Harki D. A., Kaufmann S. H., Immunodetection of human topoisomerase I-DNA covalent complexes. Nucleic Acids Res. 44, 2816–2826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiianitsa K., Maizels N., A rapid and sensitive assay for DNA–protein covalent complexes in living cells. Nucleic Acids Res. 41, e104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh E., Akopian D., Rape M., Principles of ubiquitin-dependent signaling. Annu. Rev. Cell Dev. Biol. 34, 137–162 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Tatham M. H., Jaffray E., Vaughan O. A., Desterro J. M., Botting C. H., Naismith J. H., Hay R. T., Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276, 35368–35374 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Yang M., Hsu C.-T., Ting C.-Y., Liu L. F., Hwang J., Assembly of a polymeric chain of SUMO1 on human topoisomerase I in vitro. J. Biol. Chem. 281, 8264–8274 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Matic I., van Hagen M., Schimmel J., Macek B., Ogg S. C., Tatham M. H., Hay R. T., Lamond A. I., Mann M., Vertegaal A. C. O., In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol. Cell. Proteomics 7, 132–144 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryu H., Furuta M., Kirkpatrick D., Gygi S. P., Azuma Y., PIASy-dependent SUMOylation regulates DNA topoisomerase IIα activity. J. Cell Biol. 191, 783–794 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rojas-Fernandez A., Plechanovova A., Hattersley N., Jaffray E., Tatham M. H., Hay R. T., SUMO chain-induced dimerization activates RNF4. Mol. Cell 53, 880–892 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plechanovová A., Jaffray E. G., Tatham M. H., Naismith J. H., Hay R. T., Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang A., Lyu Y. L., Lin C.-P., Zhou N., Azarova A. M., Wood L. M., Liu L. F., A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J. Biol. Chem. 281, 35997–36003 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Bard J. A. M., Goodall E. A., Greene E. R., Jonsson E., Dong K. C., Martin A., Structure and function of the 26S proteasome. Annu. Rev. Biochem. 87, 697–724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tombline G., Millen J. I., Polevoda B., Rapaport M., Baxter B., Van Meter M., Gilbertson M., Madrey J., Piazza G. A., Rasmussen L., Wennerberg K., White E. L., Nitiss J. L., Goldfarb D. S., Effects of an unusual poison identify a lifespan role for Topoisomerase 2 in Saccharomyces cerevisiae. Aging 9, 68–97 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X. L., Silver H. R., Xiong L., Belichenko I., Adegite C., Johnson E. S., Topoisomerase I-dependent viability loss in saccharomyces cerevisiae mutants defective in both SUMO conjugation and DNA repair. Genetics 177, 17–30 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bachant J., Alcasabas A., Blat Y., Kleckner N., Elledge S. J., The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell 9, 1169–1182 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Nitiss K. C., Malik M., He X., White S. W., Nitiss J. L., Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc. Natl. Acad. Sci. U.S.A. 103, 8953–8958 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weger S., Hammer E., Engstler M., The DNA topoisomerase I binding protein topors as a novel cellular target for SUMO-1 modification: Characterization of domains necessary for subcellular localization and sumolation. Exp. Cell Res. 290, 13–27 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Lee M. T., Bachant J., SUMO modification of DNA topoisomerase II: Trying to get a CENse of it all. DNA Repair 8, 557–568 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hudson J. J., Chiang S.-C., Wells O. S., Rookyard C., El-Khamisy S. F., SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nat. Commun. 3, 733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris J. R., Boutell C., Keppler M., Densham R., Weekes D., Alamshah A., Butler L., Galanty Y., Pangon L., Kiuchi T., Ng T., Solomon E., The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462, 886–890 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Larsen N. B., Gao A. O., Sparks J. L., Gallina I., Wu R. A., Mann M., Räschle M., Walter J. C., Duxin J. P., Replication-coupled DNA-protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Mol. Cell 73, 574–588.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tretyakova N. Y., Groehler A. T. IV, Ji S., DNA–protein cross-links: Formation, structural identities, and biological outcomes. Acc. Chem. Res. 48, 1631–1644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laco G. S., Pommier Y., Role of a tryptophan anchor in human topoisomerase I structure, function and inhibition. Biochem. J. 411, 523–530 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stepanov A., Nitiss K. C., Neale G., Nitiss J. L., Enhancing drug accumulation in Saccharomyces cerevisiae by repression of pleiotropic drug resistance genes with chimeric transcription repressors. Mol. Pharmacol. 74, 423–431 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eaba6290/DC1