

At least two groups of fork protection factors work downstream of two RAD51-dependent fork remodeling pathways.
Abstract
Fork reversal is a common response to replication stress, but it generates a DNA end that is susceptible to degradation. Many fork protection factors block degradation, but how they work remains unclear. Here, we find that 53BP1 protects forks from DNA2-mediated degradation in a cell type–specific manner. Fork protection by 53BP1 reduces S-phase DNA damage and hypersensitivity to replication stress. Unlike BRCA2, FANCD2, and ABRO1 that protect reversed forks generated by SMARCAL1, ZRANB3, and HLTF, 53BP1 protects forks remodeled by FBH1. This property is shared by the fork protection factors FANCA, FANCC, FANCG, BOD1L, and VHL. RAD51 is required to generate the resection substrate in all cases. Unexpectedly, BRCA2 is also required for fork degradation in the FBH1 pathway or when RAD51 activity is partially compromised. We conclude that there are multiple fork protection mechanisms that operate downstream of at least two RAD51-dependent fork remodeling pathways.
INTRODUCTION
Accurate and complete DNA replication in each cell division cycle is critical to maintain genome integrity. Fork reversal is a protective mechanism that is thought to stabilize replication forks and perhaps promote template switching in response to replication obstacles that cause fork stalling (1, 2). Fork reversal generates a nascent-nascent DNA duplex and a DNA end that resembles a double-strand break (DSB). Thus, DSB processing proteins are also engaged at stalled forks. Regulation of these proteins is needed to prevent deleterious processing. For example, in addition to promoting reversal, RAD51 prevents excessive nuclease-mediated degradation of the nascent DNA (3–5). This fork protection mechanism depends on the stabilization of a RAD51 filament and can be genetically separated from fork reversal because BRCA2 has been reported to be required for protection but not reversal (6).
A growing list of proteins including BRCA2, FANCD2, ABRO1, BOD1L, VHL, and FANCA also promote fork protection (4, 7–10). Inactivation of fork protection proteins results in nascent strand degradation by several nucleases including MRE11, EXO1, and DNA2. In addition, endonucleases like MUS81 can recognize and cleave the fork junction, thereby promoting DNA resection via a DSB intermediate and fork restart via strand invasion (11, 12). The identity of the nucleases needed to degrade the nascent strands is dependent on which fork protection protein is inactivated. For example, MRE11 is needed for degradation in BRCA2-deficient cells (4), while DNA2 is required for degradation in BOD1L- and ABRO1-deficient cells (8, 9).
In addition to RAD51, fork reversal depends on adenosine triphosphate (ATP)–dependent DNA translocases. Electron microscopy analyses of replication intermediates indicate a reduction in the frequency of reversed forks when the translocases SMARCAL1, ZRANB3, or HLTF are inactivated (13–15). Furthermore, inactivation of SMARCAL1, ZRANB3, or HLTF is reported to rescue the nascent strand degradation observed in BRCA2-deficient cells, suggesting that these proteins cooperate to generate the reversed fork that is resected when BRCA2 is inactivated (6, 11, 13, 16). SMARCAL1 and ZRANB3 are recruited to stalled forks by binding the single-strand DNA binding protein RPA and the polyubiquitinated polymerase clamp PCNA, respectively (17–22). PCNA is polyubiquitinated, in part, by HLTF, which has a ubiquitin ligase function in addition to its fork remodeling motor domain (23). Each of these translocases contains a substrate recognition domain that may provide specificity to different DNA structures (24); however, how they cooperate to promote reversal is unknown.
In addition to SMARCAL1, ZRANB3, and HLTF, FBH1 inactivation also reduces the frequency of reversed forks observed by electron microscopy (25). FBH1 contains both a helicase and an F-box domain that acts as part of a Cullin-dependent ubiquitin ligase to regulate replication fork stability (26–28). The helicase activity can catalyze reversal of model replication forks and disrupt RAD51 filaments, while the ubiquitin ligase activity can target RAD51 and alter RAD51 levels on chromatin (28–30).
The DNA damage response factor 53BP1 (TP53-binding protein 1) is a key regulator of DSB repair. 53BP1 is recruited to chromatin through an RNF8/RNF168-dependent mechanism, interacts with RIF1 and PTIP, and promotes the recruitment of the Shieldin complex to counteract DSB end resection and promote end joining repair (31). Given the important activity of 53BP1 in controlling resection at DSBs, several groups have examined its function at stalled replication forks. 53BP1 is found at replication forks in proteomic datasets and is colocalized with markers of replication in cells experiencing replication stress (32, 33). 53BP1-deficient cells are hypersensitive to replication stress, exhibit defective fork restart after an acute replication challenge, have decreased replication rates, and increased fork reversal in the absence of added replication stress (34–36). Furthermore, 53BP1 was found to protect forks from nascent strand degradation in a pathway downstream of RNF168-dependent ubiquitylation of H2A (35, 36). These experiments were performed in B cells and human osteosarcoma (U2OS) cells. However, other studies failed to observe nascent strand degradation in the same cell types when 53BP1 was inactivated (37, 38). Thus, while 53BP1 regulates resection at DSBs, its functions at replication forks remain unclear.
Here, we examined whether 53BP1 has fork protection activity in multiple cell types. We find that its importance in fork protection is cell type dependent and even within a single cell type can be variable depending on acute versus chronic inactivation. Unexpectedly, although nascent strand degradation in 53BP1-deficient cells is dependent on RAD51 and FBH1, it does not require SMARCAL1, ZRANB3, or HLTF. In addition, elevated γH2AX and replication stress hypersensitivity of 53BP1-deficient cells is dependent on FBH1, suggesting that protection of nascent DNA from nucleases after FBH1-dependent fork remodeling is a key function of 53BP1 at stalled forks. These findings led us to more broadly characterize the relationship of fork protection and fork reversal factors. We find at least two categories of fork protection mechanisms that differ based on the motor proteins needed to generate the substrate for the end-resection nucleases. Both mechanisms are dependent on RAD51, suggesting that a remodeled fork is the common intermediate. Unexpectedly, BRCA2 is required for the FBH1-dependent pathway and also in circumstances in which RAD51 function is compromised with a small-molecule inhibitor. Thus, two mechanisms of fork remodeling yield nuclease substrates that are protected by different fork protection factors, and BRCA2 has more complex functions than simply stabilizing RAD51 filaments on reversed forks.
RESULTS
53BP1 protects replication forks from nascent strand degradation in a cell type–dependent manner
To begin elucidating the activities of 53BP1 at stalled replication forks, we monitored replication using single-molecule DNA fiber assays. We depleted 53BP1 in U2OS cells with four different small interfering RNAs (siRNAs), labeled the cells with the thymidine analogs 5-Chloro-2′-deoxyuridine (CldU) followed by 5-Iodo-2′-deoxyuridine (IdU), before exposing the cells to a high dose (4 mM) of hydroxyurea (HU) for 4 hours, which largely blocks all DNA synthesis and completely stalls fork movement. We found that silencing 53BP1 with all four siRNAs caused resection of the nascent DNA, resulting in a reduced IdU/CldU ratio (Fig. 1A). In contrast, Byrum and colleagues (38) reported that stalled replication forks remain stable in 53BP1-deficient U2OS cells generated by CRISPR-Cas9. Consistent with their previous results, we also did not observe nascent strand degradation in the 53BP1 mutant cell line (#79) used in that study and verified that it lacks detectable 53BP1 expression (fig. S1A).
Fig. 1. 53BP1 protects replication forks from nascent strand degradation.
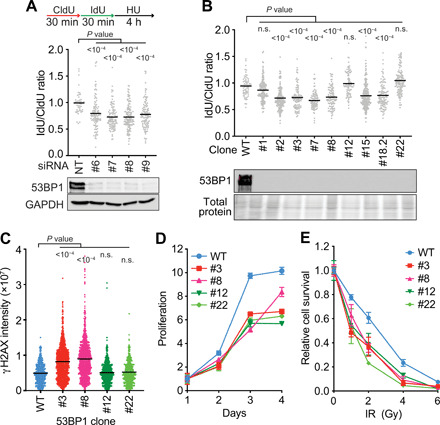
(A and B) Fork protection assays were completed by incubating cells with CldU for 30 min, followed by IdU for 30 min, and then treating with 4 mM HU for 4 hours. The lengths of CldU- and IdU-labeled DNA tracks were measured after DNA fiber spreading, and the ratio of IdU/CldU fiber lengths is plotted. All graphs are representative of at least two experiments. P values were calculated using a Kruskal-Wallis test. n.s., not significant. (A) U2OS cells were transfected with four individual siRNAs targeting 53BP1 or nontargeting (NT) siRNA and processed 72 hours after transfection. Immunoblotting was used to examine knockdown. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (B) CRISPR-Cas9–edited 53BP1 KO U2OS cells were analyzed for fork protection and compared to WT U2OS cells. (C) Quantitation of the γH2AX nuclear intensity in EdU-positive cells from the indicated 53BP1 KO cell lines. Cells were labeled with EdU for 10 min, treated with 4 mM HU for 2 hours, fixed, and stained. Each data point represents intensity in each S-phase cell. Bars are the mean, and P values were derived from a Kruskal-Wallis test. (D) Cell proliferation of the indicated cell lines (mean ± SD from n = 3). (E) Colony formation assay after exposure to the indicated dose of ionizing radiation (IR) (mean ± SD from n = 3).
Because the knockout (KO) cell line behaved differently than knockdown of 53BP1 by siRNA, we wanted to determine whether that cell line is representative of what happens when 53BP1 is inactivated by frameshift mutations at the gene level or whether there might be clonal variation. Therefore, we used CRISPR-Cas9 to make additional 53BP1 KOs in U2OS cells and then performed fork protection assays. We found that 9 of 12 clonal 53BP1 KO cell lines exhibit nascent strand degradation when treated with HU (Fig. 1B and fig. S1B). We cultured three of these cell lines for 3 months and monitored fork protection each month. The amount of degradation remained relatively constant during the time course (fig. S1B). None of these cell lines had detectable 53BP1 protein by immunoblotting, and the specific genomic mutations do not explain the differences in phenotypes (table S1).
To further characterize potential differences between the 53BP1 KO clones that maintain fork protection versus those that do not, we examined H2AX phosphorylation (γH2AX) in response to replication stress. Two cell lines that fail to maintain stable forks also have elevated γH2AX in S-phase cells, while two cell lines that have stable forks do not (Fig. 1C). However, all four cell lines have reduced proliferation rates and hypersensitivity to ionizing radiation (Fig. 1, D and E).
We also examined two pools of U2OS cells after high-efficiency transduction of two 53BP1 guide RNAs (gRNAs) into cells expressing Cas9. In both cases, we observed fork protection defects in the first month of culture that were attenuated after 2 months and absent after 3 months, although the amount of 53BP1 protein expressed remained reduced (fig. S1C). These results suggest that 53BP1 protects stalled forks from degradation in U2OS cells, but that there is clonal variation that may be due to heterogeneity in the parental cell population. There may also be an adaptation mechanism that can operate, at least in mixed populations, which reduces the fork protection defect.
To determine whether 53BP1 is needed to protect stalled forks in other cell types, we depleted 53BP1 in human embryonic kidney (HEK) 293T cells with siRNA or inactivated it with CRISPR-Cas9. Although transient depletion yielded nascent strand degradation (fig. S2A), much smaller amounts of resection were visible in clonal HEK293T KO cell lines (fig. S2B). Moreover, this fork protection defect disappeared after 3 months in culture.
We next tested 53BP1 function in hTERT-RPE1 cells and did not observe fork degradation after siRNA-mediated depletion (fig. S2C). We also used CRISPR-Cas9 to inactivate 53BP1 in hTERT-RPE1 cells. Neither pooled KO cells nor the isolated KO clones exhibit degradation in response to HU treatment (fig. S2, D and E, and table S1). We conclude that different cell types have different requirements for 53BP1 for fork protection.
SMARCAL1, ZRANB3, and HLTF are not required for fork degradation when 53BP1 is inactivated
Previous studies determined that reversed forks generated by SMARCAL1, ZRANB3, and HLTF serve as the substrate for nucleases in BRCA2-deficient cells to cause nascent DNA degradation (6, 11, 13, 16). To determine whether this is also the case in 53BP1-deficient cells, we silenced SMARCAL1, ZRANB3, or HLTF in U2OS cells cotransfected with 53BP1 siRNA. To our surprise, co-depletion of SMARCAL1, ZRANB3, or HLTF only partially reduces the degradation caused by 53BP1 silencing (Fig. 2A). We confirmed this result using 53BP1 siRNA transfected into U2OS cells in which we knocked out SMARCAL1, ZRANB3, or HLTF individually using CRISPR-Cas9 (fig. S3, A and B to D, and table S1). Furthermore, silencing 53BP1 in a SMARCAL1, ZRANB3, and HLTF triple KO (3KO) cell line also did not rescue fork degradation (Fig. 2B and table S1). We verified this result in two additional clones of 3KO cells (fig. S3E and table S1). The 3KO cells fail to express detectable SMARCAL1, ZRANB3, or HLTF (fig. S3, B to D). However, to ensure that the cell lines were not expressing some undetectable, functional fragment of one of these proteins, we also confirmed that transfecting siRNA targeting these genes into the 3KO cell line had no effect (Fig. 2B).
Fig. 2. SMARCAL1, ZRANB3, and HLTF do not generate the substrate for nascent strand degradation in 53BP1-deficient cells.
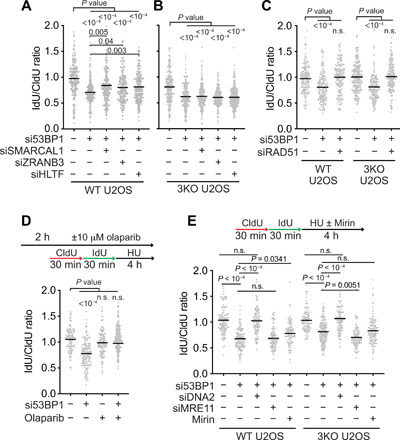
Fork protection assays and analyses were completed as in Fig. 1. (A to C) The indicated siRNAs were transfected into WT U2OS or SMARCAL1, ZRANB3, and HLTF 3KO U2OS cell lines before analyzing fork protection. (D) U2OS cells transfected with 53BP1 siRNA were treated with olaparib as indicated before analyzing fork protection. (E) WT or 3KO U2OS cells were transfected with the indicated siRNAs before analyzing fork protection. Cells were treated with mirin (50 μM) where indicated.
The inability of SMARCAL1, ZRANB3, and HLTF inactivation to fully suppress fork degradation in 53BP1-deficient cells suggests that either the degradation is partially independent of fork reversal or fork reversal leading to degradation in this genetic background is mediated by another enzyme. Because 53BP1 was previously shown to prevent fork cleavage and promote replication restart (34), we considered the possibility that SLX4-dependent nucleases like MUS81 could cleave replication forks in 53BP1-deficient cells, yielding DSBs that are then resected. However, co-depletion of MUS81 or SLX4 with 53BP1 did not rescue nascent strand degradation (fig. S4, A and B).
Because RAD51 is required for fork reversal, we determined whether inactivating RAD51 in 53BP1-deficient cells will rescue fork degradation. Silencing both RAD51 and 53BP1 in either wild-type (WT) or 3KO U2OS cells fully rescues nascent strand degradation (Fig. 2C). Poly(adenosine 5′-diphosphate–ribose) polymerase 1 (PARP1) inhibition blocks fork reversal by modulating the activity of RECQ1 (39). Treating 53BP1-silenced cells with the PARP inhibitor olaparib also rescued fork protection (Fig. 2D). Furthermore, overexpression of either WT RAD51 or an adenosine triphosphatase (ATPase) mutant K133R that forms hyperstable filaments also rescues fork degradation in the 53BP1-deficient cells (fig. S4C). In addition, the RAD51-II3A mutant that can form nucleoprotein filaments, but not perform strand exchange (40), can also rescue fork protection when overexpressed, whereas the RAD51 T131P mutant that cannot form stable filaments does not (fig. S4C). Last, inhibiting MRE11 by siRNA or treatment with the MRE11 inhibitor mirin had little or no effect, whereas DNA2 silencing rescued the nascent strand degradation similar to what has been reported for other fork protection proteins like BOD1L (Fig. 2E) (9). These results indicate that a reversed fork or another unidentified DNA2-susceptible intermediate that is dependent on RAD51 and PARP for formation and that can be stabilized by RAD51 filaments is the resection substrate in 53BP1-deficient cells.
SMARCAL1, ZRANB3, and HLTF dependency distinguishes two classes of fork protection proteins
In contrast to 53BP1, the reversal enzymes SMARCAL1, ZRANB3, and HLTF are required for fork degradation when BRCA2 is silenced, confirming previously published observations (Fig. 3, A to C, and fig. S5, A to E) (13). These results suggest a mechanistic difference between the nascent strand degradation observed in 53BP1-deficient versus BRCA2-deficient cells. A large number of other proteins act as fork protection factors. Therefore, we surveyed several to determine whether they behaved like BRCA2 or 53BP1. Nascent strand degradation in BRCA2-, FANCD2-, or ABRO1-deficient cells is dependent on the SMARCAL1, ZRANB3, and HLTF translocases (Fig. 3, A to C, and fig. S5, A to F). Inactivation of any of these translocases is sufficient to rescue the degradation in these genetic backgrounds. However, in the absence of FANCA, VHL, or BOD1L, knocking out these enzymes singly or in combination only modestly reduces nascent strand degradation (Fig. 3, A to C, and fig. S5, A to C and G to I).
Fig. 3. SMARCAL1, ZRANB3, and HLTF dependency distinguishes two classes of fork protection proteins.
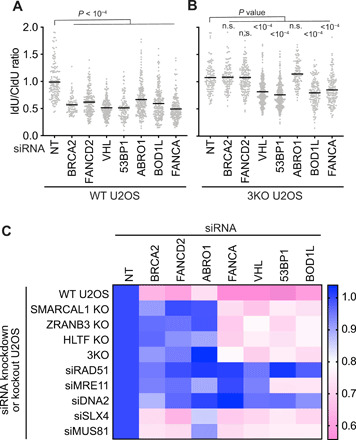
Fork protection assays and analyses were completed as in Fig. 1. (A) WT U2OS cells or (B) SMARCAL1, ZRANB3, and HLTF 3KO U2OS cells were transfected with the indicated siRNAs before analyzing fork protection. (C) Heatmap summarizing fork protection screening results. siRNA was used to silence BRCA2, FANCD2, ABRO1, FANCA, VHL, 53BP1, or ABRO1 in the indicated KO cells or cells cotransfected with the indicated siRNAs. Data in WT U2OS, SMARCAL1 KO, HLTF KO, and 3KO cell lines were n = 3 experiments in which three different clones of each of the KO cell lines were analyzed. The other data are from n = 2 experiments. The mean value was used to create the heatmap after normalizing each experiment to the nontargeting control set at a ratio of 1.0. Lower values (pink) indicate more nascent strand degradation.
Nascent strand degradation in all circumstances is suppressed by RAD51 depletion (Fig. 3C and fig. S6A). MUS81 or SLX4 silencing also only has modest effects, but DNA2 inactivation reduces fork degradation in all genetic backgrounds, and MRE11 silencing rescued in all but 53BP1- and BOD1L-deficient cells (Fig. 3C and fig. S6, B to F). These results suggest that all of these fork protection proteins may prevent the nuclease-mediated resection of a remodeled replication fork. However, the fork protection proteins segregate into two groups (BRCA2, FANCD2, and ABRO1 versus 53BP1, FANCA, BOD1L, and VHL) based on whether the SMARCAL1, ZRANB3, and HLTF translocases are needed to generate the remodeled fork substrate for the nucleases (Fig. 3C).
FBH1 is required for fork degradation when 53BP1 is inactivated
The FBH1 helicase was recently found to catalyze replication fork reversal (25), so we tested whether it might substitute for SMARCAL1, ZRANB3, and HLTF in 53BP1-deficient cells to generate the resection substrate. Consistent with this hypothesis, nascent strand degradation after 53BP1 inactivation is rescued by FBH1 silencing or by knocking out FBH1 using CRISPR-Cas9 (Fig. 4, A to C, and table S1). Complementation of FBH1 deficiency by transfection of an siRNA-resistant WT FBH1 complementary DNA (cDNA) expression vector or by viral-mediated integration of an FBH1 expression construct in FBH1 KO cells restores fork degradation (Fig. 4, B and C). FBH1 ubiquitin ligase activity is not required because expression of an FBH1 F-box mutant also promotes degradation, but a functional helicase domain is needed because the helicase mutant does not (Fig. 4, B and C). These results suggest that FBH1 uses its helicase activity to generate a fork intermediate that is degraded when 53BP1 is inactivated.
Fig. 4. FBH1 helicase activity is needed to generate the fork protection substrate for 53BP1.
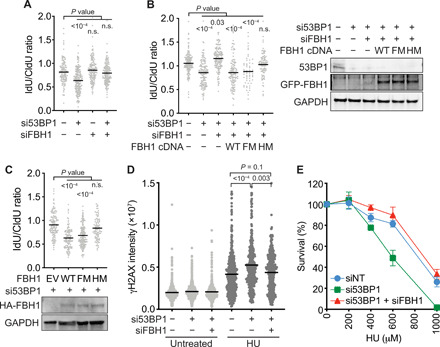
(A to C) Fork protection assays and analyses were completed as in Fig. 1. (A) WT U2OS cells were transfected with the indicated siRNAs. (B) WT U2OS cells were transfected with the indicated siRNAs and transfected with expression vectors encoding WT, F-box mutant (FM), or helicase mutant (HM) FBH1 before performing the fork protection assay. Immunoblots for 53BP1 and GFP-FBH1 are shown. (C) FBH1 KO U2OS cells were complemented with empty vector (EV), WT, FM, or HM FBH1-expressing retroviruses before depleting 53BP1 with siRNA and analyzing fork protection. HA-FBH1 protein expression was examined by immunoblotting. (D) Quantitation of the γH2AX nuclear intensity in EdU-positive cells transfected with the indicated siRNAs. Bars represent the mean, and P values were derived from a Kruskal-Wallis test. (E) U2OS cells transfected with the indicated siRNAs were treated with HU for 48 hours after plating for individual colonies. Colony number was scored after 10 days (mean ± SD, n = 3).
We also found that FBH1 inactivation reduces the elevated γH2AX caused by 53BP1 silencing in HU-treated cells (Fig. 4D). FBH1 inactivation also rescues the HU hypersensitivity of 53BP1-deficient cells (Fig. 4E). These results suggest that fork protection is a key replication stress function of 53BP1.
FBH1 was reported to be dispensable for fork degradation in BRCA2-deficient cells (41), a result that we confirmed (Fig. 5A). Furthermore, FBH1 depletion also does not rescue degradation in FANCD2- or ABRO1-deficient cells where the SMARCAL1, ZRANB3, and HLTF translocases are implicated (Fig. 5A). However, FBH1 silencing rescues fork degradation in VHL-, BOD1L-, or FANCA-deficient cells similar to its ability to rescue in 53BP1-deficient cells (Fig. 5B). This is consistent with a previous report that found that FBH1 silencing reduces nascent strand degradation when BOD1L is inactivated (9). We also confirmed that FBH1 inactivation restores fork protection to 53BP1-, VHL-, BOD1L-, or FANCA-deficient cells even when SMARCAL1, ZRANB3, and HLTF are also inactivated (Fig. 5C). Thus, FBH1, but not SMARCAL1, ZRANB3, or HLTF, is needed to form a substrate for resection nucleases in these genetic backgrounds. Because degradation in these contexts also depends on RAD51, and FBH1 catalyzes fork reversal, these results suggest that there are at least two RAD51-dependent fork reversal mechanisms that alternatively use FBH1 or the SMARCAL1, ZRANB3, and HLTF motor proteins.
Fig. 5. FBH1 is required for nascent strand degradation in 53BP1-, BOD1L-, VHL-, FANCA-, FANCC-, and FANCG-deficient cells.
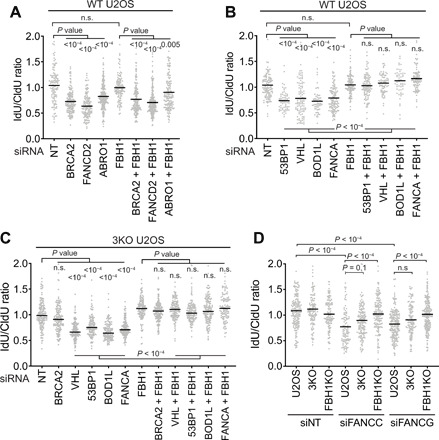
Fork protection assays were completed as in Fig. 1 after siRNA transfections in (A and B) WT U2OS or (C) SMARCAL1, ZRANB3, and HLTF 3KO U2OS cells. (D) Nontargeting (NT), FANCC, or FANCG siRNAs were transfected into WT, 3KO, or FBH1 KO U2OS cells before assessing fork protection.
FANCA is a component in Fanconi anemia (FA) core complex that ubiquitylates FANCD2 during interstrand cross-link repair. However, only the fork resection in FANCD2-deficient cells is rescued by inactivating SMARCAL1, ZRANB3, or HLTF, while inactivating FBH1 rescues fork degradation in response to FANCA silencing. We tested two additional components of the FA core complex, FANCC and FANCG, and found that they are also fork protection factors. Like FANCA, degradation in the FANCC- and FANCG-silenced cells is fully rescued by inactivating FBH1, but not SMARCAL1, ZRANB3, and HLTF (Fig. 5D). These results suggest that FANCD2 and FANCD2 ubiquitylated by the FA core complex protect different stalled forks.
BRCA2 is required to generate the fork resection substrate in the FBH1 pathway and when RAD51 function is partially compromised
If BRCA2 and 53BP1 operate in separate fork protection pathways downstream of independent fork remodeling mechanisms, then inactivating both may yield increased nascent strand degradation. To test this idea, we co-depleted BRCA2 with 53BP1 in U2OS cells. Inactivating both BRCA2 and 53BP1 did not further increase nascent strand degradation (Fig. 6A and fig. S7A). It is possible that a maximum rate of degradation may be reached with each individual deficiency or the assay may be incapable of detecting additional degradation. We also repeated the experiment in the SMARCAL1, ZRANB3, and HLTF 3KO U2OS cells, which we predicted should yield nascent strand degradation because FBH1 is available to remodel the forks, which would then be degraded in the absence of 53BP1 protection. However, we were surprised to find that co-depletion of 53BP1 and BRCA2 yielded stable nascent strands in the 3KO cells (Fig. 6A).
Fig. 6. BRCA2 is needed to generate the nascent strand degradation substrate in the FBH1 pathway and when RAD51 is partially inactivated.
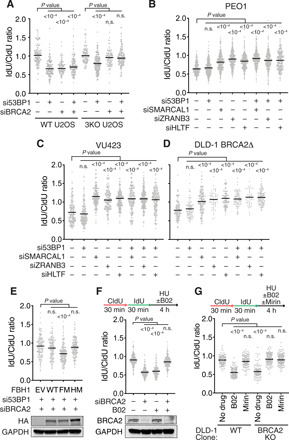
Fork protection assays were completed as in Fig. 1 in (A) WT or SMARCAL1, ZRANB3, and HLTF 3KO U2OS cells; (B) BRCA2-deficient PEO1 cells; (C) BRCA2-deficient VU423 cells; or (D) DLD-1 BRCA2 KO cells transfected with the indicated siRNAs. (E) FBH1 and HLTF double KO U2OS cells were complemented with WT, F-box mutant (FM), or helicase mutant (HM) FBH1 proteins by retrovirus infection. 53BP1 and BRCA2 were then silenced with siRNA before performing the fork protection assay. An immunoblot shows the expression levels of the HA-tagged FBH1 proteins. (F) U2OS cells transfected with BRCA2 siRNA were processed for the fork protection assay as indicated. B02 (25 μM) was included during the HU treatment where indicated. (G) DLD-1 WT or BRCA2 KO cells were treated with 25 μM B02 and/or 50 μM mirin during the HU treatment and assessed for fork protection.
We further verified this observation by co-depleting SMARCAL1, ZRANB3, or HLTF with 53BP1 in the BRCA2-deficient PEO1 cell line. Treating this cell line with HU causes nascent strand degradation because BRCA2 is not present to protect the reversed forks (Fig. 6B and fig. S7B). Inactivating SMARCAL1, ZRANB3, or HLTF individually blocks the degradation as expected. However, inactivating 53BP1 in the PEO1 cells does not cause resection (Fig. 6B). This lack of degradation is unlikely to be because 53BP1 is not important for fork protection in these cells since 53BP1 silencing in the patient-matched, BRCA2-proficient, PEO4 cells causes degradation (fig. S7C). Furthermore, inactivating SMARCAL1, ZRANB3, or HLTF in the BRCA2-deficient VU423 cells or the BRCA2 KO DLD-1 cells consistently yields stable forks even when 53BP1 is also inactivated (Fig. 6, C and D, and fig. S7, D and E). In contrast, 53BP1 inactivation causes nascent strand degradation in VU423 cells complemented with WT BRCA2 or in the BRCA2 WT DLD-1 cells (fig. S7, F and G).
Thus, neither SMARCAL1, ZRANB3, or HLTF translocase inactivation nor BRCA2 silencing alone can rescue the nascent strand degradation in 53BP1-deficient cells, but combining these genetic deficiencies or inactivating FBH1 restores fork protection. These results suggest that FBH1 cannot generate a resection substrate if SMARCAL1, ZRANB3, HLTF, and BRCA2 are also inactivated at the same time. Because our previous results show that SMARCAL1, ZRANB3, and HLTF are not needed in the FBH1 pathway, the simplest way to reconcile these findings is if BRCA2 is actually needed in the FBH1 pathway to generate the resection substrate that 53BP1 protects (fig. S8A). Because RAD51 is essential in all circumstances to generate the resection substrate, we hypothesized that BRCA2 is needed in the FBH1 pathway to promote RAD51-dependent remodeling.
To test this hypothesis and seek a mechanistic explanation, we considered whether BRCA2 acts in the FBH1 pathway to overcome the RAD51 antagonistic effects of the FBH1 ubiquitin ligase activity. This hypothesis predicts that attenuating the RAD51 antagonistic activity of FBH1 by inactivating its F-box would make BRCA2 dispensable for generating the fork degradation substrate when 53BP1 and HLTF are inactivated. To test this prediction, we generated FBH1 and HLTF double KO U2OS cell lines by CRISPR-Cas9 (table S1) and then complemented these cells with WT, F-box mutant, and helicase mutant FBH1. As predicted, in WT and helicase mutant FBH1-complemented HLTF KO cells, co-depletion of BRCA2 and 53BP1 did not cause nascent strand degradation (Fig. 6E). However, BRCA2 and 53BP1 silencing does yield degradation when the F-box mutant FBH1 is expressed (Fig. 6E). The results suggest that by helping RAD51 act at stalled replication forks, BRCA2 counteracts the ubiquitin ligase activity of FBH1 to promote fork remodeling in a RAD51-dependent but SMARCAL1-, ZRANB3-, HLTF-independent pathway.
We then determined whether there were other circumstances in which BRCA2 might be important to generate a resection substrate at a stalled fork. Because RAD51 is required for both fork reversal and fork protection, but less RAD51 function is needed for reversal (2, 41, 42), partly inactivating RAD51 using the B02 inhibitor causes fork degradation (13, 42). We repeated this experiment and observed nascent strand degradation when cells were treated with B02 or when BRCA2 was silenced (Fig. 6F). However, combining both BRCA2 siRNA and B02 yielded stable forks (Fig. 6F). We further confirmed this result in BRCA2 KO DLD-1 cells, where treatment with B02 causes fork protection and the fork degradation is SMARCAL1, ZRANB3, HLTF, and MRE11 dependent (Fig. 6G). The results suggest that the action of BRCA2 in generating a resection substrate is not limited to the FBH1-mediated fork remodeling pathway, but also extends to when RAD51 function is partially compromised (fig. S8B).
DISCUSSION
Protecting the nascent DNA at stalled replication forks is important for genome stability and cancer cell fates in response to chemotherapeutic agents and PARP inhibitors (37). 53BP1 is best studied as a DSB repair protein, and conflicting data have been published as to whether it functions in fork protection. Here, we confirmed the fork protection function of 53BP1 but found that it is context dependent. To our surprise and in contrast to BRCA2, the nascent strand degradation in 53BP1-deficient cells is fully rescued by inactivating FBH1 but not by inactivating SMARCAL1, ZRANB3, and HLTF. These results suggest that FBH1, SMARCAL1, ZRANB3, and HLTF mediate distinct fork remodeling pathways to generate resection substrates that require different fork protection proteins for stability. These results are not unique to 53BP1, and the different requirements for fork reversal motors distinguish two classes of fork protection proteins exemplified by 53BP1, FANCA, FANCC, FANCG, VHL, and BOD1L versus BRCA2, FANCD2, and ABRO1. DNA2 participates in nascent strand degradation in all genetic backgrounds tested, while MRE11 is less important in the 53BP1- and BOD1L-deficient cells.
53BP1 controls DNA end resection at DSBs to promote nonhomologous end joining repair, making it a good candidate for a fork protection protein that prevents nascent strand degradation. However, its functions in DNA replication have been inconsistent and unclear, with some studies finding no function for 53BP1 in fork protection and others finding extensive nascent strand degradation when 53BP1 is inactivated (35–38). Our data suggest that both sets of observations are correct, with cell type differences and perhaps an undefined adaptation response accounting for the different results.
We have attempted to identify the underlying differences that yield the disparate results when 53BP1 is inactivated in different cells by looking at likely factors including BRCA1, BRCA2, RAD51, RIF1, FBH1, and SHIELDIN but so far have been unsuccessful in determining a mechanism that is critical. Part of the difficulty in identifying compensatory pathways is that the mechanism of action of 53BP1 at forks remains unclear. 53BP1 can shield stretches of unreplicated DNA from processing as cells transit the cell cycle (43, 44). Whether these 53BP1 nuclear bodies are related to its fork protection activities is unknown. At DSBs, 53BP1 recruits RIF1, PTIP, and SHIELDIN to counteract resection (31). RIF1 is a fork protection factor that prevents DNA2-mediated degradation of nascent DNA just as 53BP1 does (45, 46). Furthermore, RIF1 and 53BP1 were reported to be in the same pathway to promote resistance to replication stress (34), and PTIP recruits MRE11 to replication forks (37). However, PTIP recruitment to stalled replication forks is independent of 53BP1 (37). Thus, further studies will be needed to understand the compensatory mechanisms that protect forks in some 53BP1-deficient cells. An attractive possibility is that the FBH1 remodeling pathway could be silenced in the 53BP1-deficient cells that do not exhibit fork degradation. However, RNA-sequencing experiments did not reveal any differences in FBH1 expression. In addition, the exact mechanism that 53BP1 uses to protect forks is unknown, although our studies suggest that it may be related to how BOD1L, the FANC core complex, and VHL function because they also act downstream of FBH1.
SMARCAL1, ZRANB3, HLTF, and FBH1 can all catalyze fork reversal reactions on model DNA substrates and all have been confirmed by electron microscopy to promote reversal in cells (13–15, 20, 25, 47–49). Each protein has an ATP-dependent motor domain required for the reversal reaction and different accessory domains including DNA substrate recognition domains and protein-protein interaction motifs, which presumably provide them with unique activities. Yet, in some cases, a subset of these proteins appears to function together. For example, inactivating SMARCAL1, ZRANB3, or HLTF prevents fork degradation in BRCA2-deficient cells, suggesting that they work in a concerted pathway of fork reversal (13). We find that these fork reversal enzymes are also required for nascent strand degradation in FANCD2- or ABRO1-deficient cells, and inactivating any of the enzymes yields similar results as a triple mutant, suggesting that they work in the same pathway. In contrast, even inactivating all three enzymes together is unable to fully rescue nascent strand degradation when 53BP1, BOD1L, FANCA, FANCC, FANCG, or VHL is inactivated. Reversed forks are still likely the intermediate in this nascent strand degradation pathway based on six pieces of evidence: (i) RAD51 depletion prevents resection; (ii) PARP inhibitor blocks resection; (iii) resection requires DNA2, but not SLX4 or MUS81; (iv) FBH1 inactivation blocks resection; (v) FBH1 helicase activity is needed for resection; and (vi) overexpression of RAD51 or RAD51 K133R blocks resection. Nonetheless, we cannot rule out that some other undefined structure is the intermediate. In any case, there may be some difference in the DNA or chromatin structures that are formed downstream of the SMARCAL1, ZRANB3, and HLTF versus FBH1 pathways necessitating different fork protection proteins. Alternatively, they may work in different genomic or chromatin contexts. Further studies using electron microscopy combined with analyses of protein recruitment and chromatin structures at individual stalled forks may reveal differences in DNA or chromatin structures formed by the alternative remodeling pathways.
RAD51 is not capable of reversing model replication forks by itself, and our data suggest that FBH1 can partner with it in some contexts. FBH1 has both a motor domain required for fork reversal and an F-box domain that forms part of a ubiquitin ligase that targets RAD51. The helicase, but not the F-box domain, is required to generate the substrate for resection in 53BP1-deficient cells and other genetic contexts where SMARCAL1, ZRANB3, and HLTF are not required. Although these data suggest that FBH1 promotes fork reversal in these contexts, a function for FBH1 in removing RAD51 from the nascent DNA to provide access to DNA2 could also be important as suggested previously (9).
Our data indicate that DNA2 participates in resecting the nascent DNA at least to some extent when any of the fork protection proteins are inactivated, while MRE11 is only required in some cases. MRE11 has both endonuclease and exonuclease activities that combine to remove DNA end-binding proteins and initiate resection, yielding 3′ overhangs at DSBs. DNA2 has both 5′ to 3′ and 3′ to 5′ nuclease activities that process 5′ flaps at Okazaki fragments during replication and is regulated by RPA to yield 3′ overhangs at DSBs. Yeast Dna2 also cleaves unpaired DNA strands to prevent aberrant fork reversal (50). The extensive resection of persistently stalled forks in human cells lacking fork protection factors may be the result of cycles of reversal and processing by these and other nucleases with various substrate preferences and regulation.
If the BRCA2 and 53BP1 pathways were completely separate fork protection mechanisms, then we would have expected that inactivating 53BP1 in cells in which the SMARCAL1, ZRANB3, and HLTF-BRCA2 pathway for fork protection was inactivated would yield nascent strand degradation. However, this is not the case. BRCA2 inactivation prevents nascent strand degradation when 53BP1 and any of the SMARCAL1, ZRANB3, or HLTF proteins is inactivated. Because RAD51 is essential for fork reversal in all known circumstances, and BRCA2 acts through RAD51, the simplest interpretation of these results is that BRCA2 is needed to assist RAD51 in the FBH1 pathway. Our genetic separation of function experiments with FBH1 mutants indicate that FBH1-dependent fork remodeling and degradation can occur in cells in which HLTF, BRCA2, and 53BP1 are simultaneously inactivated as long as the FBH1 ubiquitin ligase activity is also inactivated. If FBH1 retains its ubiquitin ligase activity, then this RAD51 antagonist activity could generate a requirement for BRCA2 to make the metastable RAD51 filament proposed to remodel the fork. Consistent with this idea, we also found that BRCA2 is needed to generate the resection substrate when RAD51 function is partially compromised with the RAD51 inhibitor B02. These results are consistent with the idea that the amount and functional activity of RAD51 in the cell is critical to determine whether forks undergo reversal and protection (2).
In conclusion, our results explain apparently contradictory results in the literature. Furthermore, our genetic analysis of fork protection factors and their relationship with fork remodeling proteins indicates that there are at least two distinct pathways to generate substrates for end resection at persistently stalled forks.
MATERIALS AND METHODS
Cell culture
U2OS and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 7.5% fetal bovine serum (FBS). DLD-1 and RPE-hTERT cells were cultured in RPMI1640 with 7.5% FBS. All cell lines were purchased from the American Type Culture Collection with the following exceptions: 53BP1Δ cell clone #79 was a gift from N. Mosammaparast (Washington University, St. Louis, MO, USA). VU423 and VU423 complemented with WT BRCA2 cell line and DLD-1 WT and DLD-1 BRCA2 KO cell lines were gifts from D. K. Bishop (University of Chicago, Chicago, IL, USA). PEO1 and PEO4 cell lines were gifts from A. Vindigni (Washington University, St. Louis, MO, USA). Cell lines are routinely tested for mycoplasma, and authentication was verified using short tandem repeat profiling. Details of CRISPR-Cas9–edited cell lines are in table S1. Cell proliferation was measured using the WST-1 reagent (Sigma-Aldrich).
Plasmids
pCDNA4/TO-GFP-FBH1 WT and F-box mutant were gifts from C. Sørensen. The FBH1 cDNA was cloned to pLNCX-3×HA vector, and F-box (F266A/P267A) and helicase (D698N) mutants were made by polymerase chain reaction (PCR) using the following primers: F-box mutant, AGCGCTGCTAGTGAGGTCCTGAGG (forward) and AGCAGCGCTGCAAATGTGGCTCAGT (reverse); helicase mutant, TGTGAATGAGGCCCAGGACTGC (forward) and TCATTCACAAAGATGGCGTCAAAAGAGGC (reverse).
The RAD51 II3A mutant consists of three single mutation sites—R130A, R303A, and K313A—and was generated with the following primers: R130A, TTCGCAACTGGGAAGACCCAGATCT (forward) and CAGTTGCGAATTCTCCAAACATTTCTGTGAT (reverse); R303A, ATCTGGCGAAAGGAAGAGGGGAAACCAGA (forward) and TTTCGCCAGATACAATCTGGTTGTTGATGCATG (reverse); K313A, TCTGCGCAATCTACGACTCTCCCTGTCTT (forward) and AGATTGCGCAGATTCTGGTTTCCCCTCTTC (reverse); RAD51 T131P, TTCCGACCTGGGAAGACCCAGATCT (forward) and TCCCAGGTCGGAATTCTCCAAACATTTCTG (reverse); RAD51 K133R, TGGGCGTACCCAGATCTGTCATACGCT (forward) and TGGGTACGCCCAGTTCGGAATTCTCCAAA (reverse).
Antibodies and chemicals
Antibodies and chemicals used are listed in table S2.
RNA interference
All siRNA transfections were performed using DharmaFECT reagents according to the manufacturer’s instructions. ON-TARGETplus siRNAs were purchased from Dharmacon. Experiments were completed 3 days after transfection. For siRNA sequences and the siRNA used in each experiment, please see table S2. Qiagen AllStars Negative Control Nontargeting (NT) siRNA was used in samples where a gene-selective siRNA is not indicated or to bring the total amount of siRNA to equal molar concentration when comparing samples with one versus two siRNAs.
Gene editing with CRISPR-Cas9
The pSpCas9 (BB)-2A-Puro 2 plasmid (Addgene 48139) was used to express single gRNAs (sgRNAs) for gene editing with CRISPR-Cas9. In some experiments, cells were transfected with sgRNA, trans-activating CRISPR RNA (tracrRNA) (Horizon U-002005-05), and Cas9 mRNA (Horizon CAS11860). In all cases, colonies were screened by PCR, loss of protein expression was verified by immunoblotting, and cell lines were validated by sequencing the edited alleles (table S1). The sgRNA sequences used are shown in table S2. FBH1 single or FBH1 and HLTF double KO cells were complemented with FBH1 cDNAs by retrovirus infection followed by selection with G418.
Single-molecule analysis of replication
DNA fiber spreading experiments were performed as described previously (51). Cells were labeled with 20 μM IdU followed by 100 μM CldU for 20 min each. Following stretching and fixation on glass slides, DNA was denatured in 2.5 M HCl for 80 min, washed three times with phosphate-buffered saline (PBS), and blocked in 10% goat serum/PBS with 0.1% Triton X-100 for 1 hour. DNA combing was performed according to the instructions from a Genomic Vision combing kit. Combing and fiber spreading yielded similar results.
Quantification and statistical analysis
Statistical analyses were completed using Prism. A Kruskal-Wallis test was used for experiments with more than two samples, and P values were calculated by Prism for the multiple comparisons. A two-tailed t test was used to compare two samples with normally distributed data. No statistical methods or criteria were used to estimate sample size or to include/exclude samples. Multiple siRNAs were analyzed to confirm that results were not caused by off-target effects, and at least two cell clones of CRISPR-Cas9 generated KO cells were used to ensure that results were not due to clonal variation. Statistical details of individual experiments can be found in the figure legends and in Results. All experiments were performed at least two times, and representative experiments are shown.
Immunofluorescence imaging
Cells were labeled with 5-Ethynyl-2′-deoxyuridine (EdU) for 10 min, and detergent was extracted with 0.5% Triton X-100 before fixing with 3% paraformaldehyde/2% sucrose. Slides were blocked with 10% goat serum in PBS–0.1% Triton and incubated with antibody. EdU was detected using click chemistry with an Alexa Fluor 594–conjugated azide. Immunofluorescent images were obtained and analyzed with an ImageXpress (Molecular Devices) instrument and software.
Supplementary Material
Acknowledgments
We thank. A. Vindigni, D. Bishop, N. Mosammaparast, G. Stewart, and C. Sørenson for reagents. Funding: This research was supported by grants to D.C. from the NIH (R01GM116616) and the Breast Cancer Research Foundation. Additional support came from the Vanderbilt-Ingram Cancer Center. Author contributions: W.L. and A.K. performed the experiments. W.L. and R.Z. made the KO cell lines. W.L., A.K., and D.C. analyzed the data. W.L. and D.C. conceived the project and wrote the manuscript. D.C. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data are available in the main text, Supplementary Materials, or by request. Further information and requests for resources and reagents should be directed to D.C. (david.cortez@vanderbilt.edu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc3598/DC1
REFERENCES AND NOTES
- 1.Neelsen K. J., Lopes M., Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Bhat K. P., Cortez D., RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 25, 446–453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J. A., Herrador R., Vindigni A., Lopes M., Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 208, 563–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M., Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V., Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 17, 1305–1311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mijic S., Zellweger R., Chappidi N., Berti M., Jacobs K., Mutreja K., Ursich S., Ray Chaudhuri A., Nussenzweig A., Janscak P., Lopes M., Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 8, 859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlacher K., Wu H., Jasin M., A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu S., Wu X., Wu L., Castillo A., Liu J., Atkinson E., Paul A., Su D., Schlacher K., Komatsu Y., You M. J., Wang B., Abro1 maintains genome stability and limits replication stress by protecting replication fork stability. Genes Dev. 31, 1469–1482 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgs M. R., Reynolds J. J., Winczura A., Blackford A. N., Borel V., Miller E. S., Zlatanou A., Nieminuszczy J., Ryan E. L., Davies N. J., Stankovic T., Boulton S. J., Niedzwiedz W., Stewart G. S., BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell 59, 462–477 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Espana-Agusti J., Warren A., Chew S. K., Adams D. J., Matakidou A., Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nat. Commun. 8, 2026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemaçon D., Jackson J., Quinet A., Brickner J. R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N., Vindigni A., MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarbajna S., Davies D., West S. C., Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 28, 1124–1136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taglialatela A., Alvarez S., Leuzzi G., Sannino V., Ranjha L., Huang J.-W., Madubata C., Anand R., Levy B., Rabadan R., Cejka P., Costanzo V., Ciccia A., Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 68, 414–430.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vujanovic M., Krietsch J., Raso M. C., Terraneo N., Zellweger R., Schmid J. A., Taglialatela A., Huang J.-W., Holland C. L., Zwicky K., Herrador R., Jacobs H., Cortez D., Ciccia A., Penengo L., Lopes M., Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell 67, 882–890.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai G., Kermi C., Stoy H., Schiltz C. J., Bacal J., Zaino A. M., Hadden M. K., Eichman B. F., Lopes M., Cimprich K. A., HLTF promotes fork reversal, limiting replication stress resistance and preventing multiple mechanisms of unrestrained DNA synthesis. Mol. Cell 78, 1237–1251.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolinjivadi A. M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Técher H., Baldi G., Shen R., Ciccia A., Pellegrini L., Krejci L., Costanzo V., Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell 67, 867–881.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bansbach C. E., Bétous R., Lovejoy C. A., Glick G. G., Cortez D., The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 23, 2405–2414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciccia A., Bredemeyer A. L., Sowa M. E., Terret M. E., Jallepalli P. V., Harper J. W., Elledge S. J., The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 23, 2415–2425 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postow L., Woo E. M., Chait B. T., Funabiki H., Identification of SMARCAL1 as a component of the DNA damage response. J. Biol. Chem. 284, 35951–35961 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciccia A., Nimonkar A. V., Hu Y., Hajdu I., Achar Y. J., Izhar L., Petit S. A., Adamson B., Yoon J. C., Kowalczykowski S. C., Livingston D. M., Haracska L., Elledge S. J., Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 47, 396–409 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan J., Ghosal G., Chen J., The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell 47, 410–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yusufzai T., Kong X., Yokomori K., Kadonaga J. T., The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 23, 2400–2404 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motegi A., Liaw H.-J., Lee K.-Y., Roest H. P., Maas A., Wu X., Moinova H., Markowitz S. D., Ding H., Hoeijmakers J. H. J., Myung K., Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. U.S.A. 105, 12411–12416 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poole L. A., Cortez D., Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 52, 696–714 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fugger K., Mistrik M., Neelsen K. J., Yao Q., Zellweger R., Kousholt A. N., Haahr P., Chu W. K., Bartek J., Lopes M., Hickson I. D., Sørensen C. S., FBH1 catalyzes regression of stalled replication forks. Cell Rep. 10, 1749–1757 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Chiolo I., Saponaro M., Baryshnikova A., Kim J.-H., Seo Y.-S., Liberi G., The human F-Box DNA helicase FBH1 faces Saccharomyces cerevisiae Srs2 and postreplication repair pathway roles. Mol. Cell. Biol. 27, 7439–7450 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenz A., Osman F., Folkyte V., Sofueva S., Whitby M. C., Fbh1 limits Rad51-dependent recombination at blocked replication forks. Mol. Cell. Biol. 29, 4742–4756 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fugger K., Mistrik M., Danielsen J. R., Dinant C., Falck J., Bartek J., Lukas J., Mailand N., Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. J. Cell Biol. 186, 655–663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu W. K., Payne M. J., Beli P., Hanada K., Choudhary C., Hickson I. D., FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat. Commun. 6, 5931 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Simandlova J., Zagelbaum J., Payne M. J., Chu W. K., Shevelev I., Hanada K., Chatterjee S., Reid D. A., Liu Y., Janscak P., Rothenberg E., Hickson I. D., FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J. Biol. Chem. 288, 34168–34180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Setiaputra D., Durocher D., Shieldin—The protector of DNA ends. EMBO Rep. 20, e47560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dungrawala H., Rose K. L., Bhat K. P., Mohni K. N., Glick G. G., Couch F. B., Cortez D., The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell 59, 998–1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ward I. M., Chen J., Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 276, 47759–47762 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Xu Y., Ning S., Wei Z., Xu R., Xu X., Xing M., Guo R., Xu D., 53BP1 and BRCA1 control pathway choice for stalled replication restart. eLife 6, e30523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmid J. A., Berti M., Walser F., Raso M. C., Schmid F., Krietsch J., Stoy H., Zwicky K., Ursich S., Freire R., Lopes M., Penengo L., Histone ubiquitination by the DNA damage response is required for efficient DNA replication in unperturbed S phase. Mol. Cell 71, 897–910.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Her J., Ray C., Altshuler J., Zheng H., Bunting S. F., 53BP1 mediates ATR-Chk1 signaling and protects replication forks under conditions of replication stress. Mol. Cell. Biol. 38, e00472-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ray Chaudhuri A., Callen E., Ding X., Gogola E., Duarte A. A., Lee J.-E., Wong N., Lafarga V., Calvo J. A., Panzarino N. J., John S., Day A., Crespo A. V., Shen B., Starnes L. M., de Ruiter J. R., Daniel J. A., Konstantinopoulos P. A., Cortez D., Cantor S. B., Fernandez-Capetillo O., Ge K., Jonkers J., Rottenberg S., Sharan S. K., Nussenzweig A., Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byrum A. K., Carvajal-Maldonado D., Mudge M. C., Valle-Garcia D., Majid M. C., Patel R., Sowa M. E., Gygi S. P., Harper J. W., Shi Y., Vindigni A., Mosammaparast N., Mitotic regulators TPX2 and Aurora A protect DNA forks during replication stress by counteracting 53BP1 function. J. Cell Biol. 218, 422–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berti M., Ray Chaudhuri A., Thangavel S., Gomathinayagam S., Kenig S., Vujanovic M., Odreman F., Glatter T., Graziano S., Mendoza-Maldonado R., Marino F., Lucic B., Biasin V., Gstaiger M., Aebersold R., Sidorova J. M., Monnat R. J. Jr., Lopes M., Vindigni A., Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 20, 347–354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mason J. M., Chan Y.-L., Weichselbaum R. W., Bishop D. K., Non-enzymatic roles of human RAD51 at stalled replication forks. Nat. Commun. 10, 4410 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leuzzi G., Marabitti V., Pichierri P., Franchitto A., WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 35, 1437–1451 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhat K. P., Krishnamoorthy A., Dungrawala H., Garcin E. B., Modesti M., Cortez D., RADX modulates RAD51 activity to control replication fork protection. Cell Rep. 24, 538–545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lukas C., Savic V., Bekker-Jensen S., Doil C., Neumann B., Pedersen R. S., Grøfte M., Chan K. L., Hickson I. D., Bartek J., Lukas J., 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Harrigan J. A., Belotserkovskaya R., Coates J., Dimitrova D. S., Polo S. E., Bradshaw C. R., Fraser P., Jackson S. P., Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garzón J., Ursich S., Lopes M., Hiraga S.-i., Donaldson A. D., Human RIF1-protein phosphatase 1 prevents degradation and breakage of nascent DNA on replication stalling. Cell Rep. 27, 2558–2566.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mukherjee C., Tripathi V., Manolika E. M., Heijink A. M., Ricci G., Merzouk S., de Boer H. R., Demmers J., van Vugt M. A. T. M., Ray Chaudhuri A., RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 10, 3287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bétous R., Mason A. C., Rambo R. P., Bansbach C. E., Badu-Nkansah A., Sirbu B. M., Eichman B. F., Cortez D., SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 26, 151–162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bétous R., Couch F. B., Mason A. C., Eichman B. F., Manosas M., Cortez D., Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 3, 1958–1969 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blastyák A., Hajdú I., Unk I., Haracska L., Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell. Biol. 30, 684–693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu J., Sun L., Shen F., Chen Y., Hua Y., Liu Y., Zhang M., Hu Y., Wang Q., Xu W., Sun F., Ji J., Murray J. M., Carr A. M., Kong D., The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149, 1221–1232 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Couch F. B., Bansbach C. E., Driscoll R., Luzwick J. W., Glick G. G., Bétous R., Carroll C. M., Jung S. Y., Qin J., Cimprich K. A., Cortez D., ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc3598/DC1