

The structure of oncogenic LMTK3 determines its role and functions allowing drug inhibition as a new therapeutic strategy.
Abstract
Elucidating signaling driven by lemur tyrosine kinase 3 (LMTK3) could help drug development. Here, we solve the crystal structure of LMTK3 kinase domain to 2.1Å resolution, determine its consensus motif and phosphoproteome, unveiling in vitro and in vivo LMTK3 substrates. Via high-throughput homogeneous time-resolved fluorescence screen coupled with biochemical, cellular, and biophysical assays, we identify a potent LMTK3 small-molecule inhibitor (C28). Functional and mechanistic studies reveal LMTK3 is a heat shock protein 90 (HSP90) client protein, requiring HSP90 for folding and stability, while C28 promotes proteasome-mediated degradation of LMTK3. Pharmacologic inhibition of LMTK3 decreases proliferation of cancer cell lines in the NCI-60 panel, with a concomitant increase in apoptosis in breast cancer cells, recapitulating effects of LMTK3 gene silencing. Furthermore, LMTK3 inhibition reduces growth of xenograft and transgenic breast cancer mouse models without displaying systemic toxicity at effective doses. Our data reinforce LMTK3 as a druggable target for cancer therapy.
INTRODUCTION
Protein kinases play a pivotal role in regulating intracellular signal transduction pathways involving almost every aspect of cell activity including proliferation, survival, differentiation, apoptosis, metabolism, angiogenesis, immune surveillance, and motility. Perturbation of their signaling affects their activities, which contributes to human diseases including malignancies (1–3). Targeted therapies against kinases have improved the clinical outcome of patients in the past decade. However, resistance to these treatments often develops, largely because of the aberrant activation of other kinases having a complementary or compensatory function (1, 3), and we have shown redundancy in kinase signaling in broad networks (4).
The oncogenic role of lemur tyrosine kinase 3 (LMTK3) has been established over the past years, supported by mechanistic and translational data in different tumor types and settings in vitro and in vivo (5–16). LMTK3 has been originally identified via a kinome screening as a regulator of estrogen receptor α (ERα) in breast cancer (BC) (5), able to protect it from ubiquitin-mediated proteasomal degradation. Follow-up studies have demonstrated that LMTK3 is overexpressed in more aggressive forms of BC, and it is implicated in endocrine (9) and chemotherapy resistance (10) in BC. While high LMTK3 mRNA expression has been reported to be an independent poor prognostic factor in patients with ERα+ BC (17), immunohistochemistry (IHC) analysis has further revealed that tumors overexpressing human epidermal growth factor receptor 2 (HER2) are more likely to be LMTK3 positive, while triple-negative BC (TNBC) tumors have high cytoplasmic expression of LMTK3 (8). Furthermore, the involvement of LMTK3 in invasion (12), migration (12), chromatin remodeling (13), and microRNA biogenesis (6) in BC has also been documented. Since LMTK3 has been proposed as a potential therapeutic target in BC and considering its involvement in other tumors, there is a pressing need to further decipher the signaling pathways in which LMTK3 is implicated in and identify potent, selective, and cell-permeable small-molecule inhibitors that can be used to enable pathway investigation and, in doing so, also establish onward tractability for future translational activities.
Radiolabeled biochemical assays using a positional scanning peptide library (PSPL) (18) resulted in elucidation of the LMTK3 phosphorylation consensus sequence and the subsequent identification of heat shock protein 27 (HSP27) as an in vitro phosphorylation substrate of LMTK3. Moreover, by using a stable isotope labeling by amino acids in cell culture (SILAC) quantitative phosphoproteomics approach, we uncovered various LMTK3-modulated phospho-proteins (direct and/or indirect targets) that can shed light on the biologic functions regulated by LMTK3. To aid research on LMTK3 as a therapeutic target, we report the three-dimensional x-ray crystal structure of LMTK3 alongside methods for the production and purification of the recombinant protein. Using robust in vitro and cell-based screening assay and selectivity assay combined with biophysical analyses, we identified and characterized a small-molecule adenosine 5′-triphosphate (ATP)–competitive LMTK3 inhibitor (C28) that acts by degrading LMTK3 via the ubiquitin-proteasome pathway. Our data revealed LMTK3 to be an HSP90-CDC37 (cell division cycle 37) client protein, which was able to phosphorylate CDC37 in vitro. Chemical inhibition of LMTK3 provided a notable therapeutic effect as demonstrated in a panel of 60 cancer cell lines [National Cancer Institute Developmental Therapeutics Program (NCI-DTP)] and suppressed tumor growth of BC xenograft and transgenic mice models. Together, we report the crystal structure and phosphorylation motif of LMTK3 and identify an LMTK3 inhibitor that confers cytotoxic and antitumor effects. We believe that further development and optimization of LMTK3 inhibitors based on this study could have prospective value to patients with cancer, and we provide the preclinical data to take the development of these compounds forward.
RESULTS
Crystallization and overview of the LMTK3 structure
LMTK3 shares structural and sequence similarities to the kinase domains of the transmembrane receptor epidermal growth factor receptor (EGFR also known as ERBB1 and HER1), insulin receptor (INSR), and the Janus kinase (JAK), a family of intracellular, nonreceptor tyrosine kinases (19–21). The activation loop of INSR has three tyrosine residues (22), like the kinase domain of LMTK3. Using equivalent LMTK3 residues (amino acids 130 to 413) to INSR kinase and the other kinases in this analysis, the sequence identity for EGFR, INSR, and JAK1 with LMTK3 is 26, 34, and 27%, respectively, while the sequence similarity is 45, 49, and 42%, respectively, when calculated using “Ident and Sim” (fig. S1) (23). The structure of human EGFR kinase domain in complex with the nonhydrolyzable ATP analog adenylyl-imidodiphosphate (AMP-PNP), the kinase domain of JAK1 in complex with adenosine 5′-diphosphate (ADP), and the kinase domain of INSR have been previously determined (21, 22, 24). Here, we solved the crystal structure of the LMTK3 catalytic domain (from now on referred as LMTK3cat) to 2.1-Å resolution using a recombinant protein comprising residues 134 to 444 (crystallization statistics are shown in table S1). The overall fold of the LMTK3 kinase domain (Fig. 1, A and B) was similar to that of other kinases. We find that LMTK3cat adopts an inactive conformation, with the key DYG motif (more commonly DFG motif) stabilized in the “DYG-out” conformation, while comparative studies with either JAK1 (25) [Protein Data Bank (PDB) 5KHW; Fig. 1C] or EGFR (PDB 5CNO; Fig. 1D) suggested that key features and residues of the LMTK3 kinase are all conserved. This would allow for its activation (fig. S1), following conformational changes due to phosphorylation of the activation loop and repositioning of the DYG motif, that lead to the binding and hydrolysis of ATP.
Fig. 1. Crystal structure of LMTK3.
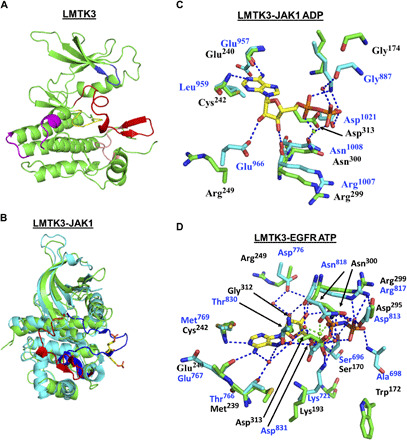
(A) Structure of the kinase domain of LMTK3 showing the main features [Protein Data Bank (PDB) 6SEQ]. Blue, the glycine-rich loop; red, activation loop; salmon, the P+1 loop. The activation loop and the P+1 loop constitute the activation segment. Yellow, the catalytic loop; magenta, the kinase insert region. (B) Superimposition of the kinase domains of LMTK3 (green) and JAK1 (cyan). The activation loop of LMTK3 is shown in red and adopts an inactive conformation. The activation loop of JAK1 is shown in blue and adopts the active state. The two JAK1 phosphotyrosine residues (Tyr1034 and Tyr1035) are shown as yellow sticks. (C) Superimposition of LMTK3 and JAK1 active site (PDB 5KHW) residues involved in ADP binding. Key residues that bind ADP in JAK1 are positioned slightly differently to those of LMTK3 because of the different catalytic states of the kinases. However, residues in JAK1 that interact with ADP are conserved within LMTK3, although two noticeable differences are seen (LMTK3 Arg249 and Cys242 replaces Glu966 and Leu959), which are unlikely to affect ATP binding. (D) Superimposition of LMTK3 and EGFR active site (PDB 5CNO) residues involved in ATP binding. A comparison with the ATP-bound form of EGFR also shows strong conservation among residues of EGFR that bind ATP and amino acid residues at equivalent positions within LMTK3 and residues that are not conserved (Ala698, Met769, and Cys773 for Trp172, Cys242, and Asp246 in LMTK3), form main-chain interactions with bound ATP. [Green, amino acid residues of LMTK3 with black residue numbers; cyan, amino acid residues of JAK1 (top) or EGFR (bottom) with blue residue numbers; red spheres, water molecules; blue dashes, hydrogen bonds; green sphere, magnesium ion; green dashes, bonds coordinating the magnesium ion.]
In LMTK3, we found that Tyr314 of the DYG motif, more commonly DFG in kinases, occupies the equivalent space of the adenine ring of ATP and thus maintains the integrity of the C-spine, a series of residues that connect the αF helix to the N-terminal lobe (fig. S2, A to D) (26, 27). Only a few other kinases in the human kinome such as leucine-rich repeat kinase 1/2 (LRRK1/2), NEK9, and VRK1/2 have a Tyr at this position. Tyrosine in this conformation is stabilized by a number of hydrogen bonds (fig. S2E). Thus, although EGFR and LMTK3 show a degree of homology, the detailed mechanisms by which they achieve an inactive state and prevent ATP binding differ in detail. The DYG motif is closely associated with the HSD motif, usually HRD for most kinases, through a series of hydrogen bonds that particularly involve the catalytic Asn300, which is normally involved in magnesium ion coordination (fig. S2F). However, within the activation loop, there are potentially three tyrosine autophosphorylation or phosphorylation activation sites at Tyr321, Tyr325, and Tyr326 (fig. S2G), which are equivalent to the INSR autophosphorylation sites Tyr1158, Tyr1162, and Tyr1163 (28). Together, the features of the LMTK3 kinase domain indicate that the critical residues required for its binding and hydrolysis of ATP are conserved (fig. S1).
Determination of the LMTK3 consensus phosphorylation motif and global analysis of the LMTK3-dependent phosphoproteome
Elucidating the preferred LMTK3 consensus phosphorylation motif is of critical importance to examine and identify in vitro LMTK3 substrates and to decipher downstream pathways regulated by LMTK3 in vivo.
Following production of LMTK3cat (fig. S3A), we performed radiolabeled kinase assays using LMTK3cat to screen a PSPL consisting of 198 peptide mixtures as previously described (18). Quantification of the peptides’ signal intensities showed that LMTK3 appears to have a strong requirement for Arg residue at positions −3 and/or −2 (Fig. 2, A and B). Moreover, we observed a reduced phosphorylation on peptides with hydrophobic residues in the −3 through to the +1 position, indicating that these residues may be deterrents to LMTK3 substrate phosphorylation. On the basis of these data, we designed an optimal LMTK3 peptide substrate (LMTK3-tide: WRRFSFCMC) and generated mutant versions after substituting key residues at different positions. In vitro kinase assays further supported the importance of the −3, −2, and +1 positions for maximal phosphorylation efficiency (Fig. 2C). Rab-coupling protein, recently described to be phosphorylated by LMTK3 at Ser435 (29), contains a similar motif (R-R-S-S).
Fig. 2. Defining the LMTK3 consensus phosphorylation motif and identifying HSP27 as an LMTK3 substrate.
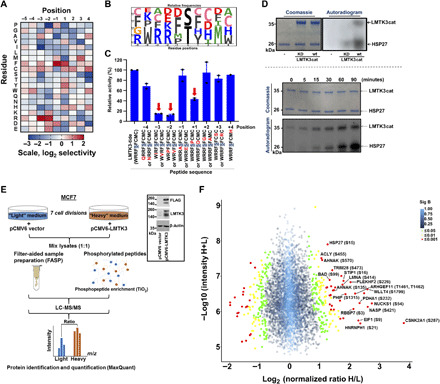
(A) A spatially 198 components arrayed PSPL was subjected to in vitro phosphorylation with active LMTK3cat. A representative image of the average log2 values of two independent experiments is shown. (B) Scaled-sequence PhosphoLogo representation of the LMTK3 consensus phosphorylation motif. The size of the letter is proportional to the signal for the corresponding amino acid at the indicated position. (C) In vitro kinase assays using wild-type (WT) LMTK3cat as source of enzyme activity and peptide variants with individual amino acid substitutions at different positions. Data shown are the average of two separate experiments (±SEM). (D) Top: In vitro kinase assay using recombinant HSP27 as a substrate and WT LMTK3cat or kinase-dead (KD) LMTK3 mutant (LMTK3cat-KD) as source of enzyme activity. Bottom: Time course in vitro kinase assay using recombinant HSP27 and LMTK3cat. (E) Schematic representation of SILAC proteomic experiment. Western blotting analysis of LMTK3 and FLAG-LMTK3 protein levels showing the transient overexpression of full-length pCMV6-LMTK3 (FLAG-tag) plasmid. m/z, mass/charge ratio. (F) Volcano scatter plot showing the log2 “normalized ratios” (H/L) against log10 “intensity” (H+L) for each characterized phosphorylated protein (phosphopeptide) following overexpression of WT-LMTK3 in MCF7 cells. Proteins are displayed in circles based on P values from significant B test. Red, P < 0.001; yellow, 0.001 > P < 0.01; green, 0.01 > P < 0.05; blue, P > 0.05.
To confirm our results, we used a PepChip microarray slide carrying 192 peptides that contain experimentally verified phosphorylation sites for different kinases and performed an in vitro kinase assay with LMTK3cat. We observed variability in the phosphorylation of specific peptides containing analogous sequences to the one predicted from the PSPL assay [Arg residues predominantly at positions −3 and/or −2, as well as a Leu or Lys residue at position −4 (L/K-R-R/X-X-S)] (fig. S3B). Among the newly identified substrates was HSP27 (also known as HSPB1), which we validated by kinase assays using full-length HSP27 (Fig. 2D and fig. S3C). We also pinpointed Ser15 and Ser82 as the main HSP27 residues phosphorylated by LMTK3 in vitro, using site-directed mutagenesis (fig. S3D), while both sites were in accordance with the identified LMTK3 phospho-motif.
There are many examples of kinase-dead (KD) mutants across various kinase families, which are typically based on disrupting residues that assist ATP’s phosphates binding/catalytic transfer. The common approach (and one that is used to determine whether the target is a true kinase or a pseudo-kinase) is to mutate Lys72 (numbered after the first crystal structure of adenosine 3′,5′-monophosphate kinase with ATP bound), which will allow ATP to still bind but will not be able to catalyze phosphorylation. Single substitution of Lys72 by Ala is often sufficient to create a KD mutant; however, also mutating Asp166, and Asp184 to Ala maximizes the pocket variation and phosphate affinity. In LMTK3, these sites were identified as Lys193, Asp295, and Asp313 by using molecular modeling studies with ATP, as shown in fig. S3 (E and F). By mutating residues Lys193, Asp295, and Asp313 (fig. S3G), we demonstrated that the kinase is catalytically dead (from now on referred as LMTK3cat-KD) as shown by its inability to phosphorylate HSP27 in vitro (Fig. 2D).
To obtain information about the LMTK3-regulated global phosphoproteomic alterations, we performed SILAC experiments (30) in combination with phosphopeptide enrichment and liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis in MCF7 BC cells (Fig. 2E) and examined the effects of LMTK3 overexpression. Of 4955 proteins that were identified in total and following normalization in respective total protein levels, we quantified 4556 potential phosphopeptides that correspond to 1718 proteins, as several proteins had more than one potential phosphorylation site. Among these phosphopeptides, 4022 phosphoserine, 515 phosphothreonine, and 19 phosphotyrosine sites were characterized (table S2). Ultimately, we uncovered different phosphorylated proteins/amino acids that were either positively or negatively affected by LMTK3, suggesting a direct and/or indirect involvement of LMTK3 during these posttranslational modifications (Fig. 2F). HSP27 was validated among the most significant hits, implying that it can also be an in vivo LMTK3 phospho-substrate. Most of the identified phospho-residues have been previously implicated in cancer, including MLLT4 (Afadin)–Ser1799, PHIP (pleckstrin homology domain interacting protein)–Ser1315, ARHGEF11 (Rho guanine nucleotide exchange factor 11)–Thr1461/T1462, PLEKHF2 (pleckstrin homology and FYVE domain containing 2)–Ser226, PDHA1 (pyruvate dehydrogenase E1 α 1 subunit)–Ser232, NUCKS1 (nuclear casein kinase and cyclin dependent kinase substrate 1)–Ser54, NASP (nuclear autoantigenic sperm protein)-Ser421, RBBP7 (RB binding protein 7, chromatin remodeling factor)–Ser3, STIP1 (stress induced phosphoprotein 1)–Ser16, and CSNK2A1 (casein kinase 2α1)–Ser287.
Together, we have determined the LMTK3 phosphorylation motif, characterized its phospho-dependent proteome in MCF7 cells, and identified HSP27 as a bona fide in vitro phosphorylation substrate of LMTK3 to be taken forward and used in our biochemical assays and cell-based experiments described below.
High-throughput homogeneous time-resolved fluorescence screening identifies an ATP-competitive inhibitor targeting LMTK3
In parallel with the aforementioned structural and functional studies and considering the oncogenic role of LMTK3, we sought to identify and examine novel inhibitors for LMTK3 by implementing the experimental pipeline described in Fig. 3. Initially, we screened a library encompassing 28,716 compounds (Charles River Discovery Research Services UK Ltd.; formerly known as BioFocus DPI Ltd.), mainly enriched for ATP analogs, to detect substrate phosphorylation using a homogeneous time-resolved fluorescence (HTRF) assay as a readout (31). Approximately ~15% of the tested compounds displayed a >50% inhibition at 20 μM concentration (Fig. 3A, phase 1). We then narrowed down our hits to n = 160 based on their potency, physicochemical, and structural properties and performed a 10-point concentration-response profiling that gave a favorable range of median inhibitory concentration (IC50) values (24 nM to 16 μM) (Fig. 3A). Eventually, 38 of the 160 most active compounds (IC50, 24 nM to 2.6 μM; table S3, A and B) were selected for our follow-up in vitro and cell-based efficacy studies.
Fig. 3. Identification of C28 as a potent inhibitor against LMTK3.
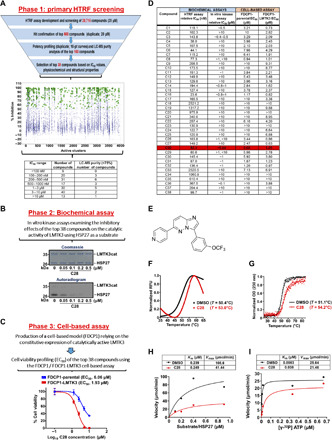
(A) Top: Experimental pipeline to identify LMTK3 inhibitors. Middle: HTRF data showing coverage of different inhibitors per active cluster. Green stars are the 868 compounds chosen for further confirmation, showing >50% mean inhibition (blue crosses are nonselected compounds). Selections were biased toward higher potency, sensible calculated physicochemical properties, and structural coverage within each cluster. Bottom: HTRF data showing the range of IC50 values and purity of the top 160 compounds. (B) The IC50 value for C28 against LMTK3cat was determined by in vitro kinase assays. (C) EC50 values for C28 in FDCP1 and FDCP1-LMTK3 cell lines. Error bars represent the means ± SD from three independent experiments. (D) Table summarizing the IC50 and EC50 values of the top 38 compounds. (E) Chemical structure of C28. (F) Characteristic thermal denaturation curves of LMTK3 (black) and LMTK3/C28 complex (red) as monitored by DSF and (G) CD spectroscopy, indicating the increased protein thermodynamic stability upon ligand binding. Tm values from DSF were determined from the maximum in the first derivative of the fluorescence with respect to the temperature, or the midpoint in the transition region by fitting a Boltzmann sigmoidal to the CD data. Experiments were performed in triplicate. DMSO, dimethyl sulfoxide; OD, optical density; RFU, relative fluorescence units. (H) Kinetic analysis of HSP27 phosphorylation by LMTK3 in the absence or presence of C28. Kinetic parameters were determined from nonlinear regression fit of the initial reaction rates as a function of HSP27 concentration to the Michaelis-Menten equation using Prism 8. (I) Kinetic analysis as a function of ATP concentration for 0.6 μM HSP27 substrate, in the absence or presence of C28. Kinetic parameters were determined from nonlinear regression fit of the initial reaction rates as a function of ATP concentration to the Michaelis-Menten equation using Prism 8.
Dose-dependent in vitro 32P γ-ATP radiolabeled kinase assays revealed variability of the inhibitory effects of the tested compounds with some of them demonstrating higher efficiency at low concentrations (<1 μM) as measured by the phosphorylation of HSP27 by LMTK3 (Fig. 3B, phase 2 and fig. S4). We then used the interleukin- 3 (IL-3)–dependent murine bone marrow–derived cell line FDCP1 (32) and engineered an LMTK3-transformed clone (FDCP1-LMTK3) that relies on the constitutive expression of catalytically active LMTK3 for its survival and proliferation (fig. S5). Using this cell-based approach, resembling the BA/F3 model that is extensively used in kinase research and drug discovery (33), we assessed the functional responses and potency of the selected inhibitors and determined their median effective concentration (EC50) values by monitoring the viability of FDCP1-parental and FDCP1-LMTK3 cell lines (Fig. 3C, phase 3).
Taking into consideration all the results summarized in Fig. 3D and considering the novelty and physicochemical properties/similarities of the top hit compounds, we focused on compound C28 (Fig. 3E and table S3B). We then assessed the ability of C28 to bind to LMTK3 by monitoring the thermal denaturation of LMTK3 and the LMTK3/C28 complex using differential scanning fluorimetry (DSF) and circular dichroism (CD) spectroscopy. As shown in Fig. 3 (F and G), both methods revealed an increase of the thermal stability of LMTK3 in the presence of C28, characteristic of ligand binding. The increase in the Tm value of LMTK3 upon C28 binding was determined at 2.6°C using DSF (50.4° ➔ 53.0°C; P = 0.001) and 3.1°C using CD (51.1° ➔ 54.2°C; P = 0.002). Only a single transition was observed in the thermal melting curves, and thermal unfolding was irreversible because of protein aggregation.
To investigate the mechanism of action of C28 (10 μM), we examined the effect of increasing substrate concentrations of HSP27 on the inhibitory activity of the compound in the presence of a constant amount of ATP. Our results revealed that the inhibitor does not affect the Km value (0.239 μM in the absence versus 0.249 μM in the presence of C28) but results in a substantially lower Vmax [108.6 μmol/min versus 41.44 μmol/min in the presence of C28 (Fig. 3H)]. The unchanged Km value with increasing concentrations of HSP27 indicates that the inhibition is substrate independent. We next examined the effect of increasing concentrations of ATP at a fixed substrate (HSP27) concentration (0.6 μM). The data from the steady-state analysis were fitted to the Michaelis-Menten equation and as shown in Fig. 3I, in the presence of C28, we observed a significant increase in the apparent Km value from 0.0053 to 0.038 μM, indicating that C28 is a competitive inhibitor.
In summary, we provide evidence of an ATP-competitive inhibitor against LMTK3, following the recent discovery of cyclic guanidine–linked sulfonamides as potential LMTK3 inhibitors (34). Given the oncogenic potential of LMTK3, identification of C28 provides a means of pharmacological inhibition as a tool for further investigating its functions, to establish a start point for development of LMTK3 inhibitors.
Selectivity profile of C28 inhibitor
To determine a more detailed profile of the selectivity of this inhibitor, we screened it against a series of 140 kinases by performing a radioactive filter binding assay (www.kinase-screen.mrc.ac.uk/services/premier-screen). We identified only 18 of 140 kinases whose activity was reduced >50% in the presence of 1 μM C28 (table S4A) of which only 4 [CLK (CDC-like kinase), 5 nM; DYRK1α (dual specificity tyrosine phosphorylation regulated kinase 1α), 6 nM; HIPK2 (homeodomain interacting protein kinase 2), 48 nM; and IRAK4 (interleukin 1 receptor associated kinase 4), 41 nM] had similarly low IC50 values to that of LMTK3 (67 nM) (Fig. 4A).
Fig. 4. Selectivity of C28 toward LMTK3.
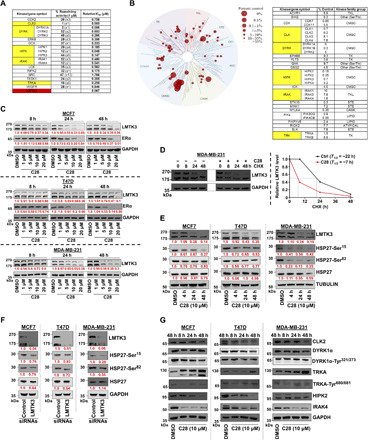
(A) Selectivity profile of C28 (1 μM) against 140 kinases using radioactive filter binding assay. The data are displayed as percent activity remaining of assay duplicates with an SD. Only kinases with >50% decrease in their activity are shown. The relative IC50 values are also presented. (B) Treespot interaction map depicting the kinome phylogenetic grouping, with kinases interacting with C28 (5 μM) represented as red circles. The larger the diameter of the circle, the higher the C28 binding affinity to the respective kinase. Kinases whose binding was inhibited by C28 to less than 10% of the control (DMSO) are shown. Lower numbers indicate most probable hits to bind to C28. Overlapping kinases identified in assays (radioactive filter binding and site-directed competition binding) are highlighted in yellow. (C) Western blots of LMTK3 and ERα in BC cell lines treated with increasing concentrations of C28 at different time points. Mean densitometry values of three independent experiments were calculated using ImageJ. GADPH, glyceraldehyde 3-phosphate dehydrogenase. (D) Effects of C28 on LMTK3 protein half-life in MDA-MB-231 cells. Cells were treated with CHX (cycloheximide) (100 μg/ml) and 10 μΜ C28 (or DMSO) for different time points. The relative LMTK3 protein levels (−/+ C28) were calculated and plotted against the time of treatment with CHX. (E) Western blots of total and phospho-HSP27 (normalized versus total) in MCF7, T47D, and MDA-MB-231 cell lines treated with 10 μΜ C28 for different time points. Mean densitometry values of three independent experiments are shown. (F) Western blots of total and phospho-HSP27 (normalized versus total) in BC cell lines treated with either control or LMTK3 small interfering RNAs (siRNAs) for 72 hours. Mean densitometry values of three independent experiments are shown. (G) Western blots of different kinases in BC cell lines treated with C28 (10 μΜ) at different time points.
Following, to further examine the specificity of C28, we used an active site–directed competition binding assay (DiscoverX KINOMEscan) and quantitatively measured the interactions between C28 and 403 purified human kinases (Fig. 4B and table S4B) (35). The S(35) selectivity index of C28 was 0.186, as measured by the percentage of the kinome inhibited below 35% of the control at this concentration [S(35) = (number of kinases with %Ctrl <35)/(number of kinases tested)]. C28 inhibited by >90% the activity of only 33 of 403 kinases, most of which (20 of 33) were different isoforms of the same proteins, highlighting an overall low promiscuity of C28 (Fig. 4B). It is worth mentioning that despite the different principles of the two assays used to examine the selectivity of the inhibitor (active site–directed competition binding assay and radioactive filter binding assay), there was considerable overlap of identified kinases targeted by C28 marked in yellow in Fig. 4 (A and B), further validating the accuracy of our results.
Treatment of different BC cell lines with increasing concentrations of C28 resulted in time- and dose-dependent degradation of LMTK3 (Fig. 4C), which was further validated following treatment with cyclohexamide, as shown by the decrease in half-life of LMTK3 in the presence of C28 (Fig. 4D).
We have previously reported that silencing of LMTK3 results in down-regulation of ERα and indeed, treatment with C28 decreased protein levels of ERα (Fig. 4C). Moreover, we also observed a reduction in the phosphorylation of HSP27 in BC cells following pharmacological (Fig. 4E) or genetic inhibition of LMTK3 (Fig. 4F), suggesting that HSP27 is also an in vivo (apart from in vitro) substrate for LMTK3. In particular, treatment with C28 for 4 hours, during which the stability of LMTK3 remained unaffected, resulted in a cell line–dependent variable decrease of HSP27 phosphorylation (Ser15 and/or Ser82), without altering the total protein levels of HSP27. A reduction of total HSP27 was noticed in MDA-MB-231 cells after 48 hours of C28 treatment, suggesting subsequent or indirect effects of the inhibitor.
Last, we also examined the effects of C28 on CLK2, DYRK1α, HIPK2, IRAK4, and TRKA (tropomyosin receptor kinase A) kinases (total levels and phospho-sites linked to their activities), which were identified as potential C28 targets from our selectivity screening. As shown in Fig. 4G, treatment of BC cell lines with 10 μM C28 for different time points led to degradation of IRAK4 and HIPK2. The level of C28 selected for these assays is above that used in the in vitro KINOMEscan assay, because CDC37 is known to antagonize the effects of ATP-competitive inhibitors of kinases (36). Previous studies suggest that mutant TRKA in human acute myeloid leukemia cells has been shown to be an HSP90-dependent kinase, subject to proteasomal degradation following HSP90 inhibition (37). It has also been demonstrated that for DYRK1A, ATP-competitive inhibitors antagonize interaction between the target kinase and kinase-specific cochaperone CDC37 in living cells (38). Thus, the assumption is that DYRK1A, in this context, is HSP90 dependent. Both IRAK4 and HIPK2 are subject to proteasomal degradation, which is blocked by MG132 treatment, but while the inhibition of HSP90 in macrophages resulted in the destabilization and degradation of IRAK-1 but not IRAK4 (39), it is as yet unclear whether HIPK2 is an HSP90 client protein. Despite the fact that these results are, in some cases, in contrast to what we have seen in this study, previous work has shown that wild-type (WT) BRAF is not HSP90 dependent (40), whereas the V600E mutant is (41). Hence, the exact specificity for HSP90 may be quite variable between different cell lines and may also depend on the stability of the protein in question within a particular cell line. In aggregate, we report the identification of a selective LMTK3 inhibitor, which appears to have a secondary selectivity toward IRAK4 and HIPK2.
LMTK3 is an HSP90-CDC37 client protein and C28 acts by promoting the proteasome-mediated degradation of LMTK3
ATP-competitive inhibitors of several different oncogenic protein kinases that depend on HSP90-CDC37 for their biological stability have been shown to promote their degradation by antagonizing the binding of the CDC37 recruitment factor to the ATP-binding site of the kinase (36). This deprives the kinases of access to the chaperone system and channels them into ubiquitylation and subsequent degradation.
It has been observed that a conserved HPNI motif within CDC37 mimics the conserved HPNI motif in kinases (42). The motif is HPNV in LMTK3 and is considered conserved and thus suggests that CDC37 may be able to recognize LMTK3. To determine whether LMTK3 is an HSP90 client kinase, we examined the effects of two HSP90 inhibitors, NVP-AUY922 and geldanamycin, on the stability of LMTK3. The HSP90-dependent CDK4 kinase (43) and HSP70, which are down-regulated or up-regulated, respectively, in response to HSP90 inhibition (44), were used as controls. As shown in Fig. 5 (A and B), increasing concentrations of the HSP90 inhibitors led to a reduction of endogenous LMTK3 protein levels and induction of HSP70, indicating that LMTK3 is an HSP90-dependent kinase. As expected, degradation of LMTK3 following NVP-AUY922 and geldanamycin treatment also resulted in the down-regulation of ERα (Fig. 5, A and B).
Fig. 5. C28 promotes proteasome-mediated degradation of LMTK3, an HSP90 client protein.
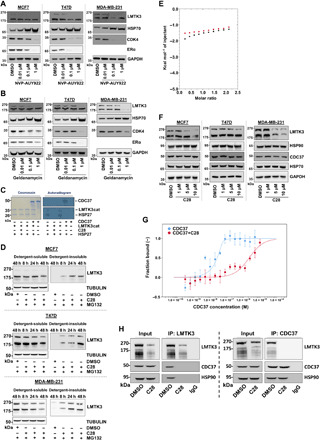
(A) Western blots of LMTK3, HSP70, and CDK4 in BC cell lines following treatment with increasing concentrations of NVP-AUY922 for 72 hours. (B) Western blots of LMTK3, HSP70, and CDK4 in BC cell lines following treatment with increasing concentrations of geldanamycin for 72 hours. (C) In vitro kinase assays using recombinant HSP27 as a substrate and LMTK3cat as source of enzyme activity, examining the effects of C28 treatment or CDC37 addition. (D) Western blots of cell lysates from BC cell lines incubated with 10 μM of MG132 for 4 hours before treatment with C28 (10 μM) for the indicated time points. LMTK3 protein levels in the detergent-soluble and detergent-insoluble fractions are presented. (E) Isothermal titration calorimetry examining the ability of C28 to bind HSP90α. Injection of 0.2 mM C28 into 20 μM human HSP90α showed no detectible heat of interaction. (F) Western blots of LMTK3, HSP90, HSP70, and CCD37 in BC cell lines following treatment with increasing concentrations of C28 for 48 hours. (G) Normalized dose-response curves comparing thermophoresis of NT647-LMTK3cat on titration of increasing concentrations of CDC37 in the presence (red, KD = 2.21 ± 1.17 μM) or absence (blue, KD = 1.29 ± 3.9 nM) of 20 μM C28. Error bars represent SE, n = 5. KD values were calculated from fitted data. (H) Left: LMTK3 was immunoprecipitated (IP) from MCF7 cells stably overexpressing LMTK3, in the presence (48 hours) or absence of C28 and the complexes were immunoblotted for LMTK3, CDC37, and HSP90. Right: CDC37 was immunoprecipitated from MCF7 cells stably overexpressing LMTK3, in the presence (48 hours) or absence of C28, and the complexes were immunoblotted for CDC37, LMTK3, and HSP90. Western blots for the respective proteins in whole cell lysates (input) were also performed.
Moreover, the ability of CDC37 cochaperone to antagonize ATP binding by protein kinase clients (36) was also confirmed using an in vitro kinase assay, where HSP27 phosphorylation by LMTK3 was inhibited in the presence of either C28 or CDC37 (Fig. 5C). LMTK3 was able to phosphorylate CDC37 in vitro, suggesting a previously unidentified role for LMTK3 within the chaperone regulatory pathway.
We then analyzed the effects of C28 on LMTK3 protein turnover following cotreatment with the proteasomal inhibitor MG132 and observed that LMTK3 was depleted from the detergent-soluble cell lysate fraction but accumulated in the insoluble fraction (Fig. 5D) indicative of a proteasomal mediated degradation, as a consequence of chaperone deprivation by C28. To rule out the possibility that the proteasome degradation of LMTK3, in response to C28 treatment, is not due to HSP90 inhibition, we tested the binding of C28 to HSP90α by isothermal titration calorimetry. Our results revealed that C28 does not bind to HSP90 (Fig. 5E). Furthermore, treatment of different BC cell lines with C28 had no effect on the protein levels of neither HSP90 nor CDC37 (Fig. 5F), further supporting that C28 is a specific LMTK3 inhibitor.
Following, to investigate the potential interaction between LMTK3 and the HSP90 cochaperone CDC37, microscale thermophoresis (MST) was used. The insect cell–expressed LMTK3cat was covalently labeled with a red NHS-647 fluorophore to allow monitoring of protein migration down an induced temperature gradient. Data quality in all experiments was determined by monitoring the fluorescence intensity (fig. S6) and raw MST data (fig. S6) throughout. With the exception of the highest CDC37 concentration used (10 μM), which was subsequently removed from the dataset, no consistent protein adsorption or aggregation was observed. As shown in Fig. 5G, titration of increasing concentrations of human CDC37 protein resulted in a dose-dependent shift in the migration of NT647-LMTK3cat, demonstrating a direct interaction between the proteins (KD = 1.29 ± 3.9 nM). On addition of C28 (20 μM), the affinity of CDC37 for LMTK3 was considerably decreased (KD = 2.21 ± 1.17 μM), demonstrating a competitive binding mechanism. Last, the endogenous association of LMTK3 with the CDC37/HSP90 complex was also confirmed in MCF7 cells following immunoprecipitation with anti-LMTK3 antibody (Fig. 5H, left) and anti-CDC37 antibody (Fig. 5H, right), while treatment with C28 abolished this interaction (Fig. 5H, both panels). Furthermore, as shown in the right panel of Fig. 5H, C28 did not affect the association between HSP90 and CDC37.
Together, these data demonstrate that LMTK3 is an HSP90-CDC37 client protein and suggest that the mechanism of action of C28 on LMTK3 is mediated via blocking the direct interaction between LMTK3 with CDC37 and therefore preventing access to the molecular chaperone HSP90, resulting in LMTK3 degradation.
C28 exhibits potent anticancer activity in different human cancer cell lines
We then investigated the potential use of C28 as an anticancer strategy by examining the viability of various BC cell lines (and one nontransformed breast cell line; MCF12A) in the presence of increasing concentrations of C28. As shown in Fig. 6A, C28 was able to inhibit the growth of BC cells with IC50 values ranging from 6.5 to 8.64 μM, while the noncancerous cell line (MCF12A) appeared to be less sensitive to C28 (IC50, 14.77 μM).
Fig. 6. C28 impairs the viability of various human cancer cell lines.
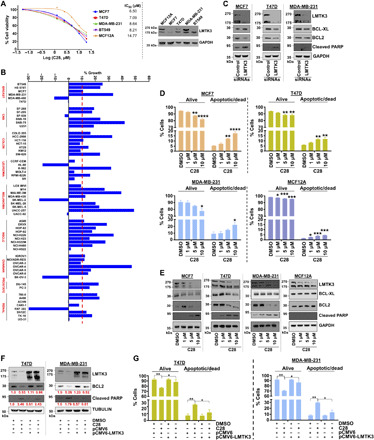
(A) Left: Viability of nontransformed and BC cell lines treated with increasing concentrations of C28 for 72 hours. The IC50 values are means from three independent experiments. Right: Western blots of LMTK3 in nontransformed and BC cell lines. (B) One-dose screening of C28 (10 μM; 24 hours) on the NCI-60 panel of tumor cell lines. The percent growth of C28-treated cells is shown. Negative values represent lethality. NSCLC, non-small cell lung cancer. (C) Western blots of BCL-XL, BCL2 and cleaved poly(ADP-ribose) polymerase (PARP) in BC cell lines following siRNA silencing of LMTK3 for 72 hours. GADPH was used as a loading control. (D) MCF7, T47D, MDA-MB-231, and MCF12A cell lines were treated with increasing concentrations of C28 for 72 hours, and the percentages of apoptotic and dead cells were analyzed by annexin V and 7-AAD staining. Results are expressed as means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Each experiment was conducted at least three times. (E) Western blots of LMTK3, BCL2, and BCL-XL and cleaved PARP in MCF7, T47D, MDA-MB-231, and MCF12A cell lines treated with increasing concentrations of C28 for 72 hours. GADPH was used as a loading control. (F) Western blots of BCL2 and cleaved PARP levels in T47D and MDA-MB-231 cell lines treated with 10 μM C28 for 72 hours followed by LMTK3 overexpression for 48 hours. Tubulin was used as a loading control. Mean densitometry values of three independent experiments are shown. (G) T47D and MDA-MB-231 cell lines were treated with 10 μM C28 for 72 hours, followed by LMTK3 overexpression for 48 hours, and the percentages of apoptotic and dead cells were analyzed by annexin V and 7-AAD staining. Results are expressed as means ± SEM. Each experiment was conducted at least two times.
We next submitted C28 to the DTP of the NCI and screened it against a panel of 60 human cancer cell lines (https://dtp.cancer.gov/discovery_development/nci-60/). At a 10 μM dose, C28 inhibited all cancer cell lines by >50% (Fig. 6B). Considering the significant growth inhibition demonstrated, the NCI decided to further evaluate C28 at five doses. The results revealed that most of the leukemia, melanoma, ovarian, and central nervous system cell lines were more resistant to C28, while renal cancer cells appeared to be more sensitive (table S5). These data demonstrate possible anticancer applicability of C28 in different tumor types.
Considering that our previous (5) and current genetic studies (Fig. 6C) have revealed an increased rate of apoptosis of BC cells following LMTK3 inhibition [small interfering RNA (siRNA)], we assessed the effects of C28 using annexin V and 7-AAD (7-aminoactinomycin D) staining. As anticipated, C28 treatment triggered apoptosis of different BC cell lines (Fig. 6D), with no effects on cellular senescence. The nontransformed MCF12A breast cell line showed a low percentage of cell death even at high concentrations of C28 and longer time points, suggesting that nontransformed breast epithelial cells may evade death while working on recuperating from C28 treatment (Fig. 6D). Western blotting demonstrated a degradation of LMTK3 levels in MCF12A but no dependence, in agreement with the cell viability/apoptosis results (Fig. 6E). As expected and consistent with a cell death apoptotic mechanism, we also observed a decrease in Bcl-xL (B-cell lymphoma-extra large) and Bcl-2 (B-cell lymphoma 2) antiapoptotic proteins and an increase in cleaved poly(ADP-ribose) polymerase (PARP) in the BC cell lines (Fig. 6E). Together, these data suggest that C28 predominantly displays its effects against cancer cells, results that require clinical exploration.
To further confirm the specificity of C28 against LMTK3 and demonstrate that the observed apoptotic effects are due to a great extent via the C28-mediated LMTK3 inhibition, we performed gain-of-function experiments overexpressing full-length LMTK3 following C28 treatment, since LMTK3 overexpression concomitant with drug exposure did not outcompete the effects of the drug. As shown in Fig. 6F, recovery of LMTK3 levels led to an increase of BCL2 and a decrease in cleaved PARP levels, in accordance with the antiapoptotic role of LMTK3, followed by an overall decrease of cell death within the short time boundaries of the experimental setup (Fig. 6G). Because of the fact that LMTK3 is ubiquitously expressed in cancer cell lines (12), it was not possible to further test the specificity of C28 in an LMTK3-null cell line model. Attempts to generate LMTK3 knockout BC cell lines using CRISPR-CAS9 were also unsuccessful, as all LMTK3-KO clones were nonviable, in line with previous reports (13).
As an extra proof-of-concept study, we looked into the potential additive effect of C28 following siRNA silencing of LMTK3. As anticipated, either genetic or pharmacological inhibition of LMTK3 led to an increase in apoptosis (fig. S7, A and B) and a decrease in cell viability (fig. S7C). When combined with siLMTK3, C28 treatment had an additive effect in the observed phenotypes and a simultaneous reduction of the residual LMTK3 protein levels. Together, these data suggest that C28 confers its anticancer functions to a great extent via LMTK3-mediated signaling, without ruling out the possibility of any off-target effects, which are invariably common with any small-molecule ATP inhibitors (45).
C28 inhibits tumor growth in orthotopic and transgenic mouse models of BC
Before testing the use of C28 in mouse models, its solubility was assessed in various formulation vehicles and dissolution conditions. We also investigated the pharmacokinetic (PK) properties of C28 in nude Balb/c mice at different doses following intravenous, intraperitoneal, and oral administration. Analysis of plasma samples by LC-MS/MS revealed that C28 (5 mg/kg), dosed intravenously, had a peak concentration (Cmax) of 1533 ng/ml (4.3 μM) at 5 min after injection, which was countered by a short half-life (T1/2 ~ 10 min), whereas lowering the dose to 1 mg/kg led to very little exposure (Cmax = 185 ng/ml). The optimal route of administration appeared to be intraperitoneally at 10 mg/kg, where Cmax was 839 ng/ml with a T1/2 of ~59 min or orally (Cmax = 632 ng/ml; T1/2 ~ 62 min) (Fig. 7A).
Fig. 7. C28 impedes tumor growth of BC mouse models.
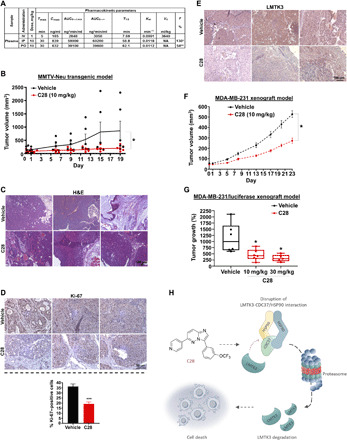
(A) PK parameters of C28 following a single intravenous (IV), intraperitoneal (IP), or oral (PO) dose administration in male BALB/c mice (n = 4 per time point). *, based on 1 mg/kg intravenous group. **, based on 5 mg/kg intravenous group. Intravenous dose of 5 mg/kg was lethal for some mice, and therefore, the dose was decreased to 1 mg/kg. (B) Tumor growth in vehicle- and C28-treated groups (n = 6 each) of MMTV-Neu transgenic mice. Unpaired t test was performed using Prism 8. Results are expressed as means ± SEM; *P < 0.05. (C) Representative images of hematoxylin and eosin (H&E) staining of tumor sections from MMTV-Neu animals treated with vehicle or C28 (10 mg/kg). Original magnification, ×10. Scale bar, 100 μm. (D) Representative IHC images of Ki-67 expression in tumor sections from MMTV-Neu animals treated with vehicle or C28 (10 mg/kg). Original magnification, ×40. Scale bar, 50 μm. The percentage of Ki-67–positive cells versus the total number of cells is shown. Data represent the average of four vehicle and five C28-treated samples. Results are expressed as means ± SEM; ***P < 0.001. (E) Representative IHC of LMTK3 expression in tumor sections from MMTV-Neu animals treated with vehicle or C28 (10 mg/kg). Original magnification, ×20. Scale bar, 100 μm. (F) Tumor growth in vehicle- and C28-treated groups (n = 14 each) of MDA-MB-231 mice xenografts. (G) Box-and-whisker plots comparing vehicle (n = 6)–treated and C28-treated (10 mg/kg, n = 6 and 30 mg/kg, n = 5) groups of MDA-MB-231–luciferase mice xenografts groups at day 21. Unpaired t test was performed using Prism 8. Results are expressed as means ± SEM; *P < 0.05. (H) Schematic model depicting the proposed mechanism of action of C28 inhibitor.
As we showed oral bioavailability of C28, we evaluated its antitumor activity in the MMTV (mouse mammary tumor virus)-Neu mammary tumor model transgenic model in which a constitutively active mouse Neu protein is expressed under control of the mouse mammary tumor virus promoter. Once tumors became palpable, mice were treated orally daily, with either vehicle control [5% dextrose/PEG400 (polyethylene glycol 400) in a 1:1 ratio] or C28 at a final concentration of 10 mg/kg (table S6A). As shown in Fig. 7B, C28 nearly completely abrogated tumor growth compared to the vehicle-treated group. Histopathologic analysis of tumor sections and other organs (lung, stomach, heart, kidney, liver, and spleen) between different groups did not show any notable differences, further demonstrating the lack of toxicity of the C28 inhibitor (Fig. 7C and fig. S8). Moreover, Ki-67 IHC analysis showed decreased proliferation following treatment with C28 (Fig. 7D). Regarding LMTK3, IHC staining revealed a reduction of LMTK3 expression in the C28-treated tumors, albeit a degree of variability was observed in certain cases (Fig. 7E). The IHC data here confirm ex vivo our on-target perturbation assays.
Next, the antitumor activity of the LMTK3 inhibitor was also evaluated using a BC xenograft mouse model where TNBC MDA-MB-231 cells were injected subcutaneously into immunocompromised mice. Once tumors were palpable, mice were divided into different groups and were treated orally daily at either control vehicle or C28 with a final concentration of 10 mg/kg (table S6B). After 23 days, treatment with C28 significantly impeded growth compared to the control group (Fig. 7F), with no changes in body weight observed (vehicle group: day 0, 21.22 g ➔ day 15, 21.42 g versus C28-group day 0, 21.77 g ➔ day 15, 21.84 g). Similar results were obtained in a separate study using luciferase-labeled MDA-MB-231 and treatment with C28 (either 10 or 30 mg/kg) (Fig. 7G and table S6C). In aggregate, our data demonstrate potent preclinical anticancer activity of C28 in vitro and a delay of tumor onset in vivo in mice models.
DISCUSSION
LMTK3 has been reported to be involved in several cancer types (5, 7, 11, 14–16). However, the lack of crystal structure has impeded an understanding of the role of this protein and its interactions with other molecules. By solving the crystal structure of inactive LMTK3, we showed that it has all the hallmarks (key features and residues) of an active protein kinase, able to be activated, to bind and hydrolyze ATP (Fig. 1 and figs. S1 and S2). In particular, the K/E/D/D signature motif plays important structural and catalytic roles and comprises residues Lys193 in the β3 strand, Glu210 in the center of the αC helix, Asp295 of the catalytic loop, and Asp313, the first residue of the activation segment. Although our crystallization experiments stabilized LMTK3 in an inactive conformation, which is common in such studies (46), we could clearly detect the catalytic activity of LMTK3 in our biochemical assays. This suggests that the insect cell protein production protocol used herein may yield a low amount (yet considerable for experimental purposes) of catalytically active protein (either phosphorylated or in an active conformation, but lacking phosphorylation) within the overall preparation, which could also explain the requirement of relatively high concentrations of LMTK3 in our in vitro kinase assays. The enzymatic activity of LMTK3 was validated following mutations at key residues Lys193, Asp295, and Asp313, which rendered the kinase inactive. In LMTK3, the activation segment DFG motif is instead DYG (residues 313 to 315). Although a DYG motif is not common, it has been reported in LRRK2 associated with Parkinson’s disease (47) and atypical protein kinase C (48). In the inactive “D313-out” state, Tyr314 is in an “in” conformation pointing past the gatekeeper residue Met239 and occupies regions of space that would overlap with the adenine ring of bound ATP (fig. S3). Design of kinase inhibitors that targets the inactive “DFG-out” conformation (type II inhibitors) offers potential of higher selectivity as compared to type I kinase inhibitors that target the active state, in addition to a profound impact on cellular activity (49). However, until now, the structure of half of all protein kinases in the human kinome has been resolved. From a total of 5200 structures of catalytic domains of human and mouse protein kinases contained in the KLIFS database, 84.5% (4393 structures) represent an active, DFG-in state, whereas the remaining 15.5% are annotated as inactive, DFG-out and atypical, DFG-out conformations (50). Nonetheless, efforts are under way to obtain a crystal structure of LMTK3 in the active state and to identify kinases that may phosphorylate tyrosine residues within the activation loop of LMTK3 [Tyr321, Tyr325, and Tyr326, which correspond to the autophosphorylation residues Tyr1158, Tyr1162, and Tyr1163 of INSR kinase (fig. S1)]. Overall, our optimized protein production protocol and the structure of LMTK3 presented herein will enable a supply of protein for further structural and functional studies and facilitate the structure-based design of inhibitors.
Considering the mounting role and involvement of LMTK3 in different malignancies and cellular processes (5–14), we attempted to uncover the LMTK3 consensus phosphorylation motif, another milestone of this study, and to identify potential in vitro and/or in vivo LMTK3 substrates. Radiolabeled assays using a PSPL comprising diverse peptide mixtures revealed a consensus sequence with a strict requirement for R residues at positions −3 and/or −2 (Fig. 2, A to C). Since LMTK3 is predicted in silico to have dual phospho-specificity [serine/threonine (S/T) and Y], a similar approach using a Y residue as a phosphoacceptor site would add more knowledge to this topic. Moreover, bearing in mind the documented and, in some cases, analogous roles of the other two isoforms (LMTK1 and LMTK2) in different diseases, it would be interesting to examine and compare the substrate specificities among all family members. Through the PSPL approach and other biochemical assays, we also identified HSP27 as a bona fide in vitro phosphorylation substrate for LMTK3 that can serve as a useful readout tool (Fig. 2D and fig. S4).
In parallel, using SILAC-based quantitative proteomics, we described the LMTK3-dependent global phosphoproteomic profile in BC cells (MCF7) and identified a large number of phosphorylated proteins that were significantly modulated by LMTK3 (Fig. 2, E and F). As anticipated, we detected certain hits closely matching the predicted LMTK3 consensus motif that have been previously reported to be implicated in different BC subtypes including BAD-S118, PRKD2-S197, RPRD2-1099, ALKBH5-S361, HSP90AB1-S452, and DDX17-S599. Additional identified phosphosites, which did not match the LMTK3 phosphorylation motif, either may represent indirect LMTK3-modulated targets or could still be direct substrates that do not fall under the canonical LMTK3 sequence as is the case for multiple kinases. Ultimately, use of a KD mutant LMTK3 construct would allow us to further establish the kinase-dependent (direct) phosphorylated targets of LMTK3. This is part of our ongoing studies. Following, to gain further insights into the global signatures of the identified LMTK3-dependent phosphorylation events, we performed Funrich analysis (www.funrich.org/) of the molecular functions, cellular components, biological processes, and pathways to which the uncovered phosphorylated proteins belong. A notable enrichment in molecular function terms related to transcription regulator activity, protein S/T kinase activity, and RNA and cytoskeletal protein binding was unveiled. Moreover, a similar distribution between cytoplasmic and nuclear cell components was observed, as anticipated, considering the universal subcellular distribution of LMTK3. Last, bioinformatics analysis of the signaling pathways and the functional protein-protein interaction networks affected by LMTK3 revealed a large number and a variety of connections further supporting the diverse and multiplex role of LMTK3 in signal transduction. Thus, the phosphorylation state of these proteins could be directly and/or indirectly modulated by LMTK3. Ultimately, the robust and comprehensive information derived from these distinct but complementary approaches (PSPL and phospho-proteomics) will assist us in deciphering and elucidating the pathways that LMTK3 is implicated in and help us map LMTK3 on the atlas of cancer signaling networks.
Alongside our structural studies, we adopted a high-throughput screening approach to identify compounds that could be promising candidates for drug development against LMTK3. To achieve this, we analyzed 28,716 small-molecule inhibitors using a wide range of biochemical, biophysical, and cell-based assays (Fig. 3). Herein, we report the first tool compound, namely C28, which binds to and inhibits LMTK3 (ATP-competitive inhibitor) with high selectivity and demonstrates effective anticancer effects in a variety of cancer cell lines and in in vivo BC mouse models, apparently sparing the normal epithelium. Cocrystallization of LMTK3 with C28 or AMPPNP has thus far not been possible, partly because of the considerably reduced yield of constitutively active LMTK3 protein in our insect cell expression system. Failure to cocrystalize with C28 suggests that perhaps it does not bind the inactive LMTK3 state that we crystalized and that it might instead target an active state conformation of the kinase. Thus, the difficulties regarding the cocrystallization studies are related to the fact that a great proportion of the protein appears to occupy a stable inactive conformation, which appears to be the preferential state for crystallization under the conditions tested to date. However, ongoing attempts to resolve these issues by stabilizing the expressed protein, or expressing activated LMTK3 protein, will in time provide sufficient material for complete screening and optimization of crystallization conditions.
Through a range of techniques, we have validated that LMTK3 has the classic characteristics of an HSP90 client protein. HSP90 is a well-characterized molecular chaperone that plays an essential role in the folding and stability of a host of cellular proteins, notably including a large subset of eukaryotic protein kinases (51, 52). Recruitment of client kinases is mediated by the HSP90 cochaperone protein CDC37/p50, which directly interacts with both the HSP90 dimer and the client kinase, bridging formation of a tetrameric complex (42, 53). In recent years, studies into the CDC37-mediated recruitment of kinases to the HSP90 system have demonstrated that the CDC37-kinase interaction is antagonized by ATP-competitive kinase inhibitors (36). From these data, we propose that C28 not only competes with ATP binding to the kinase active site but also acts to promote LMTK3 degradation through a mechanism of chaperone deprivation (Fig. 7H), as it has been previously shown for other kinases and their selective inhibitors (36). Inhibition of the CDC37 interaction deprives LMTK3 of access to the HSP90 chaperone system, resulting in kinase instability and, ultimately, degradation. Future work will be required to identify the E3 ubiquitin ligase(s) involved in this process.
CDC37 phosphorylation by LMTK3 provides an additional insight into LMTK3 function. Phosphorylation of CDC37 is a key mechanism for regulating client kinase access to the HSP90 chaperone system. In particular, phosphorylation by CK2 (54) and YES kinases (55), notably both Hsp90 client proteins have been well documented as critical modifications required for normal function. Additional work will be necessary to fully determine the importance of LMTK3-mediated CDC37 phosphorylation on the chaperone system as a whole.
The ability of C28 to abolish both the catalytic and the scaffolding functions of LMTK3 has allowed us to identify both LMTK3 phospho-substrates as well as interacting partners that depend on LMTK3’s scaffolding properties. Our ongoing medicinal chemistry strategy, using structure-activity relationship data, focuses on the optimization of potent and selective LMTK3 inhibitors with low off-target effects and suitable physicochemical properties that could preferentially block the LMTK3 kinase activity. Achieving this goal will result in the identification of kinase-dependent LMTK3 substrates, which will assist in further deciphering the role of LMTK3 in signal transduction.
Moreover, C28 had no toxicity in normal tissues (fig. S8) or body weights (tables S10 and S11) in BC mouse models, while its antiproliferative and proapoptotic effects on a nontransformed breast cell line (MCF12A) were significantly lower compared to other BC cell lines. Additional testing in other types of noncancerous cell lines will provide further validation of these findings. Given the short half-life of the drug, in vivo oral dosing at 10 mg/kg signifies that exposure levels above the IC50 and less so, above the IC90 of the drug, would be attainable for only a few hours.
In aggregate, the development of oral LMTK3 inhibitors may have the potential for broad clinical use, either as monotherapy or as a combinational therapy, [i.e., combined, for example, with aromatase inhibitors in ER+ BC in the same way the CDK4/6 inhibitors are (56, 57) and combined with the standard-of-care chemotherapy in triple-negative models]. More precisely, in the case of TNBC, despite immunotherapies being helpful at one level (58), there are no approved targeted therapies. A recent report also showed promising results regarding the use of CDK12/CDK13 inhibitors alone or in combination with the chemotherapeutic agent cisplatin (59). Therefore, on the basis of aberrant expression of LMTK3 in TNBC and work showing that genomic inhibition of LMTK3 leads to inhibition of cell proliferation, invasion, and migration (10, 12, 13), an LMTK3 inhibitor could represent an attractive candidate for clinical trials. On the other hand, since the mechanism of endocrine and chemotherapy resistance in BC still remains largely unexplained, there remains a need to treat these patients in a more focused way, for example, in the setting of progression on CDK4/6 inhibitors. On the basis of our results so far (in vitro, in vivo, and clinical data from patient specimen cohorts), inhibition of LMTK3 may be important in tamoxifen (Tam) (5, 8, 9) and doxorubicin (Dox) resensitization (10). Consequently, an LMTK3 inhibitor could be used alongside established therapies (e.g., Tam and Dox) to increase the sensitivity of tumors and/or potentially overcome resistance. Last, the subsequent down-regulation of ERα protein levels following LMTK3 inhibition suggests additional opportunities to use LMTK3 inhibitors in patients with ESR1 mutations and/or in ESR1 fusions that may be currently untargetable (60).
MATERIALS AND METHODS
LMTK3 protein expression and purification
The WT catalytic domain of LMTK3 (LMTK3cat; amino acids 134 to 444) was cloned into pOPINEneo, which confers a C-terminal 8× His tag, using InFusion technology. This plasmid was cotransfected into Spodoptera frugiperda (Sf9) cells with linearized Autographa californica baculovirus bacmid using FuGENE HD (Promega, no. E2311). Briefly, 0.5 ml of Sf9 insect cells was plated in a 24-well plate at 5 × 105 cells/ml and allowed to attach. A transfection cocktail was then made by mixing 2.5 μl of linearized bacmid with 100 to 500 ng of plasmid DNA in 50 μl of SF900III medium (Thermo Fisher Scientific, no. 12658001). To this, 1.5 μl of FuGENE HD was added and mixed, and the cocktail was incubated at room temperature for 30 min. The transfection cocktail was then added to the attached Sf9 cells in the 24-well plate. The plate was incubated at 27°C for 7 days before harvesting the supernatant that contained the P0 virus. This virus was amplified by infecting 50 ml of cells in suspension at 1 × 106 cells/ml with 100 μl of virus. Cells were incubated at 27°C for 7 days with shaking at 120 rpm. The P1 virus was harvested and filter-sterilized before use. Sf9 cells (2.5 liters) at 1 × 106 cells/ml were infected with 2.5 ml of P1 virus and incubated for 3 days at 27°C with shaking at 120 rpm. The cells were harvested by centrifugation and frozen before purification.
The pellet from 2.5 liters of Sf9 culture was defrosted and resuspended in ~100 ml lysis buffer [50 mM tris (pH 7.5), 500 mM NaCl, 30 mM imidazole, and 0.2% Tween 20] containing 5 μl Benzonase nuclease (250 U/μl; Sigma-Aldrich, no. 9025-65-4) and a 50 μl protease inhibitor cocktail (Sigma-Aldrich, no. P8849). Cells were broken by passing through a Basic Z cell disruptor at 241.32 N/mm2 before removal of the cell debris by centrifugation at 30,000g for 30 min at 4°C. After clarification, the cell lysate was filtered through a 0.2-μm filter before being applied to a 1-ml HisTrap FF column (GE Healthcare, no. 11-0004-58). After washing with 50 mM tris (pH 7.5), 500 mM NaCl, and 30 mM imidazole, the protein was eluted in 50 mM tris (pH 7.5), 500 mM NaCl, and 500 mM imidazole before being automatically applied to a HiLoad 16/60 Superdex 200 column (GE Healthcare, no. 8-9893-35) equilibrated in 20 mM tris (pH 7.5) and 200 mM NaCl. All purification steps took place at 4°C. Fractions containing the WT LMTK3 kinase domain (LMTK3cat) were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) before being combined and concentrated. On average, 1 mg of purified protein (>95% pure by SDS-PAGE) could be obtained per liter of infected Sf9 cells. The identity of the protein was confirmed by intact protein mass spectrometry (measured mass = 36806.44 Da; expected mass with 1× acetylation = 36805.13 Da). Similar protocol was used for generating green fluorescent protein (negative control) and as the mutant (KD; LMTK3cat-KD) recombinant LMTK3 protein (amino acids 134 to 444), which included the following mutations K193A, D295A, and D313A.
Crystallization, data collection, and processing
The WT catalytic domain of LMTK3 (LMTK3cat; amino acids 134 to 444) was concentrated to 3.2 mg/ml in 20 mM tris (pH 7.5) containing 200 mM NaCl and crystallized at 4°C from various optimizations of Molecular Dimensions PACT screen well F10 [100 mM bis-tris propane (pH 6.5), 20% (w/v) PEG3350 and 20 mM sodium/potassium phosphate]. Optimizations included simple dilution of the original condition and change of pH according to the method of Walter et al. (61) and use of an additive screen (Hampton Research, USA). Optimized crystals were grown in 70 mM bis-tris propane (pH 6.5), 14% (w/v) PEG-3350, 14 mM sodium/potassium phosphate, and 30% (w/v) sucrose at 4°C using the sitting drop method by mixing protein and reservoir in a 1:1 ratio. Diffraction data were collected at beamline I24, Diamond Light Source with an x-ray wavelength of 0.9686 Å and recorded using a Pilatus3 6M detector. Data were merged from four crystals to obtain a final dataset. Diffraction data were integrated using XDS (62) and scaled using AIMLESS (63). The structure was solved by molecular replacement using PHASER (64) using 1IRK as a search model. The structure was refined using Phenix (65) and BUSTER (66) and rebuilt between refinement cycles using Coot (67). Validation was performed using MolProbity (68), QCCheck, and tools within Coot and Phenix.
CDC37 protein expression and purification
BL21(DE3) cells were transformed with a pET27 vector expressing human CDC37 with a C-terminal 6× His tag. Transformed cells were grown in 3-liter LB media, and protein expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside. Protein was expressed at 20°C for 16 hours. Cells were resuspended in lysis buffer [20 mM tris (pH 8), 4°C, 100 mM NaCl, and 1 mM imidazole with protease inhibitors (Roche)] and lysed by sonication. Lysate was purified by IMAC with 60-ml Co2+ resin eluting in 500 mM imidazole. Eluted proteins were further purified by size exclusion chromatography (SEC) using a Superdex 200 26/60 column (GE Healthcare) in SEC buffer [20 mM tris (pH 7.4), 4°C, 500 mM NaCl, 1 mM dithiothreitol (DTT), and 1 mM EDTA], followed by ion-exchange chromatography using Q Sepharose (GE Healthcare, 60 ml) by gradient elution to 1 M NaCl [20 mM tris (pH 7.4), 4°C, 1 mM DTT, and 1 mM EDTA]. Fractions were analyzed by SDS-PAGE and Coomassie staining. Purified protein was concentrated to 1.5 mM and snap-frozen for storage at −80°C.
PepChip analysis
PepChip microarray slides are spotted with a duplicate set of 192 peptides that contain experimentally verified phosphorylation sites for different protein kinases and their original surrounding residues. Each peptide consists of eight to nine amino acids, of which approximately the central position corresponds to the putative phosphorylation site (Ser, Thr, or Tyr residue). For incubation of the PepChip slide, an in vitro kinase assay was performed, using 2 μg of WT LMTK3-KD mixed with 5 μl of 32P γ-ATP. The mix was brought onto a cover slide, after which the PepChip was posed over the sample and turned around, following incubation for 4 hours at 30°C in a closed wet box to prevent drying of the slide. The cover slide was rinsed off the PepChip with 50 mM tris (pH 7.5), 150 mM NaCl, and 0.02% Tween 20 (TBST), and the PepChip was then washed twice with 2 M NaCl, once with 2 M urea, twice with 10% SDS, and three times with MilliQ water in a washing tube. The intensity of phosphorylated substrates was then quantified using standard autoradiography.
Arrayed PSPL
A PSPL screening was performed as previously described (AnaSpec, catalog no. AS-62017-1, UK) (18). Briefly, each peptide contained a centrally fixed phosphoacceptor residue (equimolar mixture of Ser/Thr), while 1 of the 20 naturally occurring amino acids was fixed at one of the nine surrounding positions. The remaining positions in each peptide contained a degenerate, approximately equimolar, mixture of the 17 amino acids excluding cysteine (C), S, and T. Aliquots of each reaction were spotted onto a membrane and exposed to a phosphor storage screen. Phosphate incorporation was quantified using ImageJ. Each value was then divided by the average phosphate incorporation for all amino acids at that position to yield a selectivity value for each amino acid at that position relative to the phosphoacceptor site. An optimal LMTK3 peptide substrate was designed, and mutant versions were generated with individual amino acid substitutions at different positions (JPT, Cambridge Bioscience Limited). Radiolabeled in vitro kinase assays were performed following the manufacturer’s instructions (JPT, Cambridge Bioscience Limited).
In vitro kinase assays
32P γ-ATP in vitro kinase assays were performed in-house as we have previously described (69). The selectivity “premier screening” of C28 against a panel of 140 protein kinases was performed at the MRC International Centre for Kinase Profiling unit (www.kinase-screen.mrc.ac.uk/services/premier-screen).
SILAC phosphoproteomic experiment
To define the LMTK3-regulated phosphoproteome profile, a quantitative mass spectrometry analysis using SILAC was performed. Briefly, MCF7 cells were cultured for seven cell divisions in either R0K0 “light” medium (Primacyt, no. DMEM-14-LM014), containing unlabeled [12C6,14N4]-arginine (R) and [12C6, 14N2]-lysine (K) amino acids, or in R10K8 “heavy” medium (Primacyt, no. DMEM-15-LM015), containing labeled [13C6, 15N4]-arginine and [13C6, 15N2]-lysine. These custom Dulbecco’s modified Eagle’s medium (DMEM) media were supplemented with 10% SILAC dialyzed fetal bovine serum (Primacyt, no. D-FCS100-DS1002) and 1% l-glutamine/penicillin/streptomycin solution (Sigma-Aldrich, no. G1146). Labeled cells were then transfected with either the pCMV6 vector (Origene, no. PS100001) or with a pCMV6-LMTK3 plasmid encoding full-length LMTK3 (Origene, no. RC223140). After 24 hours, proteins were extracted and digested by filter-aided sample preparation.
HTRF chemical compound screening
The peptide phosphorylation activity of LMTK3 was measured using the proprietary biotin-tagged STK Substrate 1 (S1) from the HTRF KinEASE kit (Cisbio Bioassays, no. 62ST1PEB), according to the manufacturer’s instructions. Following analysis and optimization steps, the screening assays were carried out in low-volume, black 384-well plates (Corning Life Sciences, MA), with a 10-μl assay volume containing 3 μM ATP, 50 nM STK-S1 biotin, and 25 ng per well of LMTK3 recombinant kinase domain (LMTK3cat). After incubation at 37°C for 2 hours, the reaction was stopped with buffered EDTA, which contained the detection reagents, streptavidin-XL665, and the STK-antibody labeled with Eu3+-cryptate. The resulting TR-FRET signal, calculated as the fluorescence ratio at 665/620 nm, was read on an Envision and was proportional to the level of phosphorylation of the peptide. A library of 28,716 kinase inhibitor-biased compounds screened against LMTK3cat were initially tested at a single concentration (20 μM), and the % of inhibition of the phosphorylation of STK-S1 peptide was determined. Further hit confirmation of the top 868 compounds showing >50% mean inhibition was done (20 μM, in duplicate). Last, potency and LC-MS purity analysis of the top 160 inhibitors (>50% inhibition) was performed (10 different concentrations, in duplicate), and the IC50 values were calculated. All the 28,716 compounds used for the screening were selected considering Lipinski’s rule of five properties (175 < MW (molecular weight) ≤ 500, AlogP ≤ 5, ACC (hydrogen bond acceptors) ≤ 10, Don ≤ 5, TPSA (topological polar surface area) ≤ 140).
Kinase inhibitor competition binding assay
The selectivity profiling of C28 kinase inhibitor at 5 μM was analyzed using DiscoverX KINOMEscan competition binding assay against a panel of 456 kinases (www.discoverx.com).
Differential scanning fluorimetry
DSF was performed using Roche LightCycler 96 reverse transcription polymerase chain reaction instrument with excitation and emission wavelengths set to 533 and 572 nm, respectively. Solutions comprising 16 μl of 5.4 μM LMTK3cat in 200 mM tris buffer (pH 8.0), 200 mM NaCl, and 4 μl of 50× SYPRO orange (Sigma-Aldrich) and 0.2 μl of either dimethyl sulfoxide (DMSO) or C28 in DMSO (final concentration of 10 μΜ C28, 1% (v/v) DMSO, 4.3 μΜ LMTK3, and 10× SYPRO orange). The temperature range spanned from 25° to 80°C at a scan rate of 1°C/min. Data analysis was performed in LightCycler 96 (v1.1) software using the melting curve analysis and Tm values were determined as the first negative derivative of the fluorescence with respect to the temperature.
CD spectroscopy
CD spectroscopy was performed using a Jasco J instrument. Temperature was increased from 20° to 90°C at an increment of 1°C/min, and data points were acquired every 0.2°C by monitoring a wavelength of 230 nm. For thermal stability experiments, LMTK3cat samples of 5.4 μΜ in 200 mM tris buffer (pH 8.0) and 200 mM NaCl were treated with either DMSO 0.4% (v/v) or 8.3 μΜ C28 in DMSO (0.4%) to a total volume of 120 μl in 0.1-cm cuvettes. Data analysis was performed in GraphPad Prism (8) by fitting data in the transition region to a Boltzmann sigmoidal. Apparent Tm values were determined as the point in which the transition was 50% complete.
Proteasome inhibition assay
To test whether LMTK3 is degraded by the proteasome upon C28 exposure, cells were treated with 10 μΜ of the proteasome inhibitor MG132 (or DMSO) for 4 hours before incubation with 10 μΜ C28 (or DMSO). Cells were lysed in modified radioimmunoprecipitation assay (RIPA) buffer [50 mM tris-HCl (pH 7.5), 150 mM NaCl, 1% sodium deoxycholate, 1% NP-40 alternative, 0.02% SDS containing protease, and phosphatase inhibitors (Roche Diagnostics GmbH, no. 11697498001 and no. 4906845001)] at the indicated time points. Following centrifugation at 17,000g for 30 min, the supernatant was collected as the soluble protein fraction. For collection of the insoluble protein, the remaining pellet was resuspended in modified RIPA buffer and sonicated for 10 s at 4°C using an MSE Soniprep 150.
Isothermal titration calorimetry
Proteins were dialyzed overnight into 20 mM tris (pH 7.5) containing 5 mM NaCl and 1 mM EDTA. The heat of interaction was measured on an ITC200 (isothermal titration calorimeter 200) microcalorimeter (Microcal), with a cell volume of 200 μl, under the same buffer conditions, but containing 2% DMSO and at 30°C. Typically, aliquots of 0.2 mM C28 was injected into 20 μM intact HSP90α. Heat of dilution was determined in a separate experiment by diluting protein into buffer, and the corrected data were fitted using a nonlinear least-squares curve-fitting algorithm (Microcal Origin) with three floating variables: stoichiometry, binding constant, and change in enthalpy of interaction.
Microscale thermophoresis
Purified LMTK3cat was labeled with an NHS-647 red dye (Nanotemper Technologies) following the manufacturer’s protocol. Purified CDC37 or HSP27 (Abcam, no. ab48740) were serially diluted from 10 μM in MST buffer (Nanotemper Technologies) supplemented with 0.05% Tween 20 before addition of labeled LMTK3 (50 nM). Samples were centrifuged at 12,000g for 5 min before loading into premium coated capillaries. Where specified, C28 (20 μM, 0.4% DMSO) was added to LMTK3 before mixing with CDC37. MST measurements were taken with a Monolith NT.115 system using MO control software at 21°C using 20% excitation power and 60% MST power. Data were analyzed using MO affinity software (Nanotemper Technologies).
Animal experiments
For xenograft generation, MDA-MB-231 cells (1 × 106) were injected subcutaneously into the left and right flank of 4- to 5-week-old NSG mice (nonobese diabetic–scidIL-2Rγnull, the Jackson laboratory). Once tumors were palpable (approximately 0.4 to 0.5 cm in diameter), mice started a 3-week treatment by oral gavage daily, with one break per week. The treatment groups received either vehicle (5% dextrose/PEG400 in a 1:1 ratio) or C28. To prepare the formulation, C28 was first dissolved in the necessary volume of PEG400, vortexed for 1 min, and then sonicated for 30 min at 40°C. Next, 5% dextrose solution in water was added to the formulation under vigorous vortexing. Tumors were monitored twice weekly by caliper measurements. In total, three mice died during the treatment (C28-treated animals).
For the MDA-MB-231-Luc xenograft experiment, 2 × 106 MDA-MB-231-Luc cells were suspended in 50 μl of 1× phosphate-buffered saline (PBS) and 50 μl of Matrigel (1:1 ratio) and injected subcutaneously into the flanks of 5-week-old female NU/J homozygous mice (the Jackson laboratory, Maine, USA). MDA-MB-231-Luc cells (before injection) and the tumor implanted mice were imaged immediately after cell injection using d-luciferin (Promega, WI, USA) dissolved in PBS and injected intraperitoneally at a dose of 1.5 mg per mouse, 15 min before measuring luminescence using IVIS 100 bioluminescence/optical imaging system (Xenogen Corporation, CA, USA). Mice were monitored daily for any discomfort, tumor growth, and body weight. Once the tumors reached approximately 1 cm in size, mice were randomly divided into three groups: (i) vehicle: 5% dextrose/PEG400 25%:75%, v/v (n = 6); (ii) C28: 10 mg/kg (n = 6); and (iii) C28: 30 mg/kg (n = 5). The animals were imaged before the treatment started (day 0 of the treatment) to measure the tumor growth. Treatment was given for 21 days daily through oral gavage. Mice were imaged on days 14 and 21 after treatment. The tumor growth (%) increase for each mouse was calculated on the basis of the luminescence values at day 0 (randomization) and day 21.
MMTV-Neu mice were treated by oral gavage daily for a total of 19 days, as soon as tumors were palpable. The treatment groups received either vehicle (5% dextrose/PEG400 in a 1:1 ratio) or C28. Tumors were monitored twice weekly by caliper measurements. Tumor volumes were calculated using the following formula: V = a × b2/2, where “a” is the largest diameter and “b” is the smallest. All animal studies were performed in accordance with national and international regulations and were approved by the BRFAA (Biomedical Research Foundation of the Academy of Athens) ethical committee.
PK parameters: Calculated per oral bioavailability (F%) for C28 was 130% (based on the complete set of data from the 1 mg/kg intravenous group). Possible reasons for observing F% higher than 100% with certain compounds and PK conditions include “nonlinear PK” (metabolic saturation) at nonequal intravenous and oral doses and enterohepatic recirculation. The first possibility appears to be the case since the calculation based on the incomplete set of data from the 5 mg/kg group gives a more realistic 54% number for peroral bioavailability.
Hematoxylin and eosin and IHC staining
Paraffin-embedded sections were deparaffinized and subjected to hematoxylin and eosin staining or IHC analysis using standard techniques. Heat-induced antigen retrieval was performed with citrate buffer (pH 6) using a pressure cooker before the IHC staining. The LMTK3 antibody (ab110516) was used at a concentration of 10 μg/ml. The secondary antibody (Cell Signaling) was horseradish peroxidase–conjugated and was detected with DAB (Vector Laboratories). Stained IHC sections were counterstained with Mayer’s hematoxylin.
Chemical methods and synthesis procedures
Solvents were used as purchased including deuterated solvents for nuclear magnetic resonance (NMR) use. NMR spectra were recorded on a Varian NMR 600 (1H 600 MHz; 13C {1H} 151 MHz). Chemical shifts are reported in parts per million. Spectra are referenced to the corresponding protic solvent (1H) or signals of the solvent (13C). The progress of reactions was monitored by thin-layer chromatography using commercially available glass silica gel plates (60 Å, F254). The mobile phase was usually a solvent mixture, and the visualization was undertaken using ultraviolet (UV) light. Chromatographic purifications were carried out on an ISCO CombiFlash Rf75 or 150 PSI purification unit and gel columns. LC-MS purity analyses were undertaken using a 5-μm C18 110-Å column. Percent purity analysis was performed using a 30-min method in water/acetonitrile with 0.1% formic acid (5 min at 5%, 5 to 95% over 20 min, and 5 min at 95%) with the UV set to 254 nm. High-resolution mass spectrometry (HRMS) was carried out at the University of Sussex.
Synthesis of C28
Preparation of 6-chloro-3-(3-(trifluoromethoxy)phenyl)imidazo[1,2-b]pyridazine (C28-Int)
A mixture of 3-bromo-6-chloroimidazo[1,2-b]pyridazine (2.00 g, 8.60 mmol), potassium phosphate (5.48 g, 25.80 mmol), 3-(trifluoromethoxy)phenylboronic acid (1.86 g, 9.30 mmol), water (0.77 ml, 43.00 mmol), and anhydrous 1,4-dioxane (30 ml) was degassed under argon for 30 min. To the mixture was added [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II).CH2Cl2 (352 mg, 0.43 mmol), and the resulting mixture was heated to 60°C for 16 hours under an argon atmosphere. The resulting mixture was cooled to ambient temperature, filtered through Celite, and concentrated under reduced pressure to give a dark orange gum (3.6 g). The resulting residue was purified by automated flash column chromatography (n-hexane/ethyl acetate, 100:0 to 0:100, 100 g of SiO2). The appropriate fractions were combined and concentrated under reduced pressure to give 6-chloro-3-(3-(trifluoromethoxy)phenyl)imidazo[1,2-b]pyridazine (C28-Int) as a pale yellow solid (2.32 g, 86%). LC-MS [UV, electrospray ionization (ESI)] Rt = 20.37 min, [M-H] + mass/charge ratio (m/z) = 313.9, 96% purity. 1H-NMR (600 MHz, d6 −DMSO): δ = 8.46 (1H, s), 8.34 (1H, d, J = 9.4 Hz), 8.17 (1H, s), 8.15 (1H, d, J = 8.0 Hz), 7.69 (1H, m), 7.48 (1H, d, J = 9.5 Hz), and 7.42 (1H, d, J = 8.3 Hz).
Preparation of 6-(pyridin-4-yl)-3-(3-(trifluoromethoxy)phenyl)imidazo[1,2-b]pyridazine (C28)
A mixture of 6-chloro-3-(3-(trifluoromethoxy)phenyl)imidazo[1,2-b]pyridazine (980 mg, 3.13 mmol), 4-pyridinylboronic acid (578 mg, 4.70 mmol), cesium carbonate (3.06 g, 9.39 mmol), anhydrous 1,4-dioxane (15 ml), and water (5 ml) was degassed under argon for 30 min. To the mixture was added [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II).CH2Cl2 (128 mg, 0.157 mmol), and the resulting mixture was heated to 150°C under microwave irradiation for 1 hour. To the resulting mixture was added ethyl acetate (20 ml) and water (20 ml). The resulting biphasic mixture was separated, and the aqueous phase was extracted with ethyl acetate (3 × 20 ml). To the combined organic extracts was added 2 M aqueous HCl (10 ml), and the resulting mixture separated. The resulting organic phase was extracted with 2 M aqueous HCl (10 ml). The combined aqueous extracted were basified by the slow addition of saturated aqueous NaHCO3. To the mixture was added ethyl acetate (25 ml), and the resulting biphasic mixture separated. The aqueous phase was extracted with ethyl acetate (2 × 25 ml), and the combined organic extracts were washed with brine (50 ml), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give a dark yellow solid (1.3 g). The resulting residue was purified by automated column chromatography (ethyl acetate/methanol, 100:0 to 75:25, 45 g of SiO2) followed by reverse-phase chromatography [water (+0.1% formic acid)/acetonitrile (+0.1% formic acid), 95:5 to 5:95, 30 g of C18]. The appropriate fractions were combined and concentrated under reduced pressure to give 6-(pyridin-4-yl)-3-(3-(trifluoromethoxy)phenyl)imidazo[1,2-b]pyridazine (C28) as a bright yellow solid (910 mg, 82%). LC-MS (UV, ESI) Rt = 14.46 min, [M-H] + m/z = 356.9, 100% purity. 1H- NMR (600 MHz, d6 −DMSO): δ = 8.81 (2H, d, J = 5.7 Hz), 8.50 (1H, s), 8.43 (1H, d, J = 9.5 Hz), 8.36 (1H, s), 8.25 (1H, s), 8.11 to 8.08 (2H, m), 8.03 (1H, d, J = 9.5 Hz), 7.71 (1H, m), and 7.42 (1H, m). 13C NMR (151 MHz, d6 −DMSO): δ = 150.6, 149.2, 148.7, 142.3, 139.9, 135.0, 130.9, 130.4, 126.9, 125.2, 121.0, 120.2, 118.2, and 116.1 (one carbon not observed). HRMS (ESI-[+H]) m/z: Calculated for C18H12F3N4O 357.0958; found 357.0953.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Imperial BRC and ECMC, Action Against Cancer, The Colin McDavid Family Trust, The Helena Foundation, The Joseph Ettedgui Charitable Trust, A. Dusi, and The Cooper Family. The Wellcome Centre for Human Genetics is supported by the Wellcome Trust (grant 090532/Z/09/Z). R.J.O. and J.E.N. acknowledge support from the UK Medical Research Council (MR/K018779/1). C.P. acknowledges support from the Wellcome Trust (grant 210719/Z/18/Z). S.H.-H. and J.Sp. were funded by EPSRC (EP/P026990/1). N.S. and P.N. were funded by the Greek General Secretariat for Research and Technology and the Hellenic Foundation for Research and Innovation (HFRI) grant (236-EMPiBr). Patent on small-molecule inhibitors of LMTK3 has been filed to University of Sussex. Author contributions: G.G. conceived the project and designed the study. G.G. planned and oversaw the execution of all the work, collated and analyzed most of the data, interpreted the results, and wrote the first draft of the manuscript. A.D. and C.C. performed most of the biochemical and cell-based experiments. A.P. performed the biophysical experiments and contributed in the interpretation of the results and writing the respective parts. L.M.-H. performed the MST experiments and wrote the respective part of the manuscript. N.S., P.N., and A.K. performed the transgenic and the MDA-MB-231 xenograft mouse model and the related IHC analyses. J.H.L., S.S., G.S., and H.-J.L. generated the MDA-MB-231/Luc cell line, performed the respective xenograft mouse model, and did the subsequent data analysis. M.C.I., T.G., and V.V. performed specific cell-based experiments. N.E.C. was involved in initial discussions on the crystallography and the design of the LMTK3 baculovirus construct. N.E.C. and S.K. obtained the first crystals of LMTK3. J.E.N., R.L.O., R.J.O., and C.P. designed and performed the expression of the LMTK3 baculovirus construct. J.E.N., R.L.O., R.J.O., S.M.R., and C.P. carried out the crystallography and solved the structure of LMTK3. C.P. analyzed the crystallographic data, performed the ITC experiment, and wrote the appropriate sections. L.H.P. performed basic analysis on the LMTK3 crystal structure and contributed with constructive discussions and comments. L.Z. amplified the baculovirus expression construct and produced large-scale cultures. S.H.-H. and J.Sp. synthesized the C28 inhibitor used for the mouse studies. P.C. helped with the analysis of the SILAC data. J.St., J.Sp., C.P., and T.S. reviewed and edited the manuscript. All the authors read and approved the final manuscript. Competing interests: G.G. and J.Sp. are inventors on a UK patent application related to this work filed by University of Sussex (no. 2014407.7, filed 14 September 2020). J.St. is Editor of Oncogene and sits on SABs for Vaccitech, Heat Biologics, Eli Lilly, Replete, Alveo, Certis Oncology Solutions, Greenmantle and Benevolent AI; has consulted with Lansdowne Partners and Vitruvian; and sits on the Board of Directors for BB Biotech Healthcare Trust. G.G. is Editor of Gene Therapy in Cancer. L.H.P. and C.P. are coinventors of the HSP90 inhibitor AUY922 (luminespib) used in this study and receive royalties through the Rewards to Inventors scheme of The Institute of Cancer Research. L.H.P. is a nonexecutive director of Domainex Ltd and a scientific advisor to Turbine.AI. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc3099/DC1
REFERENCES AND NOTES
- 1.Blume-Jensen P., Hunter T., Oncogenic kinase signalling. Nature 411, 355–365 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Giamas G., Man Y. L., Hirner H., Bischof J., Kramer K., Khan K., Lavina Ahmed S. S., Stebbing J., Knippschild U., Kinases as targets in the treatment of solid tumors. Cell. Signal. 22, 984–1002 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Krause D. S., Van Etten R. A., Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 353, 172–187 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Stebbing J., Zhang H., Xu Y., Grothey A., Ajuh P., Angelopoulos N., Giamas G., Characterization of the tyrosine kinase-regulated proteome in breast cancer by combined use of RNA interference (RNAi) and stable isotope labeling with amino acids in cell culture (SILAC) Quantitative Proteomics. Mol. Cell. Proteomics 14, 2479–2492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giamas G., Filipović A., Jacob J., Messier W., Zhang H., Yang D., Zhang W., Shifa B. A., Photiou A., Tralau-Stewart C., Castellano L., Green A. R., Coombes R. C., Ellis I. O., Ali S., Lenz H. J., Stebbing J., Kinome screening for regulators of the estrogen receptor identifies LMTK3 as a new therapeutic target in breast cancer. Nat. Med. 17, 715–719 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Jacob J., Favicchio R., Karimian N., Mehrabi M., Harding V., Castellano L., Stebbing J., Giamas G., LMTK3 escapes tumour suppressor miRNAs via sequestration of DDX5. Cancer Lett. 372, 137–146 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Li Z., Wu J., Ji M., Shi L., Xu B., Jiang J., Wu C., Prognostic role of lemur tyrosine kinase 3 in postoperative gastric cancer. Mol. Clin. Oncol. 2, 756–760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stebbing J., Filipovic A., Ellis I. O., Green A. R., D’Silva T. R., Lenz H. J., Coombes R. C., Wang T., Lee S. C., Giamas G., LMTK3 expression in breast cancer: Association with tumor phenotype and clinical outcome. Breast Cancer Res. Treat. 132, 537–544 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Stebbing J., Filipovic A., Lit L. C., Blighe K., Grothey A., Xu Y., Miki Y., Chow L. W., Coombes R. C., Sasano H., Shaw J. A., Giamas G., LMTK3 is implicated in endocrine resistance via multiple signaling pathways. Oncogene 32, 3371–3380 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Stebbing J., Shah K., Lit L. C., Gagliano T., Ditsiou A., Wang T., Wendler F., Simon T., Szabó K. S., O’Hanlon T., Dean M., Roslani A. C., Cheah S. H., Lee S. C., Giamas G., LMTK3 confers chemo-resistance in breast cancer. Oncogene 37, 3113–3130 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakatsuki T., LaBonte M. J., Bohanes P. O., Zhang W., Yang D., Azuma M., Barzi A., Ning Y., Loupakis F., Saadat S., Volz N., Stintzing S., el-Khoueiry R., Koizumi W., Watanabe M., Shah M., Stebbing J., Giamas G., Lenz H. J., Prognostic role of lemur tyrosine kinase-3 germline polymorphisms in adjuvant gastric cancer in Japan and the United States. Mol. Cancer Ther. 12, 2261–2272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y., Zhang H., Lit L. C., Grothey A., Athanasiadou M., Kiritsi M., Lombardo Y., Frampton A. E., Green A. R., Ellis I. O., Ali S., Lenz H.-J., Thanou M., Stebbing J., Giamas G., The kinase LMTK3 promotes invasion in breast cancer through GRB2-mediated induction of integrin β1. Sci. Signal. 7, ra58 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Xu Y., Zhang H., Nguyen V. T. M., Angelopoulos N., Nunes J., Reid A., Buluwela L., Magnani L., Stebbing J., Giamas G., LMTK3 represses tumor suppressor-like genes through chromatin remodeling in breast cancer. Cell Rep. 12, 837–849 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Zhang K., Chen L., Deng H., Zou Y., Liu J., Shi H., Xu B., Lu M., Li C., Jiang J., Wang Z., Serum lemur tyrosine kinase-3: A novel biomarker for screening primary non-small cell lung cancer and predicting cancer progression. Int. J. Clin. Exp. Pathol. 8, 629–635 (2015). [PMC free article] [PubMed] [Google Scholar]
- 15.Lu L., Yuan X., Zhang Q., Zhang H., Shen B., LMTK3 knockdown retards cell growth and invasion and promotes apoptosis in thyroid cancer. Mol. Med. Rep. 15, 2015–2022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klug L. R., Bannon A. E., Javidi-Sharifi N., Town A., Fleming W. H., VanSlyke J. K., Musil L. S., Fletcher J. A., Tyner J. W., Heinrich M. C., LMTK3 is essential for oncogenic KIT expression in KIT-mutant GIST and melanoma. Oncogene 38, 1200–1210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asano T., Sato S., Yoshimoto N., Endo Y., Hato Y., Dong Y., Takahashi S., Fujii Y., Toyama T., High expression of LMTK3 is an independent factor indicating a poor prognosis in estrogen receptor α-Positive breast cancer patients. Jpn. J. Clin. Oncol. 44, 889–897 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Mok J., Kim P. M., Lam H. Y. K., Piccirillo S., Zhou X., Jeschke G. R., Sheridan D. L., Parker S. A., Desai V., Jwa M., Cameroni E., Niu H., Good M., Remenyi A., Ma J.-L. N., Sheu Y.-J., Sassi H. E., Sopko R., Chan C. S. M., De Virgilio C., Hollingsworth N. M., Lim W. A., Stern D. F., Stillman B., Andrews B. J., Gerstein M. B., Snyder M., Turk B. E., Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 3, ra12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrao R., Lupardus P. J., The Janus Kinase (JAK) FERM and SH2 domains: Bringing specificity to JAK-receptor interactions. Front. Endocrinol. 8, 71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S., The protein kinase complement of the human genome. Science 298, 1912–1934 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Yun C. H., Boggon T. J., Li Y., Woo M. S., Greulich H., Meyerson M., Eck M. J., Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11, 217–227 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbard S. R., Wei L., Ellis L., Hendrickson W. A., Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372, 746–754 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Stothard P., The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102–1104 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Caspers N. L., Han S., Rajamohan F., Hoth L. R., Geoghegan K. F., Subashi T. A., Vazquez M. L., Kaila N., Cronin C. N., Johnson E., Kurumbail R. G., Development of a high-throughput crystal structure-determination platform for JAK1 using a novel metal-chelator soaking system. Acta Crystallogr. F Struct. Biol. Commun. 72, 840–845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayliss R., Fry A., Haq T., Yeoh S., On the molecular mechanisms of mitotic kinase activation. Open Biol. 2, 120136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kornev A. P., Taylor S. S., Ten Eyck L. F., A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 14377–14382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meharena H. S., Chang P., Keshwani M. M., Oruganty K., Nene A. K., Kannan N., Taylor S. S., Kornev A. P., Deciphering the structural basis of eukaryotic protein kinase regulation. PLOS Biol. 11, e1001680 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hubbard S. R., Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gundry C., Marco S., Rainero E., Miller B., Dornier E., Mitchell L., Caswell P. T., Campbell A. D., Hogeweg A., Sansom O. J., Morton J. P., Norman J. C., Phosphorylation of Rab-coupling protein by LMTK3 controls Rab14-dependent EphA2 trafficking to promote cell:cell repulsion. Nat. Commun. 8, 14646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H., Xu Y., Filipovic A., Lit L. C., Koo C. Y., Stebbing J., Giamas G., SILAC-based phosphoproteomics reveals an inhibitory role of KSR1 in p53 transcriptional activity via modulation of DBC1. Br. J. Cancer 109, 2675–2684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Degorce F., Card A., Soh S., Trinquet E., Knapik G. P., Xie B., HTRF: A technology tailored for drug discovery – a review of theoretical aspects and recent applications. Curr. Chem. Genomics 3, 22–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dexter T. M., Garland J., Scott D., Scolnick E., Metcalf D., Growth of factor-dependent hemopoietic precursor cell lines. J. Exp. Med. 152, 1036–1047 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warmuth M., Kim S., Gu X. J., Xia G., Adrian F., Ba/F3 cells and their use in kinase drug discovery. Curr. Opin. Oncol. 19, 55–60 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Ortiz M. A., Michaels H., Molina B., Toenjes S., Davis J., Marconi G. D., Hecht D., Gustafson J. L., Piedrafita F. J., Nefzi A., Discovery of cyclic guanidine-linked sulfonamides as inhibitors of LMTK3 kinase. Bioorg. Med. Chem. Lett. 30, 127108 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., Morrison M. J., Pallares G., Patel H. K., Pritchard S., Wodicka L. M., Zarrinkar P. P., A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Polier S., Samant R. S., Clarke P. A., Workman P., Prodromou C., Pearl L. H., ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nat. Chem. Biol. 9, 307–312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao R., Nalluri S., Fiskus W., Balusu R., Joshi A., Mudunuru U., Buckley K. M., Robbins K., Ustun C., Reuther G. W., Bhalla K. N., Heat shock protein 90 inhibition depletes TrkA levels and signaling in human acute leukemia cells. Mol. Cancer Ther. 9, 2232–2242 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sonamoto R., Kii I., Koike Y., Sumida Y., Kato-Sumida T., Okuno Y., Hosoya T., Hagiwara M., Identification of a DYRK1A inhibitor that induces degradation of the target kinase using co-chaperone CDC37 fused with luciferase nanoKAZ. Sci. Rep. 5, 12728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Nardo D., Masendycz P., Ho S., Cross M., Fleetwood A. J., Reynolds E. C., Hamilton J. A., Scholz G. M., A central role for the Hsp90·Cdc37 molecular chaperone module in interleukin-1 receptor-associated-kinase-dependent signaling by toll-like receptors. J. Biol. Chem. 280, 9813–9822 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Li Z., Zhou L., Prodromou C., Savic V., Pearl L. H., HECTD3 mediates an HSP90-dependent degradation pathway for protein kinase clients. Cell Rep. 19, 2515–2528 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grbovic O. M., Basso A. D., Sawai A., Ye Q., Friedlander P., Solit D., Rosen N., V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. U.S.A. 103, 57–62 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verba K. A., Wang R. Y. R., Arakawa A., Liu Y., Shirouzu M., Yokoyama S., Agard D. A., Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stepanova L., Leng X., Parker S. B., Harper J. W., Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 10, 1491–1502 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Karkoulis P. K., Stravopodis D. J., Margaritis L. H., Voutsinas G. E., 17-Allylamino-17-demethoxygeldanamycin induces downregulation of critical Hsp90 protein clients and results in cell cycle arrest and apoptosis of human urinary bladder cancer cells. BMC Cancer 10, 481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J., Yang P. L., Gray N. S., Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Zhang X., Pickin K. A., Bose R., Jura N., Cole P. A., Kuriyan J., Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 450, 741–744 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu M., Bender S. A., Cuny G. D., Sherman W., Glicksman M., Ray S. S., Type II kinase inhibitors show an unexpected inhibition mode against Parkinson’s disease-linked LRRK2 mutant G2019S. Biochemistry 52, 1725–1736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H., Neimanis S., Lopez-Garcia L. A., Arencibia J. M., Amon S., Stroba A., Zeuzem S., Proschak E., Stark H., Bauer A. F., Busschots K., Jørgensen T. J. D., Engel M., Schulze J. O., Biondi R. M., Molecular mechanism of regulation of the atypical protein kinase C by N-terminal domains and an allosteric small compound. Chem. Biol. 21, 754–765 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Bhullar K. S., Lagarón N. O., McGowan E. M., Parmar I., Jha A., Hubbard B. P., Rupasinghe H. P. V., Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 17, 48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kooistra A. J., Kanev G. K., van Linden O. P. J., Leurs R., de Esch I. J. P., de Graaf C., KLIFS: A structural kinase-ligand interaction database. Nucleic Acids Res. 44, D365–D371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trepel J., Mollapour M., Giaccone G., Neckers L., Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitesell L., Lindquist S. L., HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Pearl L. H., Hsp90 and Cdc37 – a chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 15, 55–61 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Miyata Y., Nishida E., CK2 binds, phosphorylates, and regulates its pivotal substrate Cdc37, an Hsp90-cochaperone. Mol. Cell. Biochem. 274, 171–179 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Xu W., Mollapour M., Prodromou C., Wang S., Scroggins B. T., Palchick Z., Beebe K., Siderius M., Lee M. J., Couvillon A., Trepel J. B., Miyata Y., Matts R., Neckers L., Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50CDC37-AHA1 chaperone machine. Mol. Cell 47, 434–443 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slamon D. J., Neven P., Chia S., Fasching P. A., de Laurentiis M., Im S. A., Petrakova K., Bianchi G. V., Esteva F. J., Martín M., Nusch A., Sonke G. S., de la Cruz-Merino L., Beck J. T., Pivot X., Vidam G., Wang Y., Rodriguez Lorenc K., Miller M., Taran T., Jerusalem G., Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J. Clin. Oncol. 36, 2465–2472 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Sledge G. W. Jr., Toi M., Neven P., Sohn J., Inoue K., Pivot X., Burdaeva O., Okera M., Masuda N., Kaufman P. A., Koh H., Grischke E. M., Frenzel M., Lin Y., Barriga S., Smith I. C., Bourayou N., Llombart-Cussac A., MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 35, 2875–2884 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Schmid P., Adams S., Rugo H. S., Schneeweiss A., Barrios C. H., Iwata H., Diéras V., Hegg R., Im S. A., Shaw Wright G., Henschel V., Molinero L., Chui S. Y., Funke R., Husain A., Winer E. P., Loi S., Emens L. A.; IMpassion130 Trial Investigators , Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Quereda V., Bayle S., Vena F., Frydman S. M., Monastyrskyi A., Roush W. R., Duckett D. R., Therapeutic targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 36, 545–558.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Hartmaier R. J., Trabucco S. E., Priedigkeit N., Chung J. H., Parachoniak C. A., vanden Borre P., Morley S., Rosenzweig M., Gay L. M., Goldberg M. E., Suh J., Ali S. M., Ross J., Leyland-Jones B., Young B., Williams C., Park B., Tsai M., Haley B., Peguero J., Callahan R. D., Sachelarie I., Cho J., Atkinson J. M., Bahreini A., Nagle A. M., Puhalla S. L., Watters R. J., Erdogan-Yildirim Z., Cao L., Oesterreich S., Mathew A., Lucas P. C., Davidson N. E., Brufsky A. M., Frampton G. M., Stephens P. J., Chmielecki J., Lee A. V., Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann. Oncol. 29, 872–880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walter T. S., Diprose J. M., Mayo C. J., Siebold C., Pickford M. G., Carter L., Sutton G. C., Berrow N. S., Brown J., Berry I. M., Stewart-Jones G. B. E., Grimes J. M., Stammers D. K., Esnouf R. M., Jones E. Y., Owens R. J., Stuart D. I., Harlos K., A procedure for setting up high-throughput nanolitre crystallization experiments. Crystallization workflow for initial screening, automated storage, imaging and optimization. Acta Crystallogr. D Biol. Crystallogr. 61, 651–657 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabsch W., XDS. Acta Crystallogr. D, Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evans P. R., Murshudov G. N., How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.G. Bricogne, E. Blanc, M. Brandl, C. Flensburg, P. Keller, W. Paciorek, P. Roversi, A. Shariff, O. S. Smart, C. Vonrhein, T. O. Womack, BUSTER version 2.10.3. Cambridge, United Kingdom: Global Phasing Ltd. (2017).
- 67.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B. III, Snoeyink J., Adams P. D., Lovell S. C., Richardson J. S., Richardson D. C., MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giamas G., Hirner H., Shoshiashvili L., Grothey A., Gessert S., Kühl M., Henne-Bruns D., Vorgias C. E., Knippschild U., Phosphorylation of CK1delta: Identification of Ser370 as the major phosphorylation site targeted by PKA in vitro and in vivo. Biochem. J. 406, 389–398 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Melnick J. S., Janes J., Kim S., Chang J. Y., Sipes D. G., Gunderson D., Jarnes L., Matzen J. T., Garcia M. E., Hood T. L., Beigi R., Xia G., Harig R. A., Asatryan H., Yan S. F., Zhou Y., Gu X. J., Saadat A., Zhou V., King F. J., Shaw C. M., Su A. I., Downs R., Gray N. S., Schultz P. G., Warmuth M., Caldwell J. S., An efficient rapid system for profiling the cellular activities of molecular libraries. Proc. Natl. Acad. Sci. U.S.A. 103, 3153–3158 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wisniewski J. R., Zougman A., Nagaraj N., Mann M., Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Thingholm T. E., Jørgensen T. J., Jensen O. N., Larsen M. R., Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protoc. 1, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G., Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmidt S. H., Knape M. J., Boassa D., Mumdey N., Kornev A. P., Ellisman M. H., Taylor S. S., Herberg F. W., The dynamic switch mechanism that leads to activation of LRRK2 is embedded in the DFGψ motif in the kinase domain. Proc. Natl. Acad. Sci. U.S.A. 116, 14979–14988 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc3099/DC1