

Abstract
Type 2 diabetes has been regarded a complex multifactorial disease that lead to serious health complications including high cardiovascular risks. The twin cycle hypothesis postulated that both hepatic insulin resistance and dysfunction rather than death of beta (β) cell determine diabetes onset. Several studies were carried out to test this hypothesis, and all demonstrated that chronic excess calorie intake and ectopic fat accumulation within the liver and pancreas are fundamental to the development of this disease. However, these recent research advances cannot determine the exact cause of this disease. In this review, the major factors that contribute to the pathogenesis and remission of type 2 diabetes will be outlined. Importantly, the effect of disordered lipid metabolism, characterized by altered hepatic triglyceride export will be discussed. Additionally, the observed changes in pancreas morphology in type 2 diabetes will be highlighted and discussed in relation to β cell function.
Keywords: β-cell dysfunction, hepatic very low density lipoprotein triglyceride export, pancreas morphology, pathogenesis, remission, type 2 diabetes, twin cycle hypothesis
Introduction
Type 2 diabetes (T2DM) has become a global concern. According to the WHO and International Diabetes Federation reports, 425 million of the world population have diabetes, and this is expected to double in the next few decades due to the large number of people at prediabetes stage [1,2]. These figures are alarming, and would place a major burden on national health systems across world. In the UK alone, there are 3.8 million people diagnosed with diabetes, and T2DM is currently costing 10% of the National Health System budget [3].
T2DM is a slow onset disease that develops over many years of insulin resistance and progressive decline of β-cell function [4]. Despite contribution of genetic and environmental factors [5], metabolic factors are critical in determining the onset of T2DM. Excess calorie intake over many years is the factor that triggers ectopic fat storage and subsequent derangement in lipid metabolism. The latter will lead to a number of cellular processes that limit hepatic responsiveness to insulin function and decreased β-cell function. In the early years of disease development, blood glucose levels remain normal due to the high compensatory ability of the β-cell to encounter insulin resistance. Hence, it is unlikely that high glucose is the initiating factor of β-cell damage. Eventually, the continued fat-driven impairment of β-cell function will lead to the development of T2DM when approximately 40–60% of β-cell functional mass is lost [6–8].
For many years, T2DM has been considered a chronic disease, which is inevitably progressive. However, a series of studies – Counterpoint, Counterbalance and the Diabetes Remission Clinical Trial (DiRECT) – have changed this view. It has been demonstrated that long-term remission of T2DM is achievable via effective diet-induced weight loss [9–12]. The underlying pathophysiologic mechanisms that determine diabetes development are now broadly understood. The twin cycle hypothesis was published in 2008 to predict the aetiology and pathophysiology of T2DM development and reversal [13]. This was tested in several studies [9,10], and all emphasized the role of excess fat within the liver and pancreas on the pathogenesis of this disease [9,10,14]. DiRECT has clearly demonstrated that remission of T2DM is feasible and durable by dietary weight loss in the routine primary care environment [11,12]. In addition, the mechanistic studies of DiRECT confirmed and extended our previous observations related to T2DM development. It has been confirmed that a decrease in both hepatic and intrapancreatic fat is a prerequisite for diabetes remission provided that β-cell ability to recover after removal of this metabolic burden is retained [15]. Recently, the central importance of hepatic lipoprotein export on intrapancreatic fat accumulation and β-cell function was shown to be associated with both remission and redevelopment of T2DM [16]. Moreover, re-emergence of diabetes was related to increased enrichment of palmitic acid within the lipoproteins exported from the liver. This is important as palmitic acid is the obligatory product of de-novo lipogenesis (DNL) and the most toxic fatty acid to the β-cell. Further work is needed to understand the disordered lipoprotein and lipid metabolism in T2DM, especially in view of the increased risk of cardiovascular diseases. Many questions remain to be answered about lipid species, and fatty acid intermediates associated T2DM development and their relation to cardiovascular risks [17,18].
In this review, the pathogenesis of T2DM will be discussed from the perspective of the twin cycle hypothesis and its related studies. The disordered lipid metabolism related to the change in hepatic lipoprotein metabolism will be explained. Specifically, the effect of toxic lipid metabolites on β-cell function in respect to both diabetes development and remission will be discussed. Furthermore, our observations about abnormal pancreas morphology in T2DM and potential effects on the pathogenesis of this disease will be highlighted.
The twin cycle
It is over a decade since the twin cycle hypothesis, describing the route for reversibility of T2DM, was postulated [13]. It hypothesized that excess calorie intake over long term will divert excess energy storage towards the liver and other ectopic sites in the form of triglycerides. Excess lipid in the liver will decrease hepatocytes response to insulin leading to hepatic insulin resistance, thereby failing to switch off gluconeogenesis resulting in high plasma glucose and subsequently insulin levels. It was demonstrated that de-novo synthesis of fatty acids contributes largely to hepatic steatosis in human and animal models, and this is largely stimulated by insulin [19,20]. In T2DM, this will initiate the vicious cycle of hyperlipidaemia and hyperglycaemia due to a high basal insulin level. In the early years during T2DM development, β-cells respond to hepatic insulin resistance by increasing insulin secretion, raising the basal insulin level and reinforcing the liver cycle. Under these circumstances, hepatic export of very low-density lipoprotein triglycerides (VLDL-TG) will increase, pushing up the triglyceride level in circulation [21]. Subcutaneous adipose tissue provides a metabolically well tolerated fat storage area, but its capacity for storage is limited to a different extent in different individuals. In the face of increased hepatic VLDL-TG export, a personal fat threshold will be exceeded and ectopic fat accumulation will occur within the pancreas and other tissues [22]. This will initiate the pancreas cycle, whereby toxic fat metabolites will cause β-cell dysfunction in susceptible individuals. This is tolerated at the early stage of disease progress due to the compensatory ability of β-cell. However, when β-cell fail to compensate for increased loss of their functional mass, T2DM will emerge (Fig. 1).
Fig. 1.
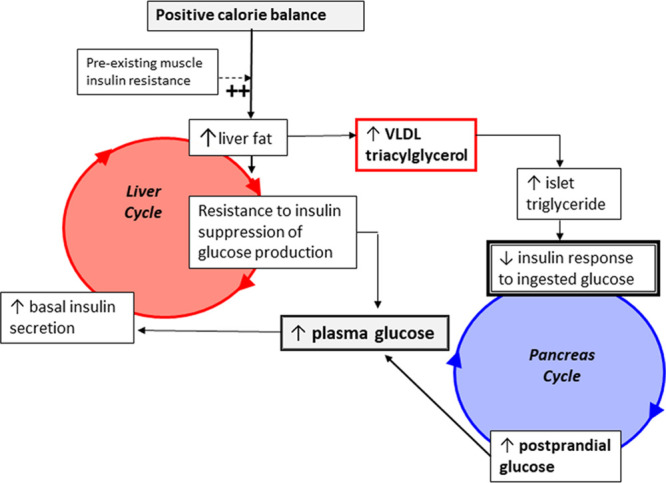
The twin cycle hypothesis of the aetiology of T2DM. Liver cycle: Prolonged exposure of excess calorie intake under pre-existed muscle insulin resistance will divert energy storage from glycogen storage into adipose tissues through activating de-novo lipogenesis (DNL) pathway. When the maximum subcutaneous fat storage capacity is reached or fat storage within the adipocytes is impaired, the plasma triglycerides level will rise and diverted into the liver. Toxic lipid intermediates from triglycerides and fatty acids metabolism will cause hepatic insulin resistance, which leads to elevated levels of fasting insulin to compensate for insulin resistance. High insulin levels will enhance DNL and enforce the liver cycle. Pancreas cycle: Excess fat accumulation within the liver leads to elevation in hepatic VLDL-TG export to other tissues. This will increase exposure of the pancreas to high triglyceride concentrations, which increase fatty acids uptake and storage within the pancreatic tissues initiating the pancreas cycle. A long-term exposure of fatty acids and related toxic metabolites including high glucose would lead to impairment in β-cell function. β-cell will overcome this stress in early years during diabetes onset by secreting more insulin. However, when 50–60% of β-cell became dysfunctional, β-cell fail to maintain normal blood glucose and T2DM will emerge. High glycaemia associated with high fasting insulinaemia will drive more DNL which will enforce both the liver and pancreas cycles. Adapted with permission from [13]. T2DM, type 2 diabetes; VLDL-TG, very low-density lipoprotein triglycerides.
Liver fat and insulin resistance
The liver is central to regulation of blood glucose through endogenous glucose production. The association between insulin resistance and nonalcoholic fatty liver disease (NAFLD) is well documented [23–26]. Although it is not clear whether NAFLD is a causative factor or a result of insulin resistance, it is widely accepted that both are related to the pathogenesis of T2DM [24,27,28]. Elucidation of the exact mechanism of hepatic insulin resistance has been a major focus for many research groups [27,29,30]. Recent data from animal and human studies highlighted the role of diacylglycerol (DAG) in activation of hepatic protein kinase Cε (PKCε), which impairs insulin signalling [31–33]. In addition, saturated fatty acids also activate toll-like receptor 4 (TLR-4) in the liver and generate ceramides potentially inhibiting insulin signalling [33–35]. However, there are contradicting reports about the role of ceramides in driving insulin resistance in human [36,37].
NAFLD is known to increase cardiovascular risks in patients with T2DM [28]. This is likely to be related to a high atherogenic profile associated with altered lipid metabolism. In our studies, almost all people with T2DM have NAFLD at varied degrees, and liver fat was normalized rapidly after weight loss, Fig.2 [9,10,15]. Importantly, this was associated with major decrease in hepatic VLDL-TG export and normalization of hepatic insulin resistance, Fig.2 [10,16].
Fig. 2.
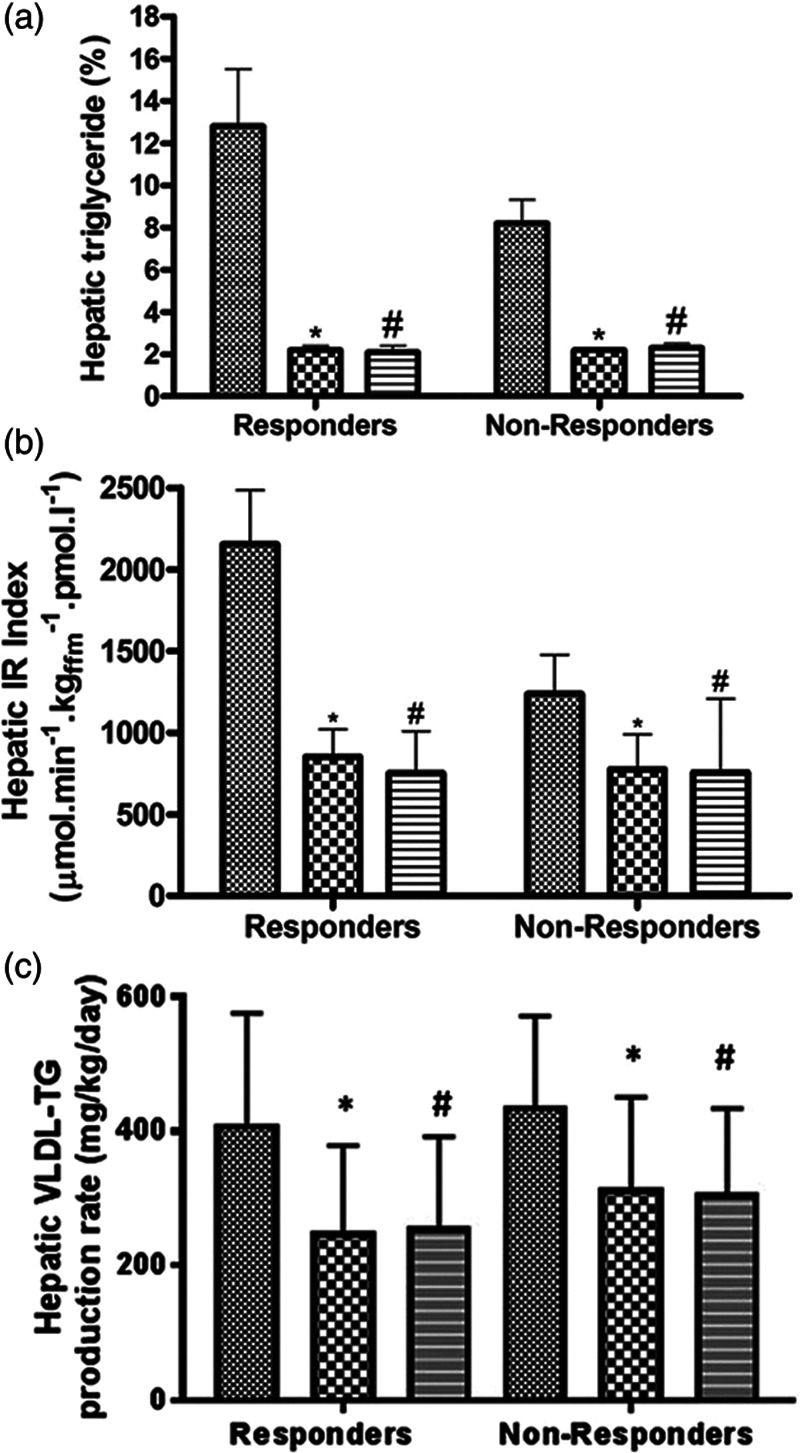
Change in liver fat, hepatic insulin resistance, and VLDL-TG production within the Counterbalance study. Hepatic triglyceride content (a), hepatic insulin resistance index (b), and hepatic VLDL1-triglyceride production (c) in those who reversed diabetes (responders: fasting blood glucose <7 mmol/L after return to isocaloric diet) and in those who failed to achieve reverse (nonresponders) at baseline (hatched bars), after VLCD (checkered bars), and after 6 months of weight maintenance (striped bars). *P < 0.05 for baseline–to–post-VLCD difference; #P < 0.05 for baseline–to–month 6 difference. Taken from [10], with permission from the American Diabetes Association. IR, insulin resistance; T2DM, type 2 diabetes; VLCD: very low calorie diet; VLDL-TG, very low-density lipoprotein triglycerides.
Polymorphisms in several genes are related to NAFLD; the PNPLA3 gene was reported to be strongly associated with NAFLD [38]. Work is currently ongoing to analyse the effect PNPLA3 polymorphism on lipid metabolism and T2DM remission within DiRECT.
Hepatic triglycerides export and lipoprotein metabolism
Another major function of the liver is to maintain lipid homeostasis. This is mainly regulated through VLDL-TG export and clearance of other lipoprotein remnants. In T2DM, lipid metabolism is abnormal, and this is a major risk factor for cardiovascular disease (CVD) development [17,18]. Disordered lipid metabolism in T2DM is characterized by overproduction of hepatic VLDL-TG [21]. This in turn is related to the expression of transcription factors that activate lipogenesis genes under elevated levels of glucose and insulin [39]. Free fatty acids derived from adipose tissue lipolysis are the major substrate for VLDL-TG production under fasting condition in healthy individuals. However, in T2DM, the contribution of DNL rises substantially [40,41]. We reported a major fall in hepatic VLDL-TG production after weight loss and this was significant only in those who achieved remission of diabetes, Fig. 3 [15,16]. This decrease was associated with sustained normalization of plasma concentration of plasma total and VLDL-specific triglycerides provided remission was maintained [16]. In those who did not achieve remission, the changes in both VLDL-TG production and plasma VLDL-TG concentration were modest (Fig. 3). On the other hand, loss of remission was associated with major rise in hepatic VLDL-TG production and plasma VLDL-TG concentration (Fig. 4), which suggest a causative effect of VLDL-TG on T2DM development although this work required to be tested in a suitable animal model to prove causality [16].
Fig. 3.
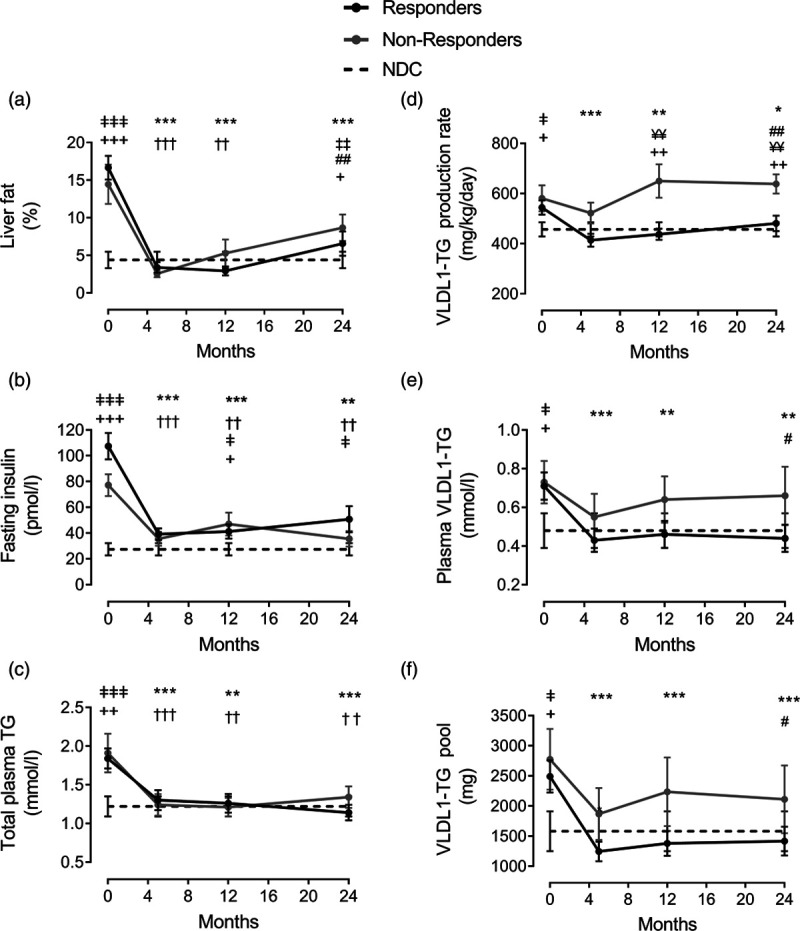
Change in lipid parameters after remission of T2DM within the DiRECT study. Liver fat (a), fasting plasma insulin (b), total plasma triglyceride (c), hepatic VLDL1-TG production (d), fasting plasma VLDL1-TG (e), and VLDL1-TG pool (f) at baseline, post weight loss (5, 12, and 24 months). Responders are presented as a solid black line, nonresponders as a solid grey line, and nondiabetic controls (NDC: measured on one occasion) as a dotted line. Data are presented as means ± SEM. Weight loss itself brought about no significant differences between responders and nonresponders at any time point. Responders vs. baseline: *P < 0.05, **P < 0.01, ***P < 0.001. Nonresponders vs. baseline: †P < 0.05, ††P < 0.01, †††P < 0.001. Responders vs. 5 months: ‡P < 0.05, ‡‡P < 0.01. Nonresponders vs. 5 months: #P < 0.05, ##P < 0.01; Responders vs. NDC: ‡P < 0.05, ‡‡P < 0.01, ‡‡‡P < 0.001; Nonresponders vs. NDC: +P < 0.05, ++P < 0.01, +++P < 0.001. Responders vs. nonresponders: ¥¥P < 0.001. Reproduced from [16] with permission from Cell Metabolism. DiRECT, Diabetes Remission Clinical Trial; T2DM, type 2 diabetes; VLDL-TG, very low-density lipoprotein triglycerides. Responders: Those who achieved remission of diabetes: HbA1c<48 mmol/mol (6.5%) and fasting blood glucose <7.0 mmol/l off all anti-diabetes medication.
Fig. 4.
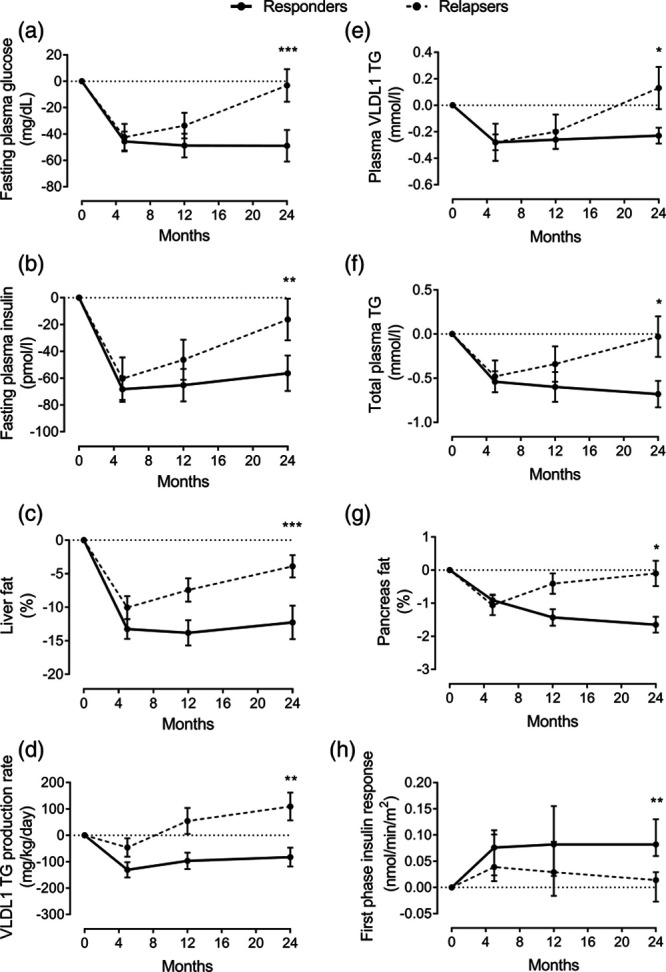
β-cell failure in response to change in lipid parameters during re-emergence of T2DM within DiRECT study. Change from baseline in fasting plasma glucose (a), fasting plasma insulin (b), liver fat (c), hepatic VLDL1-TG production (d), fasting plasma VLDL1-TG (e), total plasma triglyceride (f), intrapancreatic fat (g), and β-cell function (h) at 5 months (responders n = 38; relapsers n = 13), 12 months (n = 28/n = 13, respectively), and 24 months (n = 20/n = 13, respectively). Responders are presented as a solid black line and relapsers as a dashed line. The dotted line is the gridline at y value = 0. Paired data between baseline and each time point are presented. Data are presented as mean ± SEM except for the first phase insulin (median with IQ range) vs. 5 months in relapsers: *P < 0.05, **P < 0.01, ***P < 0.001. Taken from [16], with permission from Cell Metabolism. DiRECT, Diabetes Remission Clinical Trial; T2DM, type 2 diabetes; VLDL-TG, very low-density lipoprotein triglycerides. Responders: Those who achieved remission of diabetes: HbA1c<48 mmol/mol (6.5%) and fasting blood glucose <7.0 mmol/l off all anti-diabetes medicationRelapsers: Those who returned to diabetes state after initial remission.
ApoB and ApoE are two major lipoproteins that regulate VLDL secretion and metabolism. Liver synthesis of ApoB is essential for successful assembly and secretion of VLDL particles, and this could be a regulatory process [42]. In contract, ApoE determines the hepatic uptake of lipoprotein remnants through binding to specific receptors on the hepatocyte. ApoE is therefore critical for clearance of these highly atherogenic lipoprotein species from circulation [43]. We reported that the HDL cholesterol level increased significantly after remission of T2DM [16]. It would be therefore of interest to study the change in plasma ApoB and ApoE kinetics following remission of diabetes. Moreover, genetic polymorphisms in the ApoE gens were reported to affect lipid metabolism in Alzheimer and CVD [44,45]. The ApoE genotyping study in respect to the pathogenesis of T2DM would be of major relevance to understand lipoprotein and lipid metabolism disorders in T2DM, and this is currently underway for DiRECT study.
Adipose tissue storage
Energy from excess calorie intake has to be stored for future usage. Glycogen synthesis allows storage of glucose in the liver and muscle, and this process is regulated by insulin function [46]. When glycogen stores are filled, excess energy will be diverted as triglycerides into adipocytes, normally located in subcutaneous adipose tissues under the skin. In T2DM, hepatic and muscle insulin resistance will limit glycogen storage and will drive excess energy towards the adipose tissues [47]. Indeed, high levels of glucose and insulin can stimulate transcription factors of lipogenesis, which make this pathway dominant in T2DM [39].
The capacity of subcutaneous adipose storage is limited, and this is determined by several factors including genetics, sex, and age [48–50]. In fact, these are the major factors that determine susceptibility to develop T2DM among individuals [22]. Moreover, inflammation was reported to suppress adipose tissue capacity for expansion [51,52]. There are several alterations in metabolic processes associated with expansion of adipose tissues in T2DM. First, insulin function in suppression of lipolysis would be limited due to insulin resistance in adipose tissues [53,54]. As a result, hepatic VLDL-TG production will rise due to high fatty acids substrate coming from lipolysis of the adipose tissue. Second, excess energy in the form of VLDL-TG will be diverted into circulation to be saved into and around internal body organs [13,21]. Third, high concentrations of saturated fatty acids can elicit inflammatory response [55], which can downregulate the capacity of adipose storage and directing fat to ectopic sites [51]. Finally, metabolism of excess triglycerides within the liver and pancreas will result in toxic lipid metabolites activating cellular process that impairs both hepatocyte and β-cell function [14,15].
In addition to store energy, adipocyte produces several regulatory adipokines that can affect our metabolism [55]. Leptin and adiponectin were reported to have antidiabetic effects through regulation of glucose and fatty acids metabolism [56–58]. Moreover, the plasma-leptin-to-adiponectin ratio is considered a marker of atherogenicity in T2DM and metabolic syndrome [59,60]. In this review, white adipose tissues that compromise the majority of the total body adipose tissues were discussed. However, brown adipose tissues, which are beyond the scope of this review, have important regulatory function on body metabolism [61]. Understanding the biology of these brown adipose tissues and their role in T2DM pathogenesis may permit greater understanding.
Pancreas fat and β-cell function
The concept of fatty pancreas is becoming widely accepted, and this has been reported to be common in most pancreas-related diseases including T2DM, pancreatitis, and pancreatic cancers [15,62,63]. We reported that intrapancreatic fat is elevated in people with T2DM compared with nondiabetic controls and reversal of diabetes has always been associated with a major decrease in pancreas fat and normalization of insulin secretion [9,10,15,16,64,65]. According to the twin cycle hypothesis, excess fat is delivered to the pancreas via hepatic VLDL-TG export [13], and initial studies demonstrated that fall in VLDL-TG export was associated with a gradual decrease in intrapancreatic fat concurrent with the restoration of β-cell function [9,10]. Further studies in DiRECT confirmed those findings, and showed that changes in VLDL-TG production and intrapancreatic fat content to be related to both diabetes reversal and redevelopment, Fig. 4 [15,16]. Furthermore, we have confirmed that a specific enrichment of palmitic acid within the VLDL-TG is likely to drive these processes [16]. The deleterious effect of saturated fatty acids on β-cell function has been known for a long time [66], and several concepts to explain the lipid-induced β-cell damage were proposed including cell apoptosis and cell dedifferentiation [6,67,68]. The exact mechanism that causes β-cell dysfunction remains uncertain [6,7,69–71]. However, β-cell dedifferentiation appears to be the most likely and has become the most widely accepted to explain β-cell failure in T2DM [67,72,73]. It proposes that under metabolic conditions of excess fat and eventually glucose, β-cell is converted to an α-cell phenotype [73,74]. Conclusive data about β-cell dedifferentiation are limited, especially in human studies [73,75]. Recently, two major regulators of β-cell dedifferentiation were reported [76]. Considering the lack of β-cell–specific markers, identification of generic biomarkers for cell stress and differentiation would be useful. In this respect, growth and differentiation factor-15 (GDF-15) and fibroblast growth factor-21(FGF-21) could serve a potential candidates considering reports of their effect on lipid metabolism and nutritional stress in T2DM [77–79]. More work is needed to exclusively determine the factor (s) that cause β-cell dysfunction and recovery in T2DM.
Data from animal and human studies indicate the potential role of saturated fatty acids in inducing endoplasmic reticulum (ER) stress that lead to β-cell dysfunction [80–83]. Recent studies emphasized the role of branched-chain amino acids (BCAAs) in T2DM [84–86]. It has been reported that BCAA stimulates insulin secretion and activates the mTORC1 kinase which is related to β-cell mass and function [87]. mTORC1 is a negative regulator of autophagy, a process that known to regulate lipid metabolism [88], is reported to be regulated by calorie restriction [89,90]. β-cell autophagy was reported to be abnormal under condition of high lipids, and removing this metabolic stress restored autophagy function [91]. Several cellular mechanisms were proposed to explain β-cell dedifferentiation [75]. Clinical and metabolic studies together with other cellular and animal studies support that the process is driven by fat-induced metabolites that cause ER stress [72,81,82] leading to β-cell dysfunction. Our data confirm that β-cell damage is reversible in the early years after diabetes onset, which supports the theory of β-cell dedifferentiation rather than apoptosis. The process underlying reversal of T2DM is likely to be redifferentiation following removal of the toxic metabolic conditions after weight loss, but this remains to be determined experimentally [14]. It is possible that normalization of autophagy function after decreasing β-cell exposure to palmitic acid might contribute to β-cell redifferentiation.
Pancreas morphology
Despite its central importance to whole body metabolism, the pancreas remains one of the least studied organs. This is largely due to the complex anatomical structural and deep position within the abdomen [92]. In the past 20 years, magnetic resonance techniques have emerged as one of the most useful tools to study this organ [93]. We have successfully developed and employed techniques to study pancreas morphology and fat content in T2DM [9,65,94,95]. We found that pancreas volume is around one-third lower than normal in T2DM and the organ has very irregular borders, present soon after diagnosis and the volume appears to decrease further with increasing duration of diabetes [94,95]. It has not been established whether T2DM develops more readily in those born with a small pancreas or loss of volume is secondary to the disease process. Insulin acts as a potent growth hormone at high concentration such as experienced by parenchymal pancreas tissues by paracrine action after a meal [47,96]. Therefore, lack of local acute insulin secretion in T2DM may explain the decline in pancreas volume. In support of this notion, it was reported that pancreas volume is decreased in people diagnosed with type 1 diabetes, where local insulin secretion is completely absent [97,98]. Restoration of insulin secretion did not bring about any improvement in pancreas volume during the first 6 months after diabetes remission [95]. However, this is expected considering the long-term of insulin deficiency experienced by pancreatic tissues during T2DM onset, which is expected to be around 10 years, and a longer-term fellow-up within DiRECT revealed a significant increase in pancreas volume only in those who achieved return of insulin secretion [99]. It is notable that insulin and insulin like growth factor-1 (IGF-1) receptors share high homology, and insulin can therefore bind to the IGF-1 receptor at low affinity, which explain the trophic effects of insulin at high concentration [100]. It is possible that lack of both IGF-1 and insulin are involved in pancreatic tissue atrophy observed to happen in T2DM. Further work on the change in IGF-1 levels following T2DM remission, and how this could be related to change in pancreas volume is required.
Synthesis of information
The twin cycle hypothesis and evolving studies have changed the perception about T2DM being a long-term progressive disease. The counterpoint study demonstrated remission from T2DM for the first time, and the counterbalance identified the effect of diabetes duration on the likelihood of return to normal blood control [9,10]. Recently, DiRECT extended our previous findings [11,12]. This had changed the clinical guidelines, which now acknowledge and apply definitive weight loss in T2DM management programmes [101]. Understanding the pathophysiological processes that lead to T2DM development and reversal is crucial to control this disease. The twin cycle hypothesis was formulated after prior study of liver metabolism. It outlined the main aetiological features of T2DM and predicted the potential route for reversal [13,102]. Until now, the exact mechanism(s) that explain how T2DM is reversed after weight loss are lacking, but hepatic insulin resistance and β-cell dysfunction have now been shown to be the major determining factors for the pathogenesis of this disease [14]. There is an accumulated body of evidence from clinical and metabolic studies over the past 10-15 years [9,10,15,16] supported by other cellular and animal studies, all emphasized the deleterious effect of lipids on hepatic insulin resistance [29,32,33,103] and β-cell function [80–83].
The lipotoxic effect of fat-derived metabolites on the hepatic insulin resistance is widely accepted. Toxic lipid intermediates were derived from fatty acids metabolism including DAG and ceramides [36,103]. In the pancreas, there are conflicting opinions whether lipotoxicity or glucotoxicty is the cause of β-cell dysfunction in T2DM [69]. It is difficult to rule out the effect of glucose from the effect of fatty acids. However, it is clear that high glucose levels cannot initiate the process. Once T2DM is established, it is likely that both glucose and fatty acids could synergistically add to the metabolic stress and lead to β-cell dysfunction [70,104], and this is more likely at advanced stages during the progress of this disease. However, there has to be an initiating factor for loss of β-cell capacity before diabetes onset. The precise mechanism of fat/glucose-induced damage to the β-cell is not finally established. Apoptosis or cell death can be observed after in-vitro β-cell exposure to saturated fatty acids [66,69,105–107]. However, it has been demonstrated recently that β-cell undergoes dedifferentiation rather than apoptosis under metabolic stress [67,73,75,108,109]. Data derived from diabetes reversal after weight loss support the dedifferentiation concept and propose that the return of β-cell function can happen through the process of redifferentiation [72].
One of the major predictions of the twin cycle hypothesis is hepatic VLDL-TG being the upstream process to deliver the toxic lipid metabolites to peripheral tissues including the pancreas [13]. We and others have confirmed that hepatic VLDL-TG production is elevated in T2DM [10,13,15,21]. We also found that the change in hepatic VLDL-TG production was associated with both remission and redevelopment of T2DM [10,15,16]. It has been established for many years that prolonged β-cell exposure to saturated fatty acids is harmful [81,105,110]. Palmitic acid is predominately produced during DNL, and incubation of β-cells with relatively low concentration of palmitic acid induced ER stress [81,83,107]. In obesity and T2DM, palmitic acid was reported to initiate the inflammatory response that causes β-cell damage via TLR4-dependent pathway [110,111]. Furthermore, T2DM is associated with raised levels of inflammatory cytokines including interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), and nuclear factor kappa-B (NF-κB) [107,112,113]. Importantly, the palmitic acid content of the exported VLDL-TG was shown to decrease after remission and markedly increase in response to relapse into diabetes [16]. Taken together, these data suggest the important role of hepatic VLDL-TG in delivering toxic lipid metabolites that may initiate harmful cellular process. Further work is required to verify the causality factor of VLDL-TG on β-cell function.
The pancreas is composed of endocrine and exocrine systems, and both systems are required to maintain normal function. We have reported that pancreas morphology is abnormal in T2DM, and this was evident from the time of diagnosis [94,95]. Acinar cell mass reflects the total pancreas volume considering the small contribution of islet and ductal systems (~5%). Clinical and observational studies of the pancreas have naturally focussed on islet function itself, but the relevance of acinar cells to endocrine function has not been considered [114–117]. While intrapancreatic fat was found to increase with long diabetes duration, both β-cell function and pancreas volume were reported to decline [10,95,118] raising the question of possible causative relationships between these variables. In the ZDF rat model of T2DM, fat replacement of the acinar cells developed into fibrosis [119], and advanced fibrosis may lead to destruction of the islets and β-cell dysfunction [116,119–121]. Immunohistochemistry studies of postmortem tissues of pancreas from people with T2DM indicated that loss of β-cell function was associated with acinar cell fibrosis [122]. Plasticity and regeneration of acinar cells are well documented [123–125], and elucidation of the physiological and molecular associations of acinar cell mass regeneration is required. IGF-1 is anabolic and growth factor hormone produced mainly in the liver [126]. If the trophic effect of insulin affects pancreas volume, IGF-1 would be expected to have a greater effect due to its higher growth function ability. This is likely to be achieved in collaboration with insulin considering the affinity between insulin and IGF-1 binding protein [100]. IGF-1 has been reported to decrease in type 1 diabetes, ageing, and T2DM [127–130] where pancreas volume was reported to decline [94,97,118]. Additionally, studies on postmortem pancreas of people with T2DM have shown fibrosis in exocrine tissues associated with decline in β-cell and increase in α-cell mass [122]. Whether this morphometric changes in acinar cells are related or secondary to loss of β-cell function warrants more investigation.
Conclusion
In summary, T2DM can now be viewed as a state of excess liver and intrapancreatic fat content. The underlying pathophysiologic mechanisms that determine diabetes development and remission are partially understood. However, disordered hepatic VLDL-TG export and the associated abnormalities in lipid metabolism during excess calorie intake appear to be central to the pathogenesis of this disease. Further investigations to identify toxic lipid metabolites and the precise in-vivo mechanisms by which they lead to β-cell dysfunction are required. Heterogeneity of T2DM has been overstated and relates largely to individual capacities both for subcutaneous storage of fat and susceptibility to fat-induced β-cell dysfunction. There is major overlap in the basic pathogenesis of both pathogenesis of both CVD and T2DM. Additionally, the relevance of abnormal pancreas morphology to the pathogenesis of T2DM requires more definitive study to identify the factors that cause acinar cell loss and regeneration and how this may affect β-cell function.
Acknowledgements
I would like to acknowledge Professor Roy Taylor and all members of his wider research team over the past 12 years involved in the Counterpoint, Counterbalance, and DiRECT studies. Diabetes UK funded Counterpoint and DiRECT. Counterbalance was funded by Newcastle Biomedical Research Centre and supported by a fellowship from the Novo Nordisk Foundation.
Conflicts of interest
There is no conflict of interest.
References
- 1.Forouhi NG, Wareham NJ. Epidemiology of diabetes. Medicine. 2019; 47:22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018; 14:88–98 [DOI] [PubMed] [Google Scholar]
- 3.Iacobucci G. Diabetes prescribing in England consumes nearly 10% of primary care budget. BMJ. 2014; 349:g5143. [DOI] [PubMed] [Google Scholar]
- 4.Porta M, Curletto G, Cipullo D, Rigault de la Longrais R, Trento M, Passera P, et al. Estimating the delay between onset and diagnosis of type 2 diabetes from the time course of retinopathy prevalence. Diabetes Care. 2014; 37:1668–1674 [DOI] [PubMed] [Google Scholar]
- 5.Murea M, Ma L, Freedman BI. Genetic and environmental factors associated with type 2 diabetes and diabetic vascular complications. Rev Diabet Stud. 2012; 9:6–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003; 52:102–110 [DOI] [PubMed] [Google Scholar]
- 7.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005; 54Suppl 2S97–107 [DOI] [PubMed] [Google Scholar]
- 8.U.K. Prospective Diabetes Study Group. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes. 1995; 44:1249–1258 [PubMed] [Google Scholar]
- 9.Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011; 54:2506–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steven S, Hollingsworth KG, Al-Mrabeh A, Avery L, Aribisala B, Caslake M, et al. Very low calorie diet and 6 months of weight stability in type 2 diabetes: pathophysiological changes in responders and nonresponders. Diabetes Care. 2016; 39:158–165 [DOI] [PubMed] [Google Scholar]
- 11.Lean ME, Leslie WS, Barnes AC, Brosnahan N, Thom G, McCombie L, et al. Primary care-led weight management for remission of type 2 diabetes (DiRECT): an open-label, cluster-randomised trial. Lancet. 2018; 391:541–551 [DOI] [PubMed] [Google Scholar]
- 12.Lean MEJ, Leslie WS, Barnes AC, Brosnahan N, Thom G, McCombie L, et al. Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2019; 7:344–355 [DOI] [PubMed] [Google Scholar]
- 13.Taylor R. Pathogenesis of type 2 diabetes: tracing the reverse route from cure to cause. Diabetologia. 2008; 51:1781–1789 [DOI] [PubMed] [Google Scholar]
- 14.Taylor R, Al-Mrabeh A, Sattar N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019; 7:726–736 [DOI] [PubMed] [Google Scholar]
- 15.Taylor R, Al-Mrabeh A, Zhyzhneuskaya S, Peters C, Barnes AC, Aribisala BS, et al. Remission of human type 2 diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for β cell recovery. Cell Metab. 2018; 28:547–556 [DOI] [PubMed] [Google Scholar]
- 16.Al-Mrabeh A, Zhyzhneuskaya SV, Peters C, Barnes AC, Melhem S, Jesuthasan A, et al. Hepatic lipoprotein export and remission of human type 2 diabetes after weight loss. Cell Metab. 2020; 31:233–249.e4 [DOI] [PubMed] [Google Scholar]
- 17.Dokken BB. The pathophysiology of cardiovascular disease and diabetes: beyond blood pressure and lipids. Diabetes Spectrum. 2008; 21:160–165 [Google Scholar]
- 18.Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007; 87:507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008; 118:829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarz JM, Linfoot P, Dare D, Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr. 2003; 77:43–50 [DOI] [PubMed] [Google Scholar]
- 21.Adiels M, Olofsson SO, Taskinen MR, Borén J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008; 28:1225–1236 [DOI] [PubMed] [Google Scholar]
- 22.Taylor R, Holman RR. Normal weight individuals who develop type 2 diabetes: the personal fat threshold. Clin Sci (Lond). 2015; 128:405–410 [DOI] [PubMed] [Google Scholar]
- 23.Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999; 107:450–455 [DOI] [PubMed] [Google Scholar]
- 24.Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015; 62:S47–S64 [DOI] [PubMed] [Google Scholar]
- 25.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010; 51:679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001; 50:1844–1850 [DOI] [PubMed] [Google Scholar]
- 27.Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol. 2017; 14:32–42 [DOI] [PubMed] [Google Scholar]
- 28.Targher G, Bertolini L, Poli F, Rodella S, Scala L, Tessari R, et al. Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes. 2005; 54:3541–3546 [DOI] [PubMed] [Google Scholar]
- 29.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004; 279:32345–32353 [DOI] [PubMed] [Google Scholar]
- 30.Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature. 2019; 576:51–60 [DOI] [PubMed] [Google Scholar]
- 31.Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014; 59:713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry RJ, Peng L, Cline GW, Wang Y, Rabin-Court A, Song JD, et al. Mechanisms by which a very-low-calorie diet reverses hyperglycemia in a rat model of type 2 diabetes. Cell Metab. 2018; 27:210–217.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012; 148:852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014; 20:678–686 [DOI] [PubMed] [Google Scholar]
- 35.Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014; 20:687–695 [DOI] [PubMed] [Google Scholar]
- 36.Petersen MC, Shulman GI. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. 2017; 38:649–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galbo T, Perry RJ, Jurczak MJ, Camporez JP, Alves TC, Kahn M, et al. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc Natl Acad Sci U S A. 2013; 110:12780–12785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, Valenti L. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol. 2013; 19:6969–6978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013; 48:434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005; 115:1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014; 146:726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haas ME, Attie AD, Biddinger SB. The regulation of ApoB metabolism by insulin. Trends Endocrinol Metab. 2013; 24:391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feingold KR, Grunfeld C. Feingold KR. Introduction to lipids and lipoproteins. Endotext. 2000, South Dartmouth, MA: MDText.com, Inc. [Google Scholar]
- 44.Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013; 9:106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sofat R, Cooper JA, Kumari M, Casas JP, Mitchell JP, Acharya J, et al. Circulating apolipoprotein E concentration and cardiovascular disease risk: meta-analysis of results from three studies. PLos Med. 2016; 13:e1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen P, Nimmo HG, Proud CG. How does insulin stimulate glycogen synthesis? Biochem Soc Symp. 1978; 43:69–95 [PubMed] [Google Scholar]
- 47.Carey PE, Halliday J, Snaar JE, Morris PG, Taylor R. Direct assessment of muscle glycogen storage after mixed meals in normal and type 2 diabetic subjects. Am J Physiol Endocrinol Metab. 2003; 284:E688–E694 [DOI] [PubMed] [Google Scholar]
- 48.Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, et al. ; EPIC-InterAct Consortium; Cambridge FPLD1 Consortium. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017; 49:17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santosa S, Jensen MD. Adipocyte fatty acid storage factors enhance subcutaneous fat storage in postmenopausal women. Diabetes. 2013; 62:775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gustafson B, Nerstedt A, Smith U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat Commun. 2019; 10:2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kursawe R, Dixit VD, Scherer PE, Santoro N, Narayan D, Gordillo R, et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes. 2016; 65:610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Pelt DW, Guth LM, Wang AY, Horowitz JF. Factors regulating subcutaneous adipose tissue storage, fibrosis, and inflammation may underlie low fatty acid mobilization in insulin-sensitive obese adults. Am J Physiol Endocrinol Metab. 2017; 313:E429–E439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011; 60:2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017; 23:804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2012; 249:218–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perry RJ, Wang Y, Cline GW, Rabin-Court A, Song JD, Dufour S, et al. Leptin mediates a glucose-fatty acid cycle to maintain glucose homeostasis in starvation. Cell. 2018; 172:234–248.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, et al. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Med. 2014; 20:759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duncan BB, Schmidt MI, Pankow JS, Bang H, Couper D, Ballantyne CM, et al. Adiponectin and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes. 2004; 53:2473–2478 [DOI] [PubMed] [Google Scholar]
- 59.Satoh N, Naruse M, Usui T, Tagami T, Suganami T, Yamada K, et al. Leptin-to-adiponectin ratio as a potential atherogenic index in obese type 2 diabetic patients. Diabetes Care. 2004; 27:2488–2490 [DOI] [PubMed] [Google Scholar]
- 60.López-Jaramillo P, Gómez-Arbeláez D, López-López J, López-López C, Martínez-Ortega J, Gómez-Rodríguez A, Triana-Cubillos S. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm Mol Biol Clin Investig. 2014; 18:37–45 [DOI] [PubMed] [Google Scholar]
- 61.Schrauwen P, van Marken Lichtenbelt WD, Spiegelman BM. The future of brown adipose tissues in the treatment of type 2 diabetes. Diabetologia. 2015; 58:1704–1707 [DOI] [PubMed] [Google Scholar]
- 62.Pezzilli R, Calculli L. Pancreatic steatosis: is it related to either obesity or diabetes mellitus? World J Diabetes. 2014; 5:415–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang H, Maitra A, Wang H. Obesity, intrapancreatic fatty infiltration, and pancreatic cancer. Clin Cancer Res. 2015; 21:3369–3371 [DOI] [PubMed] [Google Scholar]
- 64.Zhyzhneuskaya A, Al-Mrabeh A, Peters C, Barnes A, Aribisala B, Hollingsworth KG, et al. Time course of normalization of functional β-cell capacity in the diabetes remission clinical trial after weight loss in type 2 diabetes. Diabetes Care. 2020; 43:813–820 [DOI] [PubMed] [Google Scholar]
- 65.Al-Mrabeh A, Hollingsworth KG, Steven S, Tiniakos D, Taylor R. Quantification of intrapancreatic fat in type 2 diabetes by MRI. PLoS One. 2017; 12:e0174660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes. 2001; 50:69–76 [DOI] [PubMed] [Google Scholar]
- 67.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012; 150:1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006; 291:E275–E281 [DOI] [PubMed] [Google Scholar]
- 69.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004; 53Suppl 1S119–S124 [DOI] [PubMed] [Google Scholar]
- 70.El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, Hardy S, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003; 144:4154–4163 [DOI] [PubMed] [Google Scholar]
- 71.Cantley J, Ashcroft FM. Q&A: insulin secretion and type 2 diabetes: why do β-cells fail? BMC Biol. 2015; 13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.White MG, Shaw JA, Taylor R. Type 2 diabetes: the pathologic basis of reversible β-cell dysfunction. Diabetes Care. 2016; 39:2080–2088 [DOI] [PubMed] [Google Scholar]
- 73.Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab. 2016; 101:1044–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun. 2014; 5:4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β-cell dedifferentiation in diabetes: recent findings and future research directions. J Endocrinol. 2018; 236:R109–R143 [DOI] [PubMed] [Google Scholar]
- 76.Son J, Ding H, Accii D, Califano A. AFF3 and BACH2 are master regulators of metabolic inflexibility, β/α-cell transition, and dedifferentiation in type 2 diabetes. bioRxiv. [Google Scholar]
- 77.Patel S, Alvarez-Guaita A, Melvin A, Rimmington D, Dattilo A, Miedzybrodzka EL, et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab. 2019; 29:707–718.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen W, Hoo RL, Konishi M, Itoh N, Lee PC, Ye HY, et al. Growth hormone induces hepatic production of fibroblast growth factor 21 through a mechanism dependent on lipolysis in adipocytes. J Biol Chem. 2011; 286:34559–34566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, Maratos-Flier E. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes. 2010; 59:2781–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Biden TJ, Boslem E, Chu KY, Sue N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol Metab. 2014; 25:389–398 [DOI] [PubMed] [Google Scholar]
- 81.Pinnick KE, Collins SC, Londos C, Gauguier D, Clark A, Fielding BA. Pancreatic ectopic fat is characterized by adipocyte infiltration and altered lipid composition. Obesity (Silver Spring). 2008; 16:522–530 [DOI] [PubMed] [Google Scholar]
- 82.Pinnick K, Neville M, Clark A, Fielding B. Reversibility of metabolic and morphological changes associated with chronic exposure of pancreatic islet beta-cells to fatty acids. J Cell Biochem. 2010; 109:683–692 [DOI] [PubMed] [Google Scholar]
- 83.Cunha DA, Hekerman P, Ladrière L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. 2008; 121:2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012; 15:606–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. 2014; 10:723–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. 2016; 22:421–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cheng Q, Beltran VD, Chan SM, Brown JR, Bevington A, Herbert TP. System-L amino acid transporters play a key role in pancreatic β-cell signalling and function. J Mol Endocrinol. 2016; 56:175–187 [DOI] [PubMed] [Google Scholar]
- 88.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009; 458:1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mattson MP, Allison DB, Fontana L, Harvie M, Longo VD, Malaisse WJ, et al. Meal frequency and timing in health and disease. Proc Natl Acad Sci U S A. 2014; 111:16647–16653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014; 19:181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zummo FP, Cullen KS, Honkanen-Scott M, Shaw JAM, Lovat PE, Arden C. Glucagon-like peptide 1 protects pancreatic β-cells from death by increasing autophagic flux and restoring lysosomal function. Diabetes. 2017; 66:1272–1285 [DOI] [PubMed] [Google Scholar]
- 92.Navarro S. A brief history of the anatomy and physiology of a mysterious and hidden gland called the pancreas. Gastroenterol Hepatol. 2014; 37:527–534 [DOI] [PubMed] [Google Scholar]
- 93.Matos C, Cappeliez O, Winant C, Coppens E, Devière J, Metens T. MR imaging of the pancreas: a pictorial tour. Radiographics. 2002; 22:e2. [DOI] [PubMed] [Google Scholar]
- 94.Macauley M, Percival K, Thelwall PE, Hollingsworth KG, Taylor R. Altered volume, morphology and composition of the pancreas in type 2 diabetes. PLoS One. 2015; 10:e0126825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Al-Mrabeh A, Hollingsworth KG, Steven S, Taylor R. Morphology of the pancreas in type 2 diabetes: effect of weight loss with or without normalisation of insulin secretory capacity. Diabetologia. 2016; 59:1753–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Poggi C, Le Marchand-Brustel Y, Zapf J, Froesch ER, Freychet P. Effects and binding of insulin-like growth factor I in the isolated soleus muscle of lean and obese mice: comparison with insulin. Endocrinology. 1979; 105:723–730 [DOI] [PubMed] [Google Scholar]
- 97.Williams AJ, Thrower SL, Sequeiros IM, Ward A, Bickerton AS, Triay JM, et al. Pancreatic volume is reduced in adult patients with recently diagnosed type 1 diabetes. J Clin Endocrinol Metab. 2012; 97:E2109–E2113 [DOI] [PubMed] [Google Scholar]
- 98.Goda K, Sasaki E, Nagata K, Fukai M, Ohsawa N, Hahafusa T. Pancreatic volume in type 1 and type 2 diabetes mellitus. Acta Diabetol. 2001; 38:145–149 [DOI] [PubMed] [Google Scholar]
- 99.Al-Mrabeh A, Zhyzhneuskaya S, Peters C, Barnes A, Hollingsworth K, Sattar N, et al. Restoration of insulin secretory function brings about recovery of pancreas volume in type 2 diabetes. Diabetologia. 2019; 62:S82–S82 [Google Scholar]
- 100.LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin-like growth factor 1. Nat Clin Pract Endocrinol Metab. 2007; 3:302–310 [DOI] [PubMed] [Google Scholar]
- 101.Davies MJ, D’Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018; 41:2669–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Taylor R. Type 2 diabetes: etiology and reversibility. Diabetes Care. 2013; 36:1047–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010; 375:2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002; 143:339–342 [DOI] [PubMed] [Google Scholar]
- 105.Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes. 2001; 50:1771–1777 [DOI] [PubMed] [Google Scholar]
- 106.Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A. 1998; 95:2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kharroubi I, Ladrière L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology. 2004; 145:5087–5096 [DOI] [PubMed] [Google Scholar]
- 108.Accili D, Talchai SC, Kim-Muller JY, Cinti F, Ishida E, Ordelheide AM, et al. When β-cells fail: lessons from dedifferentiation. Diabetes Obes Metab. 2016; 18Suppl 1117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.White MG, Marshall HL, Rigby R, Huang GC, Amer A, Booth T, et al. Expression of mesenchymal and α-cell phenotypic markers in islet β-cells in recently diagnosed diabetes. Diabetes Care. 2013; 36:3818–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012; 15:518–533 [DOI] [PubMed] [Google Scholar]
- 111.Rogero MM, Calder PC. Obesity, inflammation, toll-like receptor 4 and fatty acids. Nutrients. 2018; 10:E432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kristiansen OP, Mandrup-Poulsen T. Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes. 2005; 54Suppl 2S114–S124 [DOI] [PubMed] [Google Scholar]
- 113.Mirza S, Hossain M, Mathews C, Martinez P, Pino P, Gay JL, et al. Type 2-diabetes is associated with elevated levels of TNF-alpha, IL-6 and adiponectin and low levels of leptin in a population of Mexican Americans: a cross-sectional study. Cytokine. 2012; 57:136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bateman AC, Turner SM, Thomas KS, McCrudden PR, Fine DR, Johnson PA, et al. Apoptosis and proliferation of acinar and islet cells in chronic pancreatitis: evidence for differential cell loss mediating preservation of islet function. Gut. 2002; 50:542–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Das SL, Kennedy JI, Murphy R, Phillips AR, Windsor JA, Petrov MS. Relationship between the exocrine and endocrine pancreas after acute pancreatitis. World J Gastroenterol. 2014; 20:17196–17205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Czakó L, Hegyi P, Rakonczay Z, Jr, Wittmann T, Otsuki M. Interactions between the endocrine and exocrine pancreas and their clinical relevance. Pancreatology. 2009; 9:351–359 [DOI] [PubMed] [Google Scholar]
- 117.Piciucchi M, Capurso G, Archibugi L, Delle Fave MM, Capasso M, Delle Fave G. Exocrine pancreatic insufficiency in diabetic patients: prevalence, mechanisms, and treatment. Int J Endocrinol. 2015; 2015:595649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Saisho Y, Butler AE, Meier JJ, Monchamp T, Allen-Auerbach M, Rizza RA, Butler PC. Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin Anat. 2007; 20:933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Matsuda A, Makino N, Tozawa T, Shirahata N, Honda T, Ikeda Y, et al. Pancreatic fat accumulation, fibrosis, and acinar cell injury in the Zucker diabetic fatty rat fed a chronic high-fat diet. Pancreas. 2014; 43:735–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moran A, Brunzell C, Cohen RC, Katz M, Marshall BC, Onady G, et al. ; CFRD Guidelines Committee. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010; 33:2697–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hart NJ, Aramandla R, Poffenberger G, Fayolle C, Thames AH, Bautista A, et al. Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight. 2018; 3:98240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988; 9:151–159 [PubMed] [Google Scholar]
- 123.Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, Stoffers DA. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest. 2007; 117:971–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Murtaugh LC, Keefe MD. Regeneration and repair of the exocrine pancreas. Annu Rev Physiol. 2015; 77:229–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhou Q, Melton DA. Pancreas regeneration. Nature. 2018; 557:351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Puche JE, Castilla-Cortázar I. Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J Transl Med. 2012; 10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carroll PV, Christ ER, Umpleby AM, Gowrie I, Jackson N, Bowes SB, et al. IGF-I treatment in adults with type 1 diabetes: effects on glucose and protein metabolism in the fasting state and during a hyperinsulinemic-euglycemic amino acid clamp. Diabetes. 2000; 49:789–796 [DOI] [PubMed] [Google Scholar]
- 128.Friedrich N, Thuesen B, Jørgensen T, Juul A, Spielhagen C, Wallaschofksi H, Linneberg A. The association between IGF-I and insulin resistance: a general population study in Danish adults. Diabetes Care. 2012; 35:768–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cohen DH, LeRoith D. Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer. 2012; 19:F27–F45 [DOI] [PubMed] [Google Scholar]
- 130.Milman S, Huffman DM, Barzilai N. The somatotropic axis in human aging: framework for the current state of knowledge and future research. Cell Metab. 2016; 23:980–989 [DOI] [PMC free article] [PubMed] [Google Scholar]