

Nanoparticle-targeted down-regulation of Kv1.3 in memory T cells corrects lupus nephritis pathology in vitro and in vivo.
Abstract
Lupus nephritis (LN) is an autoimmune disease with substantial morbidity/mortality and limited efficacy of available therapies. Memory T (Tm) lymphocytes infiltrate LN kidneys, contributing to organ damage. Analysis of LN, diabetic nephropathy, and healthy donor kidney biopsies revealed high infiltration of active CD8+ Tm cells expressing high voltage-dependent Kv1.3 potassium channels—key T cell function regulators—in LN. Nanoparticles that selectively down-regulate Kv1.3 in Tm cells (Kv1.3-NPs) reduced CD40L and interferon-γ (IFNγ) in Tm cells from LN patients in vitro. Kv1.3-NPs were tested in humanized LN mice obtained by engrafting peripheral blood mononuclear cells (PBMCs) from LN patients into immune-deficient mice. LN mice exhibited features of the disease: increased IFNγ and CD3+CD8+ T cell renal infiltration, and reduced survival versus healthy donor PBMC engrafted mice. Kv1.3-NP treatment of patient PBMCs before engraftment decreased CD40L/IFNγ and prolonged survival of LN mice. These data show the potential benefits of targeting Kv1.3 in LN.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a devastating autoimmune disorder with a wide variety of clinical symptoms predominantly affecting cutaneous, musculoskeletal, cardiovascular, and respiratory systems. SLE-related complications result in more than 10,000 hospitalizations per year. Lupus nephritis (LN) occurs in up to 60% of patients with SLE and results in significant mortality and morbidity; 10 to 30% of patients with LN develop end-stage renal disease requiring dialysis or a kidney transplant (1). As of today, there are limited therapeutic options for SLE. Current available therapies comprise nonsteroid anti-inflammatory drugs, corticosteroids, and immune suppressants. Most of these therapies have serious side effects and limited efficacy. While many new therapeutics will soon enter phase 3 clinical trials, only one new biologic (Belimumab) has been approved by the Food and Drug Administration since 1955 (2). Therefore, new safe and effective treatments are in need.
SLE is characterized by significant inflammatory immune responses due to the loss of tolerance to multiple self-antigens, production of autoantibodies, and extensive infiltration of pathogenic T lymphocytes into the skin, kidney, brain, joints, and other organs (3, 4). Targeted correction of specific dysfunctional immune cells is an attractive approach for SLE treatment. T lymphocytes, and particularly memory T (Tm) cells, are important players in the pathology of SLE as they are more abundant than in healthy individuals and display an abnormal behavior (3, 5, 6). Circulating T cells of patients with SLE show a characteristic combination of hyperactivity [with high interferon-γ (IFNγ), interleukin-17 (IL-17), chemokine receptors such as CXCR3, and CD40 ligand] and anergy (with low IL-2 production) (4, 7). These are key proteins that facilitate the stimulation of B cells, infiltration into target tissues, and inflammation. T cells stimulate B cells to produce autoantibodies that result in activation of complement and deposition of immunocomplexes that lead to organ damage and eventual failure. Activated T and B cells produce proinflammatory cytokines. Furthermore, pathogenic Tm cells infiltrate organs and tissues and directly contribute to disease development (4, 8). Specific suppression of Tm cells would thus be advantageous in SLE.
Ca2+ signaling plays a critical role in many inflammatory and autoimmune diseases. Circulating T cells in SLE demonstrated altered Ca2+ signaling with a characteristic enhanced Ca2+ response to T cell receptor stimulation and, ultimately, enhanced activation of Ca2+- dependent transcription factors, such as the nuclear factor of activated T cells (NFAT) and cyclic adenosine monophosphate response element modulator, that control the expression of genes encoding many of the key proteins responsible for the altered functionality of SLE T cells (3, 5). Ca2+ signaling in T cells is generated by the concerted function of ion channels, transporters, and pumps (9–11). In particular, voltage-dependent Kv1.3 potassium channels are highly expressed in activated effector Tm cells where they control the Ca2+ influx through Ca2+ release–activated Ca2+ channels by maintaining a hyperpolarized membrane potential (9). Effector Tm cells constitute the majority of CD4+ T cells infiltrating the kidney in a mouse model of kidney disease (12). It is well established that inhibition of Kv1.3 suppresses the Ca2+ response of T lymphocytes to antigen stimulation and downstream effector functions (9, 13). Thus, Kv1.3 channels are attractive immunosuppressive therapeutic targets (14–16). The curative efficacy of K+ channel pharmacological blockers including Kv1.3 blockers was demonstrated in a model of renal disease where they decreased proteinuria and reduced the glomerular crescents, compared to placebo (17–19). Targeting Kv1.3 in Tm cells is thus attractive in SLE as this intervention will suppress multiple Ca2+-dependent mechanisms implicated in the progression of this complex disease. We have developed nanoparticles (NPs) that selectively target Tm cells and knockdown Kv1.3 channels (Kv1.3-NPs) in these cells effectively reducing Ca2+ signaling, IFNγ, and CD40L in vitro in Tm cells from healthy donors (HDs)(20, 21).
In this study, we performed a comparative analysis of kidney biopsies from patients with LN, patients with diabetic nephropathy (DN), and healthy controls and demonstrated that the kidneys of patients with LN are highly infiltrated with active CD8+ Tm cells with increased Kv1.3 expression. Furthermore, we generated humanized mice that resembled the pathology observed in patients with LN and demonstrated that selective knockdown of Kv1.3 in Tm cells suppressed proinflammatory IFNγ and CD40L and, ultimately, increased survival.
RESULTS
Kidneys of patients with LN show increased CD8+ Tm cell infiltration
Experiments were performed to determine the abundance of different immune cells in kidney biopsies from patients with stage IV LN. Kidney biopsies from living donors for renal transplant [normal kidneys (NKs)] and patients with DN were used as controls. The clinical, as well as histopathological, features of the patients with LN and DN included in the study are outlined in tables S1 and S2, respectively. We performed immunohistochemical experiments to quantify the infiltration of T lymphocytes in the kidneys of patients with LN and DN compared to NKs. We observed increased infiltration of CD8+ (Fig. 1, A and B) and CD4+ T lymphocytes (Fig. 1, A and C) in LN and DN. Notably, we observed that the ratio of CD8+ to CD4+ T cells was increased in the kidneys of patients with LN as compared to the kidneys of patients with DN (Fig. 1D). We conducted further immunofluorescence studies to evaluate the memory phenotype of the CD8+ kidney infiltrating T cells (KITs) in patients with LN. We did not study CD4+ T cells as they are a more complex population that composes also of regulatory T (Treg) cells. Immunofluorescence staining indicated a threefold higher number of memory (CD45RO+) cells in LN kidneys as compared to DN kidneys (Fig. 1, E and F). Of these memory cells, 57% were CD8+ Tm (CD8+CD45RO+) cells in LN compared to only 39% in DN. The tissue density of CD8+CD45RO+ cells was fourfold higher in LN as compared to DN (Fig. 1G). We also used the NanoString platform to profile the relative abundance of other immune cell populations at the mRNA level in LN, DN, and NK kidney biopsy tissues, which is performed by measuring the abundance of marker genes (outlined in table S3) that are stably and specifically expressed in an immune cell type (22). We did a pairwise comparison between LN versus NK and DN versus NK for the relative immune cell type abundance, and the data are presented in Fig. 2. We observed that as compared to NK, the LN and DN kidneys showed a significant increase in the total tissue-infiltrating leukocytes, cytotoxic cells, T helper 1 (TH1) cells, Treg cells, and macrophages. Furthermore, there was also an increase in CD45 cells and T lymphocytes in both LN and DN, albeit with a borderline significance for LN, with DN kidneys additionally showing an increase in CD8+ T cells, exhausted CD8+ T cells, B cells, dendritic cells, mast cells, and neutrophils. We did not observe a significant increase in CD8+ T cells in LN in the NanoString experiments. This was possibly due to the small number of samples tested and clustering of the patients with LN in two groups (high and low CD8). The latter was confirmed in the immunohistochemistry (IHC) analysis, as shown in fig. S1, where the LN patient samples of the IHC experiments that were used in the NanoString analysis are marked. However, the overall IHC analysis of 10 patients with LN (Fig. 1B and fig. S1) did not demonstrate any separation in two distinct groups based on CD8 infiltration.
Fig. 1. LN kidneys show increased infiltration with CD8+ T cells with a predominantly memory phenotype.
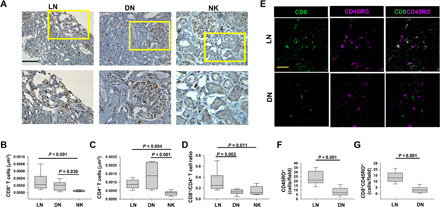
(A) IHC of CD8 (brown signal) and CD4 (pink signal) expression in representative kidney biopsies from LN, DN, and NK individuals. Scale bar, 100 μm. Magnified tissue from the highlighted area (yellow rectangles) is presented in the bottom panels. (B to D) Number of CD8+ T cells (B) and CD4+ T cells (C) in LN, DN, and NK were quantitated as described in Materials and Methods. (D) The ratio of CD8+ T cells to CD4+ T cells measured in LN kidneys, DN kidneys, and NKs. (E) Representative confocal images of kidney biopsy sections stained for CD8 (green) and CD45RO channels (magenta) in patients with LN and DN. Scale bar, 50 μm. (F and G) Number of CD45RO+ cells (F) and CD8+CD45RO+ cells (G) were quantitated as described in Materials and Methods. In (B) to (D), data are presented as box and whisker plots. The data are reported as the median (horizontal line), first (top box), and third (bottom box) quartiles in biopsy samples from 10 LN kidneys, 10 DN kidneys, and 10 NKs. In (F) and (G), the data are presented as box and whisker plots in biopsy samples from four LN kidneys and four DN kidneys where at least five fields were imaged per patient. Data in (B) to (D) were analyzed by one-way analysis of variance (ANOVA) (P < 0.001), and post hoc testing was performed by Tukey’s test, while data in (F) and (G) were analyzed by Student’s t test.
Fig. 2. Immune cell profiling of kidney biopsies from LN, DN, and healthy individuals (NK) with NanoString nCounter Autoimmune Profiling panel.
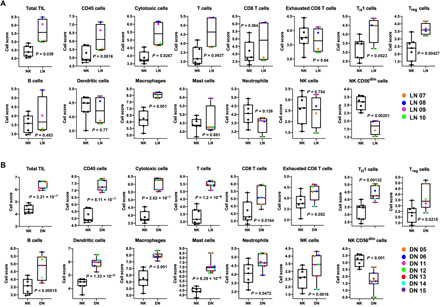
Shown here is the pairwise comparison of the abundance of the total tissue-infiltrating leukocytes (TILs) and the individual immune cell types between for (A) LN (n = 4 patients) and NK (n = 7 individuals) samples and (B) DN (n = 7 patients) and NK (n = 7 individuals) samples. The abundance of the different immune cell types (at the RNA level) in the kidney biopsies was calculated as log2 cell type scores (see Materials and Methods) and is presented as box and whisker plots. The data are reported as the median (horizontal line), first (top box), and third (bottom box) quartiles, and each symbol represents a single LN, DN, and NK individual. Statistical significance for the comparative cell type abundance was calculated using two-tailed Student’s t test. The cell scores for a specific cell type can only be compared between two groups (such as NK and LN) but do not support claims that a cell type is more abundant than another cell type within the same group.
CD8+ T cells in LN kidneys show increased cytotoxicity and proliferation
Studies have shown that infiltration by hyperactive CD8+ T cells plays a pivotal role in the kidney damage in LN (8, 23, 24). The hyperactive nature of these CD8+ T cells may due to increased Ca2+ signaling, which, in turn, is regulated by Kv1.3 channels (9). While Kv1.3 channels display an abnormal behavior in circulating SLE T cells, their expression and function in KITs are poorly understood (25–27). Kv1.3 regulates granzyme B (GrB) production and proliferation of T cells (28, 29). We performed immunofluorescence staining for Kv1.3, GrB, and the proliferation marker Ki-67 on kidney biopsy specimens from LN, DN, and NK (Fig. 3A). We observed that the surface abundance of Kv1.3 proteins within the CD8+ KITs was higher in LN and DN kidneys as compared to NK, albeit lower in LN than DN (Fig. 3B). Nevertheless, CD8+ T cells from LN kidneys had increased GrB (Fig. 3C) and Ki-67 (Fig. 3D) as compared to DN kidney and NK. These results showed that high Kv1.3 expression in LN KITs occurs with increased cytotoxicity and cell proliferation. Therefore, a therapy that blocks Kv1.3 channels in Tm cells of patients with LN could reduce the activity of KITs. Probing the potential for therapeutic agents, our laboratory has developed Kv1.3-NPs (20, 21), lipid nanovesicles enclosing small interfering RNA (siRNA) against Kv1.3 and coated with a fluorescent monoclonal antibody against the CD45RO marker of Tm lymphocytes (Fig. 4A). We conducted in vitro and in vivo experiments to test the efficacy of these NPs in SLE/LN.
Fig. 3. CD8+ T cells in LN kidneys show increased cytotoxicity and proliferation.
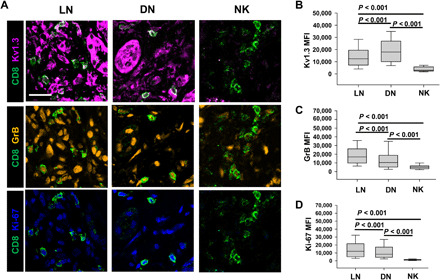
(A) Representative merged confocal images of kidney biopsy sections stained for CD8 (green), Kv1.3 (magenta), GrB (cytotoxicity marker; orange,), and Ki-67 (proliferation marker; blue) in LN, DN, and NK individuals. Scale bar, 25 μm. (B to D) Fluorescence intensities (measured as mean gray values) of Kv1.3 (B), GrB (C), and Ki-67 (D) in CD8+ T cells in kidney biopsies from LN, DN, and NK individuals. In (B) to (D), data are presented as box and whisker plots. The data are reported as the median (horizontal line), first (top box), and third (bottom box) quartiles for 675 CD8+ T cells from 10 LN kidneys, 520 cells from 10 DN kidneys, and 36 cells from 10 NK kidney biopsies. Data were analyzed by one-way ANOVA [P < 0.001 for (B) to (D)]. Post hoc testing was performed by Dunn’s test.
Fig. 4. In vitro treatment with Kv1.3-NPs decreases CD40L expression and IFNγ production in CD45RO+ T cells from patients with LN.
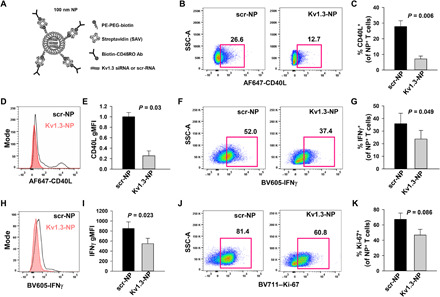
(A) Schematic representation of the structure of a lipid NP used to deliver siRNA against Kv1.3 (Kv1.3-NPs) or scramble sequence RNA (scr-NP). PE-PEG-biotin, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)-2000]; Ab, antibody. (B) Representative flow cytometry plots gated on SLE CD3+ T cells incubated overnight with either scr-NPs or Kv1.3-NPs showing CD40L expression after stimulation with thapsigargin (TG) for 3 hours. CD45RO+ population was identified by gating on the live CD3+ T cell population expressing NP fluorescence [Alexa Fluor 488 streptavidin (SAV) was used for functionalization of the NPs]. (C) Quantification of CD40L abundance in activated, scr-NP– or Kv1.3-NP–treated CD3+ T cells in five patients with SLE. (D) Representative histogram showing CD40L expression in activated NP+ CD3+ T cells from a patient with SLE incubated with either scr-NPs or Kv1.3-NPs. (E) Average geometric MFI (gMFI) of CD40L expression in activated, scr-NP– or Kv1.3-NP–treated CD3+ T cells in five patients with SLE. Data were normalized to MFI of scr-NPs. (F) Representative flow cytometry plots gated on activated NP+ CD3+ T cells from a patient with SLE incubated with either scr-NPs or Kv1.3-NPs showing IFNγ production. (G) Quantification of IFNγ induction in activated, scr-NP– or Kv1.3-NP–treated CD3+ T cells in three patients with SLE. (H) Representative histogram showing IFNγ production in activated NP+ CD3+ T cells from a patient with SLE incubated with either scr-NPs or Kv1.3-NPs. (I) gMFI of IFNγ abundance in activated, scr-NP– or Kv1.3-NP–treated CD3+ T cells in three patients with SLE. (J) Representative flow cytometry plots gated on SLE CD3+ T cells incubated overnight with either scr-NPs or Kv1.3-NPs showing Ki-67 expression. (K) Quantification of Ki-67 in activated, scr-NP– or Kv1.3-NP–treated CD3+ T cells in three patients with SLE. Bars represent means ± SEM. Data were analyzed by paired Student’s t test.
In vitro treatment with Kv1.3 NPs decreases CD40L expression and IFNγ production in Tm cells of patients with LN
T cell activation is accompanied by an increase in the cytosolic Ca2+, which activates calcineurin thus inducing NFAT nuclear translocation and downstream transcription of CD40L and inflammatory cytokines, both contributing to the pathogenesis of LN (5, 30). We have previously shown that treatment with Kv1.3-NPs selectively blocks the activation-induced increase in intracellular Ca2+, CD40L expression, and IFNγ production in Tm cells from HDs (20, 21). Here, we conducted experiments to test whether, similar to our findings in HDs, Kv1.3-NPs inhibited the expression of CD40L and IFNγ in vitro in Tm cells from patients with LN (Fig. 4) (21). CD3+ T cells isolated from patients with LN were incubated overnight with fluorescent Kv1.3-NPs. In all of the flow cytometry analyses, the proteins of interest were measured in the cells that bound and incorporated the fluorescent NPs, which were indicative of the CD45RO+ Tm population (21). We observed that the percentage of CD45RO+ LN T cells that expressed CD40L was reduced by 73% in cells treated with Kv1.3-NPs as compared to cells treated with control NPs containing scramble sequence RNA (scr-NPs; Fig. 4, B and C). The mean fluorescence intensity (MFI) of CD40L in the LN Tm cells transduced with Kv1.3-NPs was reduced by 70% as compared to scr-NP–treated controls (Fig. 4, D and E). Furthermore, Kv1.3-NPs also decreased the percentage of LN IFNγ-producing T cells by 35% (Fig. 4, F and G) and reduced the IFNγ MFI in these cells by 36% (Fig. 4, H and I). We also observed that Kv1.3-NPs brought about a 30% reduction, albeit nonstatistically significant, in the proliferating LN Tm cells (Fig. 4, J and K). Overall, these data show the in vitro efficacy of the Kv1.3-NPs in LN Tm cells.
Humanized mouse generated by engraftment of LN peripheral blood mononuclear cells exhibits LN-like phenotype
Our next step was to test the immunomodulatory effects and efficacy of the Kv1.3-NPs in vivo using a murine model for SLE. However, mouse T cells have a different ion channel composition than human T cells and differ in the way Kv1.3 regulates membrane potential and Ca2+ signaling (31). Murine T cells express other Kv channels besides Kv1.3, while in human T cells, Kv1.3 is the only Kv channel present (31, 32). We thus generated a humanized mouse model of SLE/LN. We engrafted peripheral blood mononuclear cells (PBMCs) from patients with LN in severely immunodeficient NOD/LtSz-SCID IL-2RG−/− (NSG) mice (LN mice) and evaluated the immune phenotype and kidney pathology 6 weeks after engraftment (Fig. 5A). NSG mice engrafted with HDs PBMC mice (HDs mice) and nonengrafted mice (NE mice) were used as controls. LN mice displayed an increased abundance of human CD8+, CD3+, CD4+, and CD45+ cells in the spleen (Fig. 5B), accompanied by a significantly higher CD8/CD4 ratio (Fig. 5C) and increased serum human immunoglobulin G (IgG) antibodies (Fig. 5D) as compared to HDs and NE mice. However, in LN mice, we did not see any increase in the levels of autoantibodies against double-stranded DNA (anti-dsDNA) (fig. S2). Six weeks after engraftment, we observed increased infiltration of CD3+ and CD8+ T cells in the kidneys of LN mice, while the abundance of CD38+ plasma cells and IgG in the kidneys of LN mice did not differ significantly from HDs mice (Fig. 5E and fig. S3). Furthermore, LN mice also demonstrated evidence of kidney damage such as proteinuria (Fig. 5F), and the life span of LN mice was significantly shorter than HDs and NE mice (Fig. 5G). On profiling the immune cell populations on days 2 and 7 after engraftment, we observed a significant increase in the abundance of naïve and memory populations of human CD4+ and CD8+ T cells in the spleens of LN mice at day 7, but not their bone marrow or lungs (Table 1). Furthermore, while we detected CD38+ plasma cells in the spleen on day 2, they were significantly reduced by day 7, which may explain why we did not detect any plasma cells in the kidneys at 6 weeks (fig. S3) while we observed IgG in the serum (Fig. 5D). Similar to the PBMCs from patients with LN, the splenocytes of LN mice demonstrated a robust production of IFNγ before and after stimulation, indicating that their pathological properties were maintained after engraftment (Fig. 6). We then performed immunofluorescence experiments to evaluate whether the KITs in LN mice exhibit an altered Kv1.3 phenotype compared to peripheral T cells. We observed that, compared to splenic CD8+ T cells, the CD8+ T cells from the kidneys of the same mice from which the spleens were taken showed significantly higher Kv1.3 protein abundance (Fig. 7). Overall, these LN mice demonstrated robust engraftment of donor cells and developed features resembling lupus pathology. Furthermore, LN KITs showed a Kv1.3high phenotype raising the importance of this channel as a targetable moiety to control KIT hyperfunctionality in LN. We then used this humanized LN mouse model to study whether targeted treatment of donors’ PBMCs by Kv1.3-NPs before engraftment would alter the disease development.
Fig. 5. Humanized mouse generated by engraftment of LN PBMCs exhibits LN-like phenotype.
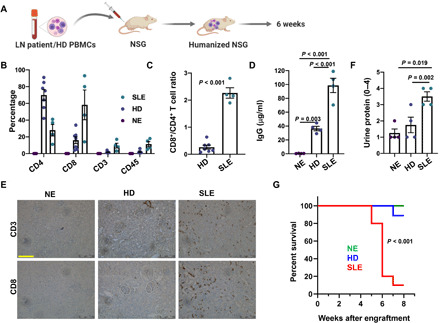
(A) Schematic of humanized mouse model generation by engraftment of a NSG mouse with PBMCs from either a patient with LN or an HDs. (B) Left: Splenic T cell abundance in LN and HDs engrafted, as well as NE, mice measured by flow cytometry 6 to 8 weeks after engraftment. Human-specific antibodies were used to detect the T lymphocyte markers presented in the figure. (C) The ratio of CD8+ T cells to CD4+ T cells measured in splenocytes from LN and HDs mice. (D) Serum IgG levels (human) measured 6 weeks after engraftment in LN, HDs, and NE mice. (E) IHC of CD3 and CD8 expression (brown signal) in kidney tissues harvested from LN, HDs, and NE mice 6 weeks after engraftment. Representative image is shown here. Scale bar, 100 μm. (F) Urine protein measured in LN and HDs engrafted, as well as NE, mice. (G) Survival in LN, HDs, and NE mice presented as Kaplan-Meier Survival curve. Significance was evaluated by a log-rank test. All experiments were performed 6 to 8 weeks after engraftment. PBMCs from three SLE/LN individuals and two HDs were used for engrafting 4 to 12 mice per group. Bars represent means ± SEM, and each symbol represents an individual mouse. In (B), the CD4, CD8, CD3, and CD45 abundances were compared between the NE, HDs, and LN groups by one-way ANOVA (P < 0.001 for all groups). Data in (C) were analyzed by Student’s t test, while data in (D) and (F) were analyzed by one-way ANOVA (P < 0.05) and post hoc testing was performed by Holm-Sidak method.
Table 1. Immune cell population in LN mice on days 2 and 7 after engraftment.
PBMCs from two patients with LN were engrafted in four NSG mice, and immune cell populations were profiled on days 2 and 7 by flow cytometry and are presented as percentages of total live cells. Naïve T cells were defined as CD3+CD45RO−CD38−FSCintermediate; Tm cells were defined as CD3+CD45RO+CD38−FScintermediate; plasma cells were defined as CD3−CD38+. Data were analyzed by Student’s t test.
| % of total live cells | |||||
| Day 2 | Day 7 | P | N | ||
| Spleen | CD4+ Tnaive | 0.17 ± 0.002 | 2.18 ± 0.001 | <0.001 | 4 |
| CD4+ Tm | 0.04 ± 0.0001 | 1.24 ± 0.001 | <0.001 | 4 | |
| CD8+ Tnaive | 0.21 ± 0.001 | 1.84 ± 0.0001 | <0.001 | 4 | |
| CD8+ Tm | 0.01 ± 0.0002 | 0.8 ± 0.011 | <0.001 | 4 | |
| CD38+ plasma | 0.87 ± 0.0007 | 0.27 ± 0.001 | <0.001 | 4 | |
| Bone marrow | CD4+ T | 0.23 ± 0.001 | 0.17 ± 0.003 | <0.001 | 4 |
| CD4+ Tm | 0.1 ± 0.001 | 0.04 ± 0.0001 | <0.001 | 4 | |
| CD8+ T | 0.23 ± 0.001 | 0.14 ± 0.002 | <0.001 | 4 | |
| CD8+ Tm | 0.03 ± 0.0002 | 0.001 ± 0.0001 | <0.001 | 4 | |
| CD38+ plasma | 0.06 ± 0.0001 | 0.07 ± 0.003 | 0.016 | 4 | |
| Lung | CD4+ T | 1.16 ± 0.007 | 1.4 ± 0.002 | <0.001 | 4 |
| CD4+ Tm | 0.25 ± 0.008 | 0.24 ± 0.0009 | <0.001 | 4 | |
| CD8+ T | 0.35 ± 0.0012 | 0.32 ± 0.001 | <0.001 | 4 | |
| CD8+ Tm | 0.11 ± 0.001 | 0.11 ± 0.002 | 1.000 | 4 | |
| CD38+ plasma | 0.12 ± 0.0009 | 0.22 ± 0.002 | <0.001 | 4 | |
Fig. 6. The ability of LN T cells to produce increased IFNγ is conserved in splenocytes of LN engrafted mice.
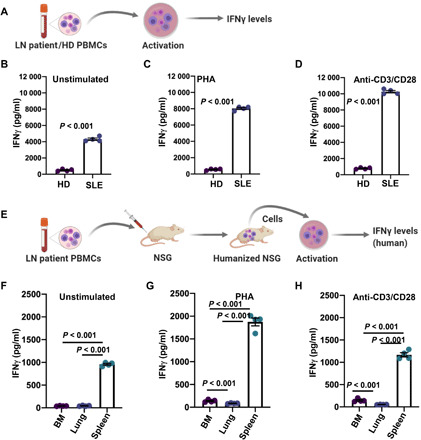
(A) Experimental protocol to measure IFNγ levels in peripheral blood T cells from patients with LN. (B to D) IFNγ levels in unstimulated (B), Phytohemagglutinin (PHA)–stimulated (C), and anti-CD3/CD28–stimulated (D) peripheral blood CD3+ T cells from four patients with LN (SLE). (E) Experimental protocol for ex vivo measurement of IFNγ levels in LN mice. Single cells from the bone marrow, lungs, and spleens of the LN mice were stimulated ex vivo, and IFNγ levels were measured. (F to H) IFNγ levels in unstimulated (F), PHA-stimulated (G), and anti-CD3/CD28–stimulated (H) single cells from the bone marrows, lungs, and spleens from four LN mice on day 7 after engraftment. The in vitro and ex vivo cell activations were performed for 48 hours. Bars represent means ± SEM, and each symbol represents an individual patient/mouse. Data in (B) to (D) were analyzed by Student’s t test, while data in (F) to (H) were analyzed by one-way ANOVA [P < 0.001 for (F), P < 0.001 for (G), and P < 0.001 for (H)] and post hoc testing was performed by Holm-Sidak method.
Fig. 7. CD8+ T cells in the kidneys of LN mice show increased Kv1.3 expression.
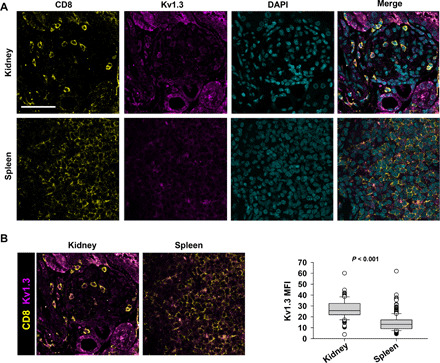
(A) Representative confocal images of kidney and spleen tissues harvested 6 weeks after engraftment from LN mice that were stained for CD8 (yellow), Kv1.3 (magenta), and nuclei [4′,6-diamidino-2-phenylindole (DAPI); cyan]. Scale bar, 50 μm. (B) Left: Merged images of CD8 and Kv1.3 channels in the kidney and spleen from LN mice showing Kv1.3 staining in the CD8+ T cells. Right: Fluorescence intensities (measured as mean gray values) of Kv1.3 in CD8+ T cells in the kidneys and spleens from LN mice. Data are presented as box and whisker plots. The data are reported as the median (horizontal line), first (top box), and third (bottom box) quartiles for 134 CD8+ T cells from two LN mice kidneys and 400 CD8+ T cells from two LN mice spleens. Significance was determined by Mann-Whitney rank sum test.
Kv1.3-NPs decrease CD40L and IFNγ ultimately increasing survival in the humanized LN mice
We pretreated PBMCs from patients with LN with either scr-NPs or Kv1.3-NPs, engrafted them in NSG mice, and then evaluated the expression of CD40L and IFNγ in splenocytes, peripheral blood T cells, and single cells from the bone marrow and lungs after stimulation ex vivo (Fig. 8A). We observed that preincubation of LN PBMCs with Kv1.3-NPs did not affect the engraftment. We measured the same number of T cells in the spleens and lungs of LN mice engrafted with untreated PBMCs and with PBMCs that were pretreated with scr-NPs and Kv1.3-NPs (fig. S4). Similar to our observations from our in vitro studies on LN T cells transduced with scr-NPs and Kv1.3-NPs (Fig. 4), we observed that the splenocytes isolated from LN mice whose engrafting PBMCs were pretreated with Kv1.3-NPs showed a reduced abundance of CD40L (Fig. 8B) and IFNγ (Fig. 8C) on day 7 after engraftment, whereas the cell proliferation was reduced although this effect was not statistically significant (Fig. 8D). However, the peripheral blood T cells and single cells isolated from the bone marrow and lungs of LN mice transduced with either scr-NPs or Kv1.3-NPs did not show any significant differences in CD40L, IFNγ, or Ki-67 (fig. S5). Furthermore, we observed that pretreatment with Kv1.3-NPs before engraftment increased the survival of LN mice by 66% (Fig. 8E). These results provide mechanistic insights into targeting Kv1.3 channels in SLE/LN and its benefits.
Fig. 8. Pretreatment of PBMCs from patients with LN with Kv1.3-NPs before engraftment decreases CD40L and IFNγ production and increases survival in the humanized LN mice.
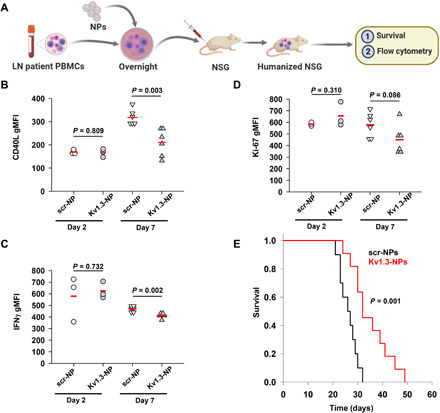
(A) PBMCs from patients with LN were preincubated overnight with either scr-NPs or Kv1.3-NPs and engrafted in NSG mice. Ex vivo flow cytometry experiments were performed on these mice, and survival was recorded in a separate set of experiments. (B to D) Fluorescence intensities of CD40L (B), IFNγ (C), and Ki-67 (D) in splenocytes of LN mice pretreated with either scr-NPs or Kv1.3-NPs before engraftment, as shown in (A). Cells were stimulated with TG for 3 hours. Shown here are fluorescence intensities measured on days 2 and 7 after engraftment. Four to six mice were engrafted per group with PBMCs from two to three patients with LN. Each symbol represents an individual mouse. Horizontal red lines represent the means for each group. Significance was calculated by Student’s t test. (E) Survival in LN mice: PBMCs from patients with LN were treated with either scr-NPs or Kv1.3-NPs before engraftment. Kaplan-Meier survival curve is shown. Data are from 10 to 11 mice per group engrafted with PBMCs from two patients with LN. Significance was calculated by a log rank test.
DISCUSSION
Several human studies have underscored the contribution of T cells, including CD8+ T cells, to the pathogenesis of LN (4, 8, 23, 24, 33–37). In this study, we showed that the kidneys of patients with LN are highly infiltrated with active CD8+ Tm cells and demonstrated, in a humanized LN mouse model, that selective down-regulation of Kv1.3 in Tm cells has beneficial effects such as suppression of proinflammatory IFNγ, CD40L, and increased host survival.
Using NanoString RNA profiling analysis, we evaluated the immune cell distribution in the kidneys of patients with LN as compared to DN and healthy kidneys. DN was chosen as disease control wherein nonimmune–mediated processes drive the nephropathy, while a Tm dysregulation plays a major role in LN kidney defect causation (4, 8, 38, 39). We observed a high infiltration of lymphocytes, cytotoxic cells, and Tm cells in both LN and DN kidneys as compared to HDs. Moreover, IHC analysis revealed that the LN kidney displayed a higher CD8+/CD4+ ratio and memory (CD45RO+) phenotype than DN. These findings are in accordance with published reports of a predominant infiltration of CD8+ T cells over CD4+ T cells in LN kidneys, features that correlate with poor treatment response and poor patient prognosis (24, 33, 36). Furthermore, a CD8+/CD4+ T cell ratio shifted in favor of CD8+ Tm cells was reported in the urine of patients with SLE, which led to the speculation that in LN effector memory CD8+ cells migrate from the peripheral blood to the kidneys and appear in the urinary sediment (6, 34). In agreement with this result, we found that more than 50% of the CD8+ KITs in patients with LN had a memory phenotype. Similar findings were reported in a recent single-cell transcriptomic analysis of LN and NK biopsies where the authors detected an abundance of cytotoxic T cells, lack of exhausted CD8+ T cells, and lack of TH1 CD4+ T cells in LN kidneys (40). In addition to the prevalence of T cells, histological studies in LN kidneys also report the presence of macrophages, B cells, and plasmablasts (40, 41). We observed an increased abundance of macrophages in LN and DN as compared to healthy kidneys but in DN, it was accompanied by a higher abundance of neutrophils, mast cells, and dendritic cells, which is in agreement with an increase in innate inflammatory cells reported in DN kidneys (39). Ultimately, LN kidney biopsies did not show exhausted CD8+ T cells, suggesting more active KITs in LN. Immunohistochemical studies confirmed the activated phenotype of CD8+ KITs in LN as these cells showed higher GrB production and cell proliferation. The presence of CD8+ T cells in the peripheral blood of patients with SLE with increased cytotoxic and IFNγ-producing capacities has been associated with the clinical presentation of LN (42).
GrB and IFNγ production are regulated by Kv1.3 channels (9, 13, 16, 28, 29). In agreement with our findings on the active state of CD8+ KITs in LN, we observed an increase in Kv1.3 channel expression in these cells as compared to HDs. However, the Kv1.3 levels in LN were lower than in DN KITs, albeit the latter showed lower functional capabilities than LN. We cannot exclude that Kv1.3 function may be higher in LN KITs as posttranslational modifications affect Kv1.3 activity. Overall, these data show a characteristic increase in Tm cells, Kv1.3, and Ca2+-dependent functions (such as IFNγ and CD40L) in KITs of patients with LN raising the possibility that a targeted down-regulation of Kv1.3 in Tm cells could ameliorate these alterations that have been associated with the manifestations of the disease (5).
Here, we showed that targeted knockdown of Kv1.3 with Kv1.3-NPs, whose selectivity and specificity were demonstrated previously by us, was effective in reducing IFNγ and CD40L of circulating Tm cells from patients with LN in vitro, thus confirming what already reported for HDs (20, 21). Testing the Kv1.3-NPs in vivo was challenging and required the development of a humanized mouse model of LN because the Kv1.3 phenotype of human T cells is different from in mice, who express other redundant Kv channels (31, 32, 43). To meet this challenge, we generated humanized LN-like mice by engrafting PBMCs from patient with LN into NSG mice. Similar models were successfully used by other groups (44). The humanized LN mice we generated demonstrated lupus-like characteristics such as increased CD8/CD4 ratio, proteinuria, and decreased survival. Furthermore, as compared to HDs mice, the kidneys of LN mice were highly infiltrated with human CD3+CD8+ T cells (34). These human CD3+ T cells from LN mice conserved the ability to produce high amount of IFNγ when stimulated ex vivo similar to T cells circulating in the blood of patients with LN (8). The CD8+ KITs in these mice showed higher Kv1.3 expression than splenocytes. Increased expression of Kv1.3 may be explained by the kidney-specific microenvironment as suggested by Chen et al. (45) that attributed increased KIT functionality in LN prone mice to reprogramming by hypoxia-inducible factor 1. However, a role for hypoxia in the increased Kv1.3 that we observed in KITs is unlikely as we have previously shown that hypoxia reduces Kv1.3 surface expression in T lymphocytes (46). Still, the increased Kv1.3 in KITs of LN mice makes this channel a relevant target for the development of new immunosuppressive therapies. However, a limitation of this model is that the splenocyte population of CD38+ plasma cells was reduced by day 7 after engraftment and, accordingly, we did not observe plasma cells infiltrating the kidneys. We observed systemic human IgG, but at levels significantly lower than those reported in patients with LN, and no detectable circulating anti-dsDNA antibodies. The lack of anti-dsDNA antibodies may be explained by the possibility that we may have only a few (or none) auto-antibody–producing plasma cells in the PBMC samples used for engraftment. We could not detect B cells in the LN kidney at late time points (6 weeks) after engraftment. However, we detected plasma cells in the spleen at early time points (2 to 7 days after engraftment), albeit declining over time and circulating human IgG at week 6 after engraftment. These factors, together with the absence of complement in the NSG mice, diminish the contribution of immunocomplexes and complement deposition to kidney damage in this mouse model (47). However, at the same time, it provides us with a unique opportunity to segregate and confirm the contribution of T cells to kidney damage similar to previously postulated (4, 8). Furthermore, we acknowledge the potential contribution of graft versus host (GvH) disease to the pathology of our LN mouse model due to engraftment of human PBMCs into the NSG mice. However, in this study, we engrafted NSG mice with equal numbers of cells from patients with either HDs or LN under the same experimental conditions, yet the LN mice demonstrated enhanced pathogenic effects. In addition, we did not observe significant weight loss in LN mice (until the end stage of the disease) or diarrhea that is a common sign of GvH. Hence, because of the controlled nature of the experiments, this mouse model was an adequate preclinical model for Kv1.3-NP testing.
Using this mouse model, we found that, similar to our observation in vitro in circulating Tm cells derived from patients with LN, Kv1.3-NPs induced a time-dependent decrease in CD40L and IFNγ expression and proliferative capacity of spleen cells. CD40L has been a therapeutic target in SLE for quite some time as it regulates T cell/B cell/dendritic cell interactions (37, 48). However, clinical trials with antibodies against CD40L have been halted because of thromboembolic complications (CD40L is present in platelets) (49, 50). Recently, a phase 2 trial in patients with SLE with an investigational anti-CD40L antibody, dapirolizumab pegol, showed promising initial results as well but was halted as it failed to meet its primary end point of a significant reduction in disease activity (51). Here, we succeeded in inhibiting CD40L expression and, concomitantly IFNγ, in Tm cells by Kv1.3 knockdown, thus achieving multiple beneficial effects through a single target and, ultimately, prolonging survival of LN mice. Dalazatide, an inhibitor of the Kv1.3 channel, has been reported to reduce IFNγ, IL-17, and tumor necrosis factor–α in CD4+ and CD8+ T cells from patients with SLE (52). Kv1.3-NPs provide the advantage over pharmacological blockers of cell-specific delivery, which limits off-target effects, and, possibly, a better safety profile due to their natural lipid–based composition (53). NPs with similar lipid composition have been previously used safely in vivo (54, 55). However, only in vivo treatment of mice with established pathology will ultimately show if these Kv1.3-NPs are of therapeutic value in SLE/LN. These are long-term studies that require carefully controlled toxicological as well as dose-response and kinetics experiments. Overall, the findings presented herein provide mechanistic insights regarding the benefits of targeted down-regulation of Kv1.3 in Tm cells in SLE.
MATERIALS AND METHODS
Experimental design
This study was designed to investigate the immune cell infiltration in LN kidneys, develop a humanized mouse model for LN, and use it to study the effects of Kv1.3-NPs in vivo. The NanoString and immunofluorescence experiments allowed us to assess the functional status of the LN CD8+ T cells, especially in the context of Kv1.3 channel function. We developed a humanized mouse model of LN by engrafting PBMCs from a patient with LN into a NSG mouse (LN mice), which was validated by staining the kidneys to study immune cell infiltration, IFNγ production, and measuring proteinuria. We then studied the effects of targeted suppression of Kv1.3 in Tm cells in the LN mice by engrafting them with PBMCs pretreated with Kv1.3-NPs (scr-NPs were used as control). We then assessed the T cell activity in vivo by measuring CD40L abundance, IFNγ production, and cell proliferation (all Kv1.3-dependent processes) in the human T cells isolated from these mice and the survival of the LN mice. These results provided us with an understanding of the functional consequences of targeting Kv1.3 channels in Tm cells in LN. The mice were randomly assigned between the control and experimental groups. The criteria for patient inclusion are described in detail below. The sample size for the experiments was selected on the basis of pilot experiments and was sufficient to detect statistical significance between the treatments; samples were not excluded, and investigators were not blinded during experiments. Statistical methods are summarized in the “Statistical analysis” section, and the P values and statistical methods used for each experiment are provided in the individual figures and legends.
Human samples
Five-micrometer-thick tissue scrolls and tissue sections prepared from formalin-fixed paraffin-embedded (FFPE) kidney biopsy specimens from 10 patients with LN in the age range of 20 to 68 years, 15 patients with DN in the age range of 32 to 63 years, and 10 living kidney donors (NKs) in the age range of 29 to 62 years were provided by the University of Cincinnati Biorepository (P = 0.17 between the age range of patients with LN and DN). The patients with LN had a confirmed diagnosis of stage IV LN based on the International Society of Nephrology/Renal Pathology Society lesion definition (56). These patients were de-identified by the biorepository, and the complete clinical information of these patients, along with a comprehensive histopathology report, were provided (see tables S1 and S2). For the in vitro and humanized mouse studies, peripheral blood was obtained from patients with de-identified SLE from the University of Cincinnati Medical Center under standard-of-care conditions in the age range of 22 to 64 years. The inclusion criteria for patients were a positive diagnosis for LN and fulfillment of the American College of Rheumatology classification criteria for SLE. Our study included 13 patients with SLE: 2 males and 11 females, 9 African-American and 4 Caucasian; 77% of whom were on dialysis. Peripheral blood was also drawn from seven age-matched (±5 years) healthy volunteers (HDs). Informed consent was obtained from all patients with SLE and HDs. Discarded blood units from Hoxworth Blood Center (University of Cincinnati) were also used as healthy controls for our study. The age and demographic information for these blood units are not available. The study protocols and the informed consent forms for all the studies were approved by the University of Cincinnati Institutional Review Board (IRB nos. 2017-3186 and 2013-1950).
Cell isolation and in vitro activation
PBMCs were isolated from whole blood by Ficoll-Paque density gradient centrifugation (GE Healthcare Biosciences) as described previously (21). CD3+ T cells were isolated from PBMCs by negative selection using the EasySep Human T Cell Enrichment Kit (STEMCELL Technologies Inc.). Cells were maintained in RPMI 1640 medium supplemented with 10% human serum, penicillin (200 U/ml), streptomycin (200 μg/ml), 1 mM l-glutamine, and 10 mM Hepes (Thermo Fisher Scientific).
T cell activation
T cells were activated using 2 μM thapsigargin (TG; MilliporeSigma) or with plate-bound mouse anti-human CD3 (10 μg/ml) and mouse anti-human CD28 (10 μg/ml) antibodies (BioLegend) as previously described (21). In experiments where IFNγ was measured using enzyme-linked immunosorbent assay (ELISA), some cells were activated with phytohemagglutinin (10 μg/ml). The activation protocol and activation times for each experiment are described in the individual figure legends.
Humanized LN mouse model generation and validation
Immunodeficient NOD/LtSz-SCID IL-2RG−/− (NSG) background mice were obtained from the Comprehensive Mouse and Cancer Core (Division of Experimental Hematology and Cancer Biology) at Cincinnati Children’s Hospital Medical Center. To generate humanized LN mice, NSG mice were intravenously engrafted with 8 × 106 PBMCs derived from patients with SLE/LN (LN mice) or healthy individuals (HDs mice), while nonengrafted mice were used as controls (NE mice) (57, 58). Eight- to 12-week-old mice of both sexes were used. All animals were matched by age and sex between the experimental groups. Animal work was approved by the Cincinnati Children’s Hospital Research Foundation’s Institutional Animal Care and Use Committee. The peripheral blood was drawn from the engrafted mice, and T cells were enriched using a Ficoll density gradient. Serum IgG was measured using the IgG (total) Human ELISA Kit (Thermo Fisher Scientific) as per the manufacturer’s instructions. Proteinuria was assessed visually using Multistix 10 SG Urine Test Strips (Siemens Medical Solutions) as per the manufacturer’s instructions. Antibodies against ds-DNA in the sera of LN, HDs, and NE mice and in the sera of patients with LN and HDs were measured by ELISA (EUROIMMUN AG) as per the manufacturer’s instructions. Mice were euthanized at times mentioned in the figure legends, and splenocytes, as well as single-cell suspension from the lungs and bone marrow, were isolated using standard isolation protocols, maintained in T cell medium, activated, and used for downstream experiments. Kidneys and spleens were harvested, fixed in 10% neutral-buffered formalin, and paraffinized, and IHC, and/or immunofluorescence staining, was performed.
Immune cell profiling with NanoString nCounter
NanoString nCounter (NanoString Technologies) was used to analyze the abundance of immune cells in the LN, DN, and NK kidney biopsy RNA samples. Total RNA was isolated using the Roche High Pure FFPET RNA isolation kit (Roche Diagnostics) from 5-μm-thick tissue scrolls obtained from FFPE kidney biopsy specimens from LN, DN, and NK individuals. RNA was quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific); RNA quality was analyzed using an Agilent 2100 Bioanalyzer (Agilent); RNA was concentrated using the RNA Clean and Concentrator kit (Zymo Corp.). The RNA samples for NanoString analysis were chosen as per the following qualitative criteria: 260:280 ratio > 1.8, 260:230 ratio > 2.0, and DV-200 (percentage of nucleotide fragments, >200 bp) > 30%. We were able to obtain RNA of appropriate quality from 4 archived paraffin-embedded LN kidney biopsies of 10 processed, from 7 DN samples of 15 processed, and from 7 NK samples of 10 processed. One hundred to 200 ng of RNA from four LN, seven DN, and seven NK individuals was analyzed using the nCounter Human Autoimmune Profiling Panel (NanoString), which included 750 autoimmune disease–related genes including genes that provided unique cell profiling data to measure the relative abundance of 14 immune cell types and 20 internal housekeeping genes. The hybridization assay was performed at NanoString on the nCounter MAX system, and raw gene expression data were supplied for further analysis. NanoString data analysis was performed by Canopy Biosciences (St. Louis, MO). For cell type profiling in LN, DN, and NK kidney biopsy samples, the nCounter Advanced Analysis 2.0 software (NanoString) was used. Briefly, raw counts were normalized using the geometric mean of internal housekeeping gene probes. Relative cell type abundance was determined with the method described by Danaher et al. (22). The method quantifies cell populations using marker genes that act as reference genes (see table S3), which are expressed stably and specifically in given cell types. The abundance estimates of the immune cell type scores were calculated in log2 scale expression of their characteristic genes.
Immunohistochemistry
Five-micrometer-thick kidney biopsy FFPE sections from LN, DN, and NK individuals and FFPE sections from the kidneys dissected from LN-, HDs-, and NE-humanized mice were stained using standard IHC staining protocols. Briefly, the slides were deparaffinized and stained with the following antibodies (all from Roche): monoclonal rabbit anti-human CD8 (clone SP57) and monoclonal rabbit–anti-human CD4 (clone SP35), monoclonal rabbit–anti-human CD3 (clone 2GV6), monoclonal rabbit–anti-human CD38 (clone SP149), and polyclonal rabbit–anti-human IgG in a Ventana BenchMark ULTRA automated IHC slide staining system (Roche). The ultraView Universal DAB Detection Kit (Roche) containing a horseradish peroxide multimer and 3,3′-diaminobenzidine tetrahydrochloride (DAB) chromogen was used for indirect detection of the primary antibody. For dual CD4 and CD8 antibody staining, indirect staining of the primary antibody was carried out with an ultraView Universal Alkaline Phosphatase Red detection kit (Roche) where alkaline phosphatase chromogen is used along with DAB. The slides were counterstained with hematoxylin, and images were obtained at ×20 magnification on a Leica DMi8 inverted microscope with Leica Application Suite X software (Leica Microsystems Inc.). At least 5 to 10 fields per slide were imaged. CD4 and CD8 cells in the kidneys were quantitated using Image J.
Immunofluorescence
For immunofluorescence experiments, 5-μm-thick FFPE sections prepared from the LN, DN, and NK kidney biopsy samples, as well as 5-μm-thick FFPE sections prepared from kidneys and spleens of LN mice, were deparaffinized, and antigen retrieval was performed on the deparaffinized samples in a vegetable steamer using 10 mM sodium citrate buffer. The samples were blocked with a solution of tris-buffered saline containing 1% bovine serum albumin and 10% (fetal bovine serum) and stained with following primary antibodies: For determining CD45RO abundance, the human kidney sections were stained with an Alexa Fluor 647–conjugated rabbit anti-human CD8 alpha antibody (clone EP1150, Abcam) and Alexa Fluor 594–conjugated mouse anti-human CD45RO antibody (clone UCHL1, BioLegend). For CD8 functionality, sections were stained with mouse monoclonal anti-human CD8 antibody (clone 4B1, Novus Biologicals), rabbit monoclonal anti-human Ki-67 (clone EPR3612, Abcam), and guinea pig polyclonal anti-human Kv1.3 (Alomone Labs). The specificity of the polyclonal Kv1.3 antibody was shown by us earlier (16). The sections were washed and incubated with the following secondary antibodies: Alexa Fluor 488 anti-mouse, Alexa Fluor 647 anti-rabbit, and Alexa Fluor 555 anti–guinea pig (Thermo Fisher Scientific). Slides were then washed and directly labeled with Alexa Fluor 405 mouse anti-human GrB (clone B18.1, Novus Biologicals). The LN mouse kidney and spleen sections were stained with mouse monoclonal anti-human CD8 and guinea pig polyclonal anti-human Kv1.3 primary antibodies and Alexa Fluor 488 anti-mouse, Alexa Fluor 647 anti-rabbit, and Alexa Fluor 555 anti–guinea pig secondary antibodies, while nuclei were stained with 4′,6-diamidino-2-phenylindole (1 μg/ml; Thermo Fisher Scientific) and mounted with coverslips using Fluoromount-G (eBiosciences, Thermo Fisher Scientific). Confocal microscopy was performed on a Zeiss 710 laser scanning confocal microscope (Zeiss GmBH) using a 40× water immersion lens at room temperature; the pinhole was set at 1 airy unit. Data were obtained using the “Multi-Track” option of the microscope to exclude the cross-talk between the channels, and >5 fields per slide were imaged. To determine the memory cell phenotype, CD45RO+ cells and CD45RO+CD8+ cells were quantitated in each field imaged using ImageJ. To measure Kv1.3 expression, cell proliferation, and cytotoxicity, the confocal images were analyzed using ImageJ as described previously (16). Briefly, in each field, a region of interest (ROI) was drawn around the CD8+ T cells. The image threshold and background were adjusted and MFIs (measured as mean gray values in ImageJ) for the Kv1.3, Ki-67, and GrB channels were measured within the ROI.
Nanoparticles
NPs were synthesized and functionalized as previously described by us (20, 21). Briefly, we obtained lipid vesicles consisting of a chloroform solution of l-α-phosphatidylcholine, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)-2000], and cholesterol (Avanti Polar Lipids Inc.) by mixing them in a 3:1:1 molar ratio, drying them under N2 gas, rehydrating the vesicles with (pH 7.4) phosphate-buffered saline, followed by sonication and extrusion (LIPEX Thermobarrel Extruder, Northern Lipids Inc.) to yield 100-nm unilamellar vesicles (NPs). We confirmed the size of these vesicles with Zetasizer Nano ZS (Malvern Instruments Inc.). The NPs were then functionalized to selectively recognize CD45RO+ Tm cells by decorating them with biotinylated anti-human CD45RO primary antibody (10 μg/ml; clone UCHL1, BioLegend) and Alexa Fluor 488–conjugated streptavidin (BioLegend). The NPs were purified on CL-4B columns (GE Healthcare Bio-Sciences AB), lyophilized for 48 hours (Labconco), and stored at −80°C. Fifty micrograms of the lyophilized NPs were reconstituted in 200 to 400 pM of either Kv1.3 siRNA (Santa Cruz Biotechnology; Kv1.3-NPs) or nontargeting scrambled sequence siRNA (Santa Cruz Biotechnology; scr-NPs) along with protamine sulfate (MilliporeSigma) at 1:5 M ratio in 100 μl of nuclease-free water. For the in vitro experiments, 3 × 105 freshly isolated T cells were mixed with 50 μl of Kv1.3-NPs or scr-NPs and incubated overnight in T cell medium. For the in vivo experiments, 6 × 106 to 8 × 106 PBMCs from patients with LN were mixed with Kv1.3-NPs or scr-NPs as described above and were engrafted intravenously in an NSG mouse. Peripheral blood T cells, splenocytes, and single cells from the bone marrow and lungs of these mice were isolated and counted.
Flow cytometry
Surface staining for flow cytometry was performed per standard flow cytometry protocols. Briefly, cells (~1 × 106 cells per condition) from patients or humanized mice were either nonstimulated or activated with TG in the presence of brefeldin A and stained with the following antibodies: Pacific Blue–conjugated anti-CD3, PE-Cy7–conjugated anti-CD45RA, and Alexa Fluor 647–conjugated anti CD40L. For intracellular staining, the cells were permeabilized using BD Cytofix/Cytoperm kit (BD Biosciences) and stained with Brilliant violet 605–conjugated mouse anti-human anti-IFNγ and Brilliant violet 711–conjugated mouse anti-human Ki-67 antibodies (all from BioLegend). Data were collected on an LSR II flow cytometer (BD Biosciences) and analyzed with FlowJo software (FlowJo LLC). For some experiments, cells were gated on the NP+ population (presence of Alexa Fluor 488 fluorescence for both Kv1.3-NP– and scr-NP–treated cell population was indicative of successful NP incorporation (21) and used for downstream analysis.
IFNγ measurements
IFNγ levels were measured using a commercially available ELISA kit (Thermo Fisher Scientific), according to the manufacturer’s instructions, in stimulated and unstimulated PBMCs isolated from patients with LN, as well in splenocytes and single cells from the bone marrow and lungs from LN mice.
Statistical analysis
Statistical analyses were performed using either Student’s t test (paired or unpaired), Mann-Whitney rank sum test (in experiments where samples failed normality), and analysis of variance (ANOVA) as indicated. Post hoc testing on ANOVA was performed by multiple pairwise comparison procedures using either Holm-Sidak, Tukey, or Dunn’s methods. Statistical analysis was performed using SigmaPlot 13.0 (Systat Software Inc.). P ≤ 0.05 was defined as statistically significant. The appropriate statistical tests along with their significance values are described in the individual figure legends.
Supplementary Material
Acknowledgments
We are grateful to the patients and HDs that participated in the study. We would like to acknowledge clinical coordinators from the Division of Nephrology, University of Cincinnati for assistance in the collection of patient samples. We would like to thank R. Jandarov (Department of Biostatistics and Bioinformatics, University of Cincinnati) for valuable advice on statistical analysis, L. C. Kottyan and J. B. Harley (Center for Autoimmune Genomics and Etiology, Cincinnati Children’s Hospital Medical Center) for assistance with pilot studies and G. Pauletti (Department of Pharmacology and Systems Physiology) for providing the Zetasizer Nano ZS. Confocal microscopy images were acquired at the Live Microscopy Core (Department of Pharmacology and Systems Physiology, University of Cincinnati). All flow cytometry experiments were performed using equipment maintained by the Research Flow Cytometry Core (Division of Rheumatology) at Cincinnati Children’s Hospital Medical Center. RNA quality control was performed at Genomics, Epigenomics, and Sequencing Core, Department of Environmental Health, University of Cincinnati. Kidney biopsy samples, biospecimens, and associated data were provided by the University of Cincinnati Biorepository, while IHC staining was performed at the Research Pathology Core, Cincinnati Children’s Hospital Medical Center. Experimental schema was drawn using BioRender. Funding: This work was funded by support from Dialysis Clinic Inc. Reserve fund, Paul Teschan Research fund, and NIH (grant R01CA95286) research grants awarded to L.C. and in part by a Cincinnati Children’s Research Foundation Academic and Research Committee Award to H.M. Flow cytometry experiments at the Research Flow Cytometry Core were supported by a grant from the NIH (grant AR070549). Stipend support for F.Z.I. was provided from the University Of Cincinnati Office Of Research’s Undergraduate Student Stipend and Research Cost Awards for Faculty-Student Collaboration 2017-18 program. H.M. was also supported by the National Center for Advancing Translational Sciences of the NIH, under award number 2KL2TR001426-05A1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Author contributions: Conceptualization: L.C. and M.K. Methodology: A.A.C. and M.K. Formal analysis: A.A.C. and M.K. Investigation: A.A.C, M.K., and F.Z.I. Resources: H.J.D., K.S.K., and H.M.; Writing (original draft): A.A.C., M.K., and L.C. Writing (review and editing): A.A.C., M.K., and L.C. Visualization: A.A.C. and M.K. Supervision: L.C. Project administration: A.A.C., M.K., and L.C. Funding acquisition: L.C. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabd1471/DC1
REFERENCES AND NOTES
- 1.Davidson A., What is damaging the kidney in lupus nephritis? Nat. Rev. Rheumatol. 12, 143–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vukelic M., Li Y., Kyttaris V. C., Novel treatments in lupus. Front. Immunol. 9, 2658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moulton V. R., Suarez-Fueyo A., Meidan E., Li H., Mizui M., Tsokos G. C., Pathogenesis of human systemic lupus erythematosus: A cellular perspective. Trends Mol. Med. 23, 615–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsokos G. C., Autoimmunity and organ damage in systemic lupus erythematosus. Nat. Immunol. 21, 605–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katsuyama T., Tsokos G. C., Moulton V. R., Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front. Immunol. 9, 1088 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopetschke K., Klocke J., Grießbach A.-S., Humrich J. Y., Biesen R., Dragun D., Burmester G.-R., Enghard P., Riemekasten G., The cellular signature of urinary immune cells in lupus nephritis: New insights into potential biomarkers. Arthritis Res. Ther. 17, 94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.J. C. Crispin, G. C. Tsokos, T cells, in Dubois’ Lupus Erythematosus and Related Syndromes, D. J. Wallace. B. H. Hahnn, Eds. (Elsevier, 2013), pp. 93–103. [Google Scholar]
- 8.Suárez-Fueyo A., Bradley S. J., Klatzmann D., Tsokos G. C., T cells and autoimmune kidney disease. Nat. Rev. Nephrol. 13, 329–343 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Cahalan M. D., Chandy K. G., The functional network of ion channels in T lymphocytes. Immunol. Rev. 231, 59–87 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panyi G., Vámosi G., Bodnár A., Gáspár R., Damjanovich S., Looking through ion channels: Recharged concepts in T-cell signaling. Trends Immunol. 25, 565–569 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Feske S., Wulff H., Skolnik E. Y., Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 33, 291–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner J.-E., Becker M., Mittrücker H.-W., Panzer U., Tissue-resident lymphocytes in the kidney. J. Am. Soc. Nephrol. 29, 389–399 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.K. G. Chandy, H. Wulff, C. Beeton, P. A. Calabresi, G. A. Gutman, M. Pennington, Kv1.3 potassium channel: Physiology, pharmacology and therapeutic indications, in Voltage-Gated Ion Channels as Drug Targets, D. J. Triggle, Ed. (Wiley-VCH Verlag GmbH & Co. KGaA, 2006), vol. 7, pp. 214–274. [Google Scholar]
- 14.Conforti L., Potassium channels of T lymphocytes take center stage in the fight against cancer. J. Immunother. Cancer 5, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conforti L., The ion channel network in T lymphocytes, a target for immunotherapy. Clin. Immunol. 142, 105–106 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Chimote A. A., Hajdu P., Sfyris A. M., Gleich B. N., Wise-Draper T., Casper K. A., Conforti L., Kv1.3 channels mark functionally competent CD8+ tumor-infiltrating lymphocytes in head and neck cancer. Cancer Res. 77, 53–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kazama I., Roles of lymphocyte Kv1.3-channels in the pathogenesis of renal diseases and novel therapeutic implications of targeting the channels. Mediators Inflamm. 2015, 436572 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C., Zhang L., Shi Y., Yi H., Zhao Y., Chen J., Pollock C. A., Chen X.-M., The KCa3.1 blocker TRAM34 reverses renal damage in a mouse model of established diabetic nephropathy. PLOS ONE 13, e0192800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang C., Pollock C. A., Chen X.-M., KCa3.1: A new player in progressive kidney disease. Curr. Opin. Nephrol. Hypertens. 24, 61–66 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Hajdu P., Chimote A. A., Thompson T. H., Koo Y., Yun Y., Conforti L., Functionalized liposomes loaded with siRNAs targeting ion channels in effector memory T cells as a potential therapy for autoimmunity. Biomaterials 34, 10249–10257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chimote A. A., Hajdu P., Kottyan L. C., Harley J. B., Yun Y., Conforti L., Nanovesicle-targeted Kv1.3 knockdown in memory T cells suppresses CD40L expression and memory phenotype. J. Autoimmun. 69, 86–93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danaher P., Warren S., Dennis L., D’Amico L., White A., Disis M. L., Geller M. A., Odunsi K., Beechem J., Fling S. P., Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer 5, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang J., Eggenhuizen P., O’Sullivan K. M., Alikhan M. A., Holdsworth S. R., Ooi J. D., Kitching A. R., CD8+ T cells effect glomerular injury in experimental anti-myeloperoxidase GN. J. Am. Soc. Nephrol. 28, 47–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Agati V. D., Appel G. B., Estes D., Knowles D. M., Pirani C. L., Monoclonal antibody identification of infiltrating mononuclear leukocytes in lupus nephritis. Kidney Int. 30, 573–581 (1986). [DOI] [PubMed] [Google Scholar]
- 25.Nicolaou S. A., Neumeier L., Steckly A., Kucher V., Takimoto K., Conforti L., Localization of Kv1.3 channels in the immunological synapse modulates the calcium response to antigen stimulation in T lymphocytes. J. Immunol. 183, 6296–6302 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicolaou S. A., Szigligeti P., Neumeier L., Lee S. M., Duncan H. J., Kant S. K., Mongey A. B., Filipovich A. H., Conforti L., Altered dynamics of Kv1.3 channel compartmentalization in the immunological synapse in systemic lupus erythematosus. J. Immunol. 179, 346–356 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicolaou S. A., Neumeier L., Takimoto K., Lee S. M., Duncan H. J., Kant S. K., Mongey A. B., Filipovich A. H., Conforti L., Differential calcium signaling and Kv1.3 trafficking to the immunological synapse in systemic lupus erythematosus. Cell Calcium 47, 19–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu L., Wang T., Gocke A. R., Nath A., Zhang H., Margolick J. B., Whartenby K. A., Calabresi P. A., Blockade of Kv1.3 potassium channels inhibits differentiation and granzyme B secretion of human CD8+ T effector memory lymphocytes. PLOS ONE 8, e54267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fung-Leung W.-P., Edwards W., Liu Y., Ngo K., Angsana J., Castro G., Wu N., Liu X., Swanson R. V., Wickenden A. D., T cell subset and stimulation strength-dependent modulation of T cell activation by Kv1.3 blockers. PLOS ONE 12, e0170102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mak A., Kow N. Y., The pathology of T cells in systemic lupus erythematosus. J. Immunol. Res. 2014, 419029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanner M. R., Beeton C., Differences in ion channel phenotype and function between humans and animal models. Front. Biosci. (Landmark Ed.) 23, 43–64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beeton C., Chandy K. G., Potassium channels, memory T cells, and multiple sclerosis. Neuroscientist 11, 550–562 (2005). [DOI] [PubMed] [Google Scholar]
- 33.McKinney E. F., Lyons P. A., Carr E. J., Hollis J. L., Jayne D. R. W., Willcocks L. C., Koukoulaki M., Brazma A., Jovanovic V., Kemeny D. M., Pollard A. J., MacAry P. A., Chaudhry A. N., Smith K. G. C., A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat. Med. 16, 586–591 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dolff S., Abdulahad W. H., van Dijk M. C. R. F., Limburg P. C., Kallenberg C. G. M., Bijl M., Urinary T cells in active lupus nephritis show an effector memory phenotype. Ann. Rheum. Dis. 69, 2034–2041 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Tilstra J. S., Avery L., Menk A. V., Gordon R. A., Smita S., Kane L. P., Chikina M., Delgoffe G. M., Shlomchik M. J., Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Invest. 128, 4884–4897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couzi L., Merville P., Deminière C., Moreau J.-F., Combe C., Pellegrin J.-L., Viallard J.-F., Blanco P., Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum. 56, 2362–2370 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Crispín J. C., Kyttaris V. C., Terhorst C., Tsokos G. C., T cells as therapeutic targets in SLE. Nat. Rev. Rheumatol. 6, 317–325 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper M. E., Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet 352, 213–219 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Tesch G. H., Diabetic nephropathy – Is this an immune disorder? Clin. Sci. 131, 2183–2199 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Arazi A., Rao D. A., Berthier C. C., Davidson A., Liu Y., Hoover P. J., Chicoine A., Eisenhaure T. M., Jonsson A. H., Li S., Lieb D. J., Zhang F., Slowikowski K., Browne E. P., Noma A., Sutherby D., Steelman S., Smilek D. E., Tosta P., Apruzzese W., Massarotti E., Dall’Era M., Park M., Kamen D. L., Furie R. A., Payan-Schober F., Pendergraft W. F. III, McInnis E. A., Buyon J. P., Petri M. A., Putterman C., Kalunian K. C., Woodle E. S., Lederer J. A., Hildeman D. A., Nusbaum C., Raychaudhuri S., Kretzler M., Anolik J. H., Brenner M. B., Wofsy D., Hacohen N., Diamond B.; Accelerating Medicines Partnership in SLE network , The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 20, 902–914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boucher A., Droz D., Adafer E., Noël L.-H., Characterization of mononuclear cell subsets in renal cellular interstitial infiltrates. Kidney Int. 29, 1043–1049 (1986). [DOI] [PubMed] [Google Scholar]
- 42.Manjarrez-Orduño N., Menard L. C., Carman J. A., Suchard S. J., Casano F., Lee D., Daouti S., Habte S., Kansal S., Jiang C., Bandyopadhyay S., Hu Y., Furie R. A., Nadler S. G., A systemic lupus erythematosus endophenotype characterized by increased CD8 cytotoxic signature associates with renal involvement. ImmunoHorizons 1, 124–132 (2017). [Google Scholar]
- 43.Ishida Y., Chused T. M., Lack of voltage sensitive potassium channels and generation of membrane potential by sodium potassium ATPase in murine T lymphocytes. J. Immunol. 151, 610–620 (1993). [PubMed] [Google Scholar]
- 44.Koboziev I., Jones-Hall Y., Valentine J. F., Webb C. R., Furr K. L., Grisham M. B., Use of humanized mice to study the pathogenesis of autoimmune and inflammatory diseases. Inflamm. Bowel Dis. 21, 1652–1673 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen P.-M., Wilson P. C., Shyer J. A., Veselits M., Steach H. R., Cui C., Moeckel G., Clark M. R., Craft J., Kidney tissue hypoxia dictates T cell–mediated injury in murine lupus nephritis. Sci. Transl. Med. 12, eaay1620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chimote A. A., Kuras Z., Conforti L., Disruption of Kv1.3 channel forward vesicular trafficking by hypoxia in human T lymphocytes. J. Biol. Chem. 287, 2055–2067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baxter A. G., Cooke A., Complement lytic activity has no role in the pathogenesis of autoimmune diabetes in NOD mice. Diabetes 42, 1574–1578 (1993). [DOI] [PubMed] [Google Scholar]
- 48.Tsokos G. C., Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Aloui C., Prigent A., Sut C., Tariket S., Hamzeh-Cognasse H., Pozzetto B., Richard Y., Cognasse F., Laradi S., Garraud O., The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int. J. Mol. Sci. 15, 22342–22364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karnell J. L., Rieder S. A., Ettinger R., Kolbeck R., Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 141, 92–103 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Furie R., Bruce I. N., Dörner T., Leon M. G., Leszczynski P., Urowitz M. B., Haier B., Jimenez T., Barbey C., Liu J., Stach C., Efficacy and safety of dapirolizumab pegol (DZP) in patients with moderately to severely active systemic lupus erythematosus (sle): A randomised, placebo (pbo)-controlled study. Ann. Rheum. Dis. 78, 775–776 (2019). [Google Scholar]
- 52.Stevens A., Yuasa M., Peckham D., Olsen C., Iadonato S., Probst P., Dalazatide, an inhibitor of the Kv1.3 channel on activated effector memory T cells, has immunotherapy potential against systemic lupus erythematosus. Lupus Science & Medicine 3, A3–A4 (2016). [Google Scholar]
- 53.Bobo D., Robinson K. J., Islam J., Thurecht K. J., Corrie S. R., Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 33, 2373–2387 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Peer D., Park E. J., Morishita Y., Carman C. V., Shimaoka M., Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science 319, 627–630 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Look M., Stern E., Wang Q. A., DiPlacido L. D., Kashgarian M., Craft J., Fahmy T. M., Nanogel-based delivery of mycophenolic acid ameliorates systemic lupus erythematosus in mice. J. Clin. Invest. 123, 1741–1749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bajema I. M., Wilhelmus S., Alpers C. E., Bruijn J. A., Colvin R. B., Cook H. T., D’Agati V. D., Ferrario F., Haas M., Jennette J. C., Joh K., Nast C. C., Noël L.-H., Rijnink E. C., Roberts I. S. D., Seshan S. V., Sethi S., Fogo A. B., Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: Clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 93, 789–796 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Wunderlich M., Chou F.-S., Link K. A., Mizukawa B., Perry R. L., Carroll M., Mulloy J. C., AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 24, 1785–1788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wunderlich M., Brooks R. A., Panchal R., Rhyasen G. W., Danet-Desnoyers G., Mulloy J. C., OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood 123, e134–e144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabd1471/DC1