

Caspase-8–dependent GSDMD activation promotes antibacterial defense; however, this pathway can also mediate TNF shock in vivo.
Abstract
Gasdermin D (GSDMD) is a pore-forming protein that promotes pyroptosis and release of proinflammatory cytokines. Recent studies revealed that apoptotic caspase-8 directly cleaves GSDMD to trigger pyroptosis. However, the molecular requirements for caspase-8–dependent GSDMD cleavage and the physiological impact of this signaling axis are unresolved. Here, we report that caspase-8–dependent GSDMD cleavage confers susceptibility to tumor necrosis factor (TNF)–induced lethality independently of caspase-1 and that GSDMD activation provides host defense against Yersinia infection. We further demonstrate that GSDMD inactivation by apoptotic caspases at aspartate 88 (D88) suppresses TNF-induced lethality but promotes anti-Yersinia defense. Last, we show that caspase-8 dimerization and autoprocessing are required for GSDMD cleavage, and provide evidence that the caspase-8 autoprocessing and activity on various complexes correlate with its ability to directly cleave GSDMD. These findings reveal GSDMD as a potential therapeutic target to reduce inflammation associated with mutations in the death receptor signaling machinery.
INTRODUCTION
Gasdermin D (GSDMD) is a pore-forming protein that has emerged as an important player during different modes of cell death including pyroptosis, apoptosis, and NETosis (1). GSDMD consists of a cytotoxic N-terminal domain (GSDMDNT) and a repressor C-terminal domain (GSDMDCT) that is connected by a flexible linker (2). Activation of GSDMD requires cleavage at position aspartate D276 in mouse (D277 in humans) within the linker region by inflammatory caspases, e.g., caspase-1, caspase-4, caspase-5, and caspase-11, which are activated in large multiprotein inflammasome complexes that assemble upon infection or cellular stress. This cleavage event liberates the cytotoxic GSDMDNT p30 fragment and enables GSDMDNT to insert into and oligomerize within the plasma membrane and initiate lytic cell death by pyroptosis (2). Active caspase-1 also cleaves the proinflammatory cytokine, pro–interleukin-1β (pro–IL-1β), into its mature active form and promotes its secretion through GSDMD pores (3, 4). Given the inflammatory nature of pyroptosis and IL-1β, it is not unexpected that the activity of GSDMD is tightly regulated. For example, repair of GSDMD pores by the endosomal sorting complexes required for transport–III machinery limits cellular cytotoxicity during inflammasome activation (5). Furthermore, GSDMD also harbors a caspase-3/7 cleavage site at position D88 in mouse (D87 in human) within the GSDMDNT, which inactivates the pore-forming ability of GSDMDNT (6). Since apoptotic caspase-3 and caspase-7 activation was previously observed upon inflammasome assembly (7) and caspase-3/7–deficient macrophages secrete more IL-1β than control cells upon NOD-like receptor protein 3 (NLRP3) inflammasome activation (8), this raises the question of whether cleavage of GSDMD at position D88 suppresses pyroptosis and IL-1β secretion in inflammasome-stimulated cells.
Recent studies have revealed that caspase-8, the initiator caspase in the extrinsic apoptosis pathway, directly cleaves GSDMD to trigger pyroptosis under specific conditions (9–11). The extrinsic apoptosis pathway is initiated following engagement of cell surface death receptors such as Fas (CD95) or tumor necrosis factor receptor 1 (TNFR1). Activation of Fas–Fas ligand (FasL) signaling triggers the assembly of a membrane-bound death-inducing signaling complex (DISC) comprising Fas-associated protein with death domain (FADD) and caspase-8. On the DISC, caspase-8 undergoes proximity-induced autoactivation, which further activates cytosolic executioners caspase-3 and caspase-7 to drive cellular apoptosis (12). Similar to Fas signaling, TNF-TNFR1 ligation in most healthy cells, including macrophages, promotes the assembly of a multiprotein complex called TNF Complex I (13, 14). In addition, in contrast to the DISC, Complex I assembly does not drive apoptosis but, instead, promotes prosurvival and proinflammatory gene expression by activating NF-κB (nuclear factor κB) and mitogen-activated protein kinase. However, when protein synthesis is inhibited, for example, upon exposure to the protein synthesis inhibitor cycloheximide (CHX), TNFR1 engagement promotes the formation of a cytosolic multiprotein complex composed of TNFR1-associated death domain protein, FADD, and caspase-8, called TNF Complex IIa, to initiate apoptosis (13, 14). When core components of TNF Complex I such as transforming growth factor β–activated kinase 1 (TAK1) or the E3 ligase cellular inhibitor of apoptosis protein 1 (cIAP1) and cIAP2 are inhibited or depleted, TNFR1 ligation, in turn, promotes the association of FADD, receptor-interacting serine/threonine kinase 1 (RIPK1), and caspase-8 to assemble into a cytosolic complex called TNF Complex IIb (14, 15). Under such conditions, RIPK1 kinase activity is required to drive caspase-8 activation (13, 14). In addition to death receptor signaling, emerging studies revealed that TLR3 or TLR4 stimulation in IAP-depleted cells can directly activate caspase-8 via a Complex IIb–like ripoptosome complex that is composed of RIPK3, RIPK1, FADD, and caspase-8 and that bypasses the strict requirement of TNFR1 and the kinase activity of RIPK1 (16–18). In keeping with this, a recent study further demonstrated that TLR stimulation in TAK1-deficient cells triggers caspase-8 activation independently of RIPK1 kinase activity (19). Two recent studies reported that injection of the bacterial effector protein YopJ, an acetyltransferase that inhibits TAK1 function during Yersinia pestis or Yersinia pseudotuberculosis infection in macrophages, triggers Complex IIb–dependent caspase-8 activation, which directly cleaves GSDMD to drive pyroptosis (10, 11). In agreement with these studies, we demonstrated that exposure of murine macrophages to a TAK1 inhibitor or a bivalent second mitochondria-derived activator of caspases–mimetic compound, AZD 5582, which targets cIAP1, cIAP2, and X-linked IAP, an essential repressor of Complex IIb and ripoptosome in innate immune cells (18, 20–22), triggers caspase-8–dependent GSDMD cleavage and pyroptosis, which is further amplified by the NLRP3 inflammasome (9–11, 17). In parallel, caspase-8 activates executioners caspase-3 and caspase-7 to initiate apoptosis, as cells that are deficient in caspase-3, caspase-7, and GSDMD are completely resistant to Complex IIb–dependent cell death (9). Of note, under conditions of TAK1 inhibition or IAP depletion and/or inhibition, caspase-3/7–dependent cleavage and inactivation of GSDMD at position D88 is essential to suppress Complex IIb–driven pyroptosis (9).
While these observations revealed exciting cross-talk between apoptosis and pyroptosis, several key questions regarding caspase-8–dependent GSDMD activation remain outstanding. One such question is the physiological impact of this pathway in vivo. An emerging number of inflammatory diseases are associated with aberrant TNF signaling (12, 23), and whether this is associated with caspase-8–dependent GSDMD activation and tissue damage is unclear and of high medical interest. Second, while GSDMD-dependent pyroptosis promotes the clearance of intracellular pathogens by exposing them to recruited phagocytes (24–26), it is unclear how and whether GSDMD activation provides host-protective immunity against extracellular pathogens such as Yersinia. Another open question is the molecular requirements for caspase-8–dependent GSDMD cleavage. Furthermore, it is uncertain whether caspase-8 activation during extrinsic apoptosis universally leads to GSDMD cleavage. If so, then this further raises the intriguing question of how apoptosis is kept immunologically silent during development and tissue homeostasis.
In this study, we report that caspase-8 dimerization, autoprocessing, and activity are required for caspase-8–dependent GSDMD cleavage. We observed the strongest caspase-8 activation on TNF Complex IIb, which correlates with robust direct GSDMD cleavage. In contrast, caspase-8 is only weakly activated under other conditions of extrinsic apoptosis and, likewise, is a weak activator of GSDMD. We further demonstrate that GSDMD activation promotes anti-Yersinia defense in vivo and provide compelling evidence that caspase-8–dependent GSDMD activation promotes TNF-induced lethality independent of inflammasome activation. While GSDMD cleavage and inactivation at position D88 do not suppress pyroptosis downstream of canonical or noncanonical inflammasome activation, they are required for optimal anti-Yersinia defense in vivo and are essential to suppress TNF-induced lethality.
RESULTS
GSDMD cleavage at position D88 does not suppress pyroptosis during canonical or noncanonical inflammasome signaling
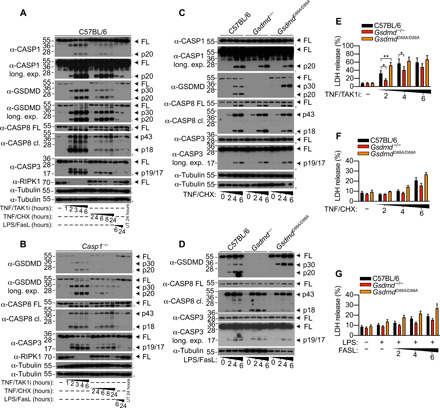
We recently demonstrated that cleavage of mouse GSDMD at position D88 by apoptotic caspase-3 and caspase-7 counteracts caspase-8–dependent pyroptosis in TNF Complex IIb–stimulated cells (9). However, whether GSDMD cleavage at position D88 plays a similar role in inflammasome-stimulated cells is unclear. To investigate this possibility, we primed wild-type (WT) or caspase-3/7 cleavage-resistant GsdmdD88A/D88A bone marrow–derived macrophages (BMDMs) with lipopolysaccharide (LPS) for 4 hours and stimulated these cells with nigericin or adenosine triphosphate (ATP), log-phase Salmonella enterica serovar Typhimurium (S. Typhimurium), or poly(deoxyadenylic-deoxythymidylic) [poly(dA:dT)] to activate the NLRP3, NLRC4 (NLR family CARD domain-containing protein 4), and AIM2 (absent in melanoma 2) inflammasome, respectively. GsdmdD88A mutation did not affect LPS-induced TNF secretion compared to WT BMDMs, indicating that GsdmdD88A mutation does not affect TLR4 signaling and is unlikely to affect inflammasome priming (fig. S1A). Canonical inflammasome activation triggered comparable levels of lactate dehydrogenase (LDH) (Fig. 1, A and B, and fig. S1, B and C) and IL-1β release (Fig. 1, C and D, and fig. S1, D and E) from WT and GsdmdD88A/D88A BMDMs, demonstrating that cleavage of GSDMD at position D88 does not suppress pyroptosis or unconventional protein secretion downstream of caspase-1 activation. In support of this, further processing of GSDMD p30 by caspase-3/7 at position D88 to generate the inactive p20 fragment was barely detectable in inflammasome-stimulated cells but readily observed in apoptotic macrophages (Fig. 1, E and F, and fig. S1F). Activation of the NLRC4 inflammasome in unprimed BMDMs was reported to promote caspase-8–dependent cell lysis (27); whether GSDMD cleavage at position D88 suppresses pyroptosis under such condition is unclear. We, therefore, activated the NLRC4 inflammasome by transfecting ultrapure flagellin into WT and GsdmdD88A/D88A BMDMs. Cytosolic flagellin triggered comparable LDH release in WT and GsdmdD88A/D88A BMDMs regardless of Pam3CSK4 priming (fig. S1G), indicating that cleavage of GSDMD at position D88 does not suppress pyroptosis upon recognition of cytosolic flagellin in macrophages, regardless of priming. Consistent with that, further cleavage of GSDMD p30 into the inactive p20 fragment was barely detectable upon flagellin transfection but readily detected in macrophages stimulated with TNF and the TAK1 inhibitor 5-z7-oxozeaenol (TAK1i) (fig. S1H). Given that inflammasome-dependent caspase-3 activation is weakly detected in WT cells but strongly observed in pyroptosis-deficient Gsdmd−/− BMDMs (Fig. 1F and fig. S1I) (28, 29), our data suggest that the kinetic of apoptotic caspase activation in pyroptosis-proficient WT macrophages is unlikely to suppress the lytic activity of GSDMDNT upon canonical inflammasome activation. We previously demonstrated that murine neutrophils resist pyroptosis upon NLRC4 inflammasome activation (30) but eventually undergo GSDMD-independent cell death at late time points (31). Given that neutrophils are highly sensitive to apoptosis, this raised the possibility that GSDMD is constitutively cleaved and inactivated by caspase-3/7 in neutrophils to subvert pyroptosis. To investigate this hypothesis, we challenged LPS-primed WT and GsdmdD88A/D88A bone marrow neutrophils with log-phase S. Typhimurium to activate the NLRC4 inflammasome. Consistent with macrophage studies, GsdmdD88A mutation had no impact on neutrophil lysis (Fig. 1G) or IL-1β release (Fig. 1H) compared to WT neutrophils. Addition of the pan-caspase inhibitor Q-VD-Oph enhanced the viability of untreated neutrophils but had no impact on neutrophil viability after 5 hours of S. Typhimurium infection, suggesting that S. Typhimurium–induced neutrophil cell death at a later time point is caspase independent (fig. S1J). To investigate whether the GsdmdD88A mutation impairs host defense in vivo, we challenged WT, GsdmdD88A/+, or GsdmdD88A/D88A littermates with S. Typhimurium intraperitoneally and measured bacterial loads in these animals. In accordance with our observations in cell culture studies, bacterial burden in the liver and spleen at 48 hours after infection was indistinguishable between GsdmdD88A/D88A and WT or heterozygous littermate controls (Fig. 1, I and J). Last, we confirmed that GsdmdD88A mutation likewise does not affect noncanonical inflammasome signaling, as delivery of cytosolic LPS triggered an equivalent level of pyroptosis in WT and GsdmdD88A/D88A BMDMs (fig. S1, K and L). Together, our data indicate that cleavage of GSDMD at position D88 does not suppress pyroptosis and cytokine secretion during canonical or noncanonical inflammasome activation in either macrophages or neutrophils.
Fig. 1. GSDMD inactivation at D88 does not suppress pyroptosis or IL-1β secretion upon canonical inflammasome activation.

(A to F) BMDMs were primed with LPS (100 ng/ml) for 4 hours and stimulated with nigericin (5 μM), ATP (2.5 mM), log-phase S. Typhimurium [multiplicity of infection (MOI) of 10], or poly(dA:dT) (1 μg/ml). Where indicated, BMDMs were treated with TNF (100 ng/ml)/TAK1i (125 nM) for 4 hours. (A and B) LDH and (C and D) IL-1β release was measured at the indicated time points. (E and F) Mixed supernatant and cell extract from (E) two C57BL/6 and two GsdmdD88A/D88A mice were examined by immunoblotting 1 hour after stimulation or (F) at the indicated time points. FL, full-length; long exp., long exposure. (G and H) Bone marrow neutrophils were primed with LPS (100 ng/ml) for 4 hours and infected with log-phase S. Typhimurium (MOI of 25). (G) LDH and (H) IL-1β release was measured 3 hours after infection. (I and J) Mice were challenged with 1 × 105 colony-forming units (CFU) of log-phase S. Typhimurium by intraperitoneal injection, and bacterial burden was quantified 48 hours later. (A) Data are represented as means + SEM of cell stimulation from three mice or (B to D, G, and H) means + SD of triplicate cell stimulation from three independent experiments. (I and J) Data are geometric means and pooled from two independent experiments. Immunoblots are representative of three independent experiments.
TNF Complex IIb but not Complex IIa or DISC enables direct cleavage of GSDMD by caspase-8
Since GSDMD inactivation at position D88 has no major function in canonical or noncanonical inflammasome–activated cells (Fig. 1 and fig. S1) but suppresses caspase-8–dependent pyroptosis (9), we focused the remainder of our study on caspase-8–dependent GSDMD activation. While several studies demonstrated that caspase-8 activated by TNF Complex IIb promotes direct GSDMD cleavage and pyroptosis (9–11), it is unclear whether caspase-8 activated in other multiprotein complexes such as the cytoplasmic TNF Complex IIa and plasma membrane–bound DISC is able to cleave and activate GSDMD. To this end, we compared GSDMD cleavage in WT or Casp1−/− BMDMs costimulated with TNF and TAK1i or CHX to induce assembly of TNF Complex IIb or IIa, respectively (13–15). Consistent with previous reports (9–11), TNF and TAK1i costimulation triggered robust cleavage of full-length GSDMD into the active p30 fragment and an appearance of the inactive p20 fragment resulting from a second inactivation cleavage by caspase-3/7 at position D88 (Fig. 2A). Given that caspase-8 can directly cleave GSDMD in TNF Complex IIb–stimulated cells (9–11), GSDMD processing was only partially reduced in Casp1−/− BMDMs upon TNF and TAK1i costimulation, as anticipated (Fig. 2A). Unexpectedly, while both caspase-1 and caspase-8 contribute to efficient GSDMD cleavage upon Complex IIb assembly, GSDMD processing in TNF- and CHX-costimulated macrophages was remarkably reduced in Casp1−/− BMDMs, indicating that caspase-1 is the dominant protease that cleaves GSDMD under conditions of TNF Complex IIa assembly (Fig. 2A) and that caspase-8–dependent GSDMD cleavage plays only a minor role under these conditions. LPS can sensitize macrophages to TNF Complex IIa/b–dependent cell death via autocrine TNF signaling. As anticipated, GSDMD cleavage was still largely caspase-1–dependent upon LPS and CHX costimulation (Fig. 2A). Since TAK1i and CHX may affect the expression of rapidly turned over proteins such as c-FLIP (cellular FLICE-like inhibitory protein) (32, 33), a known inhibitor of caspase-8 (34), we next primed WT and Casp1−/− BMDMs with TNF or LPS for 3 hours to induce c-FLIP expression before TAK1i or CHX stimulation (Fig. 2B). Consistent with earlier observations (Fig. 2A), TAK1i induced robust GSDMD processing into the active p30 and inactive p20 fragments in TNF- or LPS-primed WT BMDMs (Fig. 2B). However, GSDMD cleavage into the active p30 fragment was largely abolished in LPS- but not TNF-primed Casp1−/− BMDMs; instead, GSDMD appears as an inactive p43 fragment resulting from a single cleavage by caspase-3/7 at position D88 in LPS-primed Casp1−/− macrophages (Fig. 2B). This indicates that LPS priming suppresses caspase-8–dependent GSDMD activation under conditions of Complex IIb assembly. Consistent with costimulated cells (Fig. 2A), GSDMD cleavage upon CHX stimulation in TNF- or LPS-primed cells was strongly reduced in Casp1−/− BMDMs (Fig. 2B), indicating that caspase-1 is the dominant protease that cleaves GSDMD upon Complex IIa formation, regardless of priming.
Fig. 2. Caspase-8 activation on TNF Complex IIb is most efficient in directing GSDMD cleavage.
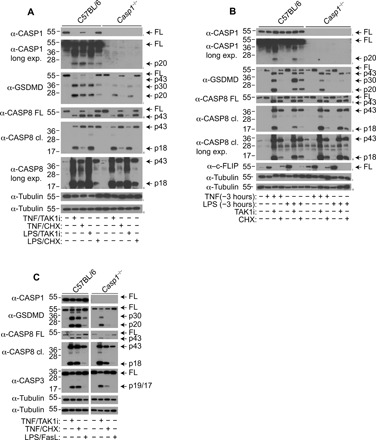
(A) BMDMs were costimulated with TNF (100 ng/ml)/TAK1i (125 nM), TNF (100 ng/ml)/CHX (10 μg/ml), LPS (100 ng/ml)/TAK1i (125 nM), or LPS (100 ng/ml)/CHX (10 μg/ml) for 6 hours, and mixed supernatant and cell extracts were analyzed by immunoblot. (B) BMDMs were primed with TNF (100 ng/ml) or LPS (100 ng/ml) for 3 hours before stimulation with TAK1i (125 nM) or CHX (10 μg/ml) for 6 hours, and mixed supernatant and cell extracts were analyzed by immunoblot. (C) BMDMs were costimulated with TNF (100 ng/ml)/TAK1i (125 nM) or TNF (100 ng/ml)/CHX (10 μg/ml) or primed with LPS (100 ng/ml) for 16 hours and treated with FasL (100 ng/ml) for 6 hours, and mixed supernatant and cell extracts were analyzed by immunoblot. (C) Blots are cropped from the same film. Immunoblots are representative of two to three independent experiments.
Next, we investigated whether caspase-8 activated upon plasma membrane–bound DISC, such as the Fas (CD95) complex, induces caspase-8–dependent GSDMD cleavage in BMDMs. Because Fas is weakly expressed in BMDMs, we first primed BMDMs with LPS or Pam3CSK4 to induce Fas expression (35), before stimulation with FasL. Since LPS but not Pam3CSK4 priming sensitized macrophages to FasL-induced apoptosis (fig. S2, A and B) and caspase-8 autoprocessing plateaus at 6 hours after FasL treatment (fig. S2C), we decided to examine GSDMD cleavage in LPS-primed macrophages at this time point. FasL induced weaker caspase-8 autoprocessing and GSDMD cleavage compared to TNF/CHX or TNF/TAK1i stimulation (Fig. 2C). Of note, FasL-induced GSDMD processing was notably reduced in Casp1−/− BMDMs (Fig. 2C), indicating that similar to Complex IIa assembly (Fig. 2, A and B), caspase-1 is the dominant protease that processes GSDMD under conditions of DISC assembly. Together, our data indicate that in BMDMs, direct cleavage of GSDMD by caspase-8 occurs mostly upon Complex IIb formation, while caspase-8 activated on Complex IIa or the DISC can only weakly do so or indirectly via activation of caspase-1 inflammasome.
Caspase-8 activity on various complexes correlates with its ability to directly cleave GSDMD
Next, we performed a time course analysis to investigate whether the difference in caspase-8 activity on various complexes might account for the enhance ability of Complex IIb to directly process GSDMD. TNF and TAK1i costimulation triggered robust caspase-8 autoprocessing and disappearance of the caspase-8 substrate RIPK1 (36) as early as 2 hours after stimulation, which correlated with GSDMD processing in both WT and Casp1−/− BMDMs (Fig. 3, A and B). In contrast, caspase-8 autoprocessing into its p18 fragment and RIPK1 cleavage was significantly weaker in TNF/CHX- and LPS/FasL-stimulated cells compared to TNF/TAK1i-treated BMDMs, even after 24 hours of stimulation (Fig. 3, A and B). These data suggest, at least in part, that the caspase activity generated by different complexes determines the ability of caspase-8 to directly cleave GSDMD during extrinsic apoptosis.
Fig. 3. Differential activation of caspase-8 Complex IIa, Complex IIb, and DISC.
(A to D) BMDMs were treated with were costimulated with TNF (100 ng/ml)/TAK1i (125 nM) or TNF (100 ng/ml)/CHX (10 μg/ml) or primed with LPS (100 ng/ml) for 16 hours and treated with FasL (100 ng/ml) for the indicated time points, and mixed supernatant and cell extracts were analyzed by immunoblot or (E to G) LDH release into the cell culture supernatant was quantified. (A to D) Immunoblots are representative of two to three independent experiments. (E to G) Data are means + SEM of pooled data from (E and F) six or (G) five independent experiments. Data were normally distributed and analyzed using a parametric t test. *P < 0.05 or **P < 0.01. UT, untreated.
Since cleavage of GSDMD at position D88 by apoptotic caspase-3/7 suppresses the prolytic properties of GSDMD upon Complex IIb formation (9), we wondered whether this cleavage event likewise suppresses pyroptosis upon TNF Complex IIa or DISC assembly. To examine this possibility, we compared cell lysis in WT, Gsdmd−/−, and caspase-3/7 cleavage-resistant GsdmdD88A/D88A BMDMs upon Complex IIa, Complex IIb, and DISC assembly. TNF/CHX and LPS/FasL stimulation triggered cleavage of full-length GSDMD into the active p30 and inactive p20 fragments in WT macrophages. As anticipated, there was an accumulation of the active GSDMD p30 fragment and a complete loss of inactive p20 fragment in GsdmdD88A/D88A cells (Fig. 3, C and D). However, accumulation of active GSDMD p30 in LPS/FasL- and TNF/CHX-stimulated macrophages had minimal impact on cellular lysis but significantly enhanced pyroptosis under conditions of TNF Complex IIb assembly (Fig. 3, E to G). Together, our data indicate that the enhanced activation of caspase-8 on Complex IIb but not Complex IIa or the DISC licenses direct GSDMD cleavage and pyroptosis and that caspase-3/7–dependent GSDMD inactivation mainly suppresses cell lysis under conditions of TNF Complex IIb assembly.
Caspase-8 autoprocessing is required for GSDMD cleavage and pyroptosis
Caspase-8 is an initiator caspase in the death receptor signaling pathway and is activated upon dimerization on multiprotein complexes such as the DISC or Complex II. Cleavage of caspase-8 between the large and small subunits stabilizes the enzymatic activity of the protease and is required for apoptotic signaling (37, 38). Uncleaved caspase-8 in complex with its enzymatically inactive homolog c-FLIP is also active, albeit with a restricted substrate repertoire and can only cleave some substrates such as RIPK3 (39). Thus, whether caspase-8 homodimerization and processing are also required for caspase-8–dependent GSDMD cleavage is unclear. To investigate this, we ectopically expressed full-length GSDMD together with an engineered version of caspase-8 in human embryonic kidney (HEK) 293T cells, in which we replaced the caspase-8 death-effector domains (DED) with an FK506-binding protein (FKBP)–dimerizable domain to control caspase-8 dimerization upon addition of the homodimerizer drug AP20187 (37). Consistent with our previous report (9), expression of caspase-8 alone was insufficient to trigger GSDMD cleavage; dimerization and autoprocessing of WT caspase-8 triggered robust cell rupture, propidium iodide (PI) uptake, and the release of cleaved GSDMD p30 fragment into the cell culture supernatant (Fig. 4A and fig. S3). By contrast, expression of catalytically inactive (C360A) or uncleavable caspase-8 (D210A/D216A/D223A/D374A/D384A) markedly reduced GSDMD cleavage, cell rupture, and release of cleaved GSDMD into the cell culture supernatant (Fig. 4A and fig. S3), indicating that the catalytic function and autoprocessing of caspase-8 are required for GSDMD cleavage and pyroptosis in an overexpression system.
Fig. 4. Caspase-8 dimerization and autoprocessing is required for GSDMD cleavage.
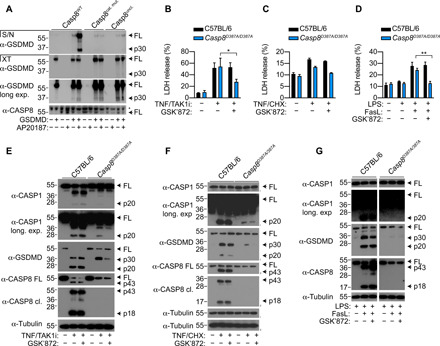
(A) HEK 293T cells were transfected with GSDMD together with WT caspase-8 (Casp8WT), caspase-8 catalytic mutant (Casp8cat. mut.), or uncleavable caspase-8 (Casp8uncl.). Vector control was transfected such that each set of constructs received equivalent amounts of DNA. Twenty-four hours after transfection, transfected cells were treated with AP20187 (dimerizer) for a further 6 hours. Cell extracts (XT) and precipitated supernatants (S/N) were analyzed by immunoblotting for GSDMD and caspase-8. (B to G) WT or caspase-8 uncleavable (D387A) Casp8D387A/D387A BMDMs were stimulated with TNF (100 ng/ml)/TAK1i (125 nM) or TNF (100 ng/ml)/CHX (10 μg/ml) or primed with LPS (100 ng/ml) for 16 hours and treated with FasL (100 ng/ml). Where indicated, cells were treated with GSK’872 (1 μM) 20 to 30 min before stimulation. LDH release was measured at (B and C) 4 hours or (D) 6 hours after stimulation. (E to G) Mixed supernatant (S/N) and cell extracts were examined by immunoblotting. (G) Blots are cropped from the same film. (E to G) Immunoblots are representative of three independent experiments. (B to D) Data are means + SEM of pooled data from four independent experiments. Data were normally distributed and analyzed using a parametric t test. *P < 0.05 or **P < 0.01.
To confirm that this is also true in primary cells, we prepared BMDMs from WT and Casp8D387A/D387A mice, which express a caspase-8 autoproteolysis mutant (D387A), and examined cell lysis and the cleavage status of GSDMD upon TNF/TAK1i, TNF/CHX, and LPS/FasL treatment. Unexpectedly, unlike in the HEK 293T system, we observed comparable levels of LDH release between WT and Casp8D387A/D387A BMDMs upon TNF/TAK1i and LPS/FasL stimulation, while TNF/CHX treatment in Casp8D387A/D387A BMDMs only induced a modest increase in cell lysis compared to untreated cells (Fig. 4, B to D). We observed a reduction of full-length caspase-8 in Casp8D387A/D387A BMDM upon extrinsic apoptosis (Fig. 4, E and G), consistent with a recent study (40). In contrast to the HEK 293T overexpression system (Fig. 4A), GSDMD processing into the active p30 fragment was largely intact in TNF/TAK1i- and TNF/CHX-stimulated Casp8D387A/D387A BMDM (Fig. 4, E and F). Because TAK1i and CHX suppresses c-FLIP expression (32, 33) and caspase-8/c-FLIP heterodimer have proteolytic activity in the absence of interdomain cleavage to suppress RIPK3-dependent necroptosis (39, 41), we reasoned that TAK1i and CHX sensitized TNF-stimulated Casp8D387A/D387A BMDMs to RIPK3-dependent necroptosis and secondary GSDMD cleavage via the MLKL–NLRP3–caspase-1 signaling axis (42, 43). The RIPK3 kinase inhibitor GSK’872 suppressed cell lysis, caspase-1 processing, and GSDMD cleavage in TNF/TAK1i- and TNF/CHX-stimulated Casp8D387A/D387A BMDMs but not in WT cells (Fig. 4, E and F). Caspase inhibition was reported to sensitize Fas-stimulated lymphocytes to necroptosis (44). In agreement with that, addition of the RIPK3 kinase inhibitor, GSK’872 suppressed cell lysis upon LPS/FasL stimulation in Casp8D387A/D387A but not WT BMDMs (Fig. 4D). Unlike Complex IIa and IIb, FasL-induced necroptosis in Casp8D387A/D387A BMDMs did not activate caspase-1 and GSDMD cleavage (Fig. 4G). Together, these data indicate that upon Complex IIb formation, caspase-8 requires homodimerization and autoprocessing to cleave GSDMD and trigger pyroptosis, while under conditions where caspase-8 autoprocessing is inhibited, Complex IIa, Complex IIb, and DISC sensitize BMDMs to RIPK3-dependent necroptosis.
GSDMD promotes TNF-induced lethality independent of the inflammasome
Having determined the molecular requirements for caspase-8–dependent GSDMD cleavage, we next sought to characterize whether caspase-8–driven GSDMD processing drives inflammatory diseases. Since RIPK1 kinase activity and caspase-8 promote GSDMD-dependent macrophage pyroptosis in vitro (9–11) and RIPK1 and caspase-8 promote TNF-induced shock in vivo (45–47), we investigated the contribution of caspase-8–dependent GSDMD activation in a mouse model of TNF-induced shock. For this, we challenged WT, Gsdmd−/−, GsdmdD88A/D88A, and Casp1−/− mice intravenously with TNF (500 μg/kg) and monitored hypothermia and lethality in these animals. As anticipated, WT animals displayed strong hypothermia and started to succumb as early as 4 hours after TNF challenge (Fig. 5, A and B). Unexpectedly, Gsdmd−/− animals were remarkably resistant to hypothermia, and, consequently, more than 75% of Gsdmd−/− animals survived at 72 hours after TNF challenge, while the same conditions triggered lethality in 60% of WT animals (Fig. 5, A and B). To confirm that the observed phenotype is robust and not a strain- or facility-specific artifact, we monitored TNF-induced lethality using a second, independently generated Gsdmd−/− mouse line (48) housed in another mouse facility. Consistent with our initial observation (Fig. 5, A and B), mice from the second Gsdmd−/− line were also significantly protected from TNF-induced lethality compared to WT animals (fig. S4A and movies S1 and S2), confirming that GSDMD confers susceptibility to TNF-induced shock. Notably, GsdmdD88A/D88A mice, which are unable to inactivate GSDMD, displayed an exquisite sensitivity to TNF challenge as they reached 100% mortality within 26 hours after TNF challenge while 40% of WT mice survived up to 72 hours after TNF challenge (Fig. 5B). This indicates that the inactivating cleavage of GSDMD at position D88 is physiologically important to suppress TNF-induced shock. Given that both caspase-1 and caspase-8 are able to activate GSDMD under conditions of TNF Complex IIb assembly (Fig. 2A) (9), we sought to investigate the contribution of caspase-1 to TNF-induced shock. In keeping with a previous report (49), Casp1−/− mice displayed the same sensitivity as WT animals to TNF-induced hypothermia and lethality (Fig. 5, A and B), indicating that GSDMD activation during TNF-induced shock in vivo can occur through caspase-8 cleavage and independently of caspase-1 or inflammasome activation.
Fig. 5. GSDMD drives TNF-induced lethality independently of inflammasomes.
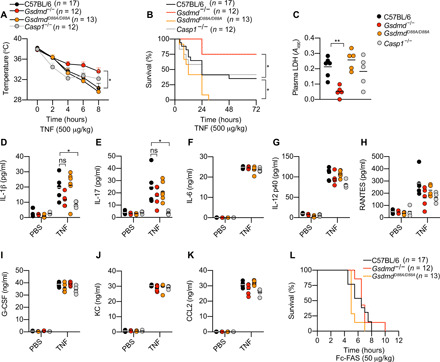
(A to K) Mice were challenged intravenously with TNF (500 μg/kg). (A) Body temperature and (B) survival were monitored over time. (C) Plasma LDH was quantified at 4 hours after TNF challenge. A490, absorbance at 490 nm. (D to K) Plasma cytokines were measured 3 hours after TNF challenge. CCL2, C-C motif chemokine ligand 2; KC, keratinocyte-derived chemokine; PBS, phosphate-buffered saline; ns, not significant; G-CSF, granulocyte colony-stimulating factor. (L) Mice were challenged intravenously with Fc-FAS (50 μg/kg) and monitored for lethality. The number of mice (n) used in each experiment are indicated. (A and B) Data are means + SEM pooled from three independent experiments, and (A) statistical analysis was performed using a two-way analysis of variance (ANOVA). (B) Survival curves were compared using log-rank Mantel-Cox test. (C to K) Data are geometric means from four to five mice per genotype and analyzed using a parametric t test. *P < 0.05; **P < 0.01.
As TNF signaling promotes proinflammatory cytokine and chemokine production in addition to cell death and GSDMD is required for unconventional protein secretion (3, 4, 50), we next determined whether cell lysis or “cytokine storm” is the principal driver of TNF-induced lethality. For this, we first measured markers of tissue damage such as LDH and alanine aminotransferase (ALT) in the plasma of TNF-challenged animals. In agreement with the survival data (Fig. 5B and fig. S4A), TNF-induced plasma LDH was significantly reduced in Gsdmd−/− but not Casp1−/− animals, compared to WT controls (Fig. 5C). Plasma LDH was modestly elevated in GsdmdD88A/D88A compared to WT controls (Fig. 5C). We also observed a similar trend for plasma ALT levels, although the difference between genotypes at this time point was not statistically significant (fig. S4B). Next, we quantified plasma cytokine levels in TNF-challenged animals. Although plasma IL-1β and IL-17 levels were significantly reduced in Casp1−/− mice compared to WT controls (Fig. 5, D and E), Casp1 deficiency did not confer any protection to TNF-induced shock compared to WT animals (Fig. 5, A and B). Since the concentration of other cytokines were comparable between WT, Gsdmd−/−, and GsdmdD88A/D88A mice (Fig. 5, D to K) and markers of tissue damage were reduced in Gsdmd−/− compared to WT animals, our data indicate that caspase-8–dependent GSDMD cell lysis and tissue damage are likely to be the major drivers of TNF-induced shock.
Last, we examined whether Gsdmd deficiency likewise protected mice from FasL-induced hepatitis since lethality in this experimental murine model is also caspase-8 dependent (39, 51). FasL injection triggered 100% lethality in WT animals by 8 hours after injection (Fig. 5L). Although Gsdmd−/− mice trended toward a slight delay in lethality compared to WT animals, this reduction was not statistically significant (Fig. 5L). Together, our data demonstrate that GSDMD confers susceptibility to TNF-induced shock, and this occurs through direct caspase-8 cleavage, independent of caspase-1 or the inflammasome.
GSDMD promotes host defense against Yersinia infection in vivo
Two recent studies reported that infection with the extracellular bacteria Y. pestis or Y. pseudotuberculosis triggers caspase-8–dependent GSDMD cleavage and macrophage pyroptosis (10, 11). While caspase-8– and RIPK1-dependent cell death are important for anti-Yersinia host defense, whether this function is mediated by GSDMD or alternative caspase-8 substrates remains unclear. To investigate this, we first examined the cleavage status of GSDMD and macrophage pyroptosis following infection with Y. pseudotuberculosis in WT, Gsdmd−/−, and GsdmdD88A/D88A macrophages. In agreement with previous reports (10, 11), Y. pseudotuberculosis infection in unprimed WT macrophages triggered cleavage of full-length GSDMD into the active p30 and inactive p20 fragments (Fig. 6A) and GSDMD-dependent pyroptosis (Fig. 6B). As anticipated, given that Yersinia infection triggers Complex IIb assembly and robust caspase-8 activation (10, 11), GsdmdD88A/D88A macrophages were 1.5-fold more susceptible to cellular lysis compared to WT BMDMs under unprimed conditions (Fig. 6B). Compared to unprimed cells, LPS priming reduced caspase-3 activation and a corresponding loss of GSDMD p30 cleavage into the inactive p20 fragment upon Y. pseudotuberculosis infection (Fig. 6A). In agreement with that, LDH release from LPS-primed cells is now comparable between WT and GsdmdD88A/D88A macrophages following Yersinia infection (Fig. 6C). Previous studies reported that Yersinia activates the caspase-1 inflammasomes in a caspase-8–dependent manner (10, 52, 53). We observed that caspase-1 autoprocessing into the p20 fragment was comparable between all genotypes tested, regardless of duration or LPS priming (Fig. 6A and fig. S5A), indicating that caspase-8–dependent caspase-1 activation does not require GSDMD. Next, to examine whether GSDMD-dependent cell lysis promotes IL-1β secretion following Y. pseudotuberculosis infection, we challenged unprimed macrophages with Y. pseudotuberculosis for 16 hours. Consistent with a previous report (11), overnight Y. pseudotuberculosis infection triggered robust IL-1β secretion in WT BMDMs (Fig. 6D). IL-1β secretion was markedly reduced in Gsdmd−/− BMDM, while GsdmdD88A/D88A mutation promoted IL-1β secretion compared to WT cells (fig. 6D), in line with the known role of GSDMD in driving unconventional cytokine secretion (3, 4, 50). Although caspase-8 can directly process IL-1β under conditions of TNF Complex IIb/ripoptosome assembly (16, 17, 20), IL-1β maturation upon Yersinia infection was largely NLRP3–caspase-1 dependent since IL-1β secretion was completely abrogated in the presence of the NLRP3-specific inhibitor MCC950 or caspase-1 deficiency (Fig. 6D and fig. S5B). In keeping with the LDH data (Fig. 6C), LPS-primed WT macrophages secreted equivalent levels of IL-1β and IL-18 upon Y. pseudotuberculosis infection (Fig. 6, E and F). Of note, while pro–IL-1β maturation appears to be completely caspase-1 dependent during Y. pseudotuberculosis infection (Fig. 6D and fig. S5B), cleavage of IL-18 into its mature, active p18 form is largely intact in Casp1−/− BMDM (fig. S5B), likely reflecting caspase-8–dependent IL-18 maturation, as previously reported (35, 54).
Fig. 6. GSDMD-dependent cell death promotes anti-Yersinia defense in vivo.
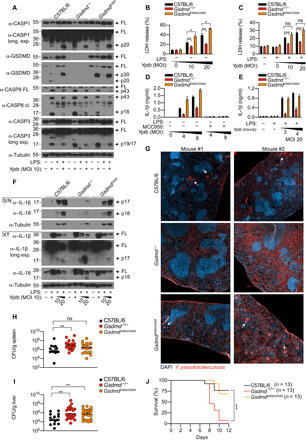
(A) Unprimed or LPS-primed (100 ng/ml; 3 hours) BMDMs were infected with Y. pseudotuberculosis (Yptb) at an MOI of 10 and for 3 hours, and mixed supernatant and cell extracts were examined by immunoblotting. (B) Unprimed or (C) LPS-primed (100 ng/ml; 3 hours) BMDMs were infected with Y. pseudotuberculosis at the indicated MOI for 3 hours, and LDH release was quantified. (D) Unprimed BMDMs were infected with Y. pseudotuberculosis at the indicated MOI, and IL-1β release was measured 16 hours after infection. Where indicated, cells were treated with MCC950 (10 μM) 20 to 30 min before infection. (E) LPS-primed (100 ng/ml; 3 hours) BMDMs were challenged with Y. pseudotuberculosis at an MOI of 20, and IL-1β release was measured at the indicated time points. (F) BMDMs were infected with Y. pseudotuberculosis with the indicated MOI for 4 hours, and precipitated supernatant and cell extracts were analyzed by immunoblot. (G to J) Mice were challenged orally with 2 × 108 CFU of Y. pseudotuberculosis, (G) immunofluorescence of infected spleen were analyzed (white arrows indicate large clusters surrounded by pyogranulomatous reaction), and (H and I) bacterial burden was quantified 5 days after infection. DAPI, 4′,6-diamidino-2-phenylindole. (J) Mouse survival was monitored for 12 days. (A and F) Immunoblots are representative of two to three independent experiments. (B, C, and E) Data are means + SEM of pooled data from three independent experiments. (D) Data are means + SD of triplicate cell stimulation and are a representative from four independent experiments. (H and I) Data are geometric means and pooled from two independent experiments. (G) Images are representative of four to five mice. (B and C) Data were normally distributed and analyzed using a parametric t test. (J) Survival curves were compared using log-rank Mantel-Cox test. (H and I) Data were analyzed using Mann-Whitney t tests. *P < 0.05, **P < 0.01, or ***P < 0.001.
Next, to determine the impact of Gsdmd deficiency and GsdmdD88A mutation during in vivo infection, we orally challenged WT, Gsdmd−/−, and GsdmdD88A/D88A mice with Y. pseudotuberculosis. In keeping with a previous report (55), immunofluorescence analysis revealed that Y. pseudotuberculosis were primarily detected large clusters surrounded by pyogranulomatous reaction in the spleen of immunocompetent WT animals (Fig. 6G, white arrows, and fig. S5C). In contrast, an extensive dissemination of individualized bacteria was commonly observed throughout the spleen of Gsdmd−/− animals (Fig. 6G and fig. S5C), which correlated with significantly higher bacterial burden in the spleen and liver (Fig. 6, H and I), increased mortality (Fig. 6J), and failure to contain bacteria within tight foci compared to WT controls. To ensure the robustness of our study, we used a second, independently generated Gsdmd−/− mouse line housed in another facility (56) and confirmed that GSDMD is indeed required for anti-Yersinia defense in vivo (fig. S5, D and E). These data suggest that GSDMD is required for the formation of pyogranuloma, presumably to restrict pathogen dissemination and replication (55). We also observed a reduction in pyogranuloma formation and the appearance of more free bacteria in GsdmdD88A/D88A spleen compared to WT controls, although this effect was not as notable compared to Gsdmd−/− mice (Fig. 6G, white arrows, and fig. S5C) and did not significantly influence splenic bacterial burden compared to WT mice (Fig. 6H). However, we consistently recovered more bacteria in the liver but not in the spleen of GsdmdD88A/D88A mice compared to WT controls (Fig. 6, H and I), indicating that excessive GSDMD-dependent pyroptosis impairs host defense in the liver. However, there was no significant difference in survival between WT and GsdmdD88A/D88A mice after 12 days of infection (Fig. 6J), indicating that GSDMD inactivation at position D88 promotes but is not an absolute requirement for host defense against Y. pseudotuberculosis in vivo.
DISCUSSION
Recent studies revealed that TAK1 inhibition or IAP depletion in LPS- or TNF-stimulated cells activates caspase-8 to cleave the pore-forming protein GSDMD and trigger cell lysis (9–11), revealing additional cross-talk between apoptosis and pyroptosis beyond the previously described role of caspase-8 in the activation of caspase-1 itself (16, 17, 52, 53). However, the molecular requirements for caspase-8–dependent GSDMD cleavage and the physiological impact of this signaling axis in vivo remain unclear. Here, we report that caspase-8 dimerization, catalytic activity, and autoprocessing are required for caspase-8–dependent GSDMD cleavage and pyroptosis. Furthermore, our study reveals that not all caspase-8–activating platforms are equivalent in their ability to elicit direct GSDMD processing. Notably, we observed that caspase-8 activated within TNF Complex IIb elicits the strongest ability to cleave GSDMD, while caspase-8 activated under conditions of TNF Complex IIa and DISC assembly can only weakly do so or promotes GSDMD cleavage indirectly by activating the inflammasome in macrophages (fig. S6). This provides a likely explanation to why GSDMD cleavage was not previously detected in TNF/CHX-stimulated HeLa cells (48). Although our data suggest that robust caspase-8 activity on Complex IIb correlates with its ability to directly cleave GSDMD, we do not exclude the possibility that other mechanisms may selectively enable Complex IIb–activated caspase-8 to cleave GSDMD. In this regard, the absolute requirement for RIPK1 kinase activity to drive Complex IIb–dependent caspase-8 activation represents the most notable difference between the various caspase-8–activating platforms and is a prime candidate. It is tempting to speculate that the kinase activity or scaffolding function of RIPK1 may directly or indirectly facilitate the recruitment of GSDMD to caspase-8 on Complex IIb. Future studies defining the cellular localization and interacting partners of GSDMD during Complex IIb–driven cell death will be of interest.
A recent study reported that Fas ligation triggered robust caspase-8–dependent GSDMD cleavage and pyroptosis in murine bone marrow–derived dendritic cells (54), whereas we found that Fas-activated caspase-8 is a weak activator of GSDMD in macrophages, in agreement with the emerging theme that cell death signaling exhibits distinct regulatory and functional properties in different myeloid cell subsets. For example, mixed lineage kinase domain–like (MLKL)–dependent NLRP3 activation in macrophages requires plasma membrane damage and potassium efflux (42, 43), while MLKL activates NLRP3 in dendritic cells in the absence of cell lysis (57). Since Fas is not strongly expressed in BMDMs (35), we speculate that higher expression of Fas in dendritic cells may sensitize these cells to caspase-8–dependent GSDMD cleavage upon DISC assembly. However, the ability of Fas-activated caspase-8 to drive GSDMD activation and pyroptosis is likely restricted to a subset of cell types in vivo, as we did not observe a significant contribution of GSDMD to FasL-induced hepatitis.
Although apoptotic caspase activation was previously observed upon inflammasome activation in WT macrophages (58), we find no evidence that cleavage of GSDMD by caspase-3/7 suppresses caspase-1– or caspase-11–dependent pyroptosis, as LDH and cytokine release from WT and GsdmdD88A/D88A macrophages was indistinguishable under all conditions tested. This suggests that excessive formation of GSDMD pores does not offer a simple explanation to why IL-1β secretion upon inflammasome activation is enhanced in caspase-3/7–deficient cells compared to WT controls (8). Instead, we speculate that cleavage of an unidentified caspase-3/7 substrate suppresses IL-1β secretion in canonical inflammasome-stimulated cells, and identification of such a substrate may have therapeutic potential against the myriad of IL-1–driven pathologies.
GSDMD-dependent pyroptosis is traditionally viewed as a host defense mechanism against intracellular pathogens since this form of cell death weakens intracellular pathogens and exposes them to neutrophil-mediated destruction (24–26). Whether GSDMD-driven mechanisms exist to protect against extracellular bacterial infection is unclear. Here, we make the unexpected finding that GSDMD activation and pyroptosis are also required to combat infection with the extracellular Gram-negative bacterium, Y. pseudotuberculosis. Optimal host defense against Y. pseudotuberculosis requires a RIPK1-driven cell death program, in which dying cells prime and activate bystander innate immune cells for optimal cytokine production that promotes bacteria restriction within a pyrogranuloma (55). Our study is consistent with such a model, as Gsdmd−/− mice were indeed more susceptible than WT animals to Y. pseudotuberculosis infection. Although caspase-8 deficiency appears to trigger a more severe Yersinia infection compared to caspase-1–deficient mice in vivo (52, 59), at this stage, it is challenging to investigate whether caspase-8– or caspase-1–driven GSDMD activation provides greater host defense because Casp8-deficient mice (e.g., Casp8/Ripk3 dKO) display severe defects in proinflammatory and antimicrobial gene transcription (60–62) and Casp8D387A mutation unleashes RIPK3-dependent necroptosis upon Y. pseudotuberculosis infection (62). Given that caspase-1 amplifies pyroptosis downstream of caspase-8 activation (9, 17), we predict that both caspase-1 and caspase-8 are required for GSDMD activation, which promotes mature IL-1β and IL-18 secretion that drive immune cell recruitment and activation at a site of infection (fig. S6). Neutrophils are an important cell type that protect against Yersinia infection (63); however, whether caspase-8–dependent GSDMD activation promotes neutrophil antimicrobial function is unclear and often overlooked. We recently demonstrated that cytosolic LPS triggers caspase-11–dependent GSDMD activation and neutrophil extracellular traps (NET) extrusion as a host defense mechanism against cytosolic Gram-negative bacterial infection (31). Since neutrophils can assemble a functional TNF Complex IIb (20), we cannot exclude the possibility that caspase-8–dependent GSDMD activation in TNF-stimulated neutrophils and subsequent NET extrusion contributes to the host-protective function of GSDMD during Y. pseudotuberculosis infection. Future studies characterizing the caspase-8 GSDMD signaling axis in neutrophils will be important.
TNF is a pleiotropic cytokine that has a profound effect on the immune system including inflammation, cell survival, and cell death. Not unexpectedly, aberrant TNF signaling is associated with an increasing number of inflammatory diseases including arthritic disease, plaque psoriasis, Crohn’s disease, and systemic inflammatory response syndrome (23). In particular, TNF-induced necroptosis has emerged as a major driver of these pathologies. These conclusions were drawn on the basis of the observation that genetic loss or pharmacological inhibition of RIPK1 or RIPK3, essential drivers of the TNF-induced RIPK1-RIPK3-MLKL necroptosis pathway, significantly reduced disease severity in a number of experimental murine models (45, 64, 65). Unexpectedly, further examination revealed that Mlkl deficiency conferred less protection than catalytic inactive RIPK1 or Ripk3−/− mice in several experimental models (66), indicating the existence of a pathological RIPK1/RIPK3-dependent, MLKL-independent pathway in vivo. Our study agrees with these observations, as we now provide compelling evidence that caspase-8–dependent GSDMD activation is a major driver for TNF-induced lethality. Of note, GSDMD activation proceeds independently of inflammasomes, indicating that pharmacological inhibition of inflammasomes is unlikely to offer any therapeutical value under conditions of TNF-induced shock (fig. S6). Since RIPK3 scaffolding function promotes full caspase-8 activation downstream of Complex IIb/ripoptosome (9, 15, 17), our data provide a possible explanation to why Ripk3−/− are more protected than Mlkl−/− from TNF-induced shock (66). We speculate that GSDMD activation may be a common feature of other TNF-driven diseases, and previous studies that concluded necroptosis as a major driver of TNF-dependent pathology exclusively using Ripk3−/− or catalytic inactive RIPK1 mice should be reevaluated using Mlkl−/− and Gsdmd−/− mice. Characterizing GSDMD function in RIPK1- or RIPK3-driven diseases may unveil previously unidentified therapeutic targets for such diseases.
METHODS
Animals
All animal experiments were performed under the guidelines and approval from the Swiss animal protection law (licenses VD3257 and VD877.9; Service des Affaires Vétérinaires, Direction Générale de l’Agriculture, de la Viticulture et des Affaires Vétérinaires, état de Vaud, Epalinges, Switzerland), the German animal protection law (license TVV57/2017), and guidelines from the National Institutes of Health (NIH) and the University of Pennsylvania Institutional Animal Care and Use Committee (protocol no. 804523). C57BL/6J, Gsdmd−/−, GsdmdD88A/D88A, Casp1−/−, and Casp8D387A/D387A (all either generated in or backcrossed to the C57BL/6 background) were previously described and housed in specific pathogen–free facilities at the University of Lausanne, Technical University Dresden, or the University of Pennsylvania (9, 48, 56, 62).
Yersinia infection
Y. pseudotuberculosis infection was performed, as previously described (55). WT Y. pseudotuberculosis strain 32777 was grown overnight with aeration at 26°C in 2xYT media. To induce a type 3 secretion system expression, overnight bacteria were diluted in fresh 2xYT media supplemented with 20 mM sodium oxalate and 20 mM MgCl2 and grown with aeration for 1 hour at 26°C followed by another 2 hours at 37°C. Log-phase bacteria were washed two to three times in warm Opti-MEM and were used to infect BMDMs at a multiplicity of infection (MOI) of 2 to 20. Cell culture plates were centrifuged at 300g for 5 min at 37°C to promote bacterial adhesion, and gentamicin (100 μg/ml) (Gibco) was added 1 hour after infection to kill extracellular bacteria. Where indicated, cells were primed with Escherichia coli O55:B5 LPS (100 ng/ml; InvivoGen) for 3 hours or treated with MCC950 (10 μM; InvivoGen) 20 to 30 min before infection. For in vivo infection, mice were fasted for 16 hours and challenged with 2 × 108 colony-forming units (CFU) of stationary-phase bacteria by oral gavage. To determine bacterial burden, mice were euthanized 5 days after infection and tissues were harvested, homogenized in 1 ml of phosphate-buffered saline (PBS), and serially diluted on LB agar.
Salmonella infection
S. Typhimurium SL1344 infection was performed, as previously described (30). Salmonella were grown overnight with aeration at 37°C in LB media. To induce a type 3 secretion system expression to activate the NLRC4 inflammasome, overnight culture was diluted in 1:40 in LB and grown for another 3 to 4 hours of aeration at 37°C in LB media. Log-phase bacteria were washed two to three times in PBS (Gibco), diluted in Opti-MEM (Gibco), and used to infect macrophages or neutrophils at an MOI of 10 to 25. Cell culture plates were centrifuged at 300g for 5 min at 37°C to synchronize infection, and gentamicin (100 μg/ml) (Gibco) was added 1 hour after infection to kill extracellular bacteria. For in vivo infection, mice were challenged with intraperitoneal injection with 1 × 105 CFU of log-phase Salmonella. To determine bacterial burden, mice were euthanized 48 hours after infection and tissues were harvested, homogenized in 1 ml of PBS, and serially diluted on LB agar.
Primary myeloid cell culture
BMDMs were differentiated in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) containing 20% 3T3 supernatant [as a source of macrophage colony-stimulating factor (M-CSF)], 10% heat-inactivated fetal calf serum (FCS) (BioConcept), 10 mM Hepes (BioConcept), penicillin/streptomycin (BioConcept), and nonessential amino acids (Gibco) and stimulated on days 7 to 9 of differentiation. Mature neutrophils were purified from murine bone marrow using anti–Ly6G-FITC (1A8 clone) and anti–FITC (fluorescein isothiocyanate) beads (Miltenyi), as previously described (30).
Inflammasome assays
Macrophages were seeded at a density of 5 × 104 cells per well in DMEM (Gibco) containing 20% 3T3 supernatant (as a source of M-CSF), 10% heat-inactivated FCS (BioConcept), 10 mM Hepes (BioConcept), penicillin/streptomycin (BioConcept), and nonessential amino acids (Gibco) a day before stimulation. Purified neutrophils were used for experiments on the day of purification and seeded at a density of 4 × 105 to 5 × 105 cells per well in Opti-MEM (Gibco). To activate canonical inflammasomes, cells were left unprimed or primed with ultrapure E. coli 055:B5 LPS (100 ng/ml; InvivoGen) or Pam3CSK4 (1 μg/ml; InvivoGen) for 4 hours in Opti-MEM and stimulated with nigericin (Sigma-Aldrich), ATP (Sigma-Aldrich), and S. Typhimurium (log phase), and transfected with poly(dA:dT) (InvivoGen) or ultrapure flagellin (InvivoGen) using polyethylenimine (Polysciences Inc.) or FuGENE HD (Promega), respectively. To activate the noncanonical inflammasome, macrophages were primed with Pam3CSK4 (1 μg/ml; InvivoGen) for 4 hours in Opti-MEM and transfected with ultrapure E. coli O111:B4 LPS (2 μg/ml) (InvivoGen) using 0.25% FuGENE HD (Promega) and centrifuged for 500g for 10 min at 37°C.
Apoptosis assay
Macrophages were seeded at a density of 5 × 104 cells per well in complete media a day before stimulation. To activate caspase-8, macrophages were stimulated in Opti-MEM (Gibco) with recombinant murine TNF (100 ng/ml; PeproTech) and CHX (10 μg/ml; Sigma-Aldrich) to activate TNF Complex IIa or TNF with the TAK1 inhibitor 5z7-oxozeaenol (125 nM; Sigma-Aldrich) to trigger TNF Complex IIb. Alternatively, macrophages were primed with E. coli O55:B5 LPS (100 ng/ml; InvivoGen) or Pam3CSK4 (1 μg/ml; InvivoGen) for 4 or 16 hours in complete media and stimulated with FC-FasL (100 ng/ml) (a gift from P. Schneider, University of Lausanne) in Opti-MEM (Gibco). To activate mitochondrial apoptosis, macrophages were stimulated with ABT-737 (500 nM; Selleckchem) and S63845 (500 nM; Selleckchem) in Opti-MEM (Gibco).
LDH release assay
LDH release into the cell culture supernatant was quantified using a TaKaRa LDH cytotoxicity detection kit (Clontech) and expressed as a percentage of total cellular LDH (100% lysis).
HEK 293T transfections
HEK 293T cells (3 × 105) were seeded in 24-well plates and transfected with a 2:1 ratio of caspase-8 to GSDMD using Lipofectamine 2000 in Opti-MEM according to the manufacturer’s instructions. Fresh complete DMEM was added to each well 6 hours after transfection. Twenty-four hours after transfection, media were replaced with serum-free media and 1 μM AP 20187 (dimerizer) was added. PI (5 μM) was added to wells for PI imaging. Bright-field images were acquired on an inverted Nikon Eclipse TE2000-U at ×20 magnification. Supernatants and cell lysates were harvested for immunoblotting 6 hours after dimerizer treatment. Dimerizable WT, catalytic inactive (C360A), or uncleavable caspase-8 (D210A/D216A/D223A/D374A/D384A) constructs were previously described and were a gift from A. Oberst (37).
Immunoblotting
Cell culture supernatant were precipitated with methanol and chloroform, as previously described, and resuspended cell extracts lysed in boiling lysis buffer [66 mM tris-Cl (pH 7.4), 2% SDS, 10 mM dithiothreitol, and NuPage LDS sample buffer; Thermo Fisher Scientific). Proteins were separated on 14% polyacrylamide gels and transferred onto nitrocellulose membrane using Trans-Blot Turbo (Bio-Rad). Antibodies for immunoblot were against GSDMD (1:3000; EPR19828, Abcam), caspase-1 p20 (1:1000; casper-1, AdipoGen), full-length caspase-8 (1:1000; 4927, Cell Signaling), cleaved caspase-8 (1:1000; 9429, Cell Signaling), caspase-3 (1:1000; 9662, Cell Signaling), pro–IL-1β (1:1000; AF-401-NA, R&D), IL-18 (1:1000; 5180r-100, BioVision), RIPK1 (1:1000; D94C12, Cell Signaling), c-FLIP (1:1000; Dave-2, AdipoGen), and alpha-tubulin (1:5000; DM1A, Abcam).
TNF shock model
Mice were injected intravenously with recombinant murine TNF (500 μg/kg) (PeproTech) in a volume of 100 μl. Control mice received an equal volume of PBS. Cytokine levels in the plasma were quantified 3 hours after TNF injection using the commercial Mouse Cytokine Array/Chemokine Array 31-Plex (Eve Technologies Corporation, Canada). Plasma LDH (Clontech) and ALT (Sigma-Aldrich) were quantified at 4 hours after TNF challenge according to the manufacturer’s instruction. Both male and female mice were used for these experiments and were age-matched. Mice rectal temperature was recorded with a rectal mouse thermometer (Braintree Scientific). Mice were monitored for 72 hours after injection and were considered dead when they succumbed to TNF-induced lethality.
FasL-induced hepatitis
Mice were injected intravenously with FC-FasL (50 μg/kg) (a gift from P. Schneider, University of Lausanne) in a volume of 100 μl. Both male and female mice were used for these experiments and were age matched. Mouse survival was monitored for up to 10 hours after injection and were considered dead when they succumbed to TNF-induced lethality.
Immunofluorescence staining of fixed tissue
Spleen from infected animals were harvested 5 days after infection and fixed directly in 1% PFA (paraformaldehyde) at 4°C overnight. Fixed tissues were washed extensively using PBS, incubated overnight in 30% sucrose at 4°C, and embedded in optimum cutting temperature blocks. Sections (8 μM) were prepared using cryostat (Thermo Fisher Scientific). Sections were blocked and incubated with anti–Y. pseudotuberculosis (1:4000; ab26120, Abcam) for 1 hour at room temperature, followed by secondary antibody (1:500; Thermo Fisher Scientific, goat anti-rabbit 594) and 4′,6-diamidino-2-phenylindole for 30 min. Immunofluorescence of fixed tissues were scanned using Zeiss Axio Scan.Z1.
Statistical analyses
Statistical analyses were performed using Prism 8 (GraphPad) software. Datasets were first analyzed for normality using Shapiro-Wilk normality test. Parametric t test was used for normally distributed datasets, while non-normally distributed datasets were analyzed using nonparametric Mann-Whitney t tests. A two-way analysis of variance (ANOVA) was used to analyze repeated measures over time. Survival curves were compared using log-rank Mantel-Cox test. Data were considered significant when P ≤ 0.05, with *P ≤ 0.05, **P ≤ 0.01, or ***P ≤ 0.001.
Supplementary Material
Acknowledgment
We thank F. Tacchini-Cottier (University of Lausanne), P. Schneider (University of Lausanne), and A. Oberst (University of Washington) for providing Casp1–/– mice, Fc-FasL, and dimerizable caspase-8 expression constructs, respectively. We also thank K. Soukup (University of Lausanne) and J. Joyce (University of Lausanne) for helping with image acquisition. Funding: This work was supported by a Marie Skłodowska-Curie Actions (MSCA) incoming fellowship (MSCA-IF-2018-838252) to K.W.C., a European Research Council grant (ERC2017-CoG-770988-InflamCellDeath) and a Swiss National Science Foundation grant (SNSF 175576) to P.B., a Swiss National Science Foundation grant (SNSF 310030_173123) to T.R., NIH grants (1R21135421 and 1R01128530) and a Burroughs Wellcome Fund Pathogenesis of Infectious Disease Award to I.E.B., and DFG-sponsored Heisenberg Professorship, SFB-TRR 205, SFB-TRR 127, and the International Research Training Group (IRTG) 2251 to A.L. Author contributions: B.D., K.W.C., and P.B. designed the experiments. B.D., K.W.C., J.P.G., E.B., D.L.R., W.T., C-A.A., E.R., T.F., and V.M. performed the experiments and analyzed the data. A.L., T.R., and I.E.B. provided essential reagents and expert advice. B.D. and K.W.C. prepared the figures. K.W.C. and P.B. conceptualized and supervised the study and wrote the manuscript, which all authors reviewed before submission. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc3465/DC1
REFERENCES AND NOTES
- 1.Chen K. W., Demarco B., Broz P., Beyond inflammasomes: Emerging function of gasdermins during apoptosis and NETosis. EMBO J. 39, e103397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broz P., Pelegrin P., Shao F., The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Evavold C. L., Ruan J., Tan Y., Xia S., Wu H., Kagan J. C., The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heilig R., Dick M. S., Sborgi L., Meunier E., Hiller S., Broz P., The gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 48, 584–592 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Ruhl S., Shkarina K., Demarco B., Heilig R., Santos J. C., Broz P., ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Taabazuing C. Y., Okondo M. C., Bachovchin D. A., Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 24, 507–514.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagulenko V., Thygesen S. J., Sester D. P., Idris A., Cridland J. A., Vajjhala P. R., Roberts T. L., Schroder K., Vince J. E., Hill J. M., Silke J., Stacey K. J., AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vince J. E., De Nardo D., Gao W., Vince A. J., Hall C., McArthur K., Simpson D., Vijayaraj S., Lindqvist L. M., Bouillet P., Rizzacasa M. A., Man S. M., Silke J., Masters S. L., Lessene G., Huang D. C. S., Gray D. H. D., Kile B. T., Shao F., Lawlor K. E., The Mitochondrial apoptotic effectors BAX/BAK Activate caspase-3 and -7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1β activation. Cell Rep. 25, 2339–2353.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Chen K. W., Demarco B., Heilig R., Shkarina K., Boettcher A., Farady C. J., Pelczar P., Broz P., Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38, e101638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orning P., Weng D., Starheim K., Ratner D., Best Z., Lee B., Brooks A., Xia S., Wu H., Kelliher M. A., Berger S. B., Gough P. J., Bertin J., Proulx M. M., Goguen J. D., Kayagaki N., Fitzgerald K. A., Lien E., Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarhan J., Liu B. C., Muendlein H. I., Li P., Nilson R., Tang A. Y., Rongvaux A., Bunnell S. C., Shao F., Green D. R., Poltorak A., Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. U.S.A. 115, E10888–E10897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feltham R., Vince J. E., Lawlor K. E., Caspase-8: Not so silently deadly. Clin. Transl. Immunology 6, e124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Micheau O., Tschopp J., Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Wang L., Du F., Wang X., TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Dondelinger Y., Aguileta M. A., Goossens V., Dubuisson C., Grootjans S., Dejardin E., Vandenabeele P., Bertrand M. J., RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20, 1381–1392 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lawlor K. E., Feltham R., Yabal M., Conos S. A., Chen K. W., Ziehe S., Grass C., Zhan Y., Nguyen T. A., Hall C., Vince A. J., Chatfield S. M., D’Silva D. B., Pang K. C., Schroder K., Silke J., Vaux D. L., Jost P. J., Vince J. E., XIAP loss triggers RIPK3- and caspase-8-driven IL-1β activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 20, 668–682 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Vince J. E., Wong W. W., Gentle I., Lawlor K. E., Allam R., O’Reilly L., Mason K., Gross O., Ma S., Guarda G., Anderton H., Castillo R., Hacker G., Silke J., Tschopp J., Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Lawlor K. E., Khan N., Mildenhall A., Gerlic M., Croker B. A., D’Cruz A. A., Hall C., Kaur Spall S., Anderton H., Masters S. L., Rashidi M., Wicks I. P., Alexander W. S., Mitsuuchi Y., Benetatos C. A., Condon S. M., Wong W. W., Silke J., Vaux D. L., Vince J. E., RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malireddi R. K. S., Gurung P., Kesavardhana S., Samir P., Burton A., Mummareddy H., Vogel P., Pelletier S., Burgula S., Kanneganti T. D., Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 217, e20191644 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen K. W., Lawlor K. E., von Pein J. B., Boucher D., Gerlic M., Croker B. A., Bezbradica J. S., Vince J. E., Schroder K., Cutting edge: Blockade of inhibitor of apoptosis proteins sensitizes neutrophils to TNF- but not lipopolysaccharide-mediated cell death and IL-1β secretion. J. Immunol. 200, 3341–3346 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Yabal M., Muller N., Adler H., Knies N., Gross C. J., Damgaard R. B., Kanegane H., Ringelhan M., Kaufmann T., Heikenwalder M., Strasser A., Gross O., Ruland J., Peschel C., Gyrd-Hansen M., Jost P. J., XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 7, 1796–1808 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Wicki S., Gurzeler U., Wei-Lynn Wong W., Jost P. J., Bachmann D., Kaufmann T., Loss of XIAP facilitates switch to TNFα-induced necroptosis in mouse neutrophils. Cell Death Dis. 7, e2422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalliolias G. D., Ivashkiv L. B., TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 12, 49–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jorgensen I., Zhang Y., Krantz B. A., Miao E. A., Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213, 2113–2128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao E. A., Leaf I. A., Treuting P. M., Mao D. P., Dors M., Sarkar A., Warren S. E., Wewers M. D., Aderem A., Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demarco B., Chen K. W., Broz P., Cross talk between intracellular pathogens and cell death. Immunol. Rev. 297, 174–193 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Lee B. L., Mirrashidi K. M., Stowe I. B., Kummerfeld S. K., Watanabe C., Haley B., Cuellar T. L., Reichelt M., Kayagaki N., ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep. 8, 3788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heilig R., Dilucca M., Boucher D., Chen K. W., Hancz D., Demarco B., Shkarina K., Broz P., Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci Alliance 3, e202000735 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuchiya K., Nakajima S., Hosojima S., Thi Nguyen D., Hattori T., Manh Le T., Hori O., Mahib M. R., Yamaguchi Y., Miura M., Kinoshita T., Kushiyama H., Sakurai M., Shiroishi T., Suda T., Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 10, 2091 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K. W., Gross C. J., Sotomayor F. V., Stacey K. J., Tschopp J., Sweet M. J., Schroder K., The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 8, 570–582 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Chen K. W., Monteleone M., Boucher D., Sollberger G., Ramnath D., Condon N. D., von Pein J. B., Broz P., Sweet M. J., Schroder K., Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 3, eaar6676 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Chang L., Kamata H., Solinas G., Luo J.-L., Maeda S., Venuprasad K., Liu Y. C., Karin M., The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIPL turnover. Cell 124, 601–613 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Kreuz S., Siegmund D., Scheurich P., Wajant H., NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 21, 3964–3973 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irmler M., Thome M., Hahne M., Schneider P., Hofmann K., Steiner V., Bodmer J. L., Schroter M., Burns K., Mattmann C., Rimoldi D., French L. E., Tschopp J., Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Bossaller L., Chiang P. I., Schmidt-Lauber C., Ganesan S., Kaiser W. J., Rathinam V. A., Mocarski E. S., Subramanian D., Green D. R., Silverman N., Fitzgerald K. A., Marshak-Rothstein A., Latz E., Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 189, 5508–5512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Y., Devin A., Rodriguez Y., Liu Z. G., Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oberst A., Pop C., Tremblay A. G., Blais V., Denault J. B., Salvesen G. S., Green D. R., Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J. Biol. Chem. 285, 16632–16642 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pop C., Fitzgerald P., Green D. R., Salvesen G. S., Role of proteolysis in caspase-8 activation and stabilization. Biochemistry 46, 4398–4407 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., Green D. R., Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newton K., Wickliffe K. E., Dugger D. L., Maltzman A., Roose-Girma M., Dohse M., Komuves L., Webster J. D., Dixit V. M., Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574, 428–431 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Pop C., Oberst A., Drag M., Van Raam B. J., Riedl S. J., Green D. R., Salvesen G. S., FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 433, 447–457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conos S. A., Chen K. W., De Nardo D., Hara H., Whitehead L., Nunez G., Masters S. L., Murphy J. M., Schroder K., Vaux D. L., Lawlor K. E., Lindqvist L. M., Vince J. E., Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. U.S.A. 114, E961–E969 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutierrez K. D., Davis M. A., Daniels B. P., Olsen T. M., Ralli-Jain P., Tait S. W., Gale M. Jr., Oberst A., MLKL activation triggers NLRP3-mediated processing and release of IL-1β independently of gasdermin-D. J. Immunol. 198, 2156–2164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J. L., Schneider P., Seed B., Tschopp J., Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Duprez L., Takahashi N., Van Hauwermeiren F., Vandendriessche B., Goossens V., Vanden Berghe T., Declercq W., Libert C., Cauwels A., Vandenabeele P., RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Polykratis A., Hermance N., Zelic M., Roderick J., Kim C., Van T. M., Lee T. H., Chan F. K. M., Pasparakis M., Kelliher M. A., Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 193, 1539–1543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaufmann T., Jost P. J., Pellegrini M., Puthalakath H., Gugasyan R., Gerondakis S., Cretney E., Smyth M. J., Silke J., Hakem R., Bouillet P., Mak T. W., Dixit V. M., Strasser A., Fatal hepatitis mediated by tumor necrosis factor TNFα requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity 30, 56–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F., Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Vanden Berghe T., Demon D., Bogaert P., Vandendriessche B., Goethals A., Depuydt B., Vuylsteke M., Roelandt R., Van Wonterghem E., Vandenbroecke J., Choi S. M., Meyer E., Krautwald S., Declercq W., Takahashi N., Cauwels A., Vandenabeele P., Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am. J. Respir. Crit. Care Med. 189, 282–291 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Monteleone M., Stanley A. C., Chen K. W., Brown D. L., Bezbradica J. S., von Pein J. B., Holley C. L., Boucher D., Shakespear M. R., Kapetanovic R., Rolfes V., Sweet M. J., Stow J. L., Schroder K., Interleukin-1β maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. 24, 1425–1433 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Ogasawara J., Watanabe-Fukunaga R., Adachi M., Matsuzawa A., Kasugai T., Kitamura Y., Itoh N., Suda T., Nagata S., Lethal effect of the anti-Fas antibody in mice. Nature 364, 806–809 (1993). [DOI] [PubMed] [Google Scholar]
- 52.Philip N. H., Dillon C. P., Snyder A. G., Fitzgerald P., Wynosky-Dolfi M. A., Zwack E. E., Hu B., Fitzgerald L., Mauldin E. A., Copenhaver A. M., Shin S., Wei L., Parker M., Zhang J., Oberst A., Green D. R., Brodsky I. E., Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7385–7390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weng D., Marty-Roix R., Ganesan S., Proulx M. K., Vladimer G. I., Kaiser W. J., Mocarski E. S., Pouliot K., Chan F. K., Kelliher M. A., Harris P. A., Bertin J., Gough P. J., Shayakhmetov D. M., Goguen J. D., Fitzgerald K. A., Silverman N., Lien E., Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. U.S.A. 111, 7391–7396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donado C. A., Cao A. B., Simmons D. P., Croker B. A., Brennan P. J., Brenner M. B., A two-cell model for IL-1β release mediated by death-receptor signaling. Cell Rep. 31, 107466 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peterson L. W., Philip N. H., DeLaney A., Wynosky-Dolfi M. A., Asklof K., Gray F., Choa R., Bjanes E., Buza E. L., Hu B., Dillon C. P., Green D. R., Berger S. B., Gough P. J., Bertin J., Brodsky I. E., RIPK1-dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. J. Exp. Med. 214, 3171–3182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rauch I., Deets K. A., Ji D. X., von Moltke J., Tenthorey J. L., Lee A. Y., Philip N. H., Ayres J. S., Brodsky I. E., Gronert K., Vance R. E., NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and -8. Immunity 46, 649–659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang T.-B., Yang S.-H., Toth B., Kovalenko A., Wallach D., Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Sagulenko V., Vitak N., Vajjhala P. R., Vince J. E., Stacey K. J., Caspase-1 is an apical caspase leading to caspase-3 cleavage in the AIM2 inflammasome response, independent of caspase-8. J. Mol. Biol. 430, 238–247 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Brodsky I. E., Palm N. W., Sadanand S., Ryndak M. B., Sutterwala F. S., Flavell R. A., Bliska J. B., Medzhitov R., A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allam R., Lawlor K. E., Yu E. C., Mildenhall A. L., Moujalled D. M., Lewis R. S., Ke F., Mason K. D., White M. J., Stacey K. J., Strasser A., O’Reilly L. A., Alexander W., Kile B. T., Vaux D. L., Vince J. E., Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 15, 982–990 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeLaney A. A., Berry C. T., Christian D. A., Hart A., Bjanes E., Wynosky-Dolfi M. A., Li X., Tummers B., Udalova I. A., Chen Y. H., Hershberg U., Freedman B. D., Hunter C. A., Brodsky I. E., Caspase-8 promotes c-Rel-dependent inflammatory cytokine expression and resistance against Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 116, 11926–11935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Philip N. H., DeLaney A., Peterson L. W., Santos-Marrero M., Grier J. T., Sun Y., Wynosky-Dolfi M. A., Zwack E. E., Hu B., Olsen T. M., Rongvaux A., Pope S. D., Lopez C. B., Oberst A., Beiting D. P., Henao-Mejia J., Brodsky I. E., Activity of uncleaved caspase-8 controls anti-bacterial immune defense and TLR-induced cytokine production independent of cell death. PLOS Pathog. 12, e1005910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westermark L., Fahlgren A., Fallman M., Yersinia pseudotuberculosis efficiently escapes polymorphonuclear neutrophils during early infection. Infect. Immun. 82, 1181–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Linkermann A., Brasen J. H., Darding M., Jin M. K., Sanz A. B., Heller J. O., De Zen F., Weinlich R., Ortiz A., Walczak H., Weinberg J. M., Green D. R., Kunzendorf U., Krautwald S., Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 110, 12024–12029 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luedde M., Lutz M., Carter N., Sosna J., Jacoby C., Vucur M., Gautheron J., Roderburg C., Borg N., Reisinger F., Hippe H. J., Linkermann A., Wolf M. J., Rose-John S., Lullmann-Rauch R., Adam D., Flogel U., Heikenwalder M., Luedde T., Frey N., RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 103, 206–216 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Newton K., Dugger D. L., Maltzman A., Greve J. M., Hedehus M., Martin-McNulty B., Carano R. A., Cao T. C., van Bruggen N., Bernstein L., Lee W. P., Wu X., DeVoss J., Zhang J., Jeet S., Peng I., McKenzie B. S., Roose-Girma M., Caplazi P., Diehl L., Webster J. D., Vucic D., RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc3465/DC1