

In-situ liquid TEM results reveal the co-existence of classical and non-classical nucleation paths during HA biomineralization.
Abstract
To treat impairments in hard tissues or overcome pathological calcification in soft tissues, a detailed understanding of mineralization pathways of calcium phosphate materials is needed. Here, we report a detailed mechanistic study of hydroxyapatite (HA) mineralization pathways in an artificial saliva solution via in situ liquid cell transmission electron microscopy (TEM). It is found that the mineralization of HA starts by forming ion-rich and ion-poor solutions in the saliva solution, followed by coexistence of the classical and nonclassical nucleation processes. For the nonclassical path, amorphous calcium phosphate (ACP) functions as the substrate for HA nucleation on the ACP surface, while the classical path features direct HA nucleation from the solution. The growth of HA crystals on the surface of ACP is accompanied by the ACP dissolution process. The discoveries reported in this work are important to understand the physiological and pathological formation of HA minerals, as well as to engineer the biomineralization process for bone healing and hard tissue repairs.
INTRODUCTION
Bioapatite crystals in the form of hydroxyapatite (HA) (1) are an important component of mammalian hard tissues. As an example, the tooth enamel has the highest concentration [~96 weight % (wt %)] of HA minerals (2), and bone tissue is made of about 70 wt % mineral (with nano-sized HA being the majority phase) (1). Dysfunctional or aberrant mineralization leads to a variety of medical problems, such as caries in teeth (2) and osteoporosis (3) related to the demineralization of HA and arteriosclerosis (4) related to the nondesired mineralization of HA. Therefore, understanding the mineralization pathway of HA has been the focus of biostudies (5). Recently, Wang and collaborators (6) used in situ liquid cell transmission electron microscopy (TEM) to observe the formation of calcium phosphate (CaP) crystal in simulated body fluid (SBF) solution. The biomineralization of the CaP crystal in SBF was confirmed to be based on particle attachment mechanism. Besides the classical direct nucleation of HA during the mineralization process (7), the nonclassical path has been studied for many years, where amorphous calcium phosphate (ACP) has been considered as a vital precursor (8, 9). The evidence for ACP being an intermediate phase during biomineralization of HA has been reported previously based on various characterization techniques (9). Among the various analyzing tools, TEM has the unique advantages of simultaneously revealing the morphology and phase information at the nanoscale. Using TEM, Pan et al. (10) reported that the ACP-to-HA transformation occurs at the surface of ACP particles. During the transformation process, the HA crystals embedded/adhered onto the ACP particles and showed no sign of crystal rotation or relocation. Onuma and Ito (11) proposed a cluster growth mechanism dominating the ACP-to-HA transformation, in which the Posner clusters (8) are the basic growth units for ACP and HA. Later on, Habraken et al. (7) reported a different mechanism that nano-sized calcium triphosphate complexes acted as prenucleation clusters to form ACP and HA crystals. In this pathway, various soluble ion–association complexes were formed by adding ions from solution and then progressed stepwise toward the composition of the ultimate calcium phosphate. Other researchers also observed that acicular HA nanocrystals grew from the interfaces between the aggregated ACP particles during the initial stage of the ACP-to-HA phase transformation (9). Time-resolved static light scattering study (12) revealed the heterogeneous ACP-to-HA transformation by rearrangement of the internal structure of ACP in protein medium solution, which is sharply different to the classical direct nucleation of HA in a simple system. Besides, cryogenic TEM (cryo-TEM) has been widely used to study the mineralization process by freezing the reaction solution at different reaction stages. Using cryo-TEM combined with cryo–electron tomography, Dey et al. (13) found that the HA mineralization process initiates on the arachidic acid monolayer in an SBF environment. They further proposed five stages for HA mineralization, namely, (i) loosely aggregated prenucleation clusters in solution, (ii) loosely aggregated prenucleation clusters on monolayer surface, (iii) densely aggregated prenucleation cluster on monolayer surface, (iv) ACP on the monolayer surface, and (v) HA on monolayer surface. However, the ACP-to-HA phase transformation details are still missing.
Despite the studies mentioned above, the pathways of HA mineralization, particularly at the early stages of nucleation and growth, are not clear (5, 9, 10, 14). It is essential to understand the overall landscape of the HA mineralization process by a technique that allows time-resolved studies of mineralization process at nanoscale. Recently, imaging liquid reactions using TEM has been reported as a powerful tool in the exploration of materials science that had been previously hidden by the thick liquid mass. The liquid cell TEM configuration is capable of real-time monitoring chemical reactions such as precipitation (15), nucleation and growth (16), and surface deposition (17) processes. Here, using this technique, we report high-resolution real-time imaging of the HA mineralization process within an artificial saliva solution. We observed that before the nucleation stage, the liquid solution undergoes liquid-liquid phase separation featuring the formation of neighboring ion-poor liquid phase and ion-rich liquid phase. ACP nanoparticles and crystalline HA nanocrystals are observed in the ion-rich liquid phase simultaneously, confirming the coexistence of classical direct nucleation and nonclassical ACP-assisted nucleation pathways for HA mineralization. For the nonclassical nucleation, HA crystals nucleate on the surface of the ACP substrate and then grow by agglomeration and coalescence mechanism. Our simulation results also confirm the thermodynamic possibility of the coexistence of classical and nonclassical nucleation during the HA mineralization.
RESULTS
Figure 1 (A and B) shows the schematic of the microfluidic liquid cell TEM experiment. Artificial saliva, a supersaturation solution of HA [saturation indices (SI) = 9.6] containing all necessary inorganic ions (table S1) present in human oral saliva, is used to induce the mineralization of HA. The existence of liquid during TEM imaging is confirmed using electron energy loss spectroscopy (EELS) analyses before and after flowing liquid into the TEM chamber, the results of which are shown in Fig. 1 (C and D). It has been accepted that a higher intensity of the plasma peak (versus the zero loss peak) during EELS analysis generally indicates a longer distance for the electron to transmit through the imaging area (18). The relative thickness, denoted by t/λ, where t is the sample thickness and λ is the inelastic mean free path of electrons, increases from 2.1 to 4.6 as deduced based on the number of unscattered electrons contributing to the zero-loss peak (versus the total number of incident electrons) (18). The fact that the relative intensity of the plasma peak is much higher when liquid solution is flowing into the chamber confirms the presence of liquid solution in the imaging region (19). Figure 1 (E to G) captures the very early stage of the prenucleation featuring liquid-liquid phase separation in artificial saliva. This separation is evident by observing the bright-contrast and dark-contrast zones within the saliva solution. The bright-contrast zones are defined as an ion-poor liquid phase, while the dark-contrast zones are ion-rich liquid phase. Such liquid-liquid phase separation has also been reported in other systems such as CaCO3 (20), Ag (21), and vanillin (22). Considering that artificial saliva is a calcium phosphate–supersaturated solution (20), we expect the dark zones to be associated with higher concentration of Ca2+ and PO43− ions and the bright zones to be associated with lower ionic concentration.
Fig. 1. The evidence of liquid-liquid phase separation within artificial saliva solution by liquid cell TEM imaging.
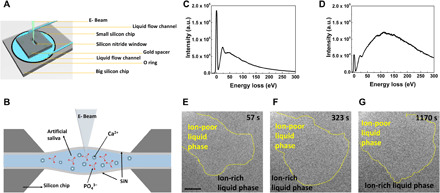
(A and B) Schematic of the in situ liquid cell, where (A) is the top view and (B) is the cross-sectional view. EELS data in (C) are from a dry silicon nitride cell and in (D) are collected from a liquid-filled cell. a.u., arbitrary units. (E to G) Time sequential TEM snapshots of the liquid-liquid separation process. The yellow dashed line marks the boundary between the ion-rich liquid phase and the ion-poor liquid phase. Scale bars, 500 nm.
Following the liquid-liquid phase separation, the formation of some nanoparticles in the ion-rich liquid phase was observed as shown in Fig. 2 (A to D). The nanoparticles precipitate only in the ion-rich liquid phase (Fig. 2, A and B) and grow to around 100 nm with time (Fig. 2A at t = 5382 s). These spherical shaped nanoparticles are confirmed to be ACP based on the selected-area electron diffraction (SAED) pattern (fig. S1B) and analytical characterization [EELS and EDS (energy dispersive x-ray spectroscopy) in fig. S2]. The ACP nanoparticles are observed to grow by two different mechanisms. For the ACP particles indicated by the white arrows, the growth appears to be dominated by the association of Posner cluster (8, 13), which is a basic unit of ACP. Other ACP nanoparticles identified by the red arrows grow through agglomeration and coalescence mechanism. This in situ observation confirms the previous discussions regarding the possibility of ACP agglomeration (7, 23). In addition to the ACP nanoparticles (Fig. 2D), there are some very small nanoparticles (~5 nm) that do not grow over time (Fig. 2, B and C). These small nanoparticles typically form in the regions of ion-rich solution but far from ACP nanoparticles. The high-resolution TEM image and corresponding fast Fourier transform (FFT) image in Fig. 2 (E and F) indicate that these small nanoparticles are crystalline HA. The direct HA nucleation from the solution as observed confirms the existence of the classical HA nucleation mechanism in this work. The observation that the precipitation of both ACP and HA nanocrystals happens within the ion-rich liquid phase (21) indicates that liquid-liquid phase separation is the first step for their nucleation (20, 22). In addition, the nucleation and growth of both ACP and HA phases are thermodynamically favorable (24).
Fig. 2. The simultaneous homogeneous nucleation of HA and ACP within the ion-rich liquid phase.
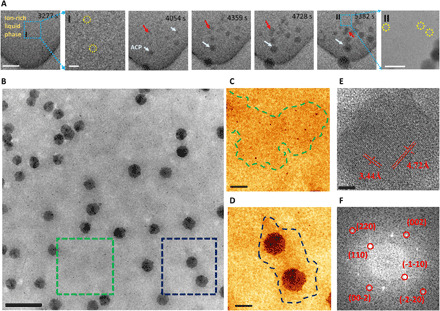
(A) Time sequential TEM images of ACP nanoparticles formed within the ion-rich liquid phase. Scale bars, 500 nm. I: Zoomed-in TEM image of the blue boxed area in (A) at 3277 s. Scale bar, 200 nm. II: Zoomed-in TEM image of the blue boxed area in (A) at 5382 s. Scale bar, 200 nm. (B) TEM images of the ion-rich liquid phase and ion-poor liquid phase. The red arrows indicate ACP particle growth by coalescence. The HA crystals are marked by yellow circles in zoomed-in images I and II. (C) Zoomed-in TEM image of the green boxed area showing some HA nuclei within the ion-rich liquid phase. (D) Zoomed-in TEM images of the blue boxed area in (B) showing two ACP nanoparticles in ion-rich liquid phase zone. The blue dashed line marks the interface between the ion-rich liquid phase and the ion-poor liquid phase. Scale bars, 500 nm (B) and 100 nm (C and D). (E) High-resolution TEM image of the HA nanoparticles. Scale bar, 5 nm. (F) FFT image of (E).
Figure 3 shows the evolution of ACP particles after their nucleation. Figure 3A shows a single ACP nanoparticle with a diameter of ~50 nm. Figure 3 (B to E) shows different stages of ACP evolution featuring the gradual heterogeneous precipitation of much smaller nanograins on the ACP particle. These nanograins are confirmed to be HA nuclei based on the SAED analysis as shown in fig. S1C, indicating a different pathway for the formation of HA that follows the intermediate ACP phase (hence, nonclassical HA nucleation process). The HA nucleation appears to have a visible image contrast in the bright-field TEM due to their higher density [3.08 g/cm3, ICDD (International Centre for Diffraction Data) file card number: 9-432] in comparison to amorphous ACP nanoparticles (density ~1.75 g/cm3) (5). The size of the ACP nanoparticles varies from 50 to 200 nm, which indicates that ACP nanoparticles grow with time until the size reaches to a certain value before HA nucleation starts. No visible coalescence or attachment of ACP nanoparticle is observed, and its growth appears to follow the classical LaMer mechanism where ionomer attachment is the dominant process (25). It is also notable that the ACP nanoparticle does not grow beyond 300 nm (same for other ACP nanoparticles), indicating that the surface energy of ACP nanoparticles offsets their volumetric Gibbs free energy (26). We can also conclude that the driving force for ACP being a template for heterogeneous HA nucleation is the relatively low activation energy required for HA nucleation on the ACP surface. The size of HAs (3 to 6 nm) heterogeneously nucleating on ACP is much smaller than the size of HAs (9 to 12 nm) directly nucleating within the ion-rich solution, confirming that the heterogeneous nonclassical nucleation is the favorable mechanism for HA nucleation. Figure 3E shows that the ACP nanoparticles deviate from a spherical shape and exhibit irregular morphology as a consequence of heterogeneous HA nucleation on the surface of ACP. With more HA nuclei appearing, the ACP substrate totally dissolves as further confirmed in Fig. 3 (F and G). It is evident that ACP dissolution happens simultaneously with the increase of the number of HA nuclei on ACP substrates. The ACP dissolution indicates that ACP is not thermodynamically stable in comparison to HA (27). It would be interesting to further study the mechanisms regarding the heterogeneous nucleation of HA on ACP while ACP is dissolved.
Fig. 3. TEM images of ACP and HA particles at different stages of ACP-to-HA transformation process.
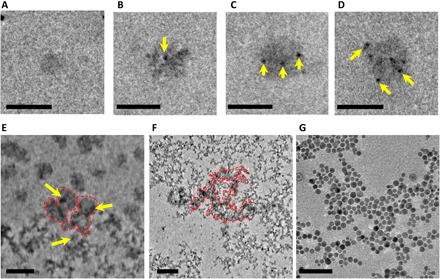
(A) Formation of small ACP nanoparticle with a diameter of ~50 nm. (B) First HA nucleus (yellow arrow) forms on ACP nanoparticle. (C) Three HA nuclei (yellow arrows) form on ACP nanoparticle. (D) Six HA nuclei form on ACP nanoparticle. Scale bars, 100 nm (A to D). (E) Part of ACP (red dashed line) has transformed to HA (yellow arrow). (F) Partial dissolution of ACP nanoparticles, where the traces of undissolved ACPs are still visible within the red dashed line region. Scale bars, 200 nm (E and F). (G) Full transition of ACPs to HA. Scale bars, 50 nm (G).
The formation of ACP from artificial saliva solution can also be confirmed by cryo-TEM results in figs. S5 and S6. To understand the beam effect on the nucleation process, cryo-TEM tests at different time and pH values were carried out. As shown in figs. S5 and S6, at different reaction time (freshly prepared solution and 24-hour aged solution) and pH values, the ACP and HA particles still coexist in the solution. The cryo-TEM result can thus exclude the possibility that the mineralization process could be notably affected by electron beam.
The growth of HA crystals at the expense of the ACP substrate is further explored in Fig. 4. We observe that such transition can have different pathways depending on the presence of hydrated solution. Figure 4A shows an individual ACP nanoparticle (~100 nm) that transforms to several HA nanoparticles (size, ~8 nm) without coalescence of HA nanoparticles. The growth of HA crystals is accompanied with the shrinking of the ACP substrate, implying that the HA crystals grow via the ACP dissolution mechanism. In BF (bright-field)–TEM mode, the crystalline HA phase appears darker than the ACP amorphous substrate with a gray (brighter) contrast. It is well known that ACP is composed of Posner cluster and 15 to 20% of water (23). Referring to the similar chemical formula of ACP [Ca9(PO4)6] and HA [Ca10(PO4)6(OH)2], one can conclude that the ionic species (such as Ca2+ and PO43−) necessary for the nucleation and growth of crystalline HA can be directly supplied from the ACP substrate at the expense of ACP mass/volume, which is observed in our work here (Fig. 4A). During the nucleation process, because the bonding effect of water is expected to be relatively weak in ACP, the dissolution of ACP happens at the ACP/solution interface, where the hexagonal packing of Posner clusters can form long-range ordered structures to generate HA nuclei (11). The locally supersaturated solution generated from dissolution of ACP is gradually dried during HA nucleation and growth. The dehydration drags laterally scattered HA nuclei to self-assemble (Fig. 4A at 108 s).
Fig. 4. Time sequential images of single ACP nanoparticle transforming to HA.
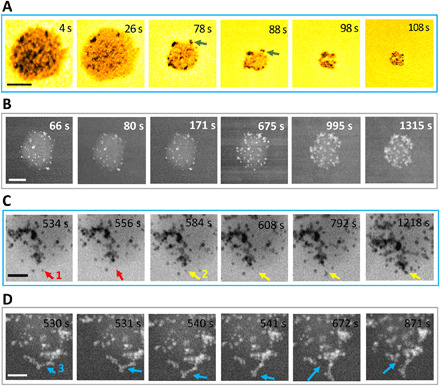
(A) TEM images showing the nucleation of HA with the dissolution of ACP in artificial saliva. Green arrows indicate that the HA crystals are attached to the surface of the ACP substrate. (B) HAADF-STEM images showing the heterogeneous nucleation of HA on the ACP substrate and the growth of HA by agglomeration and coalescence. Scale bars, 100 nm (A and B). (C) High-magnification BF-STEM images showing HA coalescence on the surface of an ACP nanoparticle. The red and yellow arrows point to HA nanoparticles that move to different locations to coalescence. Scale bar, 50 nm. (D) HAADF-STEM images show that HA nanoparticles coalescence and detach from the ACP surface. Scale bar, 50 nm. The blue arrow (location 3) indicates the position where the HA detachment happens.
Figure 4B shows another pathway for ACP-to-HA transformation, where an ACP nanoparticle is immersed in saliva solution (no dehydration). This nanoparticle is imaged using high-angle annual dark-field (HAADF) scanning TEM (STEM) mode that is sensitive to the mass density of the materials through power law equation I ~ Zα, in which I is the intensity and Z is the atomic number. The exponent α is smaller than 2 and in the range of 1.6 to 1.9 for most cases (28). In such dark-field mode, the ACP and HA nanoparticles appear with a brighter contrast compared to the surrounding liquid solution, and HA nuclei are even brighter than ACP (mean atomic number, ~15) due to the denser mass and higher Z in crystalline HA (mean atomic number, ~18). From 60 to 675 s, it is evident that the number of HA nuclei on ACP increases; from 675 to 1315 s, HA coalescence is the dominant mechanism, while the ACP substrate dissolves. Figure 4C shows the coalescence of HA nuclei on ACP with more details (movie S3). The HA nuclei indicated by the red arrow first appear at 534 s and then jump to the position labeled by red arrow in 556 s (movie S3). The yellow arrow showcases the formation of an HA particle and its attachment with large agglomerate of HA nanoparticles on the surface of the ACP substrate. The HA nucleus indicated by yellow arrow at 584-s frame is joined by three more HA nanoparticles as shown in 792-s frame. Then, at 1218 s, these HA nanoparticles attach to a large HA agglomerate. Therefore, the HA agglomeration happens when the population of HA nanoparticles covering the ACP surface increases to a certain level that the distance between adjacent HA nanoparticles reduces significantly. This enables particle-particle attraction via possible driving forces such as van der Waals (vdW) interactions, electrostatic force, and the cohesive energy release by the assemble of crystal lattice (25).
We believe that the differences in ACP dissolution rates could account for the different phenomena observed in Fig. 4 (A to C). The shrinkage (measured using diameter decrease) rate of the ACP in Fig. 4A (0.88 nm/s from 26 to 88 s) is almost 20 times higher than that of the ACP nanoparticle in Fig. 4B (0.046 nm/s). Because we already demonstrated in Fig. 3 that it is the ACP substrate that supplies the necessary monomer to support the continuous growth of HA crystals, the amount of sufficient ACP mass during HA growth is the key factor determining the growth kinetics. In Fig. 4A where the ACP substrate is quickly dissolved to be barely visible, the loss of direct ACP-HA contact prevents any further supply of monomers from supporting the coalescence of HA. In Fig. 4B, however, because the dissolution of ACP is quite slow compared to that in Fig. 4A, the coalescence of HA is thus sufficiently supported by available monomers supplied directly from the ACP substrate. The detachment of large HA clusters from the ACP surface shown in Fig. 4 (C and D) confirms that the grown HA crystals are not tightly bonded to the ACP substrate. The similar detaching behavior of large HA clusters results in loss of spherical morphology in ACPs, as previously observed in Fig. 3 (E and F). Therefore, these observations further highlight the critical role of the ACP substrate in supporting the growth of HA.
The direct nucleation and growth process of HA in artificial saliva solution points to the classical pathway (7) for the HA mineralization process (Fig. 5). The HAADF-STEM images taken from t = 8 s to t = 34 s show the nucleation of HA crystals at the vicinity of an HA cluster and their subsequent attachment to the cluster. At 8 s, there is a cluster labeled 0, which is an assembly of six HA nuclei. At t = 16 s, there is a new HA nucleus (1, yellow arrow) appearing near cluster 0, which then attaches to the cluster after 2 s. At 20 s, another HA nucleus (2, green arrow) forms, which is not directly attached to the large HA cluster but jumps around the cluster, changes the orientation, and finally makes its attachment with the cluster at t = 34 s. The growth of HA not only includes the attachment of individual nucleus to HA cluster but also exhibits cluster-to-cluster agglomeration (t = 64 to 968 s; Fig. 5). The HAADF-STEM images taken from t = 64 to 112 s showcase an example where cluster 3 marked with blue arrow attaches to another cluster nearby. Same phenomenon happens for another cluster marked as cluster 5, which initially grows by the attachment of single HA nucleus (nucleus 4, t = 123 to 136 s, red arrow) and is then followed by its attachment to an even larger HA cluster (t = 145 to 968 s).
Fig. 5. Agglomeration and coalescence of HA crystals.

Time sequential images directly show HA nucleation, agglomeration, and coalescence process. Nucleus 1 labeled by yellow arrow represents one HA nucleus attaching to a larger HA cluster. Nucleus 2 labeled by green arrow represents one HA nucleus attaching to cluster and jumping from one position to another position after its nucleation. Cluster 3 labeled by blue arrow represents one cluster attaching to another cluster. Arrows 4 and 5 show that new nuclei first attach to a small cluster and then the cluster merges to a larger cluster. Scale bar, 100 nm.
Overall, the dominant growth mechanism for HA crystals, namely, the agglomeration and coalescence mechanism, is independent of HA nucleation pathways, i.e., regardless of the classical or nonclassical nucleation. We did not observe Oswald ripening (25), island growth (29), intraparticle growth (30), or other growth mechanisms reported previously. We believe that the main driving force for the agglomeration of HA crystals is the minimization of surface energy. The total Gibbs free energy of the system (∆G) is related to the interfacial energy (γ) and the radius of the nucleus (r)
where is the Gibbs free energy of the bulk HA per unit volume (31). For the HA crystal in solution, γSL represents the solid-water interfacial energy. For the aggregated HA crystal, γSv represents the solid-vacuum interfacial energy. On the basis of literature, for most of the HA crystal planes such as (100), (110), and (004) facets, the value of γSL is a positive value, while the value for (001) is negative (31). Therefore, HA nuclei have a higher tendency to directly attach with each other when they approach each other along their high-energy facets such as (100), (110), and (004) (31). Such attachments are favorable to lower the free energy of the system. For the situation when the HA nuclei approach each other along their low-energy facets such as (001), they might not directly attach but tend to jump/rotate and adjust their orientation to allow the contact of their high-energy facets. Therefore, this could reasonably explain the observed jump and move of HA nuclei in Fig. 5 (green arrow).
To better understand the nucleation pathways of HA crystals, we used density functional theory (DFT) and the molecular dynamics (MD) calculation to understand the energy landscape (Fig. 6). In the DFT calculations, the Posner cluster is considered as the basic unit of ACP, whose formation energy can be used to represent the energy landscape describing the formation of ACP nanoparticle. The size of the Posner cluster is around 0.9 nm (8), and the hydration of Posner clusters leads to the formation of ACP nanoparticle. According to our calculations, the reaction energy to form a Posner cluster Ca9(PO4)6 (or ACP) from three Ca3(PO4)2 monomer (red curve, Fig. 6A) is −11.8 eV, which is much lower than that of the direct attachment of Ca3(PO4)2 monomer to HA crystal (−8.3 eV, black curve, Fig. 6A). Because the energy to form ACP is lower than the energy to form HA, the reaction to form the Posner cluster (or ACP) is thus thermodynamically more favorable than the attachment of monomers to form HA, indicating that forming intermediate ACP phase is an energetically favorable pathway toward HA nucleation and that the ACP-assisted HA nucleation could be the core mechanism dominating the HA mineralization process. In addition, because the reaction energy difference between ACP and HA is 3.7 eV and both of these reaction energies are negative values, the formations of ACP and HA are both likely to happen. After ACP formation in path II (Fig. 6A), the binding of a Posner cluster to the HA surface is −7.5 eV, which is less favorable than the bonding of monomer to HA surface in path I (−8.3 eV). This indicates that for the following growth stage after the ACP formation, the attachment of monomers from solution to the HA surface is probably an energetically favorable path. This could explain the observed coalescence of HA crystals on the ACP because there are enough monomers in solution to help the growth of HA crystal. In short, the DFT confirms that although both paths are possible, the ACP-assisted HA nucleation is more energetically favorable compared to the nucleation of HA directly from the solution.
Fig. 6. Simulations of HA nucleation process.
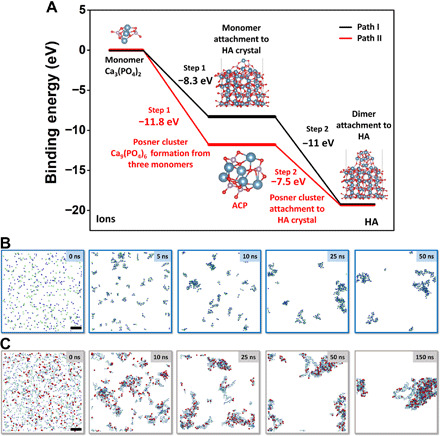
(A) DFT simulations of the binding energy needed to form HA. (B and C) MD calculation of ACP and HA formation in solution. (A) Path I: Black curve denotes the nucleation and growth pathway by directly adding Ca3(PO4)2 to HA crystal, which features two steps, with step 1 being the attachment of one Ca3(PO4)2 to HA surface and step 2 being the subsequent attachment of another two Ca3(PO4)2 monomers to the surface. Path II: Red curve denotes the mineralization process with ACP as an intermediate phase, which features two steps, with step 1 being the formation of Posner cluster Ca9(PO4)6 from three Ca3(PO4)2 as the main component of ACP and step 2 being the attachment of ACP to HA crystal surface. (B) Formation of ACP from 300 Ca2+ and 200 PO43− ions; the frames correspond to 0-, 2-, 5-, 10-, 25-, and 50-ns simulation time, respectively. Ca2+, dark blue spheres; PO43−, pale green tetrahedra. (C) Formation of HA from 1000 Ca2+, 600 PO43−, and 200 OH− ions in a 15 nm × 15 nm × 15 nm water box. The frames correspond to 0-, 5-, 10-, 25-, 25-, 50-, and 150-ns simulation time, respectively. This ion fraction yields pure HA, Ca5(OH)(PO4)3. Ca2+, dark blue spheres; PO43−, pale green tetrahedra; OH−, red bispherical rods. To emphasize the distribution of OH units (related to the HA-rich phase), the size of calcium and phosphate ions was scaled down to 60%. Scale bars, 2 nm (B and C).
To better understand the whole process, several MD calculations were carried out. On the basis of the MD results in Fig. 6 (B and C), after liquid-liquid separation to form ion-rich and ion-poor areas, the nucleation of ACP and HA can be generated in one system, which is supportive to the coexistence of the classical and nonclassical nucleation as we experimentally observed. Figure 6 (B and C) demonstrates the formation of amorphous ACP and HA clusters, respectively. In both cases, the mixtures of ions initially started to separate into ion-rich and ion-poor phases (21), followed by their aggregation into clusters and their gradual growth. Initially, oppositely charged ions started to attract each other (t = 0 to 10 ns). Next, coalescence of ion-rich phases into clusters could be seen (t = 10 ns). Eventually, small clusters aggregated to form larger amorphous assemblies of ACP or HA (>50 ns).
The main difference between ACP and HA structures lies in the presence of hydroxy units within the latter. Moreover, the time-dependent distribution of OH− groups across the clusters was inhomogeneous in space, which means that hydroxy-rich and hydroxy-poor regions could be noticed, e.g., in Fig. 6C at t = 25 to 50 ns. This observation supports the experimental results shown in Fig. 4, where small HA clusters attached to bulky ACP nanoparticles regrew into HA crystals by consuming ACP. In our simulations, hydroxy-rich phases represent HA clusters assembled on hydroxide-poor ACP. We could see that these HA regions regrew into ACP regions.
Then, we run the MD simulation of this system to obtain a longer trajectory, which would allow us to analyze the radial distribution function for hydroxide units at different simulation times. We found that the system initially favored the self-assembly of ions into hydroxy-rich and hydroxy-poor regions (fig. S7A at t = 5 to 50 ns). This could be understood as a competitive formation of HA and ACP. Eventually, the distribution of OH− units became gradually more and more homogeneous in space. This homogenization process is further analyzed in fig. S7 (B and C), which shows the radial distribution function of hydroxy group within the aggregate. To analyze the results, we used radial pair distribution function [g(r)] VMD toolkit. Parameters were set with delta(r) = 2 Å, and rmax = 35 Å. These numbers were adjusted manually; first, we did not notice a significant number of counts at r > 35 Å. Moreover, the total number of hydroxy units (21) was too scanty to obtain a smooth curve without averaging the distances; therefore, delta(r) below 2 Å gave out unclear analysis. This analysis allows us to notify that, initially, freshly aggregated ions formed hydroxy-rich and hydroxy-poor phases, where most of the hydroxy groups were close to each other. Soon after the ion aggregation (t = 5 ns), OH− groups were localized within ~3 Å from each other, which emphasized a highly localized character of HA occurrence. With time, however, the range of various distances increased, which underlined the more homogeneous distribution of hydroxy units. After ~300 ns, hydroxy groups contributed to the cluster structure in a quite uniform fashion. This observation allows us to conclude that HA clusters, represented by hydroxy-rich phases, can migrate and rearrange within the ACP cluster. Therefore, HA crystals are likely to spread out within metastable ACP particles and get converted into HA product. We can generalize these observations and state that if the number of hydroxide ions is insufficient to give out a pure HA product, HA-rich phases are likely to assemble evenly within the aggregate. Because of the presence of water molecules in the hydrated ACP structure, the conversion of ACP into HA was highly favored (see the next paragraph). To conclude, the assumption that HA seeds can consume the ACP substrate should be justified.
The MD simulation of the ion exchange between ACP and HA (fig. S8) was carried out for 1-μs time span. After analyzing this long trajectory, we found the attachment of up to 15 hydroxy groups to the cluster and a release of H2PO4− ion to the solution. The high affinity of hydroxide to calcium ions can be explained twofold: (i) by smaller dimensions of OH− anion compared to phosphate but the same overall charge and (ii) by higher local charge on hydroxyl oxygen atom (−1.32 versus −0.924). Figure S8A shows the monovalent phosphate dissociation from the HA cluster. Although only one of the three H2PO4− ions was observed to be released, it proves the concept that smaller hydroxy anions are able to replace bulky phosphate anion from the ACP cluster and to form a stable, hydroxy-rich HA as thermodynamically stable product. This observation is of a particular interest because although the concentration of OH− ions in solution is rather scanty, the growth of crystalline HA by ripening the amorphous ACP aggregates along with the conversion of hydrated water molecules is still favored. Figure S8B demonstrates the release of H2PO4− as well by providing the distance of the ion from the cluster center. Thus, at 750 ns, phosphate is still in the cluster with a constant distance of ~7.7 Å, but after 910 ns, it fluctuates between 15 and 40 Å. To summarize, the MD calculation supports the theory that after forming the ion-rich and ion-poor areas, the nucleation of HA and ACP can both happen. The release of H2PO4− ions can support the dissolution/reprecipitation mechanism.
DISCUSSION
The ACP-to-HA transformation has been reported to exhibit different pathways such as surface nucleation (14), dissolution/reprecipitation (8, 32, 33), step growth (34), and internal structure rearrangement (11, 12). Our findings discussed above support the pathway featuring ACP dissolution–HA nucleation (8, 32, 33) at the surface of ACP rather than the internal structure rearrangement within ACPs. By tracking the size of particles, it is evident that the change of particle size changed over time, which is in contrast with the proposed internal structure rearrangement mechanism (11, 12). The dissolution/reprecipitation mechanism is supported by two observations. First, the surface of ACP nanoparticles is seen to be the preferred sites for HA nucleation and growth, as shown in Fig. 4 (A and C) highlighted by the green and yellow arrows. Because the TEM image is a two-dimensional (2D) projection of 3D features, the HA crystals located along the periphery of ACP nanoparticles can be used to support our conclusion. Second, the observed jump of HA crystals and the change of their orientations on the ACP substrate indicate that the surrounding environment of HA crystals has limited physical confinement on the movement of HA crystals. It is worth mentioning that our findings are in disagreement with some ex situ studies showing no sign of ACP dissolution during HA nucleation (14). Most of these studies, based on their conventional TEM images, stated that there is no sign of ACP shrinkage during HA growth. However, we need to emphasize that ex situ TEM could not track the size changing details of the ACP substrate and the HA crystals grown on it. The dynamic details of the size changing of the ACP substrate versus HA crystals revealed in our in situ liquid TEM study reveal the growth of HA at the expense of the ACP substrate, which is thus expected to call for more attention in clarifying the role of ACP during HA mineralization.
To summarize, Fig. 7 shows the overall pathways of HA nucleation and growth based on our in situ observations. The whole biomineralization process has two paths. One is the classical nucleation path with four steps: ion supersaturation in solution (stage I), liquid-liquid separation forming an ion-poor liquid phase and ion-rich liquid phase (stage II), HA direct nucleation in the ion-rich liquid phase (stage III), and agglomeration and coalescence growth of HA (stage IV). The second path is the nonclassical nucleation path with ACP as the intermediate phase. This path is divided into six steps: ion supersaturation in solution (stage I), liquid-liquid separation forming ion-poor liquid phase and ion-rich liquid phase (stage II), formation of the Posner clusters that assemble to form ACP in ion-rich liquid phase (stage III), and dissolution of ACP at ACP/solution interface and multisite heterogeneous nucleation of HA on the surface of ACP (stage IV). There are two scenarios for the HA growth (stage V), which is distinguished by the dissolution rate of ACP. One scenario is HA self-assembly (stage V-a), with a high ACP dissolution rate. The other scenario is HA growth via agglomeration and coalescence (stage V-b) and delamination of large HA clusters from ACP (stage VI-b) with low ACP dissolution rate. In short, the HA mineralization process exhibits multisite HA nucleation, either directly in solution (classical nucleation) or indirectly on the surface of the ACP intermediate phase (nonclassical nucleation).
Fig. 7. Schematic representation of the HA crystallization pathways.
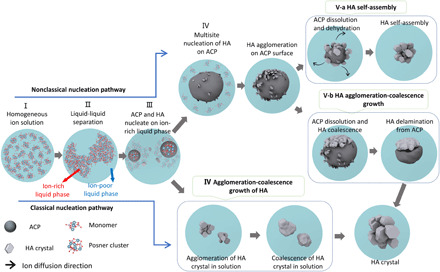
HA crystal formation can be divided into classical and nonclassical nucleation pathways. For the nonclassical nucleation pathway, there are five stages: homogeneous ions in solution (stage I), liquid-liquid separation forming ion-poor and ion-rich liquid phases (stage II), the formation of ACP and HA in ion-rich liquid phase (stage III), multisite heterogeneous nucleation of HA on the surface of ACP (stage IV), and ACP-HA phase transformation (stage V). Stage V can proceed by two different scenarios: One is HA growth only by ACP dissolution–HA reprecipitation followed by HA self-assembly (stage V-a), and the other one is the ACP dissolution–HA reprecipitation followed by HA growth via agglomeration and coalescence and then delamination from ACP (stage V-b). For the classical nucleation pathway, in stage III, the HA crystals directly nucleate from the ion-rich liquid phase. The HA crystals grow by agglomeration and coalescence (stage IV, bottom).
In conclusion, using in situ liquid TEM technique, we studied the nanoscale HA mineralization process in artificial saliva environment. The coexistence of classical and nonclassical nucleation pathways was observed. The ion-rich liquid and ion-poor liquid phase separation was detected before the HA nucleation. The nonclassical nucleation pathway features the formation of ACP as an intermediate phase during the HA mineralization process, while the classical nucleation pathway shows direct HA nucleation from the solution. For both pathways, the driving force for the growth of HA crystals is the reduction of the surface energy, which makes the growth mechanism to be agglomeration and coalescence. The growth of nonclassically nucleated HA proceeds at the surface of the ACP substrate, while ACP gradually dissolves. The dissolution rate of ACP determines the growth of HA crystals via a self-assembly mechanism or an agglomeration and coalescence mechanism. For classical HA nucleation, the growth of HA nuclei proceeds via the agglomeration and coalescence process. The findings in this work provide insights about the HA biomineralization mechanisms from the prenucleation stage up to microscale crystal formation. We clarified some ambiguities on the role of ACP, its transition to HA, and the possibilities of direct HA nucleation. The observation of both classical and nonclassical pathways indicates that more attention should be paid to the thermodynamic and kinetic parameters that can control HA biomineralization. The findings of this work should inspire further research on physiological bone formation or pathological tissue calcification mechanisms.
MATERIALS AND METHODS
Material preparation
Artificial saliva was prepared by adding potassium chloride, sodium chloride, calcium chloride dihydrate, sodium dihydrogen phosphate dihydrate, sodium sulfide nonahydrate, and sodium azide into deionized water. All the chemicals were purchased from either Fisher Scientific or Sigma-Aldrich and were used as received. Hepes (1 M; Gibco, Thermo Fisher Scientific) was used as the buffer solution to adjust the pH of artificial saliva to 6.5 to be consistent with the pH range in human saliva. The SI of the solution is 9.644 for HA (calculated by Visual MINTEQ) (10). The composition of artificial saliva and the normal range of ionic concentrations in human saliva are shown in table S1.
In situ TEM
The in situ liquid cell TEM experiment was carried out in an aberration-corrected JEOL JEM-ARM 200CF (200 kV) incorporated with the commercially available liquid TEM holder (Protochips Poseidon 500). Before assembling, the two chips were plasma-cleaned for 2 min to make the surface hydrophilic. The two–electron-beam transparent silicon nitride membranes are used to sandwich and isolate saliva solution from the vacuum environment in TEM. The microfluidic design of the liquid cell enables the flow of saliva solutions into/out of the TEM chamber. After the two chips were mounted together with their spacing controlled to be around 150 nm, artificial saliva flowed through the inner tube of the holder into the liquid cell at a rate of 300 μl/hour. All the video recordings of the subsequent reactions were taken at 30 frames/s.
The electron-beam sensitivity of the phases involved in this study should also be discussed. ACP is a very beam-sensitive material, which can decompose quickly under intense illumination. However, in our study, the silicon nitride window could protect the ACP, and the liquid flow can also dilute the electrons accumulated on ACP, which could prevent the quick decomposition of ACP. For the fully crystalized HA crystals (Figs. 2E, 3G, and 5), they are not observed to be beam sensitive. For Fig. 2, in situ liquid test, the dose rate is ~1 e/Å2 per second, which is reported as a low dose, which cannot cause rapid damage to ACP (34). The dose rate of Fig. 3 (A and B) is 17.5 to 1114 and 60 e/Å2 per second, respectively.
On the basis of reported papers, there are some radicals such as OH−, H2O2, and H* that could be generated by e-beam in irradiated solution (35). These generated radicals may cause some unwanted results. In our case, the flow mode of the liquid cell was used to minimize the electron-beam effects in the local area. The beam changes the pH of water, which may approach 4.5 under prolonged e-beam irradiation (35). However, the artificial saliva solution (pH 6.5) is a buffer solution that can adjust the pH. In addition, HA can form in a large pH range (6 to 12) (36). The formation of ACP indicated that the pH of the solution inside the liquid cell is maintained above 6 (37). Therefore, the pH change caused by e-beam will not markedly affect our result. Another way to minimize the e-beam effect in the experiment is that after observing one interesting location, the subsequent observation is carried out far away from the adjacent area. Our ACP size is consistent with that in other reports, which fall in the range of 50 to 250 nm (13), indicating that the ACP is not markedly affected by e-beam.
The silicon nitride window is an electron insulating material, which can become negatively charged after a long-time illumination by accumulate electrons. This will make the surface of silicon nitride window functional as a Langmuir monolayer with negative charge (13). This negatively charged layer helps the nucleation of HA. This may explain the fact that our reaction rate is higher than ex situ work (27). In the in situ liquid test, the nanoparticles always prefer to attach to the silicon nitride window because of the confinement effect and the attraction between nanoparticles and the surface of silicon nitride window (38). However, the confinement is not a significant concern because of the bulging of silicon nitride membrane increasing the liquid thickness to more than 1 μm (17).
Ex situ TEM
After the in situ liquid test, the silicon nitride chips are opened and then dried in the air. The dried chips are loaded on a conventional TEM holder. Ex situ TEM images and selected-area diffraction patterns were collected by JEOL JEM-3010 operated at 300 kV.
Cryo–transmission electron microscopy
The cryo-TEM is performed using Gatan cryo–transfer holder with liquid nitrogen by JEOL ARM 200 at 200 kV and JEOL 1230 TEM at 100 kV. The artificial saliva solutions with different aging time and pH are frozen using an FEI Vitrobot Mark IV plunge freezer in liquid nitrogen.
DFT calculations
The DFT calculations were calculated using Vienna Ab initio Simulation Package (VASP) (39). The geometry optimization and energy calculations were carried out using the generalized gradient approximation PBE (Perdew-Burke-Ernzerhof) functional (40) with the projector augmented wave method (41). The plane-wave basis sets up to a kinetic energy cutoff of 400 eV with a 2 × 2 × 1 K-point grid for the HA surface and Γ-point for molecular species and clusters. The convergence criterion of the total energy was set to be 1 × 10−4 eV, and all the geometries were optimized until the residual forces became less than 2 × 10−3 eV/Å.
MD calculation
The molecular structures studied were prepared in GaussView, and their atom types and charges were obtained from the CHARMM (Chemistry at Harvard Macromolecular Mechanics) force field. To minimize the influence of inert K+ counterion, the vdW radius of potassium ion was harnessed from the GROMOS (Groningen Molecular Simulation) force field calculations (42). The CHARMM general force field (43) was implemented for the bond, angle, and dihedral parameters of the ligands and solvent molecules. Nonbonding interactions between these molecules, such as a vdW attraction and a steric repulsion, were described by the Lennard-Jones (LJ) potential
| (1) |
with ε being the minimum (negative) energy of this coupling and 𝑟 being a distance where ULJ(r) has a local minimum, which are provided by the CHARMM force field. Here, the 𝑟−12 term describes the atomic repulsion, due to overlapping electron orbitals, and the 𝑟−6 term represents the vdW attractive coupling. The LJ potentials implemented in NAMD (Nanoscale Molecular Dynamics) have a cutoff distance of 1 nm. The electrostatic coupling between ions and partially charged atoms, which also belongs to nonbonding interactions, has a similar cutoff like the LJ potentials, but their long-range part is calculated by the PME method (44) in the presence of periodic boundary conditions. The MD simulations of the prepared systems were performed with NAMD (45) in an NPT (constant-temperature, constant-pressure) –ensemble at T = 298 K and P = 1 atm, using the Langevin dynamics with a damping constant of 𝛾Lang = 0.1 ps−1 and a time step of 2 fs.
ACP and HA nucleation from ions
We started with separate MD simulations of the self-assembly of ACP and HA cluster. We prepared two 15 nm × 15 nm × 15 nm water boxes with ions forming dissolved ACP and HA: Ca2+ and PO43− for ACP and Ca2+, PO43−, and OH− for HA. The systems were neutralized by using the stoichiometry of ions corresponding to their formula units, i.e., Ca3(PO4)2 for ACP and Ca5(PO4)3(OH) for HA. The ACP system was composed of 300 Ca2+ and 200 PO43− ions, which gave the concentrations equal to 0.148 M Ca2+ and 0.098 M PO43−. The HA system contained 1000 Ca2+, 600 PO43−, and 200 OH− ions, which gave the concentrations equal to 0.492 M Ca2+, 0.295 M PO43−, and 0.098 M OH−. In the experimental procedure, an artificial saliva was used as a solvent instead of pure water, but we assumed that reduced screening length by the solvated sodium, potassium, and chloride ions should not significantly influence the aggregation process in the simulation. Thus, we did not include these ions in the simulations.
Regrowth of HA from ACP substrate
To prove that HA can form by ripening of ACP, we prepared a smaller system shown in fig. S7A consisting of 210 Ca2+, 133 PO43−, and 21 OH− ions in an 8 nm × 8 nm × 8 nm water box. The concentrations of ions were equal to 0.681 M Ca2+, 0.431 M PO43−, and 0.068 M OH−. This mixture could theoretically yield an equimolar fraction of HA and ACP, i.e., half of calcium ions assembled into ACP and another half into HA.
Ion exchange simulation
Because ACP contains higher phosphate-to-calcium ratio than HA (2:3 versus 3:5), its conversion into the latter should be accompanied by a release of an excessive phosphate anion to keep the crystal or cluster neutral. As the HA formula unit contains OH− ions in its structure, this additional negative charge from hydroxy anions (during the same positive charge from calcium ions) must induce the release of phosphate ions to favorize the attraction of hydroxy groups and to keep no electrostatic charge on the final HA molecules. To check whether phosphate ions can be released, we extracted one small ACP cluster from the simulation shown in Fig. 6B. This cluster was composed of 14 Ca2+ and 9 PO43− ions, with a formula Ca14(PO4)9+. Relatively scanty charge here proves that the self-assembly of calcium and phosphate ions yields coulombically nearly neutral aggregates. Next, we harnessed this cluster to analyze whether the OH− ions can replace some of the phosphate ions, attach hydroxy groups, and finally release the phosphate anions, to maintain thus-formed HA cluster coulombically neutral.
Note that in aqueous solution at pH 6.5, free phosphate ions do not exist as a trivalent form, PO43−, but as monovalent H2PO4− and, in a smaller amount, divalent HPO42− forms. Therefore, to observe the replacement of some PO43− groups by hydroxide ions, we further corrected the system to analyze the ion exchange processes. We first converted three of PO43− anions within aforementioned Ca14(PO4)9+ cluster into H2PO4− ions to facilitate their subsequent dissociation into the phosphate form, which is stable under the aqueous experimental conditions. This step was done by attaching two hydrogen atoms to oxygen atoms, followed by forming a chemical bond and by optimizing all atom charges using CHARMM force field database. Thus-connected hydrogen atoms did not change the structure of the cluster nor interacted with other surrounding ions. Then, we added the excess of OH− ions to the water box, as well as an appropriate number of potassium counterions, which kept the system neutral. Last, the system of Ca14(PO4)6(H2PO4)37+ cluster was placed in a 4 nm × 4 nm × 4 nm water box, surrounded by randomly distributed 28 OH− and 21 K+ ions. This mixture composition can be treated as a simple example of the local conditions in the cluster soon before the conversion of ACP into HA. Although the pH 6.5 solution from the experiment was nearly neutral (pH value around 7, and thus, the concentration of pure OH− ions was negligible), we can consider that locally the contribution of hydroxide is significant. This can be explained by the fact that ACP contains water molecules hydrated by the cluster, in sharp contrast to HA. Therefore, these molecules can react with trivalent phosphate to yield di- and monovalent HPO42− and H2PO4− forms according to the reactions
| (2) |
| (3) |
The above reactions can be the reason for the presence of hydroxide ions around the cluster, despite a relatively low pH value of the reaction mixture. We justify this assumption of high local OH− anion concentration by the fact that in the final, stable HA structure, there are OH− ions assembled around calcium cations. Hence, our system is a good representation of the last step of the ACP to HA conversion before the phosphate ions are released.
Supplementary Material
Acknowledgments
Funding: We acknowledge the Research Open Access Publishing (ROAAP) Fund of the University of Illinois at Chicago for financial support towards the open access publishing fee for this article. This project was not directly funded. The efforts of R.S.Y. and K.H. were supported by the National Science Foundation (DMR award no. 1710049). T.S.’s efforts were supported by NSF CBET award no. 1803693. This work made use of instruments in the Electron Microscopy Service (Research Resources Center, UIC). This work made use of the JEOL JEM-ARM200CF in the Electron Microscopy Core of UIC’s Research Resources Center and the JEOL-JEM-ARM200CF in the NUANCE of Northwestern University. The acquisition of this instrument was supported by an MRI-R2 grant from the National Science Foundation (DMR-0959470). The cryo-TEM was done in the NUANCE Center of Northwestern University. The DFT calculations were performed using the computing resources provided by the Laboratory Computing Resource Center (LCRC) at Argonne National Laboratory. Y.Y. acknowledges funding from the Argonne National Laboratory–University of Illinois at Chicago (subcontract no. 4J-30361). A portion of this work was conducted at Argonne National Laboratory. Argonne National Laboratory is operated for DOE Office of Science by UChicago Argonne, LLC, under contract number DE-AC02-06CH11357. Author contributions: K.H. and all corresponding authors conceived the idea and discussed the detailed experimental design. K.H. ran the in situ liquid TEM experiments. K.H., A.N., Y.Y., and B.S. performed the ex situ TEM experiments and assisted with the in situ TEM experiments. C.L. ran the DFT calculation. M.S. and P.K. contributed to the MD simulation. All authors discussed the results and contributed to the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eaaz7524/DC1
REFERENCES AND NOTES
- 1.Palmer L. C., Newcomb C. J., Kaltz S. R., Spoerke E. D., Stupp S. I., Biomimetic systems for hydroxyapatite mineralization inspired by bone and enamel. Chem. Rev. 108, 4754–4783 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y.-R., Du W., Zhou X.-D., Yu H.-Y., Review of research on the mechanical properties of the human tooth. Int. J. Oral Sci. 6, 61–69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanis J. A., Melton L. J. III, Christiansen C., Johnston C. C., Khaltaev N., The diagnosis of osteoporosis. J. Bone Miner. Res. 9, 1137–1141 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Demer L. L., Tintut Y., Mineral exploration: Search for the mechanism of vascular calcification and beyond. Arterioscler. Thromb. Vasc. Biol. 23, 1739–1743 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Combes C., Rey C., Amorphous calcium phosphates: Synthesis, properties and uses in biomaterials. Acta Biomater. 6, 3362–3378 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Wang X., Yang J., Andrei C. M., Soleymani L., Grandfield K., Biomineralization of calcium phosphate revealed by in situ liquid-phase electron microscopy. Commun. Chem. 1, 80 (2018). [Google Scholar]
- 7.Habraken W. J., Tao J., Brylka L. J., Friedrich H., Bertinetti L., Schenk A. S., Verch A., Dmitrovic V., Bomans P. H., Frederik P. M., Laven J., van der Schoot P., Aichmayer B., de With G., De Yoreo J. J., Sommerdijk N. A. J. M., Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Commun. 4, 1507 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Posner A. S., Betts F., Synthetic amorphous calcium phosphate and its relation to bone mineral structure. Acc. Chem. Res. 8, 273–281 (1975). [Google Scholar]
- 9.Kim S., Ryu H.-S., Shin H., Jung H. S., Hong K. S., In situ observation of hydroxyapatite nanocrystal formation from amorphous calcium phosphate in calcium-rich solutions. Mater. Chem. Phys. 91, 500–506 (2005). [Google Scholar]
- 10.Pan H., Liu X. Y., Tang R., Xu H. Y., Mystery of the transformation from amorphous calcium phosphate to hydroxyapatite. Chem. Commun. 46, 7415–7417 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Onuma K., Ito A., Cluster growth model for hydroxyapatite. Chem. Mater. 10, 3346–3351 (1998). [Google Scholar]
- 12.Tsuji T., Onuma K., Yamamoto A., Iijima M., Shiba K., Direct transformation from amorphous to crystalline calcium phosphate facilitated by motif-programmed artificial proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 16866–16870 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dey A., Bomans P. H., Müller F. A., Will J., Frederik P. M., de With G., Sommerdijk N. A., The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nat. Mater. 9, 1010–1014 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Tao J., Pan H., Wang J., Wu J., Wang B., Xu X., Tang R., Evolution of amorphous calcium phosphate to hydroxyapatite probed by gold nanoparticles. J. Phys. Chem. C 112, 14929–14933 (2008). [Google Scholar]
- 15.He K., Nie A., Yuan Y., Ghodsi S. M., Song B., Firlar E., Lu J., Lu Y.-p., Shokuhfar T., Megaridis C. M., Shahbazian-Yassar R., In situ transmission electron microscopy explores a new nanoscale pathway for direct gypsum formation in aqueous solution. ACS Appl. Nano Mater. 1, 5430–5440 (2018). [Google Scholar]
- 16.Zheng H., Smith R. K., Jun Y.-w., Kisielowski C., Dahmen U., Alivisatos A. P., Observation of single colloidal platinum nanocrystal growth trajectories. Science 324, 1309–1312 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Song B., He K., Yuan Y., Sharifi-Asl S., Cheng M., Lu J., Saidi W. A., Shahbazian Yassar R., In situ study of nucleation and growth dynamics of Au nanoparticles on MoS2 nanoflakes. Nanoscale 10, 15809–15818 (2018). [DOI] [PubMed] [Google Scholar]
- 18.R. F. Egerton, Electron Energy-Loss Spectroscopy in the Electron Microscope (Springer US, 2011). [Google Scholar]
- 19.Holtz M. E., Yu Y., Gao J., Abruña H. D., Muller D. A., In situ electron energy-loss spectroscopy in liquids. Microsc. Microanal. 19, 1027–1035 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Wallace A. F., Hedges L. O., Fernandez-Martinez A., Raiteri P., Gale J. D., Waychunas G. A., Whitelam S., Banfield J. F., De Yoreo J. J., Microscopic evidence for liquid-liquid separation in supersaturated CaCO3 solutions. Science 341, 885–889 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Loh N. D., Sen S., Bosman M., Tan S. F., Zhong J., Nijhuis C. A., Král P., Matsudaira P., Mirsaidov U., Multistep nucleation of nanocrystals in aqueous solution. Nat. Chem. 9, 77–82 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Parimaladevi P., Kavitha C., Srinivasan K., Investigation of the effect of liquid–liquid phase separation (LLPS) on nucleation and different growth stages of vanillin and bulk growth of defect-free single crystals from aqueous solution—A new approach. CrstEngComm 16, 2565–2569 (2014). [Google Scholar]
- 23.Zhao J., Liu Y., Sun W.-b., Yang X., First detection, characterization, and application of amorphous calcium phosphate in dentistry. J. Dent. Sci. 7, 316–323 (2012). [Google Scholar]
- 24.Rabadjieva D., Gergulova R., Titorenkova R., Tepavitcharova S., Dyulgerova E., Balarew C., Petrov O., Biomimetic transformations of amorphous calcium phosphate: Kinetic and thermodynamic studies. J. Mater. Sci. Mater. Med. 21, 2501–2509 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Wang F., Richards V. N., Shields S. P., Buhro W. E., Kinetics and mechanisms of aggregative nanocrystal growth. Chem. Mater. 26, 5–21 (2014). [Google Scholar]
- 26.Cahn J. W., Hilliard J. E., Free energy of a nonuniform system. III. Nucleation in a two-component incompressible fluid. J. Chem. Phys. 31, 688–699 (1959). [Google Scholar]
- 27.Meyer J., Eanes E., A thermodynamic analysis of the amorphous to crystalline calcium phosphate transformation. Calcif. Tissue Res. 25, 59–68 (1978). [DOI] [PubMed] [Google Scholar]
- 28.Wang Z., Li Z., Park S., Abdela A., Tang D., Palmer R., Quantitative Z-contrast imaging in the scanning transmission electron microscope with size-selected clusters. Phys. Rev. B 84, 073408 (2011). [Google Scholar]
- 29.Radisic A., Ross F., Searson P., In situ study of the growth kinetics of individual island electrodeposition of copper. J. Phys. Chem. B. 110, 7862–7868 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Peng X., Manna L., Yang W., Wickham J., Scher E., Kadavanich A., Alivisatos A. P., Shape control of CdSe nanocrystals. Nature 404, 59–61 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Zhao W., Xu Z., Yang Y., Sahai N., Surface energetics of the hydroxyapatite nanocrystal–water interface: A molecular dynamics study. Langmuir 30, 13283–13292 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Seo D. S., Lee J. K., Synthesis of hydroxyapatite whiskers through dissolution–reprecipitation process using EDTA. J. Cryst. Growth 310, 2162–2167 (2008). [Google Scholar]
- 33.Bagambisa F. B., Joos U., Schilli W., Mechanisms and structure of the bond between bone and hydroxyapatite ceramics. J. Biomed. Mater. Res. 27, 1047–1055 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Lotsari A., Rajasekharan A. K., Halvarsson M., Andersson M., Transformation of amorphous calcium phosphate to bone-like apatite. Nat. Commun. 9, 4170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider N. M., Norton M. M., Mendel B. J., Grogan J. M., Ross F. M., Bau H. H., Electron–water interactions and implications for liquid cell electron microscopy. J. Phys. Chem. C 118, 22373–22382 (2014). [Google Scholar]
- 36.Liu J., Ye X., Wang H., Zhu M., Wang B., Yan H., The influence of pH and temperature on the morphology of hydroxyapatite synthesized by hydrothermal method. Ceram. Int. 29, 629–633 (2003). [Google Scholar]
- 37.Dorozhkin S. V., Amorphous calcium orthophosphates: Nature, chemistry and biomedical applications. Int. J. Mater. Chem. 2, 19–46 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Woehl T. J., Park C., Evans J. E., Arslan I., Ristenpart W. D., Browning N. D., Direct observation of aggregative nanoparticle growth: Kinetic modeling of the size distribution and growth rate. Nano Lett. 14, 373–378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999). [Google Scholar]
- 40.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Kieffer J., Mechanical degradation and viscous dissipation in B2O3. Phys. Rev. B 50, 17–29 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Straatsma T., Berendsen H., Free energy of ionic hydration: Analysis of a thermodynamic integration technique to evaluate free energy differences by molecular dynamics simulations. J. Chem. Phys. 89, 5876–5886 (1988). [Google Scholar]
- 43.Yu W., He X., Vanommeslaeghe K., MacKerell A. D. Jr., Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 33, 2451–2468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darden T., York D., Pedersen L., Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). [Google Scholar]
- 45.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kale L., Schulten K., Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eaaz7524/DC1