

Catapult-like hydrogel actuator delivers high contraction power.
Abstract
Stimuli-responsive hydrogels have large deformability but—when applied as actuators, smart switch, and artificial muscles—suffer from low work density due to low deliverable forces (~2 kPa) and speed through the osmotic pressure–driven actuation. Inspired by the energy conversion mechanism of many creatures during jumping, we designed an elastic-driven strong contractile hydrogel through storing and releasing elastic potential energy in polymer network. It can generate high contractile force (40 kPa) rapidly at ultrahigh work density (15.3 kJ/m3), outperforming current hydrogels (~0.01 kJ/m3) and even biological muscles (~8 kJ/m3). This demonstrated elastic energy storing and releasing method endows hydrogels with elasticity-plasticity switchability, multi-stable deformability in fully reversible and programmable manners, and anisotropic or isotropic deformation. With the high power density and programmability via this customizable modular design, these hydrogels demonstrated potential for broad applications in artificial muscles, contractile wound dressing, and high-power actuators.
INTRODUCTION
Stimuli-responsive hydrogels are appealing with their merits of large volume change and high water content (low solid/polymer fraction), which make the mechanical and many properties of hydrogel different from solid/dry polymers, and swell and contract via absorbing and releasing water in and out their network (i.e., osmotic-driven) upon environmental cues [e.g., temperature (1), pH (2), and light (3)]. Their excellent softness, elasticity, deformability, biocompatibility, stimulus diversity, and permeability (secure diffusion of oxygen, nutrients, metabolites, and cells enabling) have promoted their broad application in tissue engineering (4), soft electronics (5–7), miniaturized soft robotics and actuators (8–11), biomedicine (4), and mechano-therapeutic wound dressings (12). However, when it comes to building large-scale actuating systems practically, hydrogels suffer from being mechanically weak and slow, specifically low deliverable force (F, about 10−2 N or even lower) (13) and size-dependent actuation speed (e.g., hours at centimeter scale) (14, 15). The essential evaluation of high-performance actuators—high output power density—requires both a large force generated by unit mass and high actuation speed.
The energy density and power density of current typical (osmotic-driven) hydrogels are respectively ~10−2 kJ/m3 and ~10−2 W/m3, significantly lower than biological muscle (8 kJ/m3, 50 to 100 W/kg) (16). This low actuation power density of current hydrogels is rooted in the osmotic pressure–driven, diffusion-limited volume change mechanism. Under the osmotic-driven framework, actuation speed and delivered force are negatively balanced rather than acting synergistically, where one is raised by sacrificing the other. For instance, high actuation speed demands high diffusivity through the hydrogel, while high diffusivity is often linked with high porosity, which prescribes low elastic modulus and weak mechanical property of the hydrogel. Hence, this has been a long-standing dilemma, given the contractionary requirements of force and speed for material microstructures (e.g., porosity) (15, 17) or rapid in response but low in modulus (1). Furthermore, the osmotic-driven mechanism determined the maximum magnitude of the deliverable force, which is unavoidably limited by mild intrinsic hydrophobicity change. In past efforts to address this dilemma, high speed and large force have been achieved by using salt water as solvent to expedite water expulsion from gel network (18) or structurally using asymmetric hollow hydrogel structure for hydraulic actuation (13). However, altering actuation condition did not change the osmotic-driven mechanism of the material itself. Fundamentally solving the dilemma is to truly increase the unit volume/mass deliverability, i.e., output energy density (joules per cubic meter) and power density (watts per cubic meter). This necessitates a new, non-osmotic contraction mechanism with a conceptually new material design at molecular level to fundamentally break through this limit.
Jumping of many animals presents incredibly high speed and force. Mammalian skeletal muscle fibers, the natural actuators, can deliver more than 1 N force with 10 MPa modulus and show no reduction of actuation speed when bundled and thus display high work output (16, 19). They use a catapult-like energy conversion mechanism, through which potential energy is temporarily stored in elastic structures, followed by the rapid release of this energy to complete the jump (20–22). Shape memory hydrogel (SMH) as a shape-editable and recoverable material has the potential to become a carrier of energy to store and release elastic potential energy in a similar manner. However, most SMHs as actuators have low elastic moduli and/or weak reversible bonds, which are intrinsically insufficient for desirable large energy storage and high work output, so most SMHs can only recover moderate bending, folding, and twisting deformations (23, 24). Moreover, there is very little focus on the mechanical energy change of SMHs during shape fixation and restoration, which is vital for their practical applications in soft robotics, intelligent switch, and surgical materials. These all impose needs for developing strong contractile hydrogels. However, not all SMHs upon contraction can generate high force with a high energy density and a high energy standardization rate. It needs to meet the following: The hydrogel must have high toughness and be able to form strong reversible bonds to store high energy. Meanwhile, the original material should also have high elasticity to avoid excessive energy loss and thus reduce the energy conversion rate.
Here, inspired by the energy conversion mechanism in the elastic actuation of some creatures, we developed a high-power-density strong contractile hydrogel material, which can store elastic potential energy in an elastic hydrogel polymer network and subsequently release the energy, to achieve both high-force (40 kPa) and high-speed deformation (25%/min strain rate), which matched the high output work density (15.3 kJ/m3) and have even outperformed biological muscles (8 kJ/m3). The elastic-driven hydrogel (EDG) material was fabricated by fixing the deformation of a stretched elastic poly(acrylamide-co-acrylic acid)[p(AAm-co-AAc)] hydrogel through forming new bonds between carboxylic groups and Fe3+ ions in the polymer network to store elastic potential energy in the material. The EDG material could restore its original shape under ultraviolet (UV) light or in acid solution, which breaks those bonds and releases the prestored elastic potential energy. It is worth noting that, although the initial trigger of the hydrogel contraction involves the penetration of acid/light into the polymer network, it is different from conventional osmotic hydrogels whose contraction speed is fully limited by water transportation, whereas the contraction of EDG is enabled and determined by the elastic recovery after the triggering, which endows EDGs with fast and strong contract and thus high output work density. Moreover, the p(AAm-co-AAc) network of the presented EDGs has excellent elasticity and toughness and can form strong bonds with Fe3+ ions, which further increase its mechanical strength. All of these together enable materials to store and release more energy.
Furthermore, the elastic energy storing and releasing method endows the materials with a switchable mechanical property between elastic and plastic behavior. In addition, the contraction speed of the elastic actuation could be controlled on demand by adjusting the bond breaking speed. In addition, the contractive direction could be encoded simply by the prestretching direction and magnitude, allowing for diverse shape-changing behaviors from anisotropic to isotropic. In this work, we demonstrated potential functions of the EDGs to showcase their merits of programmable shape changes, large contraction force, and high elastic modulus, including expandable hydrogel tube (shrinking in transversal direction), actuator for artificial muscle, and contractile hydrogel patch for smart wound dressing. This universal design of EDGs has not only opened a new avenue to addressing the contradictory relationship between contraction force, contractive speed, and elastic modulus but also expanded the scope of responsive hydrogel applications.
RESULTS
Design of the elastic energy storing and releasing method and system
Jumping is a swift and powerful motion initiated by muscle-powered accelerations for many animals, such as frogs, kangaroos, and rabbits. The catapult mechanism first requires muscle contraction to prestore elastic potential energy via consuming chemical energy into elastic structures, followed by joint movement owing to elastic recoil by releasing the stored elastic energy to produce kinetic energy (Fig. 1A) (20–22). Moreover, this cycle of energy storing-releasing can be repeated infinitely (Fig. 1B). SMH as a shape-editable and recoverable material has the potential to become a carrier of energy to finish the storage and release of elastic potential energy in a similar manner. Inspired by this energy conversion mechanism and combined with shape memory effect of hydrogel, we developed a new class of hydrogels that can generate a large force by strong contraction. As shown in Fig. 1 (B and C), a hydrogel is mechanically stretched to accumulate elastic potential energy, and this energy is temporarily stored by forming new reversible chemical bonds within the polymer, which effectively “lock” the hydrogel at the deformed shape. The stored elastic potential energy can be released controllably in the form of kinetic energy or other potential energy by applying another stimulus to “unlock” the previously formed reversible intranetwork bonds. Such energy-releasing–induced shape restoring of an elastic network (spring recoil) can generate much larger force (F) at a higher speed (v), thus greater power (P = F • v), compared to the osmotic-driven volume change (passive network collapse) of conventional hydrogels.
Fig. 1. Conceptual scheme of the strong contractive materials based on mechanical energy storing method.
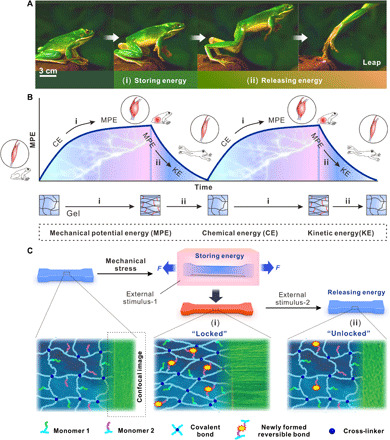
(A) Frog leaping off a rock through prestoring and subsequent releasing of elastic potential energy. (Photo credit: Purchased from vjshi.com under royalty-free license.) (B) Elastic potential energy was initially stored (red area) in elastic structures by contraction of muscle via consuming chemical energy in the body and then instantaneously releasing based on releasing (blue area) of elastic potential energy. (C) Schematics of the design strategy of shrinking materials via storing energy during stretching and then releasing the energy: storing elastic potential energy in the material by mechanically stretching it and locking it at the stretched state through forming new chemical bonds under external stimulus-1 (i). Upon external stimulus-2, the newly formed bonds break, and the prestored energy is released, resulting in rapid and powerful contraction of the material, which generates large force (ii). The green images are confocal microscopic images of the hydrogel material.
In general, the polymer chains must consist of at least two different components: one highly elastic network to minimize energy dissipation in the stretching-relaxing process to maximize the amount of energy stored and released and another smart (switchable) component to “freeze” and “restore” the tendency for elastic recoil by reversible bonding ideally with high bond force constants for effective elastic network locking. The latter is necessary as the elastic network alone cannot lock the elastic potential energy produced by mechanical stress for delayed release (fig. S2). These two components would ideally coexist or be linked through permanent bonds to form a single hydrogel network, avoiding chain sliding or energy dissipation in double network design, to maximize the energy conversion efficiency for optimum work output. Such a modular design provides two knobs for further tunability of the mechanical property and the force for material optimization and broad applications (25–29).
EDGs based on elastic energy storing and releasing
In this work, we used poly(acrylamide) (PAAm) as the elastic component (30) and poly(acrylic acid) (PAAc) as the smart component in our copolymer p(AAm-co-AAc) as an exemplary EDG. Acrylamide (AAm) and acrylic acid (AAc) monomers were mixed at different ratios and heated to be covalently cross-linked. Pure PAAm and PAAc hydrogels were used as control samples. The hydrogels were stretched mechanically and immersed in an iron (III) chloride solution. In this system, the first stimulus Fe3+ and carboxylic groups formed coordination bonds (31–34), which fixed the mechanically stretched hydrogel at its stretched geometry and stored the mechanical energy. The Fe3+-carboxylic pair was rationally selected for the following reasons: (i) its reversibility nature of the ionic (coordination) bond for realizing locking-unlocking the elastic network, (ii) its high coordination coefficient for effectively locking the network and efficiently storing maximum energy, and (iii) its versatility of dissociation with light and various molecular stimuli for diverse actuation modes. The secondary stimulus, UV light or an acid solution, destablized the coordination bonds by photoreduction (Fe3+ to Fe2+) or protonation (COO− to COOH) (Fig. 2A) (35–37). Figure 2B shows the change in length to 160% of its original length and the corresponding color change of the p(AAm-co-AAc) hydrogel film during energy storage in 0.06 M iron ion solution and then its contraction back to its original length in 1 M acid solution, whose contraction could be also achieved by UV irradiation alternatively (fig. S3).
Fig. 2. Materialization of elastic energy storing and releasing method based on reaction between carboxylic group and iron ion in p(AAm-co-AAc) hydrogels.
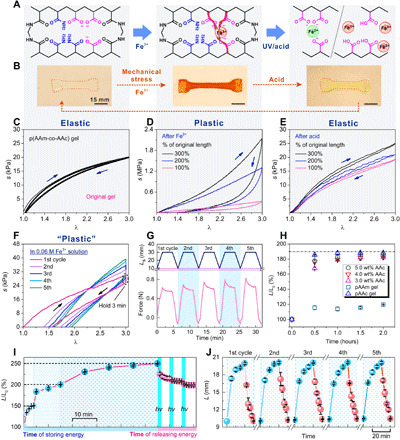
(A) p(AAm-co-AAc) hydrogel network (left) can be locked by forming new carboxylic-Fe3+ coordination (middle) and subsequently unlocked by reduction of Fe3+ ions and protonation of carboxylic acid (right). (B) Prestretched hydrogel (left) after fixation in Fe3+ solution (EDG, middle) and after contraction in 1 M acid solution (right). (Photo credit: Yanfei Ma and Mutian Hua, University of California Los Angeles.) (C to E) Stress-stretch curves of the hydrogel before (C) and after Fe3+-treatment at different prestretched ratios (percentage of the original length) (D) and after subsequent acid treatment (E), showing reversible elastic-plastic switchability. (F) Stress-stretch curves of hydrogels during Fe3+ treatment and after five loading-unloading cycles. (G) Time profiles of the loading-unloading cycles in (F), in terms of Ls (prestretched length) and force (newton). (H) Extension of hydrogels with different PAAc contents as a function of soaking time in Fe3+ solution. (I) Programmable deformability: controllable multistage energy storing and releasing of hydrogels in Fe3+ solution and under UV radiation, respectively. (J) Fully reversible energy storing in 0.06 M Fe3+ solution and releasing in 1 M acid solution of hydrogel (0.03% BIS).
The original p(AAm-co-AAc) hydrogel did not exhibit distinct hysteresis after loading-unloading over 5 cycles (at 300% uniaxial stretch) and was able to fully recover to its original length after unloading, indicating the excellent elasticity of the hydrogel (Fig. 2C and fig. S5). As a control sample, PAAm-alginate double-network hydrogels were also prepared (fig. S7). When soaked in Fe3+ solution, the p(AAm-co-AAc) hydrogel displayed hysteresis in the same loading-unloading test (holding for 3 min), and the hysteresis progressively increased over the 5 cycles, as seen in the stress-stretch curves in Fig. 2F (replotted from the data in Fig. 2G). This shows that, as the Fe3+ ions complex with the carboxylic groups on the polymer chains, these newly formed bonds effectively locked the polymer network and thus fixed the hydrogel at the prestretched length. Over the cycles, as the locking/fixation sites increased, the unloaded hydrogel was able to retain a length closer to the prestretched length (300%) from 157 to 195%, presenting more plastic behaviors and increased hysteresis.
Moreover, Fe3+ ion and carboxylic groups formed coordination bonds, which not only fixed deformation but also created a second physical network in the p(AAm-co-AAc) hydrogel, which resulted in substantially increased modulus from ~30 kPa to ~0.7 MPa (Fig. 2, C and D, and fig. S19). In addition, the mechanical behavior of the hybrid hydrogel composed of a covalently cross-linked network (elastic property) and a physical network (plastic property) depends on which network playing a leading role in the hydrogel (38). Therefore, the mechanical behavior of EDG would be a consequence of the competition between the chemical and physical network in the hydrogel. As shown in Fig. 2D, the EDG (after network locking) displayed higher extent of plastic property than the original elastic p(AAm-co-AAc) hydrogel. The EDG hydrogel retained 30% of its stretched deformation after unloading. Furthermore, the elasticity could be 100% restored after immersing in a 1 M hydrochloric acid solution for 30 min (Fig. 2E). Thus, it completed a cycle from elasticity to plasticity and eventually back to elasticity. This rational design opens up an avenue for reversibly switching a material between highly elastic and plastic.
During the process of storing mechanical energy, the ultimate fixable length of EDG directly correlates with the quantity of new bonds formed inside the polymer network. As shown in Fig. 2H, the hydrogel slices (20 mm by 10 mm by 2 mm) were stretched to 190% of the original length and fixed in a 0.06 M iron ion solution (L/L0, where L = final fixed length after stretching and Fe3+ treatment without loading and L0 = original length). The p(AAm-co-AAc) and PAAc hydrogels containing carboxylic groups exhibited a memorized temporary deformation after complexing with Fe3+ ions, and the speed of fixation depended on the concentration of carboxylic groups in the hydrogel. More carboxylic groups would offer more sites to form coordination complexes with ferric ions. The Fe3+ ions concentration in hydrogels was determined through measuring the weights of the dried hydrogel before and after bonding with Fe3+ ions. Results confirmed that the Fe3+ ions concentration increased with carboxylic group concentration in the hydrogel and that PAAc hydrogel (100% carboxylic groups) had the highest content of Fe (about 0.7 M). By contrast, the pure PAAm hydrogel (no smart bond carboxylic group) exhibited negligible extension because of swelling and lower Fe3+ ion concentrations (fig. S9).
With such controllability, the p(AAm-co-AAc) hydrogel could be programmably deformed, being fixed progressively and self-held at any desirable arbitrary lengths on demand. For example, it was first fixed at 150% of its original length, refixed at 200%, and continued at 250% by incremental prestretches. This demonstrated the controllable and multistage energy storing of the hydrogel’s deformation. Similarly, the EDG can shrink back to any desirable lengths and eventually its original length, when exposed to UV light (320 to 390 nm, 225 mW/cm2) for 1 min as the dissociation of the PAAc network begins. Upon pausing of light irradiation, the contraction of the hydrogel gradually slowed down and eventually stopped after 5 min. Shrinkage can resume when the UV light irradiation resumes (Fig. 2I). In addition, the consumed energy could be regained by reforming the coordination bonds with additional stretching to store the mechanical energy again. As shown in Fig. 2J, there is no notable decay in the energy storage and release based on the continuous measurement spanning 5 cycles, which is attributed to the outstanding elasticity of p(AAm-co-AAc) network. By contrast, for the PAAm-alginate hydrogel with relatively high plasticity and two polymer networks, its final recovery showed obvious hysteresis because of the energy dissipation during the release of stored energy, which also reduced the energy conversion rate (figs. S10 and S23). Such capability of customizable multi-stable deformation cannot be seen in conventional elastic or plastic materials. The excellent reversibility, cyclic reliability, and programmability of this chemo-mechanically modulated synthetic material (Fig. 2, I and J).
Strong contraction on demand
To quantify the elastic potential energy stored in EDG under mechanical stretching and chemical locking, the force and length were recorded in stretching processes. All mechanical tests were performed in air at room temperature using a tensile machine with a 4.5 N load cell. The hydrogels were stretched to twice or thrice their original length, and the work was calculated by integrating force-displacement curve (fig. S1). The mechanical strength of EDGs was highly sensitive to the chemical cross-linking density (fig. S4). Therefore, we measured the energy stored after 200% stretched EDGs with cross-linking densities varying from 0.02 to 0.04 weight % (wt %). As expected, the amount of elastic potential energy stored increased from 5.76 kJ/m3 to a maximum of 10.81 kJ/m3 (fig. S12A). Moreover, the 300% stretched EDG was able to store energy of 31.04 kJ/m3. As controls, unstretched hydrogels were also used (Fig. 3A).
Fig. 3. Contractive properties of EDG.
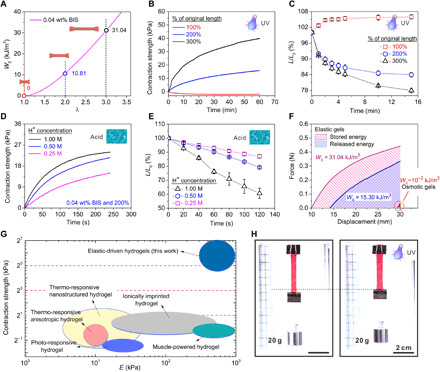
(A) Elastic energy density (WE) of EDG (0.04 wt % BIS) contraction as the function of prestretch ratio (λ). (B) EDG’s contraction strength versus UV radiation time for EDGs with different prestretch ratio. (C) Length ratio (L/L0) as a function of UV (320 to 390 nm, 225 mW/m2) radiation time was measured in water. (D and E) Contraction strength-time and length ratio-time curves of the EDG (200% energy storage, cross-linker: 0.04 wt % BIS) in 0.25, 0.5, and 1 M hydrochloric acid solution. (F) Stored energy and released energy of EDG, in comparison with the released energy of osmotic hydrogels. (G) Comparison of the contraction strength and elastic modulus between EDGs and typical osmotic hydrogels (1, 3, 14, 40, 41). (H) Photos of an EDG (300% energy storage, 0.04 wt % BIS) strip lifting a 20-g weight load upon UV light illumination. (Photo credit: Yanfei Ma and Shuwang Wu, University of California Los Angeles.)
To understand the kinetics of contractile strength, the contraction force was measured by fixing the ends of an EDG sample and then recording the forces generated by its contraction under stimulus-2, either UV light (320 to 395 nm, 17 W cm−2) or acid (0.1, 0.5, and 0.25 M). All contractile force tests were performed in water at room temperature using a tensile machine with a 0.5 N load cell. Contractile strength, i.e., force per unit cross-sectional area, was obtained and used for the contraction performance studies to eliminate the effect of sample size on the generated force. As shown in Fig. 3B, during UV radiation (320 to 395 nm, 17 W cm−2), the contraction strength rose gradually as increasing amount of Fe-carboxylic bonds were broken over time. Under the same UV exposure time, the contraction strength increased with the UV irradiation intensity (fig. S11). After 60 min of UV exposure (320 to 395 nm, 17 W cm−2), the contractile strength of the 300% EDG (stretched and fixed at 300% of the original length during energy storing) was close to 40 kPa (Fig. 3B). As expected, the samples of 200% stretch or low cross-linking density showed lower contractile strength (fig. S12B). Similar length change kinetics was observed (Fig. 3C) as in the contractile strength, verifying the dynamics. In addition, the destruction of coordinate bonds is reflected in UV-visible spectra. The 200% EDG material presented an evident absorption peak at 458 nm, implying the formation of Fe-carboxylic coordinate bonds. After exposure to the UV light, the absorption peak decreased gradually and was also accompanied by the restoration of shape (fig. S14). By contrast, the unstretched hydrogels only displayed swelling rather than contraction (negative contractile strength and increased length; Fig. 3, B and C), further proving that contractile force was induced by storing elastic potential energy.
Advantageous over conventional stimuli-responsive hydrogels, the EDG’s chemo-mechanical modulation mechanism allows for controlling the material’s response, both ratio and rate, on demand by tuning the coordination bond dissociation speed. For example, in Fig. 3 (D and E), we demonstrate that acid could controllably break the coordinate bond (via protonation of carboxylic groups) much faster than UV radiation. By altering the H+ ion concentration (0.25, 0.5, and 1 M), the response speed of EDG could be regulated from 15 kPa/240 s (contraction strength per time) or 10%/120 s (contraction percentage per time) to 24 kPa/240 s or 40%/120 s. At 1 M hydrochloric acid solution for 120 s, the EDG could contract to 60% of its stretched length and produce about 24 kPa contraction strength (Fig. 3, D and E). Using acid as the stimulus-2 enhanced the response speed to >20 times higher than using UV light and also enabled faster and complete release of the stored energy to the ground level. We could also use EDG’s color change to estimate the degree of dissociation of the coordination bonds (fig. S13 and movie S1). The sample with the 300% energy storage [0.04% N,N′-methylenebisacrylamide (BIS)] was thoroughly released by acid and brought ~0.3 N contractile force, which had a high energy density of 15.3 kJ/m3 and a high energy conversion rate of 49.29%, because the chemical energy was directly converted to mechanical work without intermediate steps of heat producing as in traditional heat engines, such as electric motor and petroleum engine (Fig. 3F). The contractions finished in ~300 s (fig. S15), which provided an average power density of ~51 W/m3. The energy density of the EDG is 1000 times higher than that of typical osmotic hydrogels (Fig. 3F and figs. S21 and S22) and even comparable to water-free polymer (39). The high output work (energy) density is particularly beneficial when there are space (or mass) limitations as in robots, implantable devices, and microelectromechanical systems (16).
For smart hydrogels, having high mechanical strength and elastic modulus are critical for expanding the scope of hydrogel applications (40). However, for conventional osmotic-driven hydrogels, the intrinsic contradiction between contraction speed and force (affected by elastic modulus and contraction ratio) strongly hampered their practical utilization (13, 40). Namely, the osmotic contraction arises from the passive collapse of a relatively relaxed polymer network upon water escape, which is solely driven by side-group reactions, while the main chains of the hydrogel provide little contractile force and even hinder the network collapse, leaving a substantial portion of the network underutilized in force generation during contraction. In addition, the slow mass transport of water molecules out of the hydrogel network further limits its collapse speed. For EDG materials, by contrast, the elastic contraction is driven by the recoil of the highly prestretched main chains of the hydrogel under large tension, not dominated or limited by mass transport, while the active restoration/actuation state of the main chains is triggered by the side-group reaction. This specific driving mechanism endowed it high elastic modulus and high stretchability simultaneously and thus a superior toughness of ~30 times higher than traditional hydrogels, such as poly(N-isopropyl acrylamide) (PNIPAm) and PAAc (movie S6 and figs. S19, S21, S24, S27, and S28). Although the initial diffusion of acidic solution into the hydrogel to trigger the hydrogel contraction is the same process as with PNIPAm and PAAc gels, the EDG materials based on elastic recoil have a greater advantage than the osmotic pressure–driven hydrogel whose volume change rate is limited by the diffusion and thus is substantially size dependent. As shown in fig. S20, the response speed of EDG (0.03 wt % BIS and 200%) in 1 M acid solution has negligible size dependency, which is substantially minimized compared to the osmotic (PAAc) hydrogels. Several key mechanical behaviors of EDG have matched and even outperformed biological muscles. The stress, strain, and energy density of EDG hydrogel were 3.7 MPa, 600%, and 15.3 kJ/m3, respectively—which were 10, 15, and 2 times higher than that of mammalian skeletal muscle (~0.35 MPa, ~40%, and 8 kJ/m3)—while owning high water content (~80%) comparable to muscles (16) and energy conversion efficiency (~49%). Although the response speed of EDG is 8 to 25 times that of osmotic hydrogel (PAAc) with the same size and polymer content, it is 600 to 1000 times less than that of muscle, which needs further improvement in subsequent work (table S4). As shown in Fig. 3 (G and F) and fig. S21, the contraction strength of the EDG hydrogel (40 kPa, 0.3 N) is 10 times (21 to 25) higher than that of osmotic hydrogels [~2 kPa; temperature-responsive (1, 40), light-responsive (3), muscle-powered (41), ionically imprinted hydrogel (14)]. In a typical case, the EDG piece (38 mm by 5.2 mm by 1.3 mm) could vertically and successfully lift up a weight of 20 g in water upon an exposure to UV light for 60 min (Fig. 3H). To simulate the above process for quantitative analysis, we used the tensile machine with a 0.5-N load cell to apply 0.2 N load on the same sample. We found that 1.79 kJ/m3 (11.70%) of the mechanical energy stored in the EDG was released in this process (fig. S17). Using a HCl solution for contraction could also bear 20 g of weight (fig. S18 and movie S2).
Anisotropicity and programmable reconfiguration
A critical factor for achieving a transition from isotropic to anisotropic deformation with hydrogels is the alignment of the material’s internal structure, which is generally synthesized by using directional stimuli, such as mechanical forces, magnetic and electric fields, and gradients of temperature and ions (42). Here, we found that the chemical fixation of the mechanically stretched EDG resulted in the formation of microcosmic fibers along the stretched direction (Fig. 4E and fig. S25). This was expected, especially for elastic materials, since the extension in the stretching direction usually leads to the contraction in other dimensions, resulting in the progressive orientation of polymer chains along the axial direction (27). As Fe3+ ions are introduced, the orientation would be fixed by the coordination bonds. After UV (320 to 390 nm, 225 mW/cm2) exposure, as coordination bonds are destroyed, the polymer network would regress, expanding in the prestretching direction and shrinking in the transversal direction.
Fig. 4. Programmable property of the EDG based on elastic energy storing and releasing method.
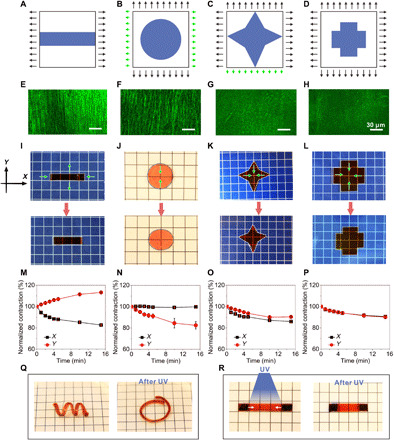
(A to D) Schematic designs, the p(AAm-co-AAc) hydrogel was encoded with the applied mechanical force. (E to H) Confocal images of the hydrogel structures after stretching and fixed. (I to L) Optical photographs of edited EDG materials before and after exposure to the UV light. (I) Contracted in x-axis direction and extend in y axis. (J) Shrunken in y-axis direction under keeping constant length in x-axis direction. (K) Three vertices (left, top, and right) contracted but the bottom remained constant. (L) Isotropic shrinkage. (Photo credit: Yanfei Ma and Mutian Hua, University of California Los Angeles.) (M to P) EDG showed contractility from anisotropic to isotropic. (Q) EDG based on elastic energy storing and releasing method could achieve 3D contraction under UV light. (R) EDG could locally shrink after local exposure to UV light. The size of the grid unit in the photo background is 6.5 mm by 6.5 mm. Photo credit: Yanfei Ma, University of California Los Angeles.
On the basis of this mechanism, we speculated that the contraction behavior of elastic actuation hydrogel could be tuned and programmed by controlling the direction, quantity, and regularity of structure fibers in the hydrogel. To realize this in our EDG material, the contractive behavior in different directions was encoded by varying the applied force’s magnitude and direction during the stretching step. We demonstrated the ability to program the mechanical domains by designing different modes of loading, specifically from two ways to three and four ways, respectively, in Fig. 4 (A to D), to tune the quantity and regularity of the polymer network fibers in the hydrogel sheets. As shown in Fig. 4A, a rectangular EDG (25 mm by 25 mm by 2 mm) was stretched and fixed to 200% of the original length along the x axis and fixed with Fe3+ ion solution. In Fig. 4B, a cylindrical-shaped sample (25 mm by 25 mm by 2 mm) was stretched and fixed to 200% along the y axis while retained unchanged in length in the x axis by applying a small force. Next, with a star-shaped EDG sheet (25 mm by 25 mm by 2 mm) in Fig. 4C, we display where both x- and y-axis directions were stretched and fixed to 200% of their original lengths, apart from the negative y-axis section that was held at original length (Fig. 4C). Last, in Fig. 4D, both x- and y-axis directions of the hydrogel (25 mm by 25 mm by 2 mm) were stretched and fixed to 200% for the cross-shaped hydrogel sheet. As a result, the quantity and regularity of the hydrogel fibers decreased with the increasing of the directionality and quantity of the force applied on the hydrogel (in Fig. 4, E to H). Experimental results showed that these structures did endow the hydrogels different contraction behaviors. As is shown in Fig. 4 (I and J), the hydrogel sheets stretched, either in x-axis direction only or in y-axis direction with x-axis length kept unchanged, and both exhibited anisotropic deformation. One contracted to 83% of the original length along the stretched direction (along length) and, meanwhile, expanded to original 113% in width; the other shrunk in y-axis direction only (84% of the original length), which resulted in the deformation from round to ellipse. The hydrogel prestretched by a pair of unbalanced forces displayed stronger contractions along the x-axis direction relative to their y axis (86 versus 90% of the original dimensions; see Fig. 4, G and K). By contrast, the hydrogel without aligned structure exhibited isotropous contraction (91% of their original length and width) as expected (Fig. 4, H and L). These observations suggest that anisotropic contraction was linked to the direction, quantity, and regularity of the hydrogel fiber. This endows the EDG material a unique capability to achieve complex shape morphing in a spatiotemporally programmable manner, unachievable with conventional osmotic hydrogels with only isotropic deformability (fig. S26). Moreover, the EDG is able to produce not only two-dimensional (2D) planer structures but also complex 3D structures and programmably reconfigure under UV light irradiation (Fig. 4Q). What is more, the EDG could also achieve local constriction function using the confined or patterned UV light irradiation (Fig. 4R).
Applications
Owning to its simplicity and versatility, the elastic energy storing and releasing system and fabrication methods for EDGs could enable several new possibilities in many areas, from soft robotics to biomedical and tissue engineering. For example, EDGs might be used as reconfigurable structures, owing to their merits of controllable anisotropic deformability, strong contraction force, and high elasticity modulus (figs. S16 and S19). EDG’s mechanical properties comparable to mammalian skeletal muscle and their similar actuation behaviors make EDG an excellent material for tissue engineering or surgery or as therapeutic devices, such as for intestine, vessels, or wind pipes. Figure 5A demonstrates that our material can be fashioned into a hydrogel tube (original length: 18.7 mm, original diameter: 4.5 mm) capable of contracting to 16 mm in length and, meanwhile, expanding to 5.2 mm in width inside a transparent plastic pipe (inner diameter: 5 mm) under UV light. Figure 5B, fig. S25, and movie S3 demonstrated that this strong contractive hydrogel material is similar to mammalian skeletal muscle—not only in terms of the similar aligned structure but also the comparable properties including stress, strain, contraction strength, work density, water content, and work efficiency (16) (table S4)—and also notably outperforms current synthetic hydrogels that have muscle-like aligned structure and comparable to mechanical performance but do not have such active contraction behavior (27, 43, 44). Furthermore, this material works like biceps to realize arm movement when mounted on a 3D printed skeleton, achieving a life-like performance including movement behavior and morphology when compared to past biomimetic hydrogel muscles from morphology (27). This demonstration also showcased good in-air operability, by simply coating the EDG with a layer of silicone oil to prevent water evaporation under UV light in ambient environment. As another example, the strong contractive material also has the potential to be used as a smart wound dressing to promote wound healing (12). Movie S4 and Fig. 5C exhibit the shrinking and healing process of a connective EDG material adhered to two parts of a cracked glass sheet (75 mm by 50 mm by 1 mm), respectively, loaded with a 20-g weight (fig. S30). The irreversible feature of the contraction endows the healing process with better stability under complex environment with varying temperature or physiological conditions. EDG could also be applied to actuate other materials by serving as the driving source. As shown in Fig. 5D, fig. S29, and movie S5, a hydrogel actuator was produced by bonding the EDG material with paper using commercial glue and was capable of generating an actuation force greater than that of an osmotic hydrogel actuator to lift a weight.
Fig. 5. Applications of EDG materials via the strong contraction force and high elasticity modulus.

(A) Contractive hydrogel tube prepared by elastic energy storing and releasing method could enlarge in the direction of tube diameter under UV light. L and D (length and diameter of the hydrogel tube). (B) Anisotropic EDG material was assembled on 3D printed arm and helps the arm realize lift. (C) Contractive material was adhered on a broken glass sheet loaded with 40-g weights and achieved the healing of the glass sheet depending on mechanical contraction. (D) Bending actuator made of EDG-paper (plastic material) bimorph structure showing strong driving force by lifting a weight (4 g), which is unachievable by the osmotic (PNIPAm) hydrogel as the control. (Photo credit: Yanfei Ma, University of California Los Angeles.)
DISCUSSION
We have demonstrated that the actuation performance and energy density of hydrogels can be greatly increased by a fundamentally elastic driving mechanism, inspired by the superior leap abilities of biological jumpers, which involves the storing and releasing of elastic potential energy at molecular level. In comparison with the commonly studied osmotic pressure–driven smart hydrogels, the elastic-driven actuation of EDGs based on the elastic energy storing and releasing method can deliver stronger and faster contraction (40 kPa, 25%/min) with better mechanical properties (high elastic modulus of 0.7 MPa and toughness of 12.7 MJ/m3). The simultaneous increase of all three properties—force, speed, and elastic modulus—fundamentally breaks the contradictory relationship between the three (13, 40), leading to a high output energy density up to 15.3 kJ/m3, which outperforms all state-of-the-art hydrogels and even biological muscles. Furthermore, the elastic energy storing and releasing method endows the hydrogel materials a unique elastic-plastic switchability and complex deformation programmability, enabling anisotropic or isotropic contraction and unprecedented multistage deformability. This modular material design composed of elastic and smart components is universal, which can be broadly applied to various elastic polymers and chemical reactions. Overall, the energy capture-return strategy solves the long-standing problem of weak or slow contraction of osmotic hydrogels and opens up an avenue for the rational design of powerful smart hydrogels with multifarious reconfiguration behaviors. Hydrous, soft, programmable, and powerful hydrogel materials, without loss of contractive speed, will improve the performance in applications such as artificial muscles, soft robots, flexible devices, and biomedical materials.
MATERIALS AND METHODS
Sample preparation and characterization
Synthesis of p(AAm-co-AAc) hydrogels
The p(AAm-co-AAc) hydrogels were synthesized via a radical polymerization. AAm (0 and 20%, weight of AAm/H2O), AAc (0, 2, 3, 4, 5, and 20%, weight ratio of AAc/H2O), ammonium persulfate (APS) (30 mg), and N,N′-methylenebisacrylamide (BIS) (0.02, 0.03, 0.04, 0.0276, and 0.33 wt % of H2O) were dissolved in 30 g of deionized water to achieve a homogeneous solution. Then, the precursor aqueous solution was poured into a glass mold that consists of two glass plates (thickness: 2 mm) and was polymerized at 60°C for 12 hours. After removing the template, the p(AAc-co-AAm) hydrogels were obtained. The codes of hydrogels and the weight fraction of each component are summarized in table S1.
Synthesis of PAAm-alginate hydrogels
Alginate was dissolved in 30 g of deionized water to get a homogeneous solution (2% weight ratio of alginate/H2O). AAm (20% weight of AAm/H2O), APS (30 mg), and BIS (0.04 wt % of H2O) were dissolved in the alginate solution. Then, the precursor aqueous solution was poured into a glass mold that consists of two glass plates (thickness: 2 mm) and was polymerized at 60°C for 12 hours. After removing the template, the PAAm-alginate hydrogels were obtained.
Preparation of the EDG materials
Hydrogel sheets were cut into a rectangle (size: 20 mm by 10 mm by 2 mm) and stretched to 200, 300, or 400% of the original length. Then, the stretched hydrogels were immersed into the 0.06 M Fe3+ solution for a different time to store mechanical energy (table S2). Last, Fe3+-loaded hydrogels were released from the mold and immersed in the deionized water for 1 hour to dissipate unstored energy.
Preparation of switchable hydrogel tube
AAm (20 wt %) (of water weight), 0.03 wt % cross-linking agent, 0.1 wt % initiator, and 2 wt % (of water weight) AAc were dissolved in 30 g of H2O as a precursor solution. Then, a 4.5-mm (diameter) iron rod was immersed into the precursor solution for 5 min. The surface of the iron rod would generate a layer of hydrogel about 0.5 mm. Next, the hydrogel tube was taken down, stretched, and fixed. Last, the shrinkable hydrogel tube was achieved.
Preparation of poly(N-isopropyl acrylamide) hydrogel
N-isopropyl AAm (NIPAm) (6.67 wt %), 2 wt % mg/ml of BIS (cross-linking agent), and 150 μl of photoinitiator Darocur (11477) initiator were dissolved in the 30-g water–dimethyl sulfoxide (DMSO) mixtures (21.9 g of DMSO and 8.1 g of water) as a precursor solution. Then, the precursor solution was poured into a PDMS mold (50 mm by 50 mm by 2 mm) and polymerized it under UV light for 10 s. The hydrogel was released from the mold and immersed into deionized water for 5 hours. Last, the PNIPAm hydrogel sheet was successfully synthesized.
Preparation of anisotropic and programmable EDG materials
The p(AAc-co-AAm) hydrogel sheet (25 mm by 25 mm by 2 mm) was stretched and fixed to 200% of the original length along the x axis or/and y axis and fixed with Fe3+ ion solution. The preparative EDG was cut into different shapes, such as rectangle, ellipse, star shape, and so on.
Mechanical measurement
Mechanical test systems with 4.5 N load cell (CellScale Biomaterials Testing) were used to perform the mechanical measurement for hydrogel materials. The crosshead velocity was 0.2 mm/s in tensile measurement and loading-unloading test. The nominal tensile stress (s), nominal tensile strain (ε), stretched times (λ), and elastic modulus (E) were calculated from the stress-stretch curve.
The repetitive storing and releasing energy of p(AAm-co-AAc) hydrogels
The p(AAm-co-AAc) hydrogels (0.03 wt % BIS) (20 mm by 10 mm by 2 mm) were stretched to 300% of the original length and immersed in 0.06 M Fe3+ solution for 20 min. The length of the hydrogel finally fixed to 200% of the original length (namely, EDG). Then, the stored energy in EDG materials was released and restored in 1 M acid solution for 10 min. Next, the restored hydrogels were immersed in ethyl alcohol for 5 min to exchange solvent and then exchange in deionized water for 5 min. This is 1 cycle. Similarly, the hydrogel continued to store and release energy again, another cycle.
Measurement of contraction force and maximum contraction work
A 0.5 N load cell was used for the mechanical test system (CellScale Biomaterials Testing) to perform contraction force (contraction strength; force per unit cross-sectional area) of the EDG. The curve of contraction force-time was achieved through keeping the displacement of the sample’s two fixtures at 20 mm (stretched to 200% of the original length) or 30 mm (300% of the original length) to record the forces under UV light (320 to 395 nm, 17 W/cm2) or acid solution (0.25, 0.5, and 1 M HCl solution). During all testing process, the sample was immersed into water to avoid additional contraction due to losing water under photo-irradiation. Maximum contraction work (releasing energy) was calculated by the force-displacement curve, for example, Fig. 3F and figs. S21 to S23. First, we clamped the EDG sample (300% energy storage, wet weight of sample: ~400 mg) on the CellScale and kept the displacement of the sample’s two fixtures at 30 mm. Then, the 1 M acid solution was used to completely and quickly release the stored energy (for 10 min). As this time, the sample would come into being high contraction force. Next, the two fixtures came back to 10 mm (original length) at the speed of 0.2 mm/s and recorded data in this process. The force-displacement curve was achieved in the contractive process. Similarly, the contraction force and work of osmotic hydrogels (PAAc gel) were measured by the CellScale Biomaterials Testing in the 1 M acid solution for 30 min. The elastic hydrogel (EDG) and osmotic hydrogel (PAAc) have the same size and monomers content.
SEM imaging
The testing samples with different cross-linking densities (0.02, 0.03, and 0.04% cross-linking agent, namely samples 6, 7, and 8) were prepared by the following procedures: (i) Sample was stretched and fixed to 200% of the original length in the 0.06 M Fe3+; (ii) the EDG samples were dried at 1 kPa for 48 hours in a freeze-drying machine. Then, the morphologies were acquired by a scanning electron microscope (SEM; Supra 40VP SEM) at a working distance of 8.9 mm and acceleration voltage of 10.0 kV. The unstretched samples were used as controls.
Confocal microscope imaging of EDG materials in wet state
To visualize the directional structure of the EDG materials, nile blue AAm was used to label. Specifically, 0.003 wt % (of water weight) nile blue AAm, 20 wt % (of water weight) AAm, 0.03 wt % BIS, 0.1 wt % APS, and 2 wt % (of water weight) AAc were dissolve in 30 g of H2O and polymerized at 60°C for 12 hours. Then, sample was stretched and fixed to 200% of the original length. A confocal microscope (Leica DMi8) was used to study the microstructures of EDG materials.
Supplementary Material
Acknowledgments
We thank our colleagues from the Department of Material Science and Engineering, University of California Los Angeles and Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences for support and assistance in the research. Funding: X.H., M.H., Y.D., and S.W. acknowledge the funding support from the NSF grant 1724526, the AFOSR grant FA9550-17-1-0311, the AFOSR award FA9550-18-1-0449, the ONR award N000141712117, and the ONR award N00014-18-1-2314. F.Z., X.P., and Y.M. thank the project of the Bureau of International Cooperation, Chinese Academy of Sciences (121B62KYSB2017009). Y.M. and F.Z. acknowledge support from the National Key Research and Development Program of China (2016YFC1100401) and the National Science Foundation of China (22032006). Author contributions: X.H. proposed the idea and supervised the research. Y.M., M.H., S.W., X.P., X.Z., F.Z., and X.H. designed the experiment. Y.M. and M.H. performed the experiments, collected the data, and prepared the figures. Y.M., M.H., and X.H. analyzed the data. Y.M., Y.D., M.H., and X.H. wrote the manuscript. All the authors discussed the manuscript. Competing interest: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabd2520/DC1
REFERENCES AND NOTES
- 1.Kim Y. S., Liu M., Ishida Y., Ebina Y., Osada M., Sasaki T., Hikima T., Takata M., Aida T., Thermoresponsive actuation enabled by permittivity switching in an electrostatically anisotropic hydrogel. Nat. Mater. 14, 1002–1007 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Beebe D. J., Moore J. S., Bauer J. M., Yu Q., Liu R. H., Devadoss C., Jo B.-H., Functional hydrogel structures for autonomous flow control inside microfluidic channels. Nature 404, 588–590 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Takashima Y., Hatanaka S., Otsubo M., Nakahata M., Kakuta T., Hashidzume A., Yamaguchi H., Harada A., Expansion–contraction of photoresponsive artificial muscle regulated by host–guest interactions. Nat. Commun. 3, 1270–1278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee K. Y., Mooney D. J., Hydrogels for tissue engineering. Chem. Rev. 101, 1869–1880 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Larson C., Peele B., Li S., Robinson S., Totaro M., Beccai L., Mazzolai B., Shepherd R., Highly stretchable electroluminescent skin for optical signaling and tactile sensing. Science 351, 1071–1074 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Bauer S., Bauer-Gogonea S., Graz I., Kaltenbrunner M., Keplinger C., Schwödiauer R., 25th anniversary article: A soft future: From robots and sensor skin to energy harvesters. Adv. Mater. 26, 149–162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keplinger C., Sun J.-Y., Foo C. C., Rothemund P., Whitesides G. M., Suo Z., Stretchable, transparent, ionic conductors. Science 341, 984–988 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Sidorenko A., Krupenkin T., Taylor A., Fratzl P., Aizenberg J., Reversible switching of hydrogel-actuated nanostructures into complex micropatterns. Science 315, 487–490 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Dong L., Agarwal A. K., Beebe D. J., Jiang H., Adaptive liquid microlenses activated by stimuli-responsive hydrogels. Nature 442, 551–554 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Ghosh A., Yoon C., Ongaro F., Scheggi S., Selaru F. M., Misra S., Gracias D. H., Stimuli-responsive soft untethered grippers for drug delivery and robotic surgery. Front. Mech. Eng. 3, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morin S. A., Shepherd R. F., Kwok S. W., Stokes A. A., Nemiroski A., Whitesides G. M., Camouflage and display for soft machines. Science 337, 828–832 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Blacklow S. O., Li J., Freedman B. R., Zeidi M., Chen C., Mooney D. J., Bioinspired mechanically active adhesive dressings to accelerate wound closure. Sci. Adv. 5, eaaw3963 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuk H., Lin S., Ma C., Takaffoli M., Fang N. X., Zhao X., Hydraulic hydrogel actuators and robots optically and sonically camouflaged in water. Nat. Commun. 8, 14230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palleau E., Morales D., Dickey M. D., Velev O. D., Reversible patterning and actuation of hydrogels by electrically assisted ionoprinting. Nat. Commun. 4, 2257 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Haraguchi K., Li H.-J., Control of the coil-to-globule transition and ultrahigh mechanical properties of PNIPA in nanocomposite hydrogels. Angew. Chem. Int. Ed. 44, 6500–6504 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Mirfakhrai T., Madden J. D. W., Baughman R. H., Polymer artificial muscles. Mater. Today 10, 30–38 (2007). [Google Scholar]
- 17.Fei R., George J. T., Park J., Grunlan M. A., Thermoresponsive nanocomposite double network hydrogels. Soft Matter 8, 481–487 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng S. Y., Shen Y., Zhu F., Yin J., Qian J., Fu J., Wu Z. L., Zheng Q., Programmed deformations of 3D-printed tough physical hydrogels with high response speed and large output force. Adv. Funct. Mater. 28, 1803366 (2018). [Google Scholar]
- 19.Godt R. E., Lindley B. D., Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J. Gen. Physiol. 80, 279–297 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Astley H. C., Roberts T. J., Evidence for a vertebrate catapult: Elastic energy storage in the plantaris tendon during frog jumping. Biol. Lett. 8, 386–389 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutz G. J., Rome L. C., Built for jumping: The design of the frog muscular system. Science 263, 370–372 (1994). [DOI] [PubMed] [Google Scholar]
- 22.Aeles J., Lichtwark G., Peeters D., Delecluse C., Jonkers I., Vanwanseele B., Effect of a prehop on the muscle-tendon interaction during vertical jumps. J. Appl. Physiol. 124, 1203–1211 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Löwenberg C., Balk M., Wischke C., Behl M., Lendlein A., Shape-memory hydrogels: Evolution of structural principles to enable shape switching of hydrophilic polymer networks. Acc. Chem. Res. 50, 723–732 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Liang R., Wang L., Yu H., Khan A., Ul Amin B., Khan R. U., Molecular design, synthesis and biomedical applications of stimuli-responsive shape memory hydrogels. Eur. Polym. J. 114, 380–396 (2019). [Google Scholar]
- 25.Choi S., Choi Y., Kim J., Anisotropic hybrid hydrogels with superior mechanical properties reminiscent of tendons or ligaments. Adv. Funct. Mater. 29, 1904342 (2019). [Google Scholar]
- 26.Kabb C. P., O’Bryan C. S., Deng C. C., Angelini T. E., Sumerlin B. S., Photoreversible covalent hydrogels for soft-matter additive manufacturing. ACS Appl. Mater. Interfaces 10, 16793–16801 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Mredha M. T. I., Guo Y. Z., Nonoyama T., Nakajima T., Kurokawa T., Gong J. P., A facile method to fabricate anisotropic hydrogels with perfectly aligned hierarchical fibrous structures. Adv. Mater. 30, 1704937 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Ye D., Yang P., Lei X., Zhang D., Li L., Chang C., Sun P., Zhang L., Robust anisotropic cellulose hydrogels fabricated via strong self-aggregation forces for cardiomyocytes unidirectional growth. Chem. Mater. 30, 5175–5183 (2018). [Google Scholar]
- 29.Sun M., Bai R., Yang X., Song J., Qin M., Suo Z., He X., Hydrogel interferometry for ultrasensitive and highly selective chemical detection. Adv. Mater. 30, 1804916 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Liu J., Yang C., Yin T., Wang Z., Qu S., Suo Z., Polyacrylamide hydrogels. II. Elastic dissipater. J. Mech. Phys. Solids 133, 103737 (2019). [Google Scholar]
- 31.Lin P., Ma S., Wang X., Zhou F., Molecularly engineered dual-crosslinked hydrogel with ultrahigh mechanical strength, toughness, and good self-recovery. Adv. Mater. 27, 2054–2059 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Li G., Gao T., Fan G., Liu Z., Liu Z., Jiang J., Zhao Y., Photoresponsive shape memory hydrogels for complex deformation and solvent-driven actuation. ACS Appl. Mater. Interfaces 12, 6407–6418 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Lin P., Zhang T., Wang X., Yu B., Zhou F., Freezing molecular orientation under stretch for high mechanical strength but anisotropic hydrogels. Small 12, 4386–4392 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Spychaj T., Schmidt B., Polymeric systems based on poly(acrylic acid) and trivalent metal cations. Macromol. Symp. 152, 173–189 (2000). [Google Scholar]
- 35.Gao Y., Wu K., Suo Z., Photodetachable adhesion. Adv. Mater. 31, e1806948 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Olsen R. A., Lee E. H., Krizekl D. T., Brownl J. C., Photochemical reduction of iron. II. Plant related factors. J. Plant Nutr. 5, 335–344 (1982). [Google Scholar]
- 37.Peng F., Li G., Liu X., Wu S., Tong Z., Redox-responsive gel–sol/sol–gel transition in poly(acrylic acid) aqueous solution containing Fe(III) ions switched by light. J. Am. Chem. Soc. 130, 16166–16167 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Ming Z., Pang Y., Liu J., Switching between elasticity and plasticity by network strength competition. Adv. Mater. 32, 1906870 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Ma M., Guo L., Anderson D. G., Langer R., Bio-inspired polymer composite actuator and generator driven by water gradients. Science 339, 186–189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia L.-W., Xie R., Ju X.-J., Wang W., Chen Q., Chu L.-Y., Nano-structured smart hydrogels with rapid response and high elasticity. Nat. Commun. 4, 2226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cvetkovic C., Raman R., Chan V., Williams B. J., Tolish M., Bajaj P., Sakar M. S., Asada H. H., Saif M. T. A., Bashir R., Three-dimensionally printed biological machines powered by skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 111, 10125–10130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sano K., Ishida Y., Aida T., Synthesis of anisotropic hydrogels and their applications. Angew. Chem. Int. Ed. 57, 2532–2543 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Fang Y., Han E., Zhang X. X., Jiang Y., Lin Y., Shi J., Wu J., Meng L., Gao X., Griffin P. J., Xiao X., Tsai H. M., Zhou H., Zuo X., Zhang Q., Chu M., Zhang Q., Gao Y., Roth L. K., Bleher R., Ma Z., Jiang Z., Yue J., Kao C. M., Chen C. T., Tokmakoff A., Wang J., Jaeger H. M., Tian B., Dynamic and programmable cellular-scale granules enable tissue-like materials. Matter 2, 948–964 (2020). [Google Scholar]
- 44.Lin S., Liu J., Liu X., Zhao X., Muscle-like fatigue-resistant hydrogels by mechanical training. Proc. Natl. Acad. Sci. U.S.A. 116, 10244–10249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun J.-Y., Zhao X., Illeperuma W. R. K., Chaudhuri O., Oh K. H., Mooney D. J., Vlassak J. J., Suo Z., Highly stretchable and tough hydrogels. Nature 489, 133–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts T. J., Marsh R. L., Probing the limits to muscle-powered accelerations: Lessons from jumping bullfrogs. J. Exp. Biol. 206, 2567–2580 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Taylor D., O’Mara N., Ryan E., Takaza M., Simms C., The fracture toughness of soft tissues. J. Mech. Behav. Biomed. Mater. 6, 139–147 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabd2520/DC1