

Brain atrophy in human epilepsy syndromes is explainable by network architecture and strongest in hub regions.
Abstract
Epilepsy is increasingly conceptualized as a network disorder. In this cross-sectional mega-analysis, we integrated neuroimaging and connectome analysis to identify network associations with atrophy patterns in 1021 adults with epilepsy compared to 1564 healthy controls from 19 international sites. In temporal lobe epilepsy, areas of atrophy colocalized with highly interconnected cortical hub regions, whereas idiopathic generalized epilepsy showed preferential subcortical hub involvement. These morphological abnormalities were anchored to the connectivity profiles of distinct disease epicenters, pointing to temporo-limbic cortices in temporal lobe epilepsy and fronto-central cortices in idiopathic generalized epilepsy. Negative effects of age on atrophy further revealed a strong influence of connectome architecture in temporal lobe, but not idiopathic generalized, epilepsy. Our findings were reproduced across individual sites and single patients and were robust across different analytical methods. Through worldwide collaboration in ENIGMA-Epilepsy, we provided deeper insights into the macroscale features that shape the pathophysiology of common epilepsies.
INTRODUCTION
Perpetual interactions among neuronal populations through the scaffold of axonal pathways promote interregional communication and shape the brain’s structural and functional network organization (1). This architecture facilitates efficient communication within the brain and may therefore be profoundly affected by pathological perturbations (2). Adopting network science can advance understanding of widespread pathophysiological effects in prevalent disorders and improve diagnostics and prognostication.
The application of neuroimaging to study common epilepsy syndromes has paradigmatically shifted from a focus on individual regions to approaches highlighting network effects, exemplified by classically defined focal epilepsies, such as temporal lobe epilepsy [TLE; (3, 4)]. While initial work focused on the mesiotemporal lobe, histopathological and neuroimaging studies increasingly detail morphological, structural, and functional compromise beyond this region (5–8), which becomes progressively more severe in patients with longer disease duration (9–12). Conversely, idiopathic generalized epilepsy (IGE), also known as genetic generalized epilepsy, has been increasingly linked to subtle degrees of structural compromise, mainly in subcortico-cortical circuits (13–16). Support for a network perspective also comes from both experimental studies in animal models and electro-clinical observations in patients showing bursts of spike and slow-wave discharges occurring simultaneously over subcortical and cortical areas (17, 18). Complementing these observations, basal ganglia atrophy as well as functional changes of the caudate and putamen have been previously noted but warrant further investigation to solidify the understanding of network disruptions in IGE (19, 20).
Initiatives such as the Human Connectome Project [HCP; (21)] provide normative structural and functional connectivity information from a large sample of healthy individuals. Studying network underpinnings of morphological abnormalities may elucidate brain-wide mechanisms in focal and generalized epilepsies. Hub regions (i.e., nodes with many connections) are a cardinal feature of brain networks and serve as relays to efficiently process and integrate information (22, 23). Their high centrality, however, also makes them vulnerable to pathological processes—the so-called nodal stress hypothesis (2). Neurodegenerative and psychiatric research has demonstrated that hubs typically show greater atrophy than locally connected peripheral nodes. This increased susceptibility to structural damage likely stems from their high metabolic activity and their association with multiple brain networks (1, 24). We anticipate that models of regional susceptibility can yield substantial advances toward our understanding of how connectome architecture configures, to some extent, gray matter atrophy in the common epilepsies.
Complementing the nodal stress hypothesis, in which patterns of cortical atrophy and hub regions appear spatially concomitant, disease epicenter mapping can identify one or more specific regions—or epicenters—whose connectivity profile may play a central role in the whole-brain manifestation of focal and generalized epilepsies (24, 25). Among common epilepsies, application of these models to TLE and IGE is justified as both syndromes have been associated with pathophysiological anomalies in mesiotemporal and subcortico-cortical networks and represent conceptual extremes of a focal to generalized continuum of epilepsy subtypes (14, 15, 26). Disease epicenter mapping in TLE and IGE may therefore identify syndrome-specific network-level substrates and provide deeper insights into how epilepsy-related atrophy patterns are anchored to the connectivity of specific structural and functional subnetworks.
The current work tested the hypothesis that brain network architecture governs whole-brain atrophy in TLE and IGE. Cortical and subcortical gray matter atrophy patterns were mapped across 19 international sites via ENIGMA-Epilepsy (27). We also leveraged the HCP dataset to derive high-resolution structural and functional normative brain networks. Two classes of network-based models tested whether, and how, healthy connectome architecture can predict regional susceptibility in the common epilepsies. Our evaluations included (i) nodal stress models, which assessed whether there is a selective vulnerability of hub regions that parallels syndrome-specific atrophy patterns, and (ii) disease epicenter mapping, which explored the influence of every brain region’s connectivity profile on the spatial distribution of gray matter atrophy in TLE and IGE. In both cases, model fit was assessed against null models with similar spatial autocorrelation (28). To demonstrate clinical relevance, we investigated whether these network-level features could predict spatial patterns of disease duration and age-related effects. We also formulated a patient-tailored adaptation of our network-based models to examine whether network-derived spatial predictors were translatable to individual patients.
RESULTS
Data samples
We studied 1021 adult patients with epilepsy (440 males, mean age ± SD = 36.72 ± 11.07 years) and 1564 healthy controls (695 males, mean age ± SD = 33.13 ± 10.45 years) from 19 centers in the international Epilepsy Working Group of ENIGMA (29). Our main analyses focused on two patient subcohorts with site-matched healthy controls: TLE with neuroradiological evidence of hippocampal sclerosis (nHC/TLE = 1418/732, 341 right-sided focus) and IGE (nHC/IGE = 1075/289). Details on subject inclusion and case-control subcohorts are provided in Materials and Methods and Table 1. Site-specific demographic information is provided in table S1. All participants were aged between 18 and 70 years.
Table 1. ENIGMA Epilepsy Working Group demographics.
Demographic breakdown of patient-specific subcohorts with site-matched controls, including age (in years), age at onset of epilepsy (in years), sex, side of seizure focus (patients with TLE only), and mean duration of illness (in years). Healthy controls from sites that did not have TLE (or IGE) patients were excluded from analyses comparing TLE (or IGE) to controls.
|
Case-control subcohorts |
Age (means ± SD) |
Age at onset (means ± SD) |
Sex (male/female) | Side of focus (L/R) |
Duration of illness (means ± SD) |
| TLE (n = 732) | 38.56 ± 10.61 | 16.07 ± 12.27 | 329/403 | 391/341 | 22.74 ± 14.06* |
| HC (n = 1418) | 33.76 ± 10.54 | – | 643/775 | – | – |
| IGE (n = 289) | 32.06 ± 10.85 | 16.84 ± 11.25 | 111/178 | – | 15.09 ± 11.70* |
| HC (n = 1075) | 31.41 ± 9.59 | – | 454/621 | – | – |
*Information available in 695 of 732 patients with TLE and 250 of 289 patients with IGE.
Cortical and subcortical atrophy in the common epilepsies
While the original ENIGMA-Epilepsy study performed a meta-analysis of statistical results submitted by the individual sites, the current study directly analyzed cortical surface and subcortical volume data in all patients and controls. Cortical thickness was measured across 68 gray matter brain regions, and volumetric measures were obtained from 12 subcortical gray matter regions and bilateral hippocampi based on the Desikan-Killiany anatomical atlas (30). Surface-based linear models compared atrophy profiles in patients relative to controls, correcting for multiple comparisons using the false discovery rate (FDR) procedure (31).
Mirroring ENIGMA-Epilepsy’s meta-analysis of summary statistics comparing neurologically healthy controls to patients with epilepsy, our mega-analysis also revealed widespread cortico-subcortical atrophy patterns in TLE and IGE syndromes. Specifically, patients with TLE showed profound atrophy in bilateral precuneus (PFDR < 4 × 10−36), precentral (PFDR < 8 × 10−36), paracentral (PFDR < 6 × 10−29), and superior temporal (PFDR < 3 × 10−14) cortices as well as ipsilateral hippocampus (PFDR < 2 × 10−199) and thalamus (PFDR < 5 × 10−64; Fig. 1A). In contrast, patients with IGE showed atrophy predominantly in bilateral precentral cortices (PFDR < 9 × 10−10) and the thalamus (PFDR < 3 × 10−6; Fig. 1B).
Fig. 1. Cortical thickness and subcortical volume in TLE and IGE.
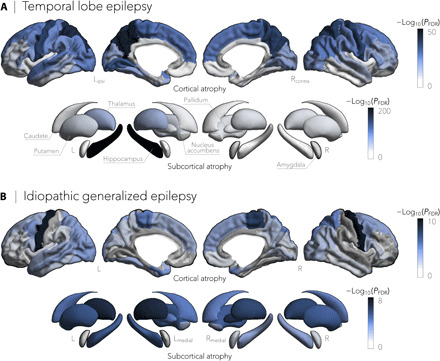
(A) Cortical thickness and subcortical volume reductions in TLE (n = 732), compared to healthy controls (n = 1418), spanned bilateral precuneus (PFDR < 4 × 10−36), precentral (PFDR < 8 × 10−36), paracentral (PFDR < 6 × 10−29), and superior temporal (PFDR < 3 × 10−14) cortices and ipsilateral hippocampus (PFDR < 2 × 10−199) and thalamus (PFDR < 5 × 10−64). (B) In contrast, gray matter cortical and subcortical atrophy in IGE (n = 289), relative to controls (n = 1075), was more subtle and affected predominantly bilateral precentral cortical regions (PFDR < 9 × 10−10) and the thalamus (PFDR < 3 × 10−6). Negative log10-transformed FDR-corrected P values are shown.
Nodal stress models predict regional susceptibility
Having established patterns of atrophy in TLE and IGE, we evaluated whether these abnormalities were associated with normative network organization. To this end, we obtained high-resolution structural (derived from diffusion-weighted tractography) and functional [derived from resting-state functional magnetic resonance imaging (fMRI)] connectivity data from a cohort of unrelated healthy adults from the HCP dataset (21). Details on subject inclusion and matrix generation are provided in Materials and Methods.
Echoing established network centrality maps in healthy individuals (22, 23), hub regions in the HCP dataset predominated in medial prefrontal, superior parietal, and angular regions (Fig. 2A). Nodal stress models, in which spatial similarity between syndrome-related atrophy patterns and degree centrality was compared through correlation analysis (and statistically assessed via nonparametric spin permutation tests; see Materials and Methods), revealed that cortical thinning in TLE implicated functional [correlation coefficient (r) = 0.69, Pspin < 0.0005] and marginally structural (r = 0.27, Pspin = 0.07) cortico-cortical hubs more strongly than nonhub regions (Fig. 2B). In contrast, in IGE, stronger relationships were observed between subcortical volume decreases and structural (r = 0.68, Pshuf < 0.01) and marginally functional (r = 0.50, Pshuf = 0.06) subcortico-cortical hubs (Fig. 2B). To verify stability, we repeated the above correlations across several graph-based nodal metrics (including pagerank centrality and eigenvector centrality) and captured virtually identical associations between atrophy patterns and network centrality measures (fig. S1).
Fig. 2. Epilepsy-related atrophy correlates with hub organization.
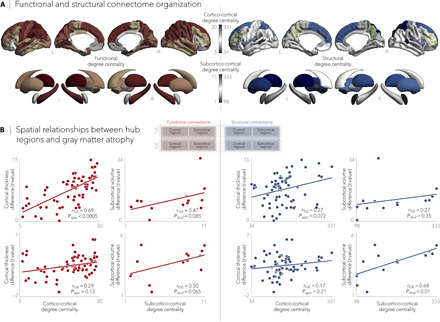
(A) Normative functional and structural network organization, derived from the HCP dataset, was used to identify hubs (i.e., regions with greater degree centrality). (B) Schematic of the figure layout is pictured in the middle. Gray matter atrophy related to node-level functional (left) and structural (right) maps of degree centrality, with greater atrophy in hub compared to nonhub regions. Stratifying findings across TLE and IGE, we observed stronger associations between cortico-cortical functional hubs and cortical atrophy patterns in TLE (Pspin < 0.0001) and between subcortical volume loss and subcortico-cortical structural hubs in IGE (Pshuf < 0.01).
TLE and IGE have distinct disease epicenters
Since hub regions are more susceptible to atrophy than nonhub regions, we next investigated whether epilepsy-related cortical thickness abnormalities were anchored to the connectivity profile of one or more brain regions. To detect syndrome-specific disease epicenters, we systematically compared every region’s functional and structural connectivity profiles to whole-brain patterns of atrophy in TLE and IGE and assessed significance of rankings using spin permutation tests. Cortical and subcortical regions were ranked in descending order based on their correlation coefficients, with highly ranked—and statistically significant—regions being identified as disease epicenters. Disease epicenters thus represented regions whose functional and structural connectivity profile spatially resembled the epilepsy-related atrophy maps (Fig. 3A).
Fig. 3. Syndrome-specific disease epicenters.
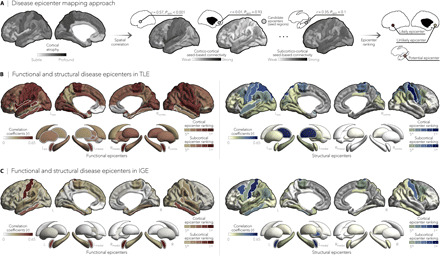
(A) Disease epicenter mapping schema. Spatial correlations between cortical atrophy patterns and seed-based cortico- and subcortico-cortical connectivity were used to identify disease epicenters in TLE and IGE. Epicenters are regions whose connectivity profiles significantly correlated with the syndrome-specific atrophy map; statistical significance was assessed using spin permutation tests. This procedure was repeated systematically to assess the epicenter value of every cortical and subcortical region, as well as in both functional and structural connectivity matrices. (B and C) Correlation coefficients indexing spatial similarity between TLE- and IGE-specific atrophy and seed-based functional (left) and structural (right) connectivity measures for every cortical and subcortical region. Regions with significant associations were ranked in descending order based on their correlation coefficients, with the first five regions identified as disease epicenters (white outline). In TLE, ipsilateral temporo-limbic cortices (functional: Pspin < 0.05, structural: Pspin < 0.1) and subcortical areas—including ipsilateral amygdala (functional: Pspin < 0.05), thalamus (functional: Pspin < 0.05, structural: Pspin < 0.01), pallidum (functional: Pspin < 0.05), putamen (functional: Pspin < 0.05), and hippocampus (functional: Pspin < 0.1)—emerged as disease epicenters. In IGE, the highest ranked disease epicenters were located in bilateral fronto-central cortices, including postcentral gyri (functional: Pspin < 0.05, structural: Pspin < 0.05), left (functional: Pspin < 0.005, structural: Pspin < 0.1) and right amygdala (functional: Pspin < 0.005), and left pallidum (structural: Pspin < 0.1). *Pspin < 0.1, n.s., nonsignificant.
In TLE, spatial correlations between atrophy maps and seed-based functional and structural connectivity profiles implicated ipsilateral temporo-limbic cortices (Pspin < 0.05) and several ipsilateral subcortical regions as disease epicenters (Pspin < 0.05; Fig. 3B). Conversely, bilateral fronto-central cortices and the amygdala emerged as epicenters in IGE (Pspin < 0.05; Fig. 3C). Although highest ranked functional and structural epicenters in TLE and IGE were not hubs themselves (as defined by the top 10% of high-degree nodes), they were significantly connected to hub regions (range of spatial correlations between epicenter-based connectivity and maps of degree centrality: rfunctional = 0.67 to 0.77, Pspin < 0.0001; rstructural = 0.27 to 0.31, Pspin < 0.09), effectively making them feeder nodes, that is, peripheral nodes that are directly linked to hubs.
Effects of age and disease duration
We adapted the above-described nodal stress and epicenter mapping models to disentangle network factors that contribute to cross-sectional markers of age and disease duration effects. Using linear models, we first examined effects of age and disease duration on morphological measures within each patient cohort, paralleling approaches from earlier work (12). Patients with TLE showed a negative effect of age on cortical thickness primarily in bilateral temporo-parietal (PFDR < 0.005) and sensorimotor cortices (PFDR < 0.01) as well as on subcortical volume mainly in ipsilateral hippocampus (PFDR < 5×10−7) and bilateral thalamus (PFDR < 0.05; Fig. 4A). Comparing these patterns to structural and functional degree centrality and epicenter maps, strongest correlations were seen with functional cortico- and subcortico-cortical hubs (rs = 0.64 to 0.65, Pshuf/spin < 0.05) and with functional and structural cortical disease epicenters (rs = 0.44 to 0.66, Pspin < 0.05; Fig. 4B). The main effect of disease duration on atrophy patterns colocalized with regions emerging from the group × age interaction (e.g., bilateral superior parietal cortex, contralateral postcentral gyrus, and ipsilateral hippocampus and thalamus, PFDR < 0.01; fig. S2A). While duration effects also showed tendencies for positive correlations with normative network centrality and epicenter values, effects were not significant (fig. S2B).
Fig. 4. Negative effects of age on cortical thickness and subcortical volume in TLE.
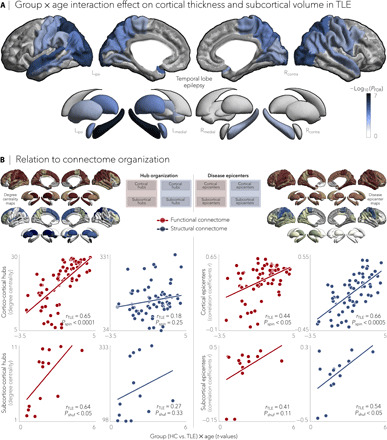
(A) Significant age-related differences on gray matter atrophy between individuals with TLE and healthy controls for all cortical and subcortical regions. Patients with TLE showed a negative effect of age on cortical thickness in bilateral temporo-parietal (PFDR < 0.005) and sensorimotor (PFDR < 0.01) cortices and on subcortical volume in ipsilateral hippocampus (PFDR < 5 × 10−7) and bilateral thalamus (PFDR < 0.05). Negative log10-transformed FDR-corrected P values are shown. (B) Schematic of the figure layout is provided in the middle. Scatterplots depict relationships between the age-related effects and functional (red) and structural (blue) maps of degree centrality (left) and disease epicenter (right). Significant associations were observed between age-related effects and every hub and epicenter measures, with the exception of structural subcortical degree centrality, suggesting a role of connectome organization on age-related effects in TLE.
In IGE, there were no significant effects of aging (fig. S3A) or disease duration (fig. S4A) on cortical and subcortical atrophy measures nor of correlations with degree centrality and epicentral maps (figs. S3B and S4B). There was a trend toward the negative effects of aging and duration of illness on gray matter atrophy, particularly affecting bilateral temporo-parietal and fronto-central cortices (Puncorr < 0.05), as well as right amygdala (Puncorr < 0.1). Exploring the effects of age at onset on morphological abnormalities yielded virtually identical findings in TLE (fig. S5A) and IGE (fig. S5B), independently.
Patient-tailored atrophy modeling
We adjusted our network-based models to assess whether normative connectivity organization configures atrophy patterns in individual patients. To do so, we first correlated patient-specific gray matter atrophy maps with normative centrality metrics. We subsequently identified each patient’s structural and functional disease epicenters by keeping brain regions whose healthy connectivity profiles significantly correlated with the patient’s atrophy map.
Although this was expected to result in lower sensitivity due to the increased heterogeneity in atrophy patterns, we observed similar associations between individualized atrophy maps and cortical degree centrality as seen in the group-level analysis. Notably, most stable associations in patients with TLE were seen between functional cortico-cortical hubs and cortical atrophy (Pspin < 0.05 in 22.4% of individuals with TLE), whereas most stable relations in patients with IGE were observed between structural cortico-cortical hubs and cortical atrophy (Pspin < 0.05 in 15.2% of individuals with IGE; Fig. 5A). Similarly, epicenter results were consistently seen in individual patients, with ipsilateral temporo-limbic and subcortical regions most often emerging as epicenters in TLE, whereas bilateral fronto-central regions, including sensorimotor cortices, were most often identified as epicenters in IGE (Fig. 5B).
Fig. 5. Patient-tailored atrophy modeling.

(A) Patient-specific associations between degree centrality (denoting hub distribution) and individualized atrophy maps showed high stability between functional cortico-cortical hubs and cortical atrophy in TLE (Pspin < 0.05 in 22.4% of patients) and high stability between structural cortico-cortical hubs and cortical atrophy in IGE (Pspin < 0.05 in 15.2% of patients). (B) We identified patient-specific structural and functional disease epicenters by keeping brain regions whose connectivity profiles significantly correlated with the patient’s atrophy map (Pspin < 0.05). In TLE, ipsilateral temporo-limbic regions and subcortical areas (including the hippocampus) were most often identified as epicenters of gray matter atrophy, whereas in IGE, bilateral fronto-central (including sensorimotor cortices) and subcortical areas most often emerged as disease epicenters. Disease epicenters in individual patients strongly resembled those seen across the group as a whole.
Findings were robust across a range of sensitivity analyses
Dominant patterns of atrophy in bilateral precuneus (PFDR < 2 × 10−25), superior parietal (PFDR < 3 × 10−24), and precentral (PFDR < 5 × 10−22) cortices as well as ipsilateral hippocampus (PFDR < 8 × 10−125) and thalamus (PFDR < 6 × 10−37; fig. S6A) were observed when comparing left and right TLE cohorts separately to controls. Left TLE additionally showed gray matter reductions in ipsilateral lateral and mesiotemporal cortices (PFDR < 2 × 10−15) and the bilateral thalamus (PFDR < 6 × 10−23). This distinction did not affect the relationship between atrophy patterns and functional cortico-cortical hub distribution (rLTLE = 0.61, Pspin < 0.005; rRTLE = 0.72, Pspin < 0.0001; fig. S6B). Functional and structural epicenters in both left and right TLE were localized in mesiotemporal and sensorimotor cortices (fig. S6C), but differed across subcortical areas, with the ipsilateral hippocampus emerging as putative disease epicenter in left, but not right, TLE (rfunctional = 0.38, Pspin = 0.05; rstructural = 0.30, Pspin = 0.09). Although negative effects of age on cortical thickness in bilateral visual (PFDR < 0.001) and superior temporal (PFDR < 0.005) cortices were common to both left and right TLE, right TLE showed more widespread group × age interaction effects, particularly affecting bilateral parietal cortex (PFDR < 0.01) and the ipsilateral hippocampus (PFDR < 3×10−8), amygdala (PFDR < 0.001), and thalamus (PFDR < 0.005). Nevertheless, associations between age-related effects and hub and disease epicenter distributions remained virtually identical in left and right TLE (fig. S6D).
Syndrome-specific cortical and subcortical atrophy maps were consistent across sites and similar to those obtained from the multisite aggregation for both TLE (fig. S7A) and IGE (fig. S7B). The highest stability in TLE was observed for correlations of atrophy with functional cortico- and subcortico-cortical degree centrality (means ± SD: r = 0.33 ± 0.24 and means ± SD: r = 0.43 ± 0.26, respectively; fig. S7C). Conversely, stability in IGE was the highest for correlations with structural subcortico-cortical degree centrality (means ± SD: r = 0.28 ± 0.22; fig. S7D). As observed in the multisite findings, site-specific ipsilateral temporo-limbic regions and subcortical areas were most often identified as disease epicenters in TLE (fig. S7E), whereas bilateral fronto-central cortices, amygdala, and thalamus most frequently emerged as disease epicenters in IGE (fig. S7F).
Our findings were not affected by varying specific matrix thresholds: Across the range of possible thresholds, we observed highly consistent associations between atrophy patterns and (i) functional cortico-cortical hubs (TLE) and structural subcortico-cortical hubs (IGE; fig. S8A) and (ii) functional and structural epicenter-based connectivity profiles (fig. S8B). Moreover, we observed comparable associations between morphological abnormalities and hub (fig. S9A) and epicenter (fig. S9, B and C) distributions obtained from a higher-resolution functional parcellation [Schaefer 400 parcels (32)].
DISCUSSION
Capitalizing on the largest multisite epilepsy neuroimaging dataset to date, we tested the hypothesis that gray matter atrophy patterns in TLE and IGE are related to the brain’s connectome architecture. First, we found that hub regions were overall more susceptible to epilepsy-related atrophy in both TLE and IGE, with cortical hubs most affected in TLE and subcortico-cortical hubs most vulnerable in IGE. These morphological abnormalities were anchored to the connectivity profiles of distinct regions, pointing to temporo-limbic disease epicenters in TLE and fronto-central cortices epicenters in IGE. Assessing negative effects of age on atrophy further revealed a dichotomy between TLE and IGE, with a stronger influence of connectome architecture on how the disease unfolds in TLE. Using a patient-tailored adaptation of our network-based models, we confirmed that relationships between atrophy and normative connectivity organization were translatable to individual patients. Findings were highly consistent across sites and methodologies, suggesting robustness and generalizability.
Our study extends prior research by revealing shared and distinct network descriptors of atrophy seen in TLE and IGE (29). Using a mega-analytic approach, we showed that patients with TLE presented with multi-lobar atrophy, affecting fronto-parietal cortices, the hippocampus, and the thalamus, extending into lateral temporal and mesiotemporal cortices in left, but not right, TLE. In contrast, patients with IGE presented with more subtle atrophy in the sensorimotor cortex and thalamus, despite having “normal” clinical MRIs (29, 33). While prior works in histology, electrophysiology, and neuroimaging have provided qualitative descriptions of disease, here, we developed network-based models to systematically examine the selective vulnerability of brain regions in the common epilepsies. This approach revealed that epilepsy-related atrophy patterns could be predicted from connectome-derived information, suggesting a vulnerability of functional cortical hubs to more pronounced atrophy in TLE, regardless of seizure focus laterality, and an increased susceptibility of structural subcortical hubs in IGE. Albeit less pronounced, similar trends were also observed for an increased vulnerability of structural cortical hubs in TLE and functional subcortical hubs in IGE. This implies that TLE and IGE—despite showing syndrome-specific atrophy patterns—may share common pathophysiological features that result in greater atrophy severity in hub regions. Irrespective of cortical or subcortical involvement, the vulnerability of hub regions to gray matter atrophy can be partly ascribed to their disproportionate number of connections and diffuse effect on structural and functional networks (2). Thus, a targeted hub attack, as in the case of epilepsy-related atrophy, may fragment and segregate the network, favoring recurrent seizure activity in TLE and IGE (34, 35).
To elucidate how gray matter atrophy targets hub regions in TLE and IGE, we tested the hypothesis that the underlying connectivity of specific regions—or epicenters—constrains syndrome-specific patterns of atrophy. Each epilepsy syndrome harbored distinct epicenters reflecting its own pathophysiology: Temporo-limbic cortical regions, ipsilateral to the seizure focus, emerged as epicenters of atrophy in TLE, whereas fronto-central cortices, specifically sensorimotor areas, were identified as disease epicenters in IGE. Although the hippocampus is often considered central to seizure generation and propagation in TLE (36), it was only identified as a disease epicenter in left TLE, likely due to its strong connections to lateral temporal and mesiotemporal cortex—regions of high atrophy in left, but not right, TLE. Marked damage in other mesiotemporal and limbic structures such as the entorhinal, piriform, and temporo-polar cortex has also been observed in subgroups of TLE and may thus contribute to variable disease expression within the broader umbrella of TLE (5, 37). Moreover, syndrome-specific epicenters were not hub regions themselves but were found to be functionally and structurally connected to hubs via feeder connections, suggesting differential mechanisms through which atrophy may spread to hub regions in TLE and IGE. Our findings are compatible with a model where epilepsy-related atrophy initially targets syndrome-specific epicenters and subsequently propagates to hub regions, which are generally more susceptible than nonhubs due to a combination of hyperactivation, increased metabolic stress, and being at the crossroads of multiple brain networks (2). Future longitudinal studies, however, will be needed to provide causal support.
Similar to the hub susceptibility findings, our epicenter approach revealed a differential sensitivity of functional and structural connectivity to TLE- and IGE-specific pathophysiology. Specifically, network associations to functionally derived cortical hubs and disease epicenters were stronger in TLE, whereas associations to structurally derived subcortical hubs and disease epicenters were most significant in IGE. Diffusion MRI is particularly sensitive to strong long-range fiber bundles and direct monosynaptic structural connections but has, unlike resting-state functional MRI, limitations in resolving short-range intracortical and spatially distributed polysynaptic cortical systems (38). In this context, resting-state functional MRI allows for the detection of functional connectivity in the absence of direct structural connections and may thus be more informative about polysynaptic configurations (38). Functionally derived disease epicenters in TLE preferentially detected network associations in polysynaptic temporo-limbic areas, whereas structural epicenters converged in superior temporal and sensorimotor cortices. In contrast, disease epicenters in IGE appeared stable across both functional and structural connectomes, likely reflecting network alterations in regions with direct structural connections.
Taking advantage of large patient cohorts with heterogeneous duration of epilepsy and a wide age range, we could infer the effect of duration of illness on gray matter atrophy and separate out negative age-related effects in epilepsy from that of normal aging. In TLE, age × group interaction findings affected atrophy in temporo-parietal and sensorimotor cortices, as well as the ipsilateral hippocampus and thalamus, as has been previously noted (9–12). Our findings suggest that age-related pathophysiological processes follow the spatial distribution of hub and epicentral regions in temporal lobe, but not idiopathic generalized, epilepsy. We thus speculate that connectome architecture exerts a strong influence on the spread of epilepsy-related cortical damage in patients with TLE. Mechanisms underlying age-related effects on atrophy in TLE remain incompletely understood but may relate to a combination of factors, including seizure-related excitotoxicity and interictal epileptic activity (39), adverse effects from antiepileptic drugs (40), and psychosocial impairments (41). Age-related and disease duration effects on atrophy findings in IGE—and links to network centrality measures and disease epicenters—were less conclusive. More subtle effects on brain structure in IGE may reflect a less severe disease trajectory compared to focal epilepsy syndromes and may also arise from the intrinsic heterogeneity across generalized syndromes. It is also possible that generalized epileptiform discharges and seizures diffuse the negative consequences of recurrent pathophysiological activity and that no single neuronal population bears the brunt of this activity.
Several sensitivity analyses suggested that our findings were not affected by differences in scanners or sites or graph theoretical metrics. Site and scanner effects were mitigated for the most part using ComBat, a post-acquisition statistical batch normalization process used to harmonize between-site and between-protocol morphological variations (42). Associations between normative network features and morphological abnormalities, as well as the identification of disease epicenters, were also consistently observed in each site independently. Repeating our analyses across different graph theoretical metrics, matrix densities, and parcellation resolutions yielded virtually identical findings, indicating method invariance of our results. While our big data effort was predominantly based on group-level analyses, findings were replicated using patient-specific atrophy maps. Further developed, patient-tailored atrophy models may justify optimism in translating insights from large-scale inference to individual patients, with potential to improve personalized prognostics, treatment monitoring, and epilepsy surgery planning.
We wish to emphasize that our work was made possible by open-access consortia such as the ENIGMA Epilepsy Working Group (29) and the HCP (21). Although HCP provides a benchmark dataset for structural and functional connectomes in healthy adults, connectivity patterns obtained from this dataset may misrepresent the altered networked architecture typically observed in individuals with epilepsy (13, 43). Exploiting individualized structural and functional connectome information in patients therefore seems to be the logical next step to improve patient-tailored atrophy models, and ongoing and future initiatives to share epilepsy connectome data may facilitate this work. Higher-resolution cortical and subcortical parcellations, together with individualized connectivity data, may help to resolve the discrepancy between functional and structural findings observed in TLE and IGE. Similarly, increased resolution of subcortical data will help identify putative disease epicenters of subcortical volume loss and ultimately deepen our understanding of epilepsy syndromes characterized by widespread subcortical atrophy, such as IGE, or by profound hippocampal and thalamic atrophy, as in TLE. Last, the cross-sectional nature of these datasets limited the investigation of effects of disease progression to between-subject effects. Age- and disease-related effects reported here may also reflect an initial precipitating insult (e.g., prolonged febrile seizures or traumatic brain injury) and not per se the progression of the disease. Similarly, cross-sectional designs are not properly tailored to track gray matter loss over time nor its association to clinical features (12). Longitudinal imaging studies that follow both patients and controls are needed to dissociate progressive pathological changes from normal aging. Whether within-patient changes in cortical and subcortical atrophy are conditioned by network organization also remains an exciting open question and awaits further investigation, ideally in prospective and large-scale collaborative follow-up studies across the spectrum of common epilepsies (12). We hope that our study lays a foundation for future longitudinal studies to model the predictive path of atrophy in newly diagnosed patients and further results in network-based clinical tools that can help to understand the pathophysiology of common epilepsies.
MATERIALS AND METHODS
ENIGMA participants
We studied 1021 adults with epilepsy (440 males, mean age ± SD = 36.72 ± 11.07 years) and 1564 healthy controls (695 males, mean age ± SD = 33.13 ± 10.45 years) obtained from the Epilepsy Working Group of ENIGMA (29). Epilepsy specialists at each center diagnosed patients according to the seizure and syndrome classifications of the International League Against Epilepsy. Inclusion of adults with TLE was based on the combination of electroclinical features and MRI findings typically associated with underlying hippocampal sclerosis. Inclusion of adults with IGE was based on the presence of tonic-clonic, absence, or myoclonic seizures with generalized spike-wave discharges on electroencephalography. We excluded participants with a progressive disease (e.g., Rasmussen’s encephalitis), malformations of cortical development, tumors, or prior neurosurgery. Local institutional review boards and ethics committees approved each included cohort study, and written informed consent was provided according to local requirements.
Mega-analysis of cortical thickness and subcortical volumetric data
All participants underwent structural T1-weighted MRI scans at each of the 19 participating centers, with scanner descriptions and acquisition protocols detailed elsewhere (29). Images were independently processed by each center using the standard ENIGMA workflow. In brief, models of cortical and subcortical surface morphology were generated with FreeSurfer 5.3.0 (44). On the basis of the Desikan-Killiany anatomical atlas (30), cortical thickness was measured across 68 gray matter brain regions, and volumetric measures were obtained from 12 subcortical gray matter regions (bilateral amygdala, caudate, nucleus accumbens, pallidum, putamen, and thalamus) and bilateral hippocampi. Data were harmonized across scanners and sites and statistically corrected for age, sex, and intracranial volume using ComBat—a batch-effect correction tool that uses a Bayesian framework to improve the stability of the parameter estimates (42).
Cortical thickness and volumetric measures were z-scored relative to site-matched pooled controls and sorted into ipsilateral/contralateral to the focus. Surface-based linear models compared atrophy profiles in patients relative to controls using SurfStat, available at http://mica-mni.github.io/surfstat. Findings were corrected for multiple comparisons using the FDR procedure (31).
Spatial permutation tests
The intrinsic spatial smoothness in two given brain maps may inflate the significance of their spatial correlation (28). We thus assessed statistical significance of these spatial correlations using spin permutation tests. This framework generates null models of overlap between cortical maps by projecting the spatial coordinates of cortical data onto the surface spheres, applying randomly sampled rotations (10,000 repetitions), and reassigning connectivity values (28). The empirical (i.e., original) correlation coefficients are then compared against the null distributions determined by the ensemble of correlation coefficients comparing spatially permuted cortical maps. To compare spatial overlap between subcortical maps, we used a similar approach with the exception that subcortical labels were randomly shuffled as opposed to being projected onto spheres.
HCP participants and MRI preprocessing
We selected a group of unrelated healthy adults (n = 207, 83 males, mean age ± SD = 28.73 ± 3.73 years, range = 22 to 36 years) from the HCP dataset (21). HCP data were acquired on a Siemens Skyra 3T and included (i) T1-weighted images [magnetization-prepared rapid gradient echo sequence, repetition time (TR) = 2400 ms, echo time (TE) = 2.14 ms, field of view (FOV) = 224 × 224 mm2, voxel size = 0.7 mm3, 256 slices], (ii) resting-state fMRI [gradient-echo echo-planar imaging (EPI) sequence, TR = 720 ms, TE = 33.1 m, FOV = 208 × 180 mm2, voxel size = 2 mm3, 72 slices], and (iii) diffusion MRI (spin-echo EPI sequence, TR = 5520 ms, TE = 89.5 ms, FOV = 210 × 180, voxel size = 1.25 mm3, b-value = 1000/2000/3000 s/mm2, 270 diffusion directions, 18 b0 images).
HCP data underwent the initiative’s minimal preprocessing (45). In brief, resting-state fMRI data underwent distortion and head motion corrections, magnetic field bias correction, skull removal, intensity normalization, and were mapped to MNI152 space. Noise components attributed to head movement, white matter, cardiac pulsation, arterial, and large vein–related contributions were automatically removed using FIX (FMRIB’s ICA-based X-noiseifier) (46). Preprocessed time series were mapped to standard gray ordinate space using a cortical ribbon-constrained volume-to-surface mapping algorithm and subsequently concatenated to form a single time series. Diffusion MRI data underwent b0 intensity normalization and correction for susceptibility distortion, eddy currents, and head motion. As with the ENIGMA-Epilepsy dataset, high-resolution functional and structural data were parcellated according to the Desikan-Killiany atlas (30).
Functional and structural connectivity matrix generation
Normative functional connectivity matrices were generated by computing pairwise correlations between the time series of all 68 cortical regions and between all subcortical and cortical regions; negative connections were set to zero. Subject-specific connectivity matrices were then z-transformed and aggregated across participants to construct a group-average functional connectome.
Normative structural connectivity matrices were generated from preprocessed diffusion MRI data using MRtrix3 (47). Anatomically-constrained tractography was performed using different tissue types derived from the T1w image, including cortical and subcortical gray matter, white matter, and cerebrospinal fluid (48). Multishell and multitissue response functions were estimated (49), and constrained spherical deconvolution and intensity normalization were performed (50). The initial tractogram was generated with 40 million streamlines, with a maximum tract length of 250 and a fractional anisotropy cutoff of 0.06. Spherical-deconvolution informed filtering of tractograms (SIFT2) was applied to reconstruct whole-brain streamlines weighted by the cross-sectional multipliers (51). Reconstructed streamlines were mapped onto the 68 cortical and 14 subcortical (including hippocampus) regions to produce subject-specific structural connectivity matrices. The group-average normative structural connectome was defined using a distance-dependent thresholding procedure, which preserved the edge length distribution in individual patients (52), and was log transformed to reduce connectivity strength variance. Hence, structural connectivity was defined by the number of streamlines between two regions (i.e., fiber density).
Nodal stress models
Nodal stress models were derived from spatial correlation analyses between cortical and subcortical syndrome-specific atrophy profiles and normative weighted degree centrality maps. Weighted degree centrality was used here to identify structural and functional hub regions by computing the sum of all weighted cortico-cortical and subcortico-cortical connections for every region (higher degree centrality denotes a hub region). Because different centrality measures can describe different topological roles in brain networks, we replicated the spatial similarity analyses across other nodal metrics, including (i) pagerank centrality [proportional to the number of steps (or time) spent at each node] and (ii) eigenvector centrality (multiple of the sum of adjacent centralities; i.e., takes into account nodes that are connected to other highly central nodes). To avoid bias in choosing an arbitrary threshold and zeroing potentially useful information, these analyses were carried out on unthresholded connectivity matrices.
Mapping disease epicenters
Syndrome-specific epicenters were identified by spatially correlating every region’s healthy functional and structural connectivity profiles to whole-brain patterns of cortical atrophy in TLE and IGE (i.e., group-level atrophy maps obtained from surface-based linear models comparing these patient cohorts to controls). This approach was repeated systematically across the whole brain, assessing the statistical significance of the spatial similarity of every region’s functional and structural connectivity profiles to syndrome-specific atrophy maps with spatial permutation tests. Cortical and subcortical epicenter regions were then identified whether their connectivity profiles significantly correlated with the disease-specific cortical atrophy map; regions with significant associations were ranked in descending order based on their correlation coefficients, with highly ranked regions representing disease epicenters. Regardless of its atrophy level, a cortical or subcortical region could potentially be an epicenter if it is (i) strongly connected to other high-atrophy regions and (ii) weakly connected to low-atrophy regions. Moreover, disease epicenters also do not necessarily represent hub regions but may rather be connected to them (i.e., so-called feeder nodes that directly link peripheral nodes to hubs). As with the nodal stress models, we eliminated matrix thresholding to ensure that connectivity to every brain region was accounted for, thus allowing detection of disease epicenters in areas with subthreshold atrophy. Similarly, and because of the low granularity of the subcortical atlas (i.e., one value per subcortical structure), we restricted our epicenter approach to cortico-cortical and subcortico-cortical connectivity. A schematic of the cortical and subcortical disease epicenter mapping approach is displayed in Fig. 3A.
To relate disease epicenter findings to the nodal stress model, we performed spatial correlations between epicenter-based connectivity profiles (i.e., whole-brain connectivity seeding from the epicenter region) and maps of cortical degree centrality (showing the spatial distribution of hub regions); strong correlations indicated that disease epicenters were functionally and structurally connected to cortical hub regions. This analysis was individually performed on the two highest ranked functional and structural disease epicenters in TLE and IGE.
Age- and duration-related effects
To study the negative effects of age on gray matter atrophy profiles, we first built linear models that included a group and age main effect term and a group × age interaction term (11). We then evaluated age-related differences on cortical thickness and subcortical volume between patients and controls by testing the significance of the interaction term. Linear models independently assessed the effects of duration of epilepsy and age of onset on cortical thickness and subcortical volume measurements in each patient cohort. We then compared these age- and duration-related effect maps to the spatial distributions of hub regions and disease epicenters.
Patient-tailored atrophy modeling
Cortical thickness and subcortical volume data in patients were z-scored relative to healthy controls to generate individualized atrophy maps and were subsequently compared to normative network centrality metrics as in the above analysis. Patient-specific atrophy maps were also used to identify each patient’s structural and functional disease epicenters by keeping the cortical and subcortical regions whose normative connectivity profiles significantly correlated with the patient’s atrophy map. Significance testing for patient-specific epicenters were computed with spin permutation tests with 1000 repetitions.
Reproducibility and sensitivity analyses
Reproducibility in left and right TLE
As seizure focus lateralization may differentially affect the distribution of gray matter atrophy (29), we repeated the nodal stress, disease epicenter, and age-related analyses in left (nLTLE = 391) and right (nRTLE = 341) TLE independently.
Reproducibility across sites
To address reproducibility of our findings across different sites, we repeated the nodal stress models and disease epicenter analyses in each individual site that provided at least five participants per diagnostic group (nTLE/HC = 16 sites, nGE/HC = 10 sites).
Stability across matrix thresholds
To verify that results were not biased by the choice of a particular threshold, we repeated the network model analyses across the range of possible matrix thresholds (i.e., top 1 to 100%, with increments of 1%). Functional and structural (i) cortico-cortical and subcortico-cortical degree centrality maps and (ii) epicenter-based connectivity profiles (obtained from the highest ranked disease epicenter in each patient cohort) were derived from thresholded connectivity matrices (computed iteratively over all thresholds) and spatially correlated to the cohort-specific atrophy maps. Significance testing of these correlations was assessed via spin permutation tests with 10,000 repetitions.
Stability across a higher resolution parcellation
In a separate analysis, functional and structural connectivity matrices were parcellated according to the Schaefer atlas that divides the cortex into 400 functional areas (32). Because cortical thickness and subcortical volume data were only available and shared on the basis of the Desikan-Killiany atlas (30), degree centrality and epicenter-based connectivity profiles were first computed on Schaefer 400 and subsequently averaged within each original 68 cortical regions to allow for spatial correlations with the atrophy maps.
Supplementary Material
Acknowledgments
We would like to express gratitude to the open science initiatives that made this work possible: (i) The ENIGMA-Epilepsy consortium and (ii) the Human Connectome Project (principal investigators: D. Van Essen and K. Ugurbil; 1U54MH091657) funded by the 16 NIH institutes and centers that support the NIH Blueprint for Neuroscience Research and by the McDonnell Center for Systems Neuroscience at Washington University. Funding: This work was partly undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. The work was also supported by the Epilepsy Society, UK. We are grateful to the Wolfson Trust and the Epilepsy Society for supporting the Epilepsy Society MRI scanner. UNICAMP was supported by FAPESP (São Paulo Research Foundation, Brazil) grant 2013/07559-3: the Brazilian Institute of Neuroscience and Neurotechnology (BRAINN). The Florey Institute acknowledges research funding from the National Health and Medical Research Council (NHMRC) of Australia (program grant 1091593, practitioner fellowship 1060312) and the support from the Victorian Government and, in particular, the funding from the Operational Infrastructure Support Grant. The UNAM centre was funded by UNAM-DGAPA (IB201712, IG200117) and Conacyt (Programa de Laboratorios Nacionales; 181508, 1782). The Bern Research Centre was funded by the Swiss National Science Foundation (grant 180365). This research was supported in part by the Science Foundation Ireland Research Frontiers Programme award (08/RFP/GEN1538). Core funding for ENIGMA was provided by the NIH Big Data to Knowledge (BD2K) program under consortium grant U54 EB020403 (to P.M.T.), by the ENIGMA World Aging Center (R56 AG058854; to P.M.T.), and by the ENIGMA Sex Differences Initiative (R01 MH116147; to P.M.T.). S. Lar. acknowledges funding from Fonds de la Recherche du Québec–Santé (FRQ-S) and the Canadian Institutes of Health Research (CIHR). J.R. was supported by the CIHR. S.S.K. was funded by the U.K. Medical Research Council (grant numbers MR/S00355X/1 and MR/K023152/1). P.S. developed this work within the framework of the DINOGMI Department of Excellence of MIUR 2018-2022 (legge 232 del 2016). T.J.O. acknowledges funding support from the NHMRC and RMH Neuroscience Foundation. S.J.A.C. and M.P.R. were funded by the U.K. Medical Research Council (programme grant MR/K013998/1). E.A. was funded by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie (grant agreement no. 750884). S.B.V. was funded by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. A.B. and N.B. were supported by FRQ-S and CIHR (MOP-57840, MOP-123520). C.R.M. acknowledges funding from the NIH (NINDS R01NS065838 and R21 NS107739). B.C.B. acknowledges research funding from the SickKids Foundation (NI17-039), the Natural Sciences and Engineering Research Council of Canada (NSERC; Discovery-1304413), CIHR (FDN-154298), Azrieli Center for Autism Research (ACAR), an MNI-Cambridge collaboration grant, salary support from FRQ-S (Chercheur-Boursier), and the Canada Research Chairs (CRC) Program. Author contributions: Study conceptualization: S. Lar., B.C.B., C.R.M., A.L., M.E.C., N.B., A.B., and S.M.S. Analysis: S. Lar. and B.C.B.; individual study sites represented by other coauthors provided preprocessed MRI data and clinical specifiers. ENIGMA Epilepsy Leadership: C.D.W., S.M.S., C.R.M., and P.M.T. Writing: S. Lar. and B.C.B.; revised and approved by other listed coauthors. Competing interests: F.C. serves on the editorial boards of Neurology, Epilepsia, Epilepsy Research, Frontiers in Neurology, and Arquivos de Neuropsiquiatria and receives grant research support from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Pesquisa (CNPq) Brazil. F.C. has also received honoraria for consulting and speaking from UCB Pharma; United Medical, Brazil; Eurofarma, Brazil; and Zodiac Pharma, Brazil. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and in the references cited. Additional data related to this paper are freely available for download (https://github.com/MICA-MNI/ENIGMA).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc6457/DC1
REFERENCES AND NOTES
- 1.Avena-Koenigsberger A., Misic B., Sporns O., Communication dynamics in complex brain networks. Nat. Rev. Neurosci. 19, 17–33 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Fornito A., Zalesky A., Breakspear M., The connectomics of brain disorders. Nat. Rev. Neurosci. 16, 159–172 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Engel J. Jr., Thompson P. M., Stern J. M., Staba R. J., Bragin A., Mody I., Connectomics and epilepsy. Curr. Opin. Neurol. 26, 186–194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richardson M. P., Large scale brain models of epilepsy: Dynamics meets connectomics. J. Neurol. Neurosurg. Psychiatry 83, 1238–1248 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Bernhardt B. C., Bernasconi A., Liu M., Hong S.-J., Caldairou B., Goubran M., Guiot M. C., Hall J., Bernasconi N., The spectrum of structural and functional imaging abnormalities in temporal lobe epilepsy. Ann. Neurol. 80, 142–153 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Concha L., Kim H., Bernasconi A., Bernhardt B. C., Bernasconi N., Spatial patterns of water diffusion along white matter tracts in temporal lobe epilepsy. Neurology 79, 455–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDonald C. R., Hagler D. J. Jr., Ahmadi M. E., Tecoma E., Iragui V., Gharapetian L., Dale A. M., Halgren E., Regional neocortical thinning in mesial temporal lobe epilepsy. Epilepsia 49, 794–803 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Labate A., Cerasa A., Aguglia U., Mumoli L., Quattrone A., Gambardella A., Voxel-based morphometry of sporadic epileptic patients with mesiotemporal sclerosis. Epilepsia 51, 506–510 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Coan A. C., Appenzeller S., Bonilha L., Li L. M., Cendes F., Seizure frequency and lateralization affect progression of atrophy in temporal lobe epilepsy. Neurology 73, 834–842 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Galovic M., van Dooren V. Q. H., Postma T. S., Vos S. B., Caciagli L., Borzì G., Rosillo J. C., Vuong K. A., de Tisi J., Nachev P., Duncan J. S., Koepp M. J., Progressive cortical thinning in patients with focal epilepsy. JAMA Neurol. 76, 1230–1239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernhardt B. C., Worsley K. J., Kim H., Evans A. C., Bernasconi A., Bernasconi N., Longitudinal and cross-sectional analysis of atrophy in pharmacoresistant temporal lobe epilepsy. Neurology 72, 1747–1754 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caciagli L., Bernasconi A., Wiebe S., Koepp M. J., Bernasconi N., Bernhardt B. C., A meta-analysis on progressive atrophy in intractable temporal lobe epilepsy: Time is brain? Neurology 89, 506–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z., Larivière S., Xu Q., Vos de Wael R., Hong S.-J., Wang Z., Xu Y., Zhu B., Bernasconi N., Bernasconi A., Zhang B., Zhang Z., Bernhardt B. C., Community-informed connectomics of the thalamocortical system in generalized epilepsy. Neurology 93, e1112–e1122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernhardt B. C., Rozen D. A., Worsley K. J., Evans A. C., Bernasconi N., Bernasconi A., Thalamo–cortical network pathology in idiopathic generalized epilepsy: Insights from MRI-based morphometric correlation analysis. Neuroimage 46, 373–381 (2009). [DOI] [PubMed] [Google Scholar]
- 15.O’Muircheartaigh J., Vollmar C., Barker G. J., Kumari V., Symms M. R., Thompson P., Duncan J. S., Koepp M. J., Richardson M. P., Abnormal thalamocortical structural and functional connectivity in juvenile myoclonic epilepsy. Brain 135, 3635–3644 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caciagli L., Wandschneider B., Xiao F., Vollmar C., Centeno M., Vos S. B., Trimmel K., Sidhu M. K., Thompson P. J., Winston G. P., Duncan J. S., Koepp M. J., Abnormal hippocampal structure and function in juvenile myoclonic epilepsy and unaffected siblings. Brain 142, 2670–2687 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blumenfeld H., From molecules to networks: Cortical/subcortical interactions in the pathophysiology of idiopathic generalized epilepsy. Epilepsia 44 (suppl. 2), 7–15 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Avoli M., Gloor P., Interaction of cortex and thalamus in spike and wave discharges of feline generalized penicillin epilepsy. Exp. Neurol. 76, 196–217 (1982). [DOI] [PubMed] [Google Scholar]
- 19.Moeller F., Siebner H. R., Wolff S., Muhle H., Boor R., Granert O., Jansen O., Stephani U., Siniatchkin M., Changes in activity of striato–thalamo–cortical network precede generalized spike wave discharges. Neuroimage 39, 1839–1849 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Keller S. S., Ahrens T., Mohammadi S., Möddel G., Kugel H., Bernd Ringelstein E., Deppe M., Microstructural and volumetric abnormalities of the putamen in juvenile myoclonic epilepsy. Epilepsia 52, 1715–1724 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Van Essen D. C., Ugurbil K., Auerbach E., Barch D., Behrens T. E. J., Bucholz R., Chang A., Chen L., Corbetta M., Curtiss S. W., Della Penna S., Feinberg D., Glasser M. F., Harel N., Heath A. C., Larson-Prior L., Marcus D., Michalareas G., Moeller S., Oostenveld R., Petersen S. E., Prior F., Schlaggar B. L., Smith S. M., Snyder A. Z., Xu J., Yacoub E.; WU-Minn HCP Consortium , The Human Connectome Project: A data acquisition perspective. Neuroimage 62, 2222–2231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo X.-N., Ehmke R., Mennes M., Imperati D., Castellanos F. X., Sporns O., Milham M. P., Network centrality in the human functional connectome. Cereb. Cortex 22, 1862–1875 (2012). [DOI] [PubMed] [Google Scholar]
- 23.van den Heuvel M. P., Sporns O., Network hubs in the human brain. Trends Cogn. Sci. 17, 683–696 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Zhou J., Gennatas E. D., Kramer J. H., Miller B. L., Seeley W. W., Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 73, 1216–1227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shafiei G., Markello R. D., Makowski C., Talpalaru A., Kirschner M., Devenyi G. A., Guma E., Hagmann P., Cashman N. R., Lepage M., Chakravarty M. M., Dagher A., Mišić B., Spatial patterning of tissue volume loss in schizophrenia reflects brain network architecture. Biol. Psychiatry 87, 727–735 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Weng Y., Larivière S., Caciagli L., Vos de Wael R., Rodríguez-Cruces R., Royer J., Xu Q., Bernasconi N., Bernasconi A., Thomas Yeo B. T., Lu G., Zhang Z., Bernhardt B. C., Macroscale and microcircuit dissociation of focal and generalized human epilepsies. Commun. Biol. 3, 244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sisodiya S. M., Whelan C. D., Hatton S. N., Huynh K., Altmann A., Ryten M., Vezzani A., Caligiuri M. E., Labate A., Gambardella A., Ives-Deliperi V., Meletti S., Munsell B. C., Bonilha L., Tondelli M., Rebsamen M., Rummel C., Vaudano A. E., Wiest R., Balachandra A. R., Bargalló N., Bartolini E., Bernasconi A., Bernasconi N., Bernhardt B., Caldairou B., Carr S. J. A., Cavalleri G. L., Cendes F., Concha L., Desmond P. M., Domin M., Duncan J. S., Focke N. K., Guerrini R., Hamandi K., Jackson G. D., Jahanshad N., Kälviäinen R., Keller S. S., Kochunov P., Kowalczyk M. A., Kreilkamp B. A. K., Kwan P., Lariviere S., Lenge M., Lopez S. M., Martin P., Mascalchi M., Moreira J. C. V., Morita-Sherman M. E., Pardoe H. R., Pariente J. C., Raviteja K., Rocha C. S., Rodríguez-Cruces R., Seeck M., Semmelroch M. K. H. G., Sinclair B., Soltanian-Zadeh H., Stein D. J., Striano P., Taylor P. N., Thomas R. H., Thomopoulos S. I., Velakoulis D., Vivash L., Weber B., Yasuda C. L., Zhang J., Thompson P. M., McDonald C. R.; ENIGMA Consortium Epilepsy Working Group , The ENIGMA-Epilepsy working group: Mapping disease from large data sets. Hum. Brain Mapp. 10.1002/hbm.25037 , (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander-Bloch A. F., Shou H., Liu S., Satterthwaite T. D., Glahn D. C., Shinohara R. T., Vandekar S. N., Raznahan A., On testing for spatial correspondence between maps of human brain structure and function. Neuroimage 178, 540–551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whelan C. D., Altmann A., Botía J. A., Jahanshad N., Hibar D. P., Absil J., Alhusaini S., Alvim M. K. M., Auvinen P., Bartolini E., Bergo F. P. G., Bernardes T., Blackmon K., Braga B., Caligiuri M. E., Calvo A., Carr S. J., Chen J., Chen S., Cherubini A., David P., Domin M., Foley S., França W., Haaker G., Isaev D., Keller S. S., Kotikalapudi R., Kowalczyk M. A., Kuzniecky R., Langner S., Lenge M., Leyden K. M., Liu M., Loi R. Q., Martin P., Mascalchi M., Morita M. E., Pariente J. C., Rodríguez-Cruces R., Rummel C., Saavalainen T., Semmelroch M. K., Severino M., Thomas R. H., Tondelli M., Tortora D., Vaudano A. E., Vivash L., von Podewils F., Wagner J., Weber B., Yao Y., Yasuda C. L., Zhang G., Bargalló N., Bender B., Bernasconi N., Bernasconi A., Bernhardt B. C., Blümcke I., Carlson C., Cavalleri G. L., Cendes F., Concha L., Delanty N., Depondt C., Devinsky O., Doherty C. P., Focke N. K., Gambardella A., Guerrini R., Hamandi K., Jackson G. D., Kälviäinen R., Kochunov P., Kwan P., Labate A., McDonald C. R., Meletti S., O’Brien T. J., Ourselin S., Richardson M. P., Striano P., Thesen T., Wiest R., Zhang J., Vezzani A., Ryten M., Thompson P. M., Sisodiya S. M., Structural brain abnormalities in the common epilepsies assessed in a worldwide ENIGMA study. Brain 141, 391–408 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desikan R. S., Ségonne F., Fischl B., Quinn B. T., Dickerson B. C., Blacker D., Buckner R. L., Dale A. M., Maguire R. P., Hyman B. T., Albert M. S., Killiany R. J., An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31, 968–980 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Methodol. 57, 289–300 (1995). [Google Scholar]
- 32.Schaefer A., Kong R., Gordon E. M., Laumann T. O., Zuo X.-N., Holmes A. J., Eickhoff S. B., Yeo B. T. T., Local-global parcellation of the human cerebral cortex from intrinsic functional connectivity MRI. Cereb. Cortex 28, 3095–3114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wandschneider B., Centeno M., Vollmar C., Symms M., Thompson P. J., Duncan J. S., Koepp M. J., Motor co-activation in siblings of patients with juvenile myoclonic epilepsy: An imaging endophenotype? Brain 137, 2469–2479 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larivière S., Vos de Wael R., Paquola C., Hong S.-J., Mišić B., Bernasconi N., Bernasconi A., Bonilha L., Bernhardt B. C., Microstructure-informed connectomics: Enriching large-scale descriptions of healthy and diseased brains. Brain Connect. 9, 113–127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larivière S., Weng Y., Vos de Wael R., Royer J., Frauscher B., Wang Z., Bernasconi A., Bernasconi N., Schrader D. V., Zhang Z., Bernhardt B. C., Functional connectome contractions in temporal lobe epilepsy: Microstructural underpinnings and predictors of surgical outcome. Epilepsia 61, 1221–1233 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Cohen A. S., Lin D. D., Quirk G. L., Coulter D. A., Dentate granule cell GABAA receptors in epileptic hippocampus: Enhanced synaptic efficacy and altered pharmacology. Eur. J. Neurosci. 17, 1607–1616 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartolomei F., Khalil M., Wendling F., Sontheimer A., Régis J., Ranjeva J.-P., Guye M., Chauvel P., Entorhinal cortex involvement in human mesial temporal lobe epilepsy: An electrophysiologic and volumetric study. Epilepsia 46, 677–687 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Honey C. J., Sporns O., Cammoun L., Gigandet X., Thiran J. P., Meuli R., Hagmann P., Predicting human resting-state functional connectivity from structural connectivity. Proc. Natl. Acad. Sci. U.S.A. 106, 2035–2040 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernasconi A., Bernasconi N., Natsume J., Antel S. B., Andermann F., Arnold D. L., Magnetic resonance spectroscopy and imaging of the thalamus in idiopathic generalized epilepsy. Brain 126, 2447–2454 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Pardoe H. R., Berg A. T., Jackson G. D., Sodium valproate use is associated with reduced parietal lobe thickness and brain volume. Neurology 80, 1895–1900 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cascino G. D., Temporal lobe epilepsy is a progressive neurologic disorder: Time means neurons! Neurology 72, 1718–1719 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Fortin J.-P., Cullen N., Sheline Y. I., Taylor W. D., Aselcioglu I., Cook P. A., Adams P., Cooper C., Fava M., McGrath P. J., McInnis M., Phillips M. L., Trivedi M. H., Weissman M. M., Shinohara R. T., Harmonization of cortical thickness measurements across scanners and sites. Neuroimage 167, 104–120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan V. L., Rogers B. P., Sonmezturk H. H., Gore J. C., Abou-Khalil B., Cross hippocampal influence in mesial temporal lobe epilepsy measured with high temporal resolution functional magnetic resonance imaging. Epilepsia 52, 1741–1749 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dale A. M., Fischl B., Sereno M. I., Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage 9, 179–194 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Glasser M. F., Sotiropoulos S. N., Wilson J. A., Coalson T. S., Fischl B., Andersson J. L., Xu J., Jbabdi S., Webster M., Polimeni J. R., Van Essen D. C., Jenkinson M.; WU-Minn HCP Consortium , The minimal preprocessing pipelines for the Human Connectome Project. Neuroimage 80, 105–124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salimi-Khorshidi G., Douaud G., Beckmann C. F., Glasser M. F., Griffanti L., Smith S. M., Automatic denoising of functional MRI data: Combining independent component analysis and hierarchical fusion of classifiers. Neuroimage 90, 449–468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tournier J.-D., Smith R., Raffelt D., Tabbara R., Dhollander T., Pietsch M., Christiaens D., Jeurissen B., Yeh C.-H., Connelly A., MRtrix3: A fast, flexible and open software framework for medical image processing and visualisation. Neuroimage 202, 116137 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Smith R. E., Tournier J.-D., Calamante F., Connelly A., Anatomically-constrained tractography: Improved diffusion MRI streamlines tractography through effective use of anatomical information. Neuroimage 62, 1924–1938 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Jeurissen B., Tournier J.-D., Dhollander T., Connelly A., Sijbers J., Multi-tissue constrained spherical deconvolution for improved analysis of multi-shell diffusion MRI data. Neuroimage 103, 411–426 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Tournier J.-D., Calamante F., Connelly A., Robust determination of the fibre orientation distribution in diffusion MRI: Non-negativity constrained super-resolved spherical deconvolution. Neuroimage 35, 1459–1472 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Smith R. E., Tournier J.-D., Calamante F., Connelly A., SIFT2: Enabling dense quantitative assessment of brain white matter connectivity using streamlines tractography. Neuroimage 119, 338–351 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Betzel R. F., Griffa A., Hagmann P., Mišić B., Distance-dependent consensus thresholds for generating group-representative structural brain networks. Netw. Neurosci. 3, 475–496 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc6457/DC1