

Abstract
Human transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), causative pathogen of the COVID-19 pandemic, exerts a massive health and socioeconomic crisis. The virus infects alveolar epithelial type 2 cells (AT2s), leading to lung injury and impaired gas exchange, but the mechanisms driving infection and pathology are unclear. We performed a quantitative phosphoproteomic survey of induced pluripotent stem cell-derived AT2s (iAT2s) infected with SARS-CoV-2 at air-liquid interface (ALI). Time course analysis revealed rapid remodeling of diverse host systems, including signaling, RNA processing, translation, metabolism, nuclear integrity, protein trafficking, and cytoskeletal-microtubule organization, leading to cell cycle arrest, genotoxic stress, and innate immunity. Comparison to analogous data from transformed cell lines revealed respiratory-specific processes hijacked by SARS-CoV-2, highlighting potential novel therapeutic avenues that were validated by a high hit rate in a targeted small molecule screen in our iAT2 ALI system.
Keywords: pneumocytes, infection, SARS-CoV-2, COVID-19, mass spectrometry, phosphoproteomics, pathways, pathogenesis, antivirals, time course
Graphical Abstract
Hekman et al. describe how a layer of primary stem cells (iAT2s) recapitulating lung biology responds to infection with SARS-CoV-2. They compare their work to previous studies with immortalized cell lines. Their data predict what effect the virus has on a lung cell and which drugs may slow infection.
Introduction
SARS-CoV-2 is a highly infectious virus responsible for the ongoing coronavirus disease 2019 (COVID-19) pandemic (Zhu et al., 2020a). The viral genome encodes at least 27 proteins (Zhu et al., 2020a), including 4 structural (spike [S], envelope [E], membrane [M], nucleocapsid [N]), 15 nonstructural, and 8 auxiliary proteins. These proteins interact with host factors to modulate host responses (Gordon et al., 2020; Stukalov et al., 2020). The main receptor of SARS-CoV-2 is angiotensin-converting enzyme 2 (ACE2), which is expressed on the surface of target cells, including lung airway and alveolar epithelia. Other host factors, such as transmembrane serine protease 2 (TMPRSS2), prime viral entry (Hoffmann et al., 2020).
In the distal lung, SARS-CoV-2 appears to preferentially infect alveolar epithelial type 2 cells (AT2s), which express ACE2 and TMPRSS2 (Hou et al., 2020; Sungnak et al., 2020). AT2s are facultative progenitors of lung alveoli, where they regenerate the epithelium following injury and secrete pulmonary surfactant, stored in lamellar bodies, reducing surface tension. While other cell types and organs are targeted by SARS-CoV-2 (Wichmann et al., 2020), morbidity and mortality in COVID-19 largely result from alveolar injury (Carsana et al., 2020), manifested as acute respiratory distress syndrome (ARDS) in severe disease. As AT2 injury is central to COVID-19 pathogenesis, there is an urgent need to delineate the mechanisms of SARS-CoV-2-driven lung pathology.
Primary AT2s are difficult to maintain in culture, but human induced pluripotent stem cell-derived alveolar epithelial type 2 cells (iAT2s) have been developed and extensively characterized (Hurley et al., 2020; Jacob et al., 2017, 2019). iAT2s are capable of robust self-renewal while faithfully maintaining an AT2-like transcriptional program when cultured at air-liquid interface (ALI) (Abo et al., 2020). Identification of host functions impacted in AT2s can reveal the mechanisms SARS-CoV-2 utilizes for propagation, providing targets to counter lung injury.
Comparison of the SARS-CoV-2 and SARS-CoV replication kinetics in Vero E6 cells showed that progeny virus production of both viruses plateaued by about 14 h post-infection (hpi) (Ogando et al., 2020). The earliest stages of infection include viral entry and release of the viral genome into the cytoplasm (1 hpi), followed by initiation of viral RNA translation and processing of the viral replicase polyproteins (1 to 3 hpi) and formation of double membrane vesicles harboring viral replication-transcription complexes (RTCs) (around 3 to 6 hpi) (Fehr and Perlman, 2015; Paul and Bartenschlager, 2013). RTCs are the sites of viral genome amplification and synthesis of subgenomic transcripts (Snijder et al., 2020). Viral RNA and proteins accumulate throughout the replication cycle, which is completed by viral egress via the ER-Golgi intermediate compartment, followed by transport of viral particles to the plasma membrane and release (8 hpi and later) (Fehr and Perlman, 2015). SARS-CoV virions can form as early as 3 hpi and are released from infected cells for days (Stertz et al., 2007).
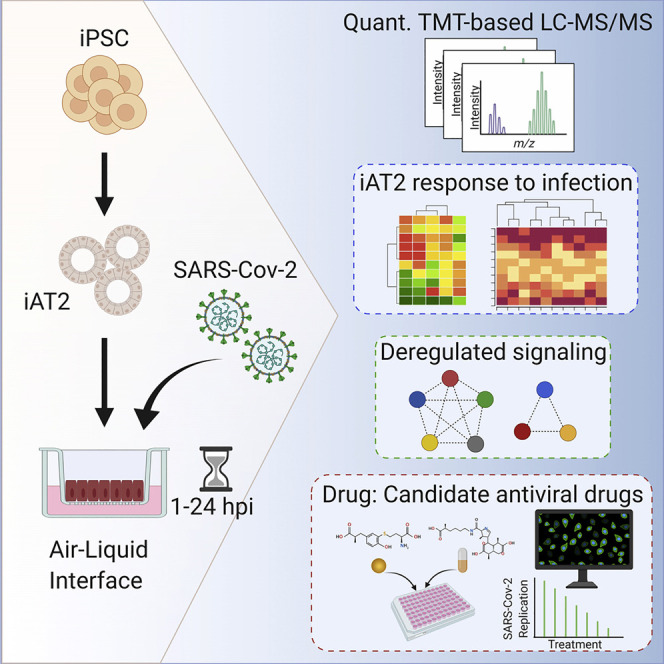
Molecular profiling of SARS-CoV-2-infected cell lines such as Vero E6 (immortalized African green monkey kidney cells) (Bouhaddou et al., 2020), tumor-derived human Caco-2 (Bojkova et al., 2020; Klann et al., 2020), and lung basal carcinoma A549 (Stukalov et al., 2020) has revealed pathways co-opted by the virus, but relevance to human lung pathobiology is limited. To provide a more pertinent understanding, we performed a deep quantitative temporal phospho/proteomic analysis to quantify cytopathologic changes induced by SARS-CoV-2 infection in iAT2s at four time points (1, 3, 6, and 24 hpi), with a focus on early events following viral entry. Specifically, we cultured iAT2s at ALI, a model that accurately reflects key aspects of pulmonary biology (Abo et al., 2020). We have shown that the iAT2 ALI cultures are permissive to SARS-CoV-2 infection and release infectious viral particles preferentially from the apical surface (Huang et al., 2020).
Our systematic analysis established a rapid and multi-faceted response of iAT2s to SARS-CoV-2, including disruption of potentially druggable pathways. Comparisons to analogous studies of SARS-CoV-2 infection of Vero E6 (Bouhaddou et al., 2020), Caco-2 (Bojkova et al., 2020), and A549 (Stukalov et al., 2020) cancer cells revealed differences contributing to the unique respiratory pathology in COVID-19. Using an integrative framework, we predicted and validated novel targets to intercept COVID-19 pathogenesis and offer these results as a community resource (http://www.bu.edu/dbin/cnsb/covid/).
Results
Pathophysiological Model of Lung Infection
We generated iAT2s from the human iPSC line SPC2-ST-B2 (Hurley et al., 2020) via our previously published lung directed differentiation protocol, followed by sorting cells that express a tdTomato fluorescent reporter targeted to one allele of the AT2-specific surfactant protein-C (SFTPC) locus (Figure 1 A, top; Hurley et al., 2020; Jacob et al., 2017). This reporter allowed isolation of >95% purity AT2-like cells with no loss of phenotype (Jacob et al., 2017).
Figure 1.

Phospho/Proteomic Profiling of Human iAT2s after SARS-CoV-2 Infection
(A) (Top) Schematic of 3D alveolospheres and iAT2 ALI cultures. Apical media was removed for 7 days before infection with SARS-CoV-2; DE, definitive endoderm; AFE, anterior foregut endoderm. Representative confocal IFA images (10×) of iAT2s expressing tdTomato from endogenous SFTPC locus. (Bottom) iAT2 ALI cultures were infected with SARS-CoV-2 (MOI = 5) for indicated times with parallel mock-treated controls. Representative staining (20×) of DNA (Hoechst, blue) and viral N (green) indicating infection.
(B) Total protein from replicate SARS-CoV-2-infected and mock-treated iAT2s was analyzed by quantitative LC-MS/MS.
(C) Replicate measurements were normalized and filtered (<1% FDR), resulting in high reproducibility (r = PCC). Venn diagram shows number of identified cellular/viral proteins/phosphosites subject to downstream analysis.
iAT2s were initially cultured as 3D alveolospheres (Jacob et al., 2017) before seeding on transwell inserts to generate ALI cultures (Abo et al., 2020) to allow for infection of the cells from the apical surface. Bulk and single-cell RNA sequencing (Abo et al., 2020; Huang et al., 2020) confirmed expression of the AT2 program (SFTPA1/2, SFTPC/D, PGC) and ACE2 and TMPRSS2 at frequencies commensurate with primary AT2s (Abo et al., 2020). To elucidate the host systems impacted by SARS-CoV-2, we infected iAT2 ALI cultures with a multiplicity of infection (MOI) of 5 (STAR Methods). To synchronize infection, virions were bound to the apical surface for 1 h at 16°C before transfer to 37°C to initiate internalization (Saeed et al., 2010). An infectivity rate of ∼20% by 24 hpi was evident by immunofluorescence analysis (IFA) (Figure 1A, bottom), consistent with previous findings (Huang et al., 2020).
Quantitative Temporal Mass Spectrometry Analysis of Infected iAT2s
Total protein from replicate ALI culture wells (∼5 million cells/time point) was extracted, trypsinized, and analyzed by precision mass spectrometry (MS) to quantify changes in the proteome and phosphoproteome relative to respective mock-infected controls. To enhance signal and minimize sampling bias, the early (1–6 hpi) samples (low amounts of viral replication) were processed separately from the later 24 hpi time point (abundant viral replication) (Figure 1B; STAR Methods). In total, we quantified 8,471 proteins (Figure 1C; Table S1), including eight viral proteins and 14,289 phosphosites (>0.7 localization likelihood) on 2 SARS-CoV-2 and 2,703 host phosphoproteins.
Despite TMT measurement compression (Karp et al., 2010), following normalization and correction (STAR Methods), reproducible and significant (|log2-fold change| > 0.25, FDR < 0.05) changes were seen in 2,872 proteins (Table S1), including AT2 markers crucial to surfactant function, such as SFTPA2. In parallel, we identified 4,688 differential (|log2 FC| > 0.25, FDR < 0.05) phosphosites mapping onto 1,166 unique lung proteins across all time points (Table S1), reflecting distinct sample clusters (Figure S1). Many correspond to regulators of pulmonary cell function, including protein kinases, phosphatases, adapters, and transcription factors (Table S1). These results permitted in-depth differential pathway analysis and functional modeling (Figure 1C). Coherent patterns emerged from the infection time course (Figure 2 A), reflecting significant functional enrichments (Figure 2B). By 24 hpi, eight distinct viral proteins (S, M, N, Orf3a/7a/8/9b, and multiple components mapping to polyprotein Rep1a) were detected (Figure 2C). The same viral proteins (and Nsp6) were seen in SARS-CoV-2-infected Caco-2 cells (Bojkova et al., 2020), indicating abundant expression of the structural proteins and preferential protein production via proteolytic processing of Rep1a (Zhang et al., 2020).
Figure 2.

Host Protein Alterations in Infected iAT2s
(A) Clusters depicting protein abundance at 1, 3, and 6 hpi.
(B) Dysregulated (FDR < 0.05) functional modules, including (bottom) U7 snRNA and HDAC-associated complexes.
(C) Volcano plot showing differential protein abundance at 24 hpi.
(D) IFA of mock-treated and SARS-CoV-2-infected iAT2s probed for PCNA (positive cells highlighted with arrows).
(E) Overlay of differential host proteins (clusters from A) onto a SARS-CoV-2 viral-host PPI network (Gordon et al., 2020) highlighting iAT2 targets (squares) of viral effectors (hexagons); hues reflect cluster assignment.
Host Proteome Impacted by Viral Infection
To characterize the initial iAT2 host responses to infection, we applied supervised clustering to the early time points (1–6 hpi), revealing four distinct groupings corresponding to immediate (∼1 hpi), early (∼3 hpi), and intermediate/late (3–6 hpi) waves of host protein expression (Figure 2A). Cluster 1, associated with a prompt response to viral entry (peak ∼1 hpi), was enriched with factors previously tied to viral infection (e.g., influenza life cycle, adj. FDR = 4.49E−32) and immunity, including markers of TRAF6-mediated cytokine induction, activation of NF-κB, and C-type lectin receptors (e.g., ITCH, ATF2, IKBKB) (Figure 2B; Table S2). MAVS, an adaptor activated by RIG I-like receptors, was downregulated, consistent with a previous report of SARS-CoV Orf9b-mediated degradation (Shi et al., 2014), suggesting that this function is retained in SARS-CoV-2. In addition, the serine-threonine kinase LATS1, WRN, and insulin-like growth factor binding protein 4 (IGFBP4), which negatively regulate cell proliferation, were upregulated (Figure 2C).
Regulation of translation initiation by mammalian target of rapamycin (mTOR) was altered immediately following viral entry, leading to a striking change in host protein abundance by 3–6 hpi (Figures 2A and 2B). Remodeling of host protein synthesis was evident in altered levels of 40S (e.g., RPS6/11/26) and 60S (e.g., RPL6/12/19) ribosomal subunits, and other multi-protein complexes linked to growth (Table S1). In contrast, expression of multiple components of the mitochondrial 28S ribosome increased markedly at 3–6 hpi (Figure 2B), presumably to accommodate energetic demands for viral replication.
Clusters 2 to 4 (3–6 hpi) were enriched for proteins linked to regulation of cell proliferation, such as cell cycle control (e.g., mitosis), including dysregulation of the checkpoint kinase regulator ATR and cell death effectors CASP7/8 (Figure 2A) and the mitotic checkpoint regulator BUB1B (Table S1), suggesting cell cycle arrest. In line with this observation, IFA revealed markedly decreased PCNA expression in SARS-CoV-2-infected iAT2s (Figures 2D and S2), indicative of virus-induced interphase arrest.
Transcriptome and Proteome Responses
Since viruses co-opt host cell transcriptional and translational machinery, we compared the iAT2 protein profiles of viral infection to complementary RNA-seq-based mRNA expression at 24 hpi (Huang et al., 2020) (Figure S2). While some processes showed high correlation, including elevated MAPK and IFN signaling and downregulated N-linked glycosylation and fatty acid metabolism, discordant functional terms predominated the comparison, including uncoupled cell cycle control, vesicle-mediated transport, and chromosomal organization (Figure S2; Table S2), suggesting differential mechanisms by which the virus mediates differential control of host systems.
Molecular Scaffold to Interpret Lung Cell Responses
SARS-CoV-2 encodes proteases, polymerases, and other effectors that interact with host factors. Though not validated in infected cells, putative cellular binding partners of individual, ectopically expressed viral proteins in transformed cells were recently reported (Gordon et al., 2020; Stukalov et al., 2020). Such static measurements may not accurately recapitulate viral-host interactome dynamics in an alveolar cell infection setting. To characterize which putative viral-host protein-protein interactions (PPI) likely occur in iAT2s during infection, we overlaid our iAT2 proteomic time course data onto a molecular association network recently reported for SARS-CoV-2 (Gordon et al., 2020) (Table S1), allowing us to deduce the kinetics of these associations.
Striking differences in the time course profiles of many putative host targets of different viral effectors were observed in infected iAT2s (Figure 2E). For example, a significant fraction of the interacting lung proteins was impacted by the virus at 3–6 hpi (Figure S3), as viral replication ramps up, implying that some of these changes result from viral effector binding. Examples include reductions in the levels of chromatin remodeling factors bound by viral E (e.g., BRD2/4), host ion transport factors (ATP6AP1/13A3) that associate with Nsp6, centrosomal proteins (NINL, CEP135/350) bound by Nsp13, and host acetyltransferase HDAC2 targeted by the viral protease Nsp5. Viral-mediated destabilization may disrupt centrosome-microtubule network reorganization, contributing to the cell cycle arrest seen by 24 hpi.
Conversely, host proteins upregulated in response to SARS-CoV-2 infection are more likely to form stable functional units with viral effectors (Figure 2E). For instance, several interactors (GGH, NPC2, OS9, FKBP7/10) of viral Orf8 upregulated in infected iAT2s are implicated in protein maturation in the ER, suggesting roles in viral particle formation. Likewise, upregulated interactors of viral Nsp13 (GCC2, GOLGA2/B1) map to Golgi components relevant to virion assembly, maturation, and egress (Schoeman and Fielding, 2019).
SARS-CoV-2 Proteins Phosphorylated by Host Enzymes
We identified phosphosites on viral membrane (M) and nucleocapsid (N) by 24 hpi (Table S1). N was heavily modified on nine unique phosphosites (Figure 3 B), clustered in a linker region between the RNA-binding (RBD) and dimerization domains, while phosphosites on M included S213/S214 in the C-terminal cytoplasmic domain. Previous reports showed that N phosphorylation by GSK3B on serine-arginine (S-R)-rich motifs is important for coronavirus replication (Wu et al., 2009) and suggested that SARS-CoV-2 N is a putative target of casein kinase 2 (CK2) (Gordon et al., 2020). CK2 substrate motifs comprise acidophilic (+1/−1 position) or proline residues (+1 position) (Goel et al., 2018). Consistently, we found two canonical proline-directed motifs (S79/S206) and one acidophilic motif (S23) on N as likely direct substrates.
Figure 3.

Phosphoproteomic Profiling Reveals Dysregulated Pathways
(A) Bar-plot of differential phosphosites (1–6 hpi).
(B) Domain structure of SARS-CoV-2 N (top) and M (bottom) showing identified phosphosites.
(C) Structural models of phospho-CNSKA2 (S197) complexed with viral N (S79).
(D) Clustering of phosphosite abundance changes.
(E) Enriched pathways and processes.
(F) Up- or downregulated kinases (KSEA).
(G) IFA of phosphogamma-H2AX (green) and viral N (red) in infected versus mock-treated iAT2 (DAPI counterstain). Greyscale images show exclusively phospho-γ-H2AX localization in iAT2s (number foci per nucleus shown at right, p value < 0.05, Wilcoxon rank-sum test).
(H) 3D models of phosphorylated RPS6KB1 (S441/T444/S447), CAMKD (T287/S319), PAK2 (S197/S209), EGFR (S991/T993), and CK1 (S171) complexed with DDX21.
(I) Major classes of proteins/domain interactions impacted by phosphorylation.
(J) Phospho-dependent interactors excluding kinases.
To verify this, we performed in vitro kinase assays with purified N, CK2, and GSK3B (see STAR Methods) and analyzed the products via MS, confirming N phosphorylation by GSK3B on three sites (S176, S180, T391) (Table S1), two of which (S176, S180) match the expected S-R-rich consensus, suggesting a role for GSK3B in SARS-CoV-2 replication. We also confirmed CK2 phosphorylates N on S23 and S410.
We generated simulation-based 3D structures to visualize phosphorylation of N. Our model shows that S79 maps to the interface of the RNA binding domain in the N-terminal region of N proposed to tetramerize (Figure 3C), suggesting a role in multimerization. Since both SARS-CoV and SARS-CoV-2 N have been shown to undergo liquid-liquid phase separation (LLPS) to facilitate viral assembly (Perdikari et al., 2020), with RNA sequestering influenced by N phosphorylation (Chang et al., 2013), we explored whether phosphorylation of SARS-CoV-2 N by GSK3B or CK2 modulated LLPS using an in vitro phase separation assay (STAR Methods). Phosphorylation by CK2 elicited a strong increase in droplet formation at concentrations of N < 1 mM. In contrast, phosphorylation by GSK3B greatly reduced LLPS by N, increasing the concentration at which LLPS occurs by 20-fold (400 nM to 10 mM) (Figure S4). These data suggest that SARS-CoV-2 N phosphorylation by host kinases modulates phase separation, impacting RNA assembly and packaging.
Global Alterations in iAT2 Cell Signaling
Supervised clustering of differential host phosphoproteins (Figure 3A) at early times revealed four temporally regulated groupings: immediate (cluster 1; 1 hpi), early (cluster 2; 3 hpi), and intermediate/late (clusters 3 and 4; 3–6 hpi) responses of iAT2s to SARS-CoV-2 infection (Figure 3D).
Pathway analysis (Figure 3E) revealed enriched (FDR < 0.05) processes linked to infection, such as viral RNA synthesis and export of ribonucleoproteins, as an immediate response to SARS-CoV-2 entry, along with eukaryotic translation initiation factor eIF4G1 and other regulators of host protein synthesis. Other prominent responses peaking at 1 hpi suggested disruption of the nuclear envelope, nuclear export, and chromatin remodeling (e.g., NCOR2, p300-CBP, HDAC1/2 complexes), suggesting that viral effectors interfere with host gene expression and post-transcriptional processing by targeting nuclear functions surprisingly early in the replication cycle.
Intermediate pathways associated with clusters 3 and 4 (Figure 3D; Table S2) centered on RNA processing (e.g., splicing, 3′ end processing), cell proliferation/survival (e.g., apoptosis), and protein synthesis (e.g., mTOR signaling), indicating remodeling of host post-transcriptional programs by 6 hpi, coincident with viral replication.
SARS-CoV-2 Targets Ser/Thr/Tyr Kinases in iAT2s
A motif-based assessment identified host kinases potentially mediating differential phosphorylation in the SARS-CoV-2 replication cycle. Kinases with activities predicted (FDR < 0.05) to be highly responsive to infection included CSNK1E, CDK2, and EEF2K in the immediate (1 hpi) response to viral entry and activation of RPS6KA3, CDK1/2, and MAPK14 by 3–6 hpi (Figure 3F).
While elevated CDK1/2 activity is characteristic of actively proliferating cells (Satyanarayana and Kaldis, 2009), it is implicated in homologous recombination-mediated DNA repair (Hentges et al., 2014), pointing to DNA damage signaling in infected iAT2s consistent with ATR upregulation (Figure 2A). To assess this, we performed IFA and observed markedly increased phosphorylation of foci formed by the DNA repair marker γ-H2AX in infected iAT2s compared to controls (p < 0.05, Wilcoxon rank-sum test) (Figure 3G). Such observations corroborate previous reports that coronaviruses activate ATR to induce cell cycle arrest (Xu et al., 2011).
In late infection (24 hpi), we predicted significant activation of CAMK2G, RPS6KB2, CSNK1E, PNCK, and to a lesser extent mTOR (p < 0.06) (Figure S2; Table S1), and downregulation of CDK2/5, MAP2K1, AURKA, ROCK2, ERBB2, and SRC (Table S1). Collectively, these kinases are essential signaling hubs controlling host cell growth, proliferation, and metabolism.
We identified conserved phosphosites, such as in kinase activation loops, that directly reflect catalytic status or other well-characterized functions missed by enrichment criteria. Specifically, we found RPS6KB1 (S441/T444/S447), CAMK2D (T287), PAK2 (PAK2 T197/209), and CDK1 (hypophosphorylation of inhibitory T14/Y15) as potentially hyperactivated in infected iAT2s (Figure 3H; Table S1). Notably, CAMK2D interacts with SARS-CoV Nsp3 (Ma-Lauer et al., 2016) and is implicated in deregulation of innate antiviral immunity. Conversely, epidermal growth factor receptor (EGFR) was hypophosphorylated on S991/T993, which is linked to receptor internalization and downregulation (Figure 3H).
To investigate the impact of phosphorylation, we modeled PPIs in the vicinity of differential phosphosites. The most frequently occurring interactions are dominated by kinase associations (e.g., GSK3, MAPL, CK1) mediating phosphorylation of a motif (e.g., DDX21-CK1 association, wherein CK1 phosphorylates DDX21 on Ser171) (Figure 3I). A diverse set of motif-domain associations were also predicted to be regulated by virus-induced changes in phosphorylation (Figure 3J). These include 14-3-3 domains that bind to specific phosphoserine/threonine-containing motifs on proteins involved in nuclear transport.
Remodeling of Cell Growth by SARS-CoV-2
Since our phospho/proteomic data indicated SARS-CoV-2-induced disruption of RPSK6B1 (Figures 3E and 3H), a key mediator of mTOR-dependent translation (Roux et al., 2007), we performed immunoblotting to confirm pathway activation (Figure 4 A). We probed for RPS6 S240/S244, targets of RPS6K (Roux et al., 2007), and PI3-AKT T308, which integrates with mTOR to control translational initiation (Saxton and Sabatini, 2017).
Figure 4.

Validation Analyses
(A) Immunoblotting of lysates from mock and SARS-CoV-2-infected iAT2 ALI at 24 hpi. Probes indicated (beta-actin loading control).
(B) (Left) Hyperphosphorylation of SRSF proteins 24 hpi. (Right) Position and relative change in phosphosites.
(C) 3D model of hyperphosphorylated sites (S199/S197/S204/S211) on SRSF9 in infected iAT2s relative to controls.
(D) Schematic of splicing events impacted by infection.
(E) Analyses of RNA-seq data (Huang et al., 2020) showing virus-induced splicing alterations.
(F) Functional annotations of differential spliced gene products.
(G) Ratio of spliced to unspliced transcripts for select mRNAs in mock or infected iAT2s. For CLK1, ratio of splicing with/without exon 4 (E4) inclusion shown. Bars represent mean (±SD) from 3 biological replicates; ∗p < 0.1; ∗∗p < 0.01; ∗∗∗p < 0.001, t test.
(H) EM images of nuclear envelope (white arrows), ER (red; double-points indicate extended ER), ribosomes (yellow), and nucleus (N) in mock and infected iAT2s (scale bar = 500 nm; insets magnified 4×).
(I) IFA (40×) and quantification (±SD) of γ-tubulin (594 nm) and viral N (488 nm) in control and infected iAT2s (counterstained with DAPI). Scale bar = 10 mm.
Our immunoblotting data confirmed an increase in RPS6 and AKT phosphorylation in infected iAT2s (Figure 4A). Consistent with AKT signaling impinging on the host translational machinery (Ruggero and Sonenberg, 2005), we observed a marked increase in phosphorylation of AKT S129, a phosphosite reported to enhance AKT signaling (Di Maira et al., 2005), and of the AKT substrate YBX1 (Figure 4A), on a site (S102) known to attenuate its translational repressor function (Evdokimova et al., 2006).
To further substantiate our findings, we probed phosphorylation of translational repressor 4E-BP1, which negatively regulates cap-dependent mRNA translation by binding to eIF4E and dissociating the eIF4G-eIF4E complex (Wang et al., 2019). Notably, we observed reduced levels of non-phosphorylated 4E-BP1 T46 in SARS-CoV-2-infected cells, implying repression of 4EBP1 (Lekmine et al., 2004) and in turn increased translation (Figure 4A).
Clusters 3/4 were enriched for kinases such as PAK2, PRKCD, STK3, CAMK2D, EGFR, and MAPK1 connected to growth regulation (Figure 3D). MAPK activates RPS6KB1 and 4E-BP1 by phosphorylating and inactivating the TSC2 complex to relieve inhibition on mTOR (Ma et al., 2005). In line with this, we detected activating phosphorylation on MAPK1 at T185 (Goetz et al., 2014) by 6 hpi (Table S1), reflecting increased catalytic activity, which we confirmed by immunoblotting (Figure 4A). Additionally, we observed hyperphosphorylation of CAMK2D at T287 (Figure 3F), implicated in apoptotic signaling (Toko et al., 2010), coincident with hyperphosphorylation and upregulated kinase activity of PAK2 (Figure 3H), another apoptotic target (Chan et al., 1999). In turn, EGFR was hyperphosphorylated on S991 (Table S1), which is linked to downregulated pro-survival signaling (Tong et al., 2009).
Hyperphosphorylation of eIF2S2 at Ser2, a target of CK2 that putatively regulates initiation complex formation (Welsh et al., 1994), persisted 24 hpi (Table S1). Likewise, infected iAT2s showed increased phosphorylation of PDCD4, which blocks pre-initiation complex assembly and disrupts eIF4A helicase activity (Dorrello et al., 2006). RPS6KB1-mediated hyperphosphorylation of S71 promotes binding to BTRC ubiquitin ligase, causing PDCD4 degradation and translational activation (Dorrello et al., 2006). Our phosphoproteomic data therefore highlight degradation of PDCD4 as another potential mTOR-dependent mechanism by which SARS-CoV-2 redirects host translation.
We likewise observed hyperphosphorylation of S44/S101/S102/S105 of HMGA2 by 3–6 hpi (Figure 3D), an architectural DNA-binding factor implicated in cell proliferation (Yu et al., 2013), DNA damage signaling (Hentges et al., 2014), and antiviral defenses (Du et al., 1993). HMGA2 S44 is a known target of CDK1, which regulates its DNA binding activity (Schwanbeck et al., 2000). Phosphorylation of C-terminal HMGA2 sites (S101/S102/S105) may alter DNA binding to regulate host gene expression (Sgarra et al., 2009).
SARS-CoV-2 Infection Rewires Host mRNA Splicing
We detected increased MAPK1 and SRPK1 activity and CLK1 levels by 3-6 hpi (Table S1), suggesting SARS-CoV-2-induced changes in splicing. SR-protein kinases (SRPK) family members (DYRKs, MAPKs, and Cdc2-like kinases/CLKs) phosphorylate Ser/Arg (SR)-repeat domain-containing proteins to enhance RNA binding and recruitment of cofactors to alternative splice sites (Blaustein et al., 2005; Gui et al., 1994; Nayler et al., 1997). We captured (Table S1) virus-induced hyperphosphorylation of S51 in SRPK1 and S494/S497 in SRPK2 that stimulate kinase activity toward SR proteins (Mylonis and Giannakouros, 2003). S497 is a target of RPS6KB1 (Mylonis and Giannakouros, 2003), which is activated in infected iAT2s (Figure 4A). Coincident with virus-induced dysregulation, we detected hyperphosphorylation of multiple SR-proteins (Figure 4B). For instance, SRSF9 was hyperphosphorylated at four N-terminal sites (Figure 4C) linked to alternative splicing (Naro and Sette, 2013).
To assess the impact on host splicing, we screened RNA-seq datasets of control and SARS-CoV-2-infected iAT2 samples (STAR Methods) for alternative splicing events triggered by SR phosphorylation (Figure 4D). Large effects were observed on exon cassette (EC) inclusion 24 hpi (Figures 4D and 4E), with ∼200 exons displaying decreasing inclusion or intron retention (IR) (Figure 4F; Table S2), potentially impacting diverse host functions.
We performed RT-PCR to directly evaluate processing of FERMT3, CCNL1, DOCK4, RBM5, FMR1, NPR2, and CLK1 transcripts displaying IR defects, and observed differential splicing of retained introns in infected cells (Figure 4G). Besides reduced IR in CLK1, increased inclusion of EC 4 of CLK1 was seen, a stress signal that activates CLK1 (Ninomiya et al., 2011). These data are consistent with the SR hyperphosphorylation we detected in infected iAT2s (Table S1). Collectively, these results imply that SARS-CoV-2 primes viral replication via altered RNA processing, which disrupts AT2 gene expression.
SARS-CoV-2 Disrupts Nuclear Integrity
While viral replication occurs in the cytoplasm, the observed RNA splicing defects imply that host nuclear functions are impacted by SARS-CoV-2 infection. Consistent with this, we detected extensive phosphorylation of lamins (LMNA/B1/B2) suggestive of nuclear lamina disruption (de Castro et al., 2017), which we confirmed by transmission electron microscopy (Figure 4G). While the nuclear envelope of the mock-infected cells was intact, we observed considerable changes in SARS-CoV-2-infected iAT2s 24 hpi, in which nuclear envelopes appeared distended, while the quantity of ER in close proximity was greatly increased and studded with ribosomes.
We also observed increased phosphorylation of major centrosomal proteins, such as CEP170 (S135/S138) at 6 hpi and CEP131 (S414/S416/S417) at 24 hpi (Table S1). The phosphorylation of these proteins, the fact that their phosphorylation by Polo-like kinases PLK1/4 is tightly controlled during mitosis (Denu et al., 2019), and the lack of other markers of mitosis point to aberrant regulation and suggest that SARS-CoV-2 remodels the centrosome-microtubule system. To test this, we analyzed the intracellular distribution of the centrosomal marker γ-tubulin by IFA. While a distinct single centrosomal focus was observed in mock-infected cells, SARS-CoV-2-infected iAT2s exhibited dispersed cytoplasmic γ-tubulin foci (Figure 4H), suggestive of centrosome fragmentation that would disrupt mitotic programs. Additionally, consistent with the elevated expression of CEP152 detected at 3–6 hpi (Table S1), IFA revealed multiple foci of CEP152 in SARS-CoV-2-infected iAT2s (Figure S5), pointing to virus-induced centrosomal amplification and disruption of microtubule organization.
We found evidence of dysregulation of receptor-mediated signaling at 1–3 hpi (Ackermann et al., 2020), including VEGF and PDGFR, and alterations in factors linked to cell junction assembly and tight junction organization. The latter are mediated by lung-specific proteins such as Claudin 7/18, LAMB3, PAK2, and PARD3 crucial to the integrity of the alveolar epithelium (Zhou et al., 2018). Likewise, we observed hyperphosphorylation of pS316 of STK3 (Table S1) in the infected cells, another key target of PLK1 in the Hippo signaling cascade linked to centrosomal function and cell cycle control, as well as epithelial cell polarity and cell-cell junctions (Chen et al., 2019).
Functional Networks Associated with SARS-CoV-2 Infection
To explore alveolar pathways and processes altered by SARS-CoV-2 infection, we performed enrichment analyses on our differential protein and phosphoprotein profiles. We merged significant results (FDR < 0.1; Table S2) into higher-level modules based on shared components (Merico et al., 2010) (Figure 5 A). Metabolic pathways were prominently impacted, including alterations in fatty acid metabolism, mitochondrial respiration, and pyrimidine nucleotide biosynthesis (Table S1 and S2). For instance, we observed upregulation of the rate-limiting pyrimidine biosynthesis enzyme DHODH in infected iAT2s, whereas enzymes linked to fatty acid (ACSL1/3/4, HADHA) and oxidative respiration (ETC I/IV) were downregulated. These results suggest major reprogramming of host metabolism to favor energy and biomass synthesis to support viral replication (Munger et al., 2008).
Figure 5.

Time-Resolved Host Cell Responses to SARS-CoV-2 Infection
(A) Enrichment map of iAT2 processes and pathways (nodes) altered by infection (red: upregulated/positive; blue: downregulated/negative), grouped and scaled according to shared (edges) and number (size) of components. Quadrants delineate the four-infection time points (1 to 24 hpi); iAT2-specific pathways are outlined in red, while black indicates conserved/generic responses.
(B) Comparative analyses of SARS-CoV-2-infected iAT2s (this study), Caco-2 (Bojkova et al., 2020), A549 (Stukalov et al., 2020), and Vero E6 cells (Bouhaddou et al., 2020); positive and negative enrichment of functional annotations based on normalized enrichment scores (NES).
(C) Heatmap of differential host pathways/processes common to all four infection studies.
Several phospho/proteomic studies of SARS-CoV-2-infected immortalized cell lines were reported recently, including Vero E6 (Bouhaddou et al., 2020), Caco-2 (Bojkova et al., 2020), and A549 cells (Stukalov et al., 2020). These cell types are fundamentally distinct from differentiated alveolar epithelial cells. By considering only factors with a consistently significant (FDR < 0.05, |log2 FC| > 0.25) response to infection, notably few host proteins were differentially regulated by SARS-CoV-2 across all four cell lines (Table S2; Figure S5). These map to TNF production (HSPB1, MIF), cellular RNA polymerase regulation (ITGA3, PRKDC, SUB1, AKR1B1), and response to dsRNA (MAVS, CAV1), suggesting that cellular context dominates the host response. One example unique to iAT2s is Claudin-18a, a marker of alveolar epithelial cells, which decreased significantly by 3–6 hpi (Table S1), as did desmoplakin, critical for desmosomal integrity. Changes to key junctional proteins in infected iAT2s suggest disruption of apical tight junctions, which is relevant to the loss of alveolar epithelial barrier function and pulmonary edema observed in COVID-19 ARDS (Teuwen et al., 2020).
Strikingly, after merging all phospho/proteomic enrichments prior to matching (Table S2), only 71 pathways were altered across all four studies (Figure 5B). These included disrupted cell cycle, nucleic acid metabolism, and immune signaling (Figure 5C). Unepectedly, A549 cells showed predominant downregulation, whereas Vero E6 showed upregulation of these components. Dozens of pathways were preferentially impacted in infected iAT2s (Figure 5A), including dysregulation of tight junction organization and lipid metabolism, providing a contextual viral signature in distal lung epithelial cells.
Functional Network Inference of Antivirals
To explore the therapeutic potential of our data, we examined the network of differential iAT2 host phospho/proteins to identify potential points of vulnerability. In particular, we solved a Steiner tree problem (STAR Methods), connecting the largest number of differential features (leaves) to upstream regulators (connectors) (Figure 6 A). We then inferred druggable linchpins (Tuncbag et al., 2016) with available compounds within virally responsive association subnetworks for each time point (Table S2), as illustrated by a subnetwork centered on the recently purported antiviral target PRKCA (Figure 6B).
Figure 6.

Network-Based Inference and Drug Testing
(A) iAT2 response subnetworks connecting differential AT2 proteins at 24 hpi.
(B) Drug candidate PRKCA connects differential host proteins, pointing to connector nodes (linchpins) as prime antiviral targets.
(C) Compared to randomized data, connectors (Steiners) are enriched for druggability (p < 10−4, empirical random sampling).
(D) Connectors also enriched for host proteins targeted by SARS-CoV-2 (Gordon et al., 2020).
(E) Overrepresentation of connectors as drug target candidates.
(F) Top-ranked candidates (based on number of SARS-CoV-2 targets) enriched for antiviral drug targets (DT), including some (mTOR) reported as essential (E) to host cell viability.
(G) Quantitative IFA of viral N (green) in infected versus control Vero E6 cells and iAT2s treated with indicated compounds or vehicle (DMSO) (counterstained with DAPI) prior to imaging at 10× and 30× mag); scale bars represent 200 mm in Vero E6, 100 mm in iAT2s. Percentage (±SD) of N-positive cells normalized to DMSO shown below.
(H) Heatmap depicting antiviral efficacy in Vero E6 and iAT2s of compounds that are not cytotoxic (>50% cell loss) at indicated concentrations (gray bars).
(I) Structural models showing validated antivirals docked to iAT2 targets.
Random sampling showed linchpins were significantly (p < 0.0001) more actionable (Figure 6C). Strikingly, these network hubs were enriched for interaction partners of SARS-CoV-2 effector proteins (Gordon et al., 2020) (Figure 6D) and for highly connected viral targets (Figure 6E), reinforcing their relevance to viral replication. As a corollary, we ranked iAT2 linchpins based on frequency of links to host proteins (Figure 6F). Intriguingly, most of these nodes were non-differential in other infected cell lines such as Caco-2.
To check their functional relevance, we cross-referenced linchpins against ∼150 antiviral compounds with reported efficacy against SARS-CoV-2 (Bouhaddou et al., 2020; Stukalov et al., 2020) versus 101 control compounds. Most iAT2 candidate targets were indicated as promising re-purposing leads (Table S2), whereas few were essential for host survival (Wang et al., 2015).
Different Antiviral Responses of iAT2 and Vero E6 Cells
We leveraged our data-driven targets to select candidate small molecule inhibitors to test for anti-SARS-CoV-2 activity in iAT2s. We chose compounds from ∼7,000 approved drugs and leads in various stages of preclinical and clinical trials in the Drug Repurposing Hub as well as other compounds that mapped to linchpins in our inferred network (p < 10−19, Fisher’s) in vicinity of known SARS-CoV-2 PPI (p < 0.005) in the iAT2 infection platform. Giving preference for medications targeting kinases and other actionable enzymes further along in clinical testing, we prioritized 31 small molecule inhibitors for testing (Key Resources Table). Drug testing was initially performed in Vero E6 cells, a cell line frequently used for anti-SARS-CoV-2 drug screening (Figure S6). The resulting efficacy and cytotoxicity data informed a smaller-scale, secondary screening protocol for iAT2s.
Of the inhibitors tested, only KN-93 (selective inhibitor of calcium/calmodulin-dependent protein kinase II/CAMK2) and tubercidin potently inhibited SARS-CoV-2 replication in both Vero E6 and iAT2s (Figures 6G and 6H). Although the role of CAMK2 in infection is not well characterized, multiple coronavirus proteins have been shown to interact with CAMK2 (Ma-Lauer et al., 2016; V’kovski et al., 2019), highlighting it as a novel SARS-CoV-2 target. The efficacy of tubercidin may reflect its structural similarity to remdesivir, a known antiviral targeting the SARS-CoV-2 replication complex (Wang and Yang, 2020).
Robust inhibitors of SARS-CoV-2 replication in Vero E6 showing reduced efficacy in iAT2s include dorsomorphin (RPS6KA1), SB-415286 (GSK3B/RPS6KB1), vandetanib (EGFR/VEGFA), and VE-822 (ATM/ATR/MTOR) (Figure 6H; Table S2). Dorsomorphin (Gordon et al., 2020) and VE-822 (Garcia et al., 2020) were recently reported as inhibitors of SARS-CoV-2 replication, but neither was tested in primary-like lung epithelial cells. Conversely, brequinar (DHODH) and axitinib (PLK4), which were modest antivirals in Vero E6 cells, showed respectable antiviral activity in iAT2s.
Strikingly, other inhibitors which failed to inhibit SARS-CoV-2 replication in Vero E6 cells were potent in iAT2s (Table S2). Four (levofloxacin, FRAX486, losmapimod, AZ20) exhibiting <10% viral reduction in Vero E6 displayed more than a log reduction in iAT2s (Figures 6G and 6H). Levofloxacin, a quinolone antibiotic used to treat pneumonia, inhibits topoisomerase TOP2A. FRAX486, a PAK2 inhibitor, was recently reported to inhibit SARS-CoV-2 in human Huh7.5 cells, but not in Calu-3 adenocarcinoma cells (Dittmar et al., 2020). Losmapimod, a MAP kinase inhibitor not previously identified as a SARS-CoV-2 antiviral, is currently in a phase 3 clinical trial as an anti-inflammatory therapeutic (Grimes and Grimes, 2020). Finally, AZ20, a selective ATR inhibitor, highlights a novel target for SARS-CoV-2 inhibition directly relevant to human pulmonary cells.
To gain insight into the molecular mechanisms underlying cell-type/species-specific differences in drug activity, we structurally modeled the compounds bound to active sites of their respective targets in Vero E6 cells (Chlorocebus sabaeus) and iAT2s (Homo sapiens). In line with our results that KN-93 efficiently blocked SARS-CoV-2 replication in both cell lines, we did not observe any structural differences in the predicted KN-93 binding pockets of human and C. sabaeus CAMK2A (Figure 6I). In contrast, we identified amino acid differences in the predicted levofloxacin binding site of TOP2A (490 isoleucine in human substituted for methionine in the C. sabaeus protein). Since methionine is bulkier than isoleucine, steric differences could account for species-specific differences in compound activity. While we did not find major structural differences in the MAPK14 pocket for losmapimod, differences around the pocket might cause an allosteric hindrance to compound engagement (Figure S6).
Discussion
SARS-CoV-2 infection disrupts molecular processes required for normal lung homeostasis leading to impaired pulmonary function. Elucidating which cellular pathways the virus hijacks in a native alveolar context is paramount for understanding pathogenesis and devising treatments. To this end, we combined human primary-like AT2 cells with phospho/proteomic time course analysis that demonstrate diverse host responses to infection of alveolar epithelial cells. Our approach mapped iAT2 responses during early infection, before viral particle formation, and later stages characterized by dominant viral replication. Our data suggest a dynamic disease signature that evolves as the virus disrupts host programs and rewires modules (Figure 7 ).
Figure 7.

Depiction of Viral Perturbations to Alveolar Type 2 Cells by SARS-CoV-2
Top: Lung pathobiology caused by SARS-CoV-2, with histologic sections of COVID-19 patient lung biopsies stained with H&E and cytokeratin AE1/AE3 showing diffuse alveolar damage, sloughed pneumocytes, and focal hyalin membrane material (200× mag). Bottom: Viral-dysregulated iAT2 pathways, processes, proteins, phosphosites, and validated drugs/targets.
Our study is an advance over recent work (Bouhaddou et al., 2020; Stukalov et al., 2020) as our iAT2 ALI model captures the unique biology of alveolar type 2 cells implicated in SARS-CoV-2 infection and acute respiratory failure in COVID-19 (Bradley et al., 2020). These cells produce surfactants essential to lung homeostasis and gas exchange, which are severely impaired in ARDS. Notably, we uncovered 2,109 dysregulated proteins not reported in surveys of infected Caco-2 (Bojkova et al., 2020), Vero E6 (Bouhaddou et al., 2020), or A549 adenocarcinoma cells (Stukalov et al., 2020) (Table S2). Differences in overall proteome architecture and responses of iAT2s compared to undifferentiated, immortalized cell lines reflects their highly specialized function and biochemical properties, highlighting AT2-specific signatures of cytopathogenesis (Figure 7). While infection rates of iAT2s (∼20% ± 5%) are lower compared to highly permissive cell lines like Caco-2 (Bojkova et al., 2020), our ALI cultures establish apical-basal cell polarity as occurs during infection in vivo (Abo et al., 2020; Huang et al., 2020), providing insights into the early, cell-intrinsic responses of a pathophysiologically pertinent cell type.
SARS-CoV-2-infected iAT2s had increased eIF2S1 S49/S52 phosphorylation (Figure S5), linked to reduced translation of 5′-capped mRNA. This is consistent with our previous observation of eIF2S1 phosphorylation in SARS-CoV-infected cells which did not affect viral mRNA translation (Krähling et al., 2009). SARS-CoV Nsp1 engages the host machinery and antagonizes mRNA translation to favor viral protein synthesis (Thoms et al., 2020). Consistent with this, we identified inhibitors of mTOR and MAPK as potent antivirals in iAT2s (Figures 6H and 6I). We also noted hyperphosphorylation of MST kinase and upregulation of LATS1 and MOB1A by 24 hpi, implicating Hippo pathway signaling (Figure 7).
Several lines of evidence point to growth arrest and apoptosis in infected iAT2s. First, we found hyperphosphorylation of EGFR at S991 (Figure 3F), a site linked to internalization. Second, we observed a DNA damage response in infected iAT2s, characterized by increased ATR kinase by 6 hpi, hypoactive cell cycle kinases, and direct phosphoproteomic evidence of activated pro-apoptotic kinase CAMK2D, suggesting that SARS-CoV-2 disrupts multiple signaling modules to cause AT2 growth arrest and apoptosis, potentially contributing to pulmonary necrosis (Carsana et al., 2020).
The increase in nuclear envelope-associated ER and attached ribosomes indicate a site not only for viral protein synthesis and replication, but also for virus-induced damage. Electron microscopy studies showed distinct membrane alterations (Ogando et al., 2020), but why the nuclear envelope is compromised is unclear. Reflecting a loss in nuclear integrity, we observed splicing defects in infected iAT2s. Influenza virus delocalizes host spliceosomal components, impacting mRNA maturation (Dubois et al., 2014), while other studies noted disruption of splicing of transcripts encoding antiviral proteins (Ashraf et al., 2019). Given our findings, it is possible that hyperphosphorylation of splicing factors by 3–6 hpi is aimed at suppressing this antiviral response.
We noted aberrant expression and phosphorylation of key centrosomal proteins, such as CEP131/152/170, primary PLK1/4 targets, and observed destabilized centrosome-microtubule assembly in SARS-CoV-2-infected iAT2s. Volasertib and axitinib, inhibitors of PLK1 and 4, respectively, showed viral inhibition in iAT2s, confirming a role in SARS-CoV-2 replication. Exploring the mechanism by which SARS-CoV-2 promotes centrosomal abnormalities and the impact of PLK-inhibition in attenuating this process is warranted.
We leveraged our dataset to discover antiviral drug targets by identifying signaling linchpins. In addition to tubercidin, which targets SARS-CoV-2 replicase, we identified 5 drugs inhibiting viral replication by >90% in iAT2s, of which 4 showed no or weak efficacy (<10% inhibition) in Vero E6 cells (Figures 6G and 6I). Since we performed our screen, a phase 3 clinical trial for treatment of COVID-19 was initiated with losmapimod, a MAP kinase inhibitor and potent antiviral in iAT2s that did not show antiviral activity in Vero E6 (Figure S6). Structural modeling suggested a potential molecular mechanism underlying the species specificity, highlighting the need for disease-relevant models. Potentially potent inhibitors of viral replication may be overlooked if antiviral screens continue to be performed in physiologically irrelevant cell lines.
Limitations
Our use of a simplified, single-cell-type system (iAT2s) does not capture the functional heterogeneity of the distal lung.
Our model represents a mixed population of infected and uninfected cells. Whether the responses we detected are cell intrinsic and concordant between infected versus adjacent iAT2s is unclear. Despite repeated attempts, we were unable to sort viable ACE2+ iAT2s before infection for analysis. Nevertheless, ample evidence indicates that host responses occur primarily in infected cells. First, disruption of the nuclear lamina was seen only in infected cells with detectable viral particles. Second, IFA showed that SARS-CoV-2-induced changes in markers of the mitotic/cell cycle, translation, and DNA damage responses occurred predominantly (pH2AX, PCNA, pS6) or exclusively (γTub) in infected cells (Figures 3G, 4I, and S7). Together, these results demonstrate that most, if not all, changes identified by our MS reflect infected cell phenotypes.
Initial injury of infected iAT2s leads to cytokine secretion, inducing secondary effects in neighboring cells. Since cells communicate during the course of infection, understanding the contribution of uninfected cells is crucial to achieving a comprehensive view of pathogenic changes induced by SARS-CoV-2. The changes we discovered in apical tight junctions (e.g., Claudin-18) likely perturb epithelial cell-cell tethering in infected alveoli (LaFemina et al., 2014). Regarding the iAT2 drug screens, we are aware of limitations stemming from testing only a few compound concentrations. While we cannot exclude the remote possibility of an effect of the SFTPC reporter used to ensure purity and minimize iAT2 heterogeneity, this screen was meant to show the utility of our data for identifying antivirals.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SARS-CoV-2 nucleocapsid (N) | Sino Biological | Cat # 40143-R019; RRID: AB_2827977 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit | Jackson ImmunoResearch | Cat # 711-545-252; RRID: AB_3213584 |
| anti-y-tubulin | Sigma | Cat # T5326; RRID: AB_532292 |
| anti-SARS-CoV-N | Rockland | Cat # 200-401-A50; RRID: AB_828403 |
| anti-mouse IgG AlexaFluor647 | Invitrogen | Cat # A32787; RRID: AB_2762830 |
| anti-rabbit IgG AlexaFluor 488 | Invitrogen | Cat # A-11008; RRID: AB_143165 |
| Non-p-4ebp1 | Cell Signaling | Cat # 4923; RRID: AB_659944 |
| 4EBP1 | Cell Signaling | Cat # 9644; RRID: AB_2097841 |
| p-RPS6 | Cell Signaling | Cat # 2211; RRID: AB_331679 |
| RPS6 | Santa Cruz | Cat # sc-74459; RRID: AB_1129205 |
| Phospho-CaMKII(b/g/d) | Invitrogen | Cat # PA5-34663; RRID: AB_2827977 |
| CAMKII-d | Invitrogen | Cat # PA5-37833; RRID: AB_2554441 |
| p-ERK1/2 | Cell Signaling | Cat # 9101; RRID: AB_331646 |
| ERK1/2 | Cell Signaling | Cat # 4696; RRID: AB_390780 |
| LATS1 | Cell Signaling | Cat # 3477; RRID: AB_2133513 |
| p-YAP | Cell Signaling | Cat # 13008; RRID: AB_2650553 |
| p-TAZ | Cell Signaling | Cat # 59971; RRID: AB_2799578 |
| YAP/TAZ | Cell Signaling | Cat # 8418; RRID: AB_10950494 |
| p-YBX1 | Cell Signaling | Cat # 2900; RRID: AB_2219273 |
| YBX1 | Santa Cruz | Cat # sc-101198; RRID: AB_2219288 |
| p-AKT | Cell Signaling | Cat # 9275; RRID: AB_329828 |
| AKT | Cell Signaling | Cat # 9272; RRID: AB_329827 |
| p-mTOR | Cell Signaling | Cat # 2971; RRID: AB_330970 |
| p-mTOR | Cell Signaling | Cat # 2974; RRID: AB_2262884 |
| mTOR | Invitrogen | Cat # PA5-34663; RRID: AB_2552015 |
| Alexa Fluor 488 Goat anti-Mouse Secondary Antibody | ThermoFisher | Cat # A-11029; RRID: AB_2534088 |
| anti-SARS-CoV-2 Nucleocapsid (N) | Sino Biological | Cat # 40143-MM05; RRID: AB_2827973 |
| Bacterial and Virus Strains | ||
| SARS-CoV-2 isolate USA_WA1/2020 | WRCEVA | USA_WA1/2020 |
| Biological Samples | ||
| Human lung sections – post mortem | Boston Medical Center | Boston University IRB Protocol H-37859 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| WAY-600 mTOR inhibitor | MedChemExpress | HY-15272 |
| WYE-125132 mTOR inhibitor | MedChemExpress | HY-10044 |
| TMTpro™ 16plex Label Reagent | Thermo | A44520 |
| Pierce Trypsin Protease, MS Grade | Thermo | 90058 |
| aclarubicin | Enzo | BML-AW8655-0005 |
| alisertib | Selleck | S1133 |
| axitinib | Selleck | S1005 |
| AZ20 | Selleck | S7050 |
| AZD5363 | Selleck | S8019 |
| bosutinib | Selleck | S1014 |
| brequinar | Broad Institute∗ | PMID: 27641501∗ |
| danusertib | Selleck | S1107 |
| dorsomorphin | Selleck | S7840 |
| ellagic-acid | Selleck | S1327 |
| emricasan | Selleck | S7775 |
| FRAX486 | Selleck | S7807 |
| KN-93 | Selleck | S7423 |
| levofloxacin | Selleck | S1940 |
| losmapimod | Selleck | S7215 |
| NVP-BEZ235 | Selleck | S1009 |
| PCI-27483 | Cayman | 21334 |
| PFI-3 | Selleck | S7315 |
| RK-33 | Selleck | S8246 |
| Roflumilast | Selleck | S2131 |
| SB-415286 | Selleck | S2729 |
| sirolimus | Selleck | S1039 |
| SRPIN340 | Selleck | S7270 |
| teriflunomide | Selleck | S4169 |
| TG-003 | Selleck | S7320 |
| TIC10 | Selleck | S7963 |
| tideglusib | Selleck | S2823 |
| tubercidin | Selleck | S8095 |
| vandetanib | Selleck | S1046 |
| VE-822 | Selleck | S7102 |
| volasertib | Selleck | S2235 |
| Critical Commercial Assays | ||
| Pierce™ Quantitative Colorimetric Peptide Assay | Thermo | 23275 |
| Deposited Data | ||
| Raw MS/MS data and search results | This Paper | ProteomeXchange - PRIDE: PXD020183 |
| Human Proteome, all canonical reviewed sequences | Swiss-Prot, uniprot.org | Downloaded 2020-02-10 |
| SARS-CoV-2 Proteome, all Swiss-Prot and TrEMBL sequences | Uniprot.org | Downloaded 2020-05-03 |
| Imaging and processed data | This Paper | https://doi.org/10.17632/vhm7zh5ssp.2 |
| Experimental Models: Cell Lines | ||
| iAT2 Type 2 Pneumocytes | CReM, Boston University | SPC2-ST-B2 |
| Vero E6 | ATCC | CRL-1586 |
| Oligonucleotides | ||
| CCNL_F_1 | IDT | TGAACGTAATCAAACCCTGGTTCA |
| CCNL-Int_R_3 | IDT | CCTCTGCACTTCAGCCCATG |
| CCNL1_R_2 | IDT | TTGCATCTACCTTGCAGCTAGAGC |
| DOCK4_F_1 | IDT | TCCTCCTCACTGTCCTCACAAGC |
| DOCK4_int_R_3 | IDT | AAAGCCTAGGCACGCTGCAC |
| DOCK4_R_2 | IDT | TCACTCGGCTTCACCTAATGTGA |
| FERMT3_F_1 | IDT | ATCTACCTGCGGTGCCAGGA |
| FERMT3_R_2 | IDT | TCCAGCGAAAGTTCAAGGCC |
| RBM5_F_1 | IDT | TAAGCCGTGGTTTCGCCTTC |
| RBM5_R_2 | IDT | TGGTGATTCAAGGAAAGCACATTG |
| NPR2_F_1 | IDT | GGCATGGCCTTTCTCCACAA |
| NPR2_R_2 | IDT | GAACCCCTTGCCAACCACAG |
| FMR1_F_1 | IDT | TGTAGCAGGGGAATCCCAGAA |
| FMR1_R_2 | IDT | CAGCATCGCTAATGCCACTGTT |
| CLK1_F_1 | IDT | TGAACCTGGACATCGCCA |
| CLK1_int_R_3 | IDT | GGTTGGCTCCTAATTGACATTCCA |
| CLK1_R_2 | IDT | GGAAAAGTTGTGGAGTGCATCG |
| Software and Algorithms | ||
| MaxQuant (1.6.7.0) | Max Planck Institute https://maxquant.org/ | 1.6.7.0 |
| Cytoscape (3.8.0) | Cytoscape Consortium https://cytoscape.org/ | 3.8.0 |
| CellProfiler Analyst | Broad Institute https://cellprofiler.org/cp-analyst | 2.2.1 |
| CellProfiler | Broad Institute https://cellprofiler.org/ | 3.1.9 |
| QuPath | Centre for Cancer Research & Cell Biology https://qupath.github.io/ |
v0.2.0 |
| Other | ||
| Orbitrap Exploris 480 Mass Spectrometer | Thermo | Exploris 480 |
| Q-Exactive HF-X Mass Spectrometer | Thermo | HF-X |
| Agilent 1100 Series HPLC | Agilent | 1100 HPLC |
| Electron Microscope with TVIPS F216 CMOS camera | Philips | CM12 |
| Cytation 1 Cell Imaging Multi-Mode Reader | BioTek | BTCYT1V |
| Nikon Ti2 Eclipse and Photometrics Prime BSI camera | Nikon | Nikon Ti2 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Emili (aemili@bu.edu).
Materials Availability
Reagents or materials used in this work may be requested from the Lead Contact by signing a completed material transfer agreement. Pluripotent stem cell lines described in this study are available from the CReM repository at the Boston University School of Medicine. More information regarding these lines can be found at http://www.bu.edu/dbin/stemcells/.
Data Code and Availability
The mass spectrometry proteomics data (including the files for viewing the annotated spectra) have been deposited to the ProteomeXchange Consortium (Deutsch et al., 2020) via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset PRIDE: PXD020183.
Experimental Model and Subject Details
Propagation of SARS-CoV-2
Vero E6 (ATCC CRL-1586) were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (50 U/mL), and streptomycin (50 mg/mL). For infection studies, the cell culture media was supplemented with 2% FBS. SARS-CoV-2 stocks (isolate USA_WA1/2020, GenBank Accession number MN985325; kindly provided by CDC’s Principal Investigator Natalie Thornburg and the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA)) were used to infect Vero E6 cells and propagate the virus. To remove confounding cytokines and other factors, viral stocks were purified by ultracentrifugation through a 20% sucrose cushion at 80,000xg for 2 h at 4°C (Olejnik et al., 2017). Pelleted virions were suspended in PBS. SARS-CoV-2 titers were determined in Vero E6 cells by 50% tissue culture infectious dose (TCID50) assay. All work with infectious SARS-CoV-2 was performed in the Biosafety Level 3 and 4 facilities of the National Emerging Infectious Diseases Laboratories, Boston University following approved SOPs.
iAT2 air-liquid interface (ALI) culture
The human induced pluripotent stem cell (iPSC) line, SPC2-ST-B2, engineered to carry a tdTomato reporter targeted to the endogenous SFTPC locus (Hurley et al., 2020), underwent directed differentiation to generate iPSC-derived alveolar epithelial type 2 cells (iAT2s) in 3D Matrigel cultures using methods we have previously published (Jacob et al., 2017; Jacob et al., 2019). Briefly, to establish pure cultures of iAT2s, cells were sorted by flow cytometry to isolate SFTPCtdTomato+ cells on day 41 of differentiation, and subsequently maintained through serial passaging as self-renewing monolayered epithelial spheres by plating in Matrigel (Corning) droplets at a density of 400 cells/μl. The cultures were fed every other day with a defined serum-free distal lung maintenance media composed of base media (cSFDM) supplemented with 3 uM CHIR99021, 10 ng/mL KGF, 50 nM dexamethasone, 0.1 mM cAMP, and 0.1 mM IBMX, referred to as “CK+DCI” media and as described previously (Jacob et al., 2019). iAT2 culture quality and purity were monitored at each passage by flow cytometry, where > 80% of cells expressing SFTPCtdTomato was observed over time, as shown previously (Hurley et al., 2020; Jacob et al., 2019).
To establish air-liquid interface (ALI) cultures, single cell suspensions of iAT2s were prepared as described recently (Abo et al., 2020). Briefly, iAT2s were plated on Transwell inserts at a density of 520,000 live cells/cm2. Two days after plating, apical media was aspirated to initiate ALI culture, and basolateral CK+DCI media was replenished 3 times per week thereafter.
SARS-CoV-2 infection of iAT2 ALI cultures
SARS-CoV-2 infections of iAT2s were performed as previously described (Huang et al., 2020). Briefly, iAT2s plated in ALI culture for 7-21 days were infected with SARS-CoV-2 at the indicated MOIs or mock-infected by adding 100 μl inoculum to the apical chamber. Inoculum was removed after 1 h at 37°C. For proteomic analyses the cells were infected with SARS-CoV-2 at an MOI of 5 and incubated at 16°C for one h before being returned to 37°C (0 h time point post-infection) to allow for attachment of the viral particles and to synchronize infection. At the indicated times post-infection, cells were processed for various analyses described below.
Method Details
Immunofluorescence analysis
For immunofluorescence staining, iAT2s cultured on Transwell inserts were fixed in 10% formalin for 30 min to 12 h and stained, mounted, and imaged as previously described (Huang et al., 2020). Primary antibodies used include SARS-CoV nucleocapsid (N) protein (rabbit polyclonal, 1:2500, Rockland Immunochemicals, 200-401-A50) which cross-reacts with SARS-CoV-2 N (Thi Nhu Thao et al., 2020), anti-g-tubulin (mouse monoclonal, 1:10,000, Sigma, T5326), anti-pro-SFTPC (mouse monoclonal, 1:500, Santa Cruz, sc-518029), and anti-dsRNA antibodies (mouse monoclonal, 1:2, Millipore, 3361) and anti-phospho-g-H2AX (mouse monoclonal, 1:1000, Sigma-Aldrich, 05-636). PCNA antibodies were a generous gift from Dr. Yoshinari Takasaki (Juntendo University School of Medicine, Japan) and used for immunostaining as described before (Xiao et al., 2007). Secondary antibodies used include (Alexa Fluor 488 AffiniPure donkey anti-rabbit IgG (H+L), (1:500, Jackson ImmunoResearch, 711-545-252), anti-rabbit IgG AlexaFluor 488 (1:500, Jackson ImmunoResearch, 711-545-152), Alexa Fluor 647 AffiniPure donkey anti-rabbit IgG (H+L) (Jackson ImmunoResearch, 711-605-152), and anti-mouse IgG AlexaFluor647, (1:500, Invitrogen, A32787). Following secondary antibody incubation, cells were washed again and DNA counterstained using the Hoechst dye (1:500, Life Technologies) or DAPI.
Slides were imaged either on the Zeiss LSM 800 confocal microscope (Zeiss) using the Zen 2012 SP1 (64-bit, black edition) software (Carl Zeiss, USA) or on the Olympus BX61 microscope equipped with a charge-coupled device camera (ORCA, Hamamatsu) and a spinning disc confocal attachment (Olympus DSU). Images were acquired on the Olympus BX61 microscope using a 60X1.42NA or 100X1.4NA objective lens. Images were deconvolved using the Autoquant constrained iterative deconvolution software in Metamorph. Signal intensity and numbers were quantified using ImageJ.
Human COVID-19 autopsy specimens, lung tissue sections, and IHC analysis
Autopsied lung tissue (FFPE) was freshly cut into 5 μm sections, protease 3 (Ventana, 760-2020) treated, heated to induce epitope retrieval with alkaline CC1 buffer (Ventana, 950-124), and stained on a Ventana Benchmark Ultra (Ventana Medical Systems, Tucson, AZ, USA) with AE1/AE3 (Ventana, 760-2135). Parallel sections were stained with H&E and AE1/AE3 to distinguish sloughed pneumocytes from immune cells. Visualization with DAB was performed via Optiview detection (Ventana, 760-700). All tissues from patients with COVID-19 were collected post-mortem at the time of autopsy at Boston Medical Center with consent from next-of-kin, and the IRB of Boston University determined that this study did not constitute human subject research. Control samples without infection were collected from healthy tissue adjacent to a lung tumor, with IRB approval under protocol H-37859.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed according to methods detailed in (Huang et al., 2020). Briefly, iAT2 ALI cultures seeded on Transwell inserts were infected with SARS-CoV-2 at a MOI of 5 or mock-infected. At 24 hpi, cells were fixed in 10% formalin for 6 h, post-fixed in 1.5% osmium tetroxide (Polysciences), stained in 1.5% uranyl acetate (Electron Microscopy Sciences, EMS) and embedded samples were thin sectioned (70 nm). Grids were stained in 4% aqueous uranyl acetate followed by lead citrate and imaged on a Philips CM12 Electron Microscope operated at 100kV. Images were recorded on a TVIPS F216 CMOS camera with a pixel size of 0.85-3.80 nm per pixel.
In vitro kinase and phase-separation assays
The pET28a vector expressing recombinant 6xHis tagged SARS-CoV-2 nucleocapsid (N) was transformed in SoluBL21 competent cells (Genlantis). Bacteria from four 1L cultures (grown at 37°C for ∼3 h to A600 ∼0.5 then switched to 16°C, induced with 0.5mM IPTG and grown overnight) were pelleted. The bacteria were resuspended in RBP buffer A (10mM HEPES pH7.4, 300mM NaCl, 10mM Imidazole) containing 2mM MgCl2, 32μg/mL Lysozyme (Sigma), 4μg/mL DNaseI, 200μM PMSF and a tablet of protease inhibitor cocktail (Pierce), then lysed 6 times in liquid N2 and a tepid water bath. The lysate was clarified by centrifugation at 10,000rcf for 10 min, diluted to ∼250ml in lysis Buffer A then loaded by circulating pump to a cobalt chloride-charged, Buffer A equilibrated 5ml HiTrap HP Chelating column (GE). The column was then washed and eluted using an AKTApure FPLC (GE) using a gradient of Buffer A and RBP Buffer B (10mM HEPES pH7.4, 1M NaCl, 300mM Imidazole). Eluted His-tagged N protein was further purified by HiPrep 26/60 S-200 size exclusion column (GE) (in 10mM HEPES pH7.4, 150mM NaCl) then concentrated by Amicon Ultra 15ml 30kDa MWCO centrifugal filters (Sigma). Concentration of N was determined by nanodrop A280, then aliquoted, snap frozen and stored at −80°C until use. Purified His-N (200μg) was phosphorylated with 1000 units of CSNK2A2 (Thermo; PV3993) or GSK3 (NEB; P6040) for 4 h at 30°C (in 10mM HEPES, pH7.4, 150mM NaCl, 1mM MgCl2, 1mM DTT, 0.01% Tween 20, and 200μM ATP (NEB; P0756S). Unphosphorylated N was incubated 4 h at 30°C in the same buffer but no kinase. His-N was then re-purified using His60 Ni Superflow beads (Takada) in spin columns (Pierce). Reactions and washes were performed in 10mM HEPES, pH7.4, 150mM NaCl, 0.01% Tween 20, then eluted in the same buffer supplemented with 1M Imidazole. The eluted protein was concentrated and buffer exchanged into 10mM HEPES, pH7.4, 150mM NaCl, 0.01% Tween 20 using Amicon Ultra 0.5ml 30kDa MWCO centrifugal filters (Sigma).
Phase separation reactions were performed using indicated concentrations of naive recombinant viral N protein or in vitro phosphorylated (p.N) N in 10mM HEPES pH7.4, 100mM NaCl, 5% PEG (polyethylene glycol-8000), and 200ng/μl total murine brain RNA. Total murine brain RNA was collected from snap frozen whole hemispheres dissected from isofluorane euthanized, PBS perfused C57Bl6 mice. RNA was extracted by tissue lysis in QIAzol reagent followed the Lipid tissue RNeasy minikit protocol (QIAGEN). Reactions were mixed in PCR tubes, then pipetted to wells on microscope slides made by SecureSeal Imaging Spacers (SS10X6.35; Grace Bio-labs), coverslipped then inverted (allowing forming droplets to settle by gravity onto coverslips) and incubated at 24°C for 2 h. Droplets in imaging spacers were imaged at 63x on a Zeiss AxioObserver A1 microscope using differential interference contrast (DIC).
Sample preparation for proteomics and phosphoproteomics analysis
At the indicated times post-infection, SARS-CoV-2- and mock-infected iAT2s were dissociated using Accutase (STEMCELL Technologies) and briefly centrifuged to obtain the cell pellets. The cells were lysed and the virus inactivated by re-suspending the cell pellets in roughly 5 packed cell volumes (p.c.v) of GuHCl lysis buffer (6 M GuHCL, 100 mM Tris pH 8.0, 40 mM chloroacetamide, 10 mM TCEP) supplemented with phosphatase inhibitor cocktail (Roche) followed by heating to 100°C for 10 min. Lysates were sonicated with a Branson probe sonicator and were then quantified via Bradford assay. The samples were diluted with 100 mM Tris, pH 8.5 buffer to lower the GuHCl concentration to 0.75 M. Lysate proteins were then digested by adding trypsin (Pierce) at a 1:50 ratio (enzyme: protein, w/w) and incubating the samples overnight at 37°C with shaking. Trypsinization was terminated with the addition of TFA to below pH 3 and the peptide digests were desalted via reversed-phase C18 columns (Sep-Pak, Waters) with a wash buffer of 0.1% TFA and elution buffer of 60% acetonitrile. The desalted peptides were then quantified with a Quantitative Colorimetric Peptide Assay (Pierce). Each sample comprising one hundred micrograms peptides was TMT-labeled with TMTPro 16plex reagents (ThermoFisher, cat. # A44520) as per the manufacturer’s protocol. Because there were more samples than could be multiplexed in one TMT set, balanced batches were produced, and a common pool of all samples was also labeled and added to each batch. Each batch of labeled peptides was again desalted on C18 columns prior to basic reversed-phase fractionation.
TMT-labeled peptides were fractionated via basic reversed-phase chromatography on the Agilent 1200 series HPLC instrument equipped with the XBridge Peptide BEH C18 column (130Å, 3.5 μm, 4.6 mm X 250 mm, Waters Corporation). Prior to loading peptides, the C18 column was washed with 100% methanol and equilibrated with Buffer A (0.1% ammonium hydroxide and 2% acetonitrile). Peptides were injected via the autosampler and eluted from the column using a gradient of mobile phase A (2% ACN, 0.1% NH4OH) to mobile phase B (98% ACN, 0.1% NH4OH) over 48 min at a flow-rate of 0.4 mL/minute. The 96 fractions collected were orthogonally concatenated into either 8 (early time points) or 16 (24 hpi) pooled fractions. Five percent of each fraction was aliquoted and saved for global proteomic profiling and the remaining 95% of peptides were used for phosphopeptide enrichment using Fe-NTA magnetic beads (CubeBiotech) (Leutert et al., 2019). Briefly, the fractionated peptides were dried and resuspended in binding buffer (80% Acetonitrile and 01% TFA). Before being added to the peptides, the Fe-NTA beads were washed three times with binding buffer. Peptides were then incubated with the Fe-NTA bead slurry for 30 min on a gyrotator at 4°C. Bound phosphopeptides were washed three times with binding buffer, after which phosphopeptides were serially eluted twice by adding 200 μL of elution buffer (50% acetonitrile and 2.5% ammonium hydroxide) to the beads. Both phosphopeptide eluates corresponding to an orthogonal fraction were combined prior to drying in a speedvac.
For sequencing in vitro kinase assay proteins, 10 μg of N or N plus kinase in 2.5ul kinase buffer was reduced and alkylated (40 mM chloroacetamide, 10 mM TCEP), and the samples diluted in 20 μl 100 mM TEAB pH 8.5 buffer prior to digestion with 1 μg Trypsin (Pierce) overnight at 37°C on a thermoxier. Trypsinization was terminated with the addition of FA to 1% final concentration and the peptide digests were desalted via Ziptip C18 (Millipore) prior to LC/MS/MS analysis.
Mass spectrometry analysis
Multiplexed phosphopeptide fractions from each time point were resuspended in mobile phase A solvent (2% acetonitrile and 0.1% formic acid) to be analyzed on the Q-Exactive HF-X mass spectrometer (ThermoFisher Scientific). Every two neighboring fractions of the 16 24 h fractions were pooled to optimize the use of instrument time. The mass spectrometer was interfaced to the Easy nanoLC1200 HPLC system (ThermoFisher Scientific). Briefly, the phosphopeptides were first loaded onto a reverse-phase nano-trap column in mobile phase A, (75 mm i.d. × 2 cm, Acclaim PepMap100 C18 3 mm, 100 Å, ThermoScientific) and separated over an EASY-Spray column, (ES803A, Thermo Scientific) using a gradient (6% to 19% over 58 min, then 19% to 36% over 34 min) of mobile phase B (0.1% formic acid, 80% acetonitrile) at a flow rate of 250 nL/min. The mass spectrometer was operated in positive ion mode with a capillary temperature of 275°C and a spray voltage of 2100 V. All data was acquired with the mass spectrometer operating in data dependent acquisition (DDA) mode. Precursor scans were acquired at a resolution of 120,000 FWHM with a maximum injection time of 120 ms in the Orbitrap analyzer. The six most abundant ions, with charge states 32, were selected for fragmentation by HCD (NCE 29%) and analyzed at a resolution of 45,000 FWHM with a maximum injection time of 250 ms. Peptides for protein abundance measurements were analyzed the same way save for a few exceptions: The Orbitrap Exploris 480 equipped with FAIMS (alternating cv = −50, cv = −70) was used (except for the 24 h fractions, which went with the HF-X), the LC gradient was longer (6% to 17% over 77 min, then 17% to 36% over 45 min), and the DDA method chose the 12 most abundant ions for fragmentation (Exploris 480 NCE 33%) at a maximum inject time of 60ms.
For the kinase assay, a targeted m/z list for phosphopeptides from N was produced by ProteinProspector 6.2.1. Briefly, the Nucleoprotein primary sequence (SwissProt, P0DTC9) was digested in-silico to produce putative tryptic peptides with S, T, and Y phosphorylation variable modifications, up to 1 missed cleavage, and charge states 2-6. The final list was composed of 410 possible m/z features from phospho-N. Samples were injected onto a nano-trap column and resolved over an EasySpray column as described above. Data was acquired on an Exploris 480 (Thermo Fisher Scientific) mass spectrometer equipped with FAIMS running dual compensation voltages at −50V and −70V. Every duty cycle was split between untargeted fast scans at 15,000 resolution (max inject 60ms) analyzing the 6 most abundant ions with a fragmentation trigger at 4e5, and phospho-N targeted scans at 60,000 resolution (max inject 400ms) targeting only the two most abundant ions that also were represented the inclusion list but with a much lower trigger for HCD at 3e5.
Quantification and Statistical Analysis
Analysis of raw mass spectrometry data
All acquired MS/MS spectra were searched against the Uniprot human complete proteome FASTA database downloaded on 2020-02-10, and the SARS-CoV-2 proteome downloaded from SwissProt on 2020-05-03 using the MaxQuant software (Version 1.6.7.0) that integrates the Andromeda search engine. TMT reporter ion quantification was performed using MaxQuant with default settings. Briefly, enzyme specificity was set to trypsin and up to two missed cleavages were allowed. Cysteine carbamidomethylation was specified as fixed modification whereas oxidation of methionine and N-terminal protein acetylation were set as variable modifications. For phosphopeptides serine, threonine and tyrosine phosphorylation were specified as variable modifications. Precursor ions were searched with a maximum mass deviation of 4.5 ppm and fragment ions with a maximum mass deviation of 20 ppm. Peptide and protein identifications were filtered at 1% FDR using the target-decoy database search strategy (Elias and Gygi, 2007). Proteins that could not be differentiated based on MS/MS spectra alone were grouped to protein groups (default MaxQuant settings). A threshold Andromeda score of 40 and a threshold delta score of 8 was applied to phosphopeptides. The MaxQuant output files designated “Phospho(STY)sites” and “proteinGroups” were used for data normalization and other statistical analysis using in-house generated scripts in the R environment.
Raw files acquired from LC-MS runs of the kinase assays were analyzed with MaxQuant (v 1.6.14.0) by searching against the human and SARS-CoV-2 proteomes with the settings designated above. Annotated mass spectra for identified phosphopeptides were produced with PDV 1.6.2 (Wang et al., 2018).
Data Analysis and Pathway Enrichment
Bioinformatic analysis was performed using R: A language and environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria) (http://www.R-project.org), version 3.6.1. The MaxQuant tables of stringently identified (FDR < 1%) protein groups and phosphosite feature intensities were log transformed. For the 24 hpi time point, the data were normalized by loess, while for the 1, 3, and 6 hpi time points, quantile normalization and a batch correction were applied across the two TMT sets using the ComBat algorithm from the sva R package; post batch correction data was used for all analysis. Quantified features at 1, 3, and 6 hpi (proteins for proteomics, phosphosites for phosphoproteomics) were standardized (Kumar and Futschik, 2007) and grouped into clusters using an unsupervised k-means clustering approach (Gu et al., 2016). The groups were split to aid visualization. For functional enrichment of proteins and phosphosites based on clustering at the 1, 3, and 6 hpi time points, Enrichr was used with the Reactome database of pathways. For differential analysis, the LIMMA (Ritchie et al., 2015) R package was used to fit a linear model accounting for the infection versus mock condition at each time point. Moderated t tests were corrected with the Benjamini-Hochberg method for false discovery rate (FDR). Gene set enrichment analysis was performed using the fgsea R package (Sergushichev, 2016) using curated gene libraries (Merico et al., 2010) using a ranked list where the gene rank is defined as -log(p value)∗sign(log2 fold change) (Reimand et al., 2019). In the case of duplicate proteins or phosphosites mapping to a common gene symbol, the one with the highest absolute value rank was retained. Kinase enrichment analysis was performed with KEA2 (Lachmann and Ma’ayan, 2009) where regulated phosphosites (p value £ 0.05, |logFC|30.25) were split into 2 categories, Upregulated (logFC 3 0.25) or Downregulated (logFC £ –0.25), and with the KSEAapp R package (Casado et al., 2013) where phosphosites were ranked as for the GSEA analysis. Publicly available data were downloaded from the referenced publications as processed feature matrixes for enrichment analysis as described for the GSEA analysis above.
Study comparisons were made at the pathway level from the common gene set library, as well as at the protein level by collapsing to Gene Symbols. To find pathways pronounced in iAT2s relative to other studies, enriched pathways were ranked by difference between minimum time point FDR in iAT2s compared to the other 3 cell lines. Statistical significance of 4,704 pathways that are enriched in at least one time point in iAT2s were compared across the 4 cell-lines: iAT2 (this study), A549 (Stukalov et al.; 2020), Caco-2 (Bojkova et al.; 2020), Vero E6 (Bouhaddou et al.; 2020). For each pathway, the minimum FDR within any time point in the 3 external cell lines (A549, Caco-2, Vero E6) was subtracted from the corresponding minimum FDR in the iAT2s. The larger the magnitude of the resultant value, the greater the difference in statistical significance of observed pathway regulation between the iAT2s and the other cells. Negative magnitudes are suggestive of iAT2-specific enrichments and vice versa for positive magnitudes. Pathways that were significantly enriched in all studies were defined as the set of 71 pathways that were significantly enriched (FDR < 0.1) in at least one time point in all four cell lines.
Western blotting
Briefly, the iAT2s were lysed and the virus inactivated by re-suspending the cell pellets in 100 μL of urea lysis buffer (9 M urea, 20 mM HEPES pH 8.0) followed by heating to 100°C for 10 min. Twenty five micrograms of protein lysate was run on a 12% SDS-PAGE Stain-Free gel (Bio-Rad). Stain-free gels were imaged using a ChemiDoc (BioRad) to ensure equal loading before transferring to Nitrocellulose using Turbo TransBlot. Membranes were blocked in 5% milk, 0.02% sodium azide in TBST at room temperature for 1 h. Primary antibodies were incubated overnight at 4°C while rocking in 5% normal horse serum in TBS. Blots were washed 3 times in TBST at room temperature. Secondary antibodies (Immun-Star HRP, BioRad) were incubated with membranes for 1 h at room temperature in 5% normal horse serum in TBS before washing 3 times in TBST. Chemiluminescent detection was performed using Pierce PicoPlus ECL detection reagent and imaged using BioRad ChemiDoc.
Alternative Splicing Analysis
Alternative splicing was analyzed using vast-tools v2.2.2 (Tapial et al., 2017) in combination with VastDB version VDB2_20.12.19 using option–IR_version 1. Changes were deemed significant if they were greater than 10 dPSI/dPIR and the expected minimum changes was different from zero at with p > 0.95 according to vast-tools’ diff module. Events were filtered requiring a minimum of 10 reads per event and a balance score (quality score 4) of ‘OK’ or ‘B1’ for alternative exons or > 0.05 for intron retention events in at least 2 of 3 replicates. Gene ontology enrichment analysis was conducted with FuncAssociate (Berriz et al., 2003) using backgrounds of all genes in the dataset that contained splicing events of the same type that survived filtering.
To perform experimental validation of splicing events, cDNA was prepared from total RNA (0.6 mg) of Mock-treated or SARS-CoV-2 infected iAT2s by reverse transcription using the SuperScript IV Reverse Transcription kit (Thermo Fisher) and random hexamer primers. Next, cDNA was used as template in a 25 μL PCR reaction with Q5 High-Fidelity DNA Polymerase (New England Biolabs) along with the oligonucleotides listed in the Key resources table. PCR reactions were resolved in 2.5% agarose gels. Intensity of the bands were quantified using the ImageJ software. Background signal was subtracted from the peak signal. Efficiency of splicing was quantified as a ratio of spliced to unspliced PCR products.
Drug targets
Utilizing DrugBank version 5.1.6 (Wishart et al., 2006), we collected annotated drugs that target proteins that were differentially expressed or phosphorylated (FDR < 0.05) in response to viral infection at each time point. Given that differential proteins in each time point translate to differing sets of drugs, we focused on a final set of 62 drugs that hit targets in every time point, translating to a set of 1,622 candidate drug targets.
We also collected a set of 87 drugs that were screened for SARS-CoV-2 viral inhibition from (Bouhaddou et al., 2020). In particular, 46 drugs with a ratio of CC50 (cell viability) to IC50 (viral inhibition) of 2 were deemed promising, while the remaining 41 drugs were considered non-promising. Furthermore, we added 11 effective and 5 ineffective drugs to these drug sets (Stukalov et al., 2020) based on their shared values for cell viability and virus growth. In total, such drug sets translate into 173 promising and 124 non-promising drug targets.
Steiner Forest Problem to define network connectors as candidate drug targets
In a directed weighted molecular interaction network, we considered a set of ‘prized genes’ (e.g., differentially expressed or phosphorylated genes) with a non-zero score in a network of weighted molecular interactions. To solve the Steiner Forest problem we prune away proteins and interactions until a forest of trees remains that simultaneously maximizes the sum of the gene prizes and the sum of the edge weights, creating a forest of trees that connects as many prized host proteins that are connected through as few non-prized nodes as possible. The prizes on a protein reflect its importance, indicating its chance to be selected in the optimal forest. As a consequence, all leafs (i.e., terminal nodes) in the optimal forest are prized proteins. Edge weights take values between 0 and 1, indicating that edges with higher weights are more likely to be selected in the optimal forest. More formally, given a weighted graph G(V, E) with node set V, edge set E, a function p(v) that assigns a prize to each node v ∈ V, a function w(e) that assigns a weight to each edge e ∈ E, we seek to find a forest F(VF, EF) that minimizes the objective function
where kv is the number of interactions gene v is involved in, c(e) = 1-w(e) and b is a scaling factor, that affects the number of prized nodes included in the optimal forest. Furthermore, m is a parameter that penalizes hub nodes with high number of interactions while k is the number of trees in the forest, and w is a parameter that controls the number of trees (Tuncbag et al., 2013). Prior to computing the optimal forest, a dummy node is attached to a subset of the nodes in graph G. Once the optimization is complete, the dummy node and all of its artificial edges are removed to reveal a forest where each tree root was connected to the dummy node. To solve the Steiner tree problem, we utilized the Omics Integrator software package using a message-passing algorithm (Tuncbag et al., 2016).
As the Steiner forest algorithm requires an input network, we assembled a directed molecular network of 57,179 edges between 11,642 host protein nodes. In particular, we accounted for 43,364 transcription factor-target interactions as of ENCODE (Gerstein et al., 2012), 19,287 kinase-substrate interactions as of PhosphoSitePlus (Hornbeck et al., 2015), 2,961 phosphatase-substrate as of the HuPho database (Liberti et al., 2013) and 5,979 signaling interactions as of the Signor database (Licata et al., 2020). For each interaction e in the input network, the Steiner forest problem requires the assignment of a weight or probability w(e), suggesting that edges with low cost c(e) = 1 – w(e) are more likely to be selected in the optimal forest. Given a directed edge e = (x, y), where the node x is the tail or the source of the interaction and node y is the head, we defined that the weighting function is the reciprocal of the outdegree of x as . Consequently, edges that involve tail genes with high outdegree will be more likely to be removed during the Steiner forest optimization. Such a step allowed us to more effectively penalize hub nodes, indicating higher confidence that edges selected in the optimal forest are more specific to a viral response instead.
As for a set of priced genes, we considered genes that were significantly differentially expressed or phosphorylated in each time step after infection if their corresponding FDR < 0.05. To generate four networks that show how interactions evolve in a cohesive manner, we assigned uniform prizes to host proteins that were differentially expressed or phosphorylated (FDR < 0.05) in iAT2s up to the given time point post infection, allowing us to treat all proteins in the same way as their differential expression or phosphorylation states varies in each time point.
Structural modeling of proteins and protein-drug interactions
Wherever possible, X-ray structures of the proteins in question were used for modeling. Short fragments of missing residues were rebuilt and phosphosites were added using CHARMM-GUI (Jo et al., 2008). Long missing regions were rebuilt using MODELER (Eswar et al., 2007). The complex of SARS-COV-2 nucleocapsid and casein kinase II subunit alpha’ (CSNK2A2) was constructed based on superposition of several structural templates (PDB Ids 6VYO, 6WZO, 2WEL). Sequence alignments between target and template sequences were done with HHsuite (Steinegger et al., 2019), and MODELER was used to build the final homology model. All unstructured highly phosphorylated sites were mapped to functional sites based on the Eukaryotic Linear Motif resource for Functional Sites in Proteins (ELM) (Kumar et al., 2020). For all classes of functional sites that were affected by the phosphorylation, the total number of affected site instances belonging to that class was computed, and the interacting protein/protein family for the class was determined based on known binding partners of that ELM site family. The three-state (alpha helix, beta sheet, extended conformation) secondary structure prediction for the highly phosphorylated sites was done using PSIPRED (Jones, 1999). Mapping to Pfam (El-Gebali et al., 2019) entries was done using the Pfam annotation available for Uniprot sequences.
The models of ligand interactions with their protein targets for KN-93, levofloxacin, AZ20, FRAX486 and losmapimod were built using LigTBM - a high accuracy template-based docking approach (Alekseenko et al., 2020; Kotelnikov et al., 2020).
Small molecule inhibitor testing
1x104 Vero E6 cells were seeded per well of 96-well plates in DMEM growth medium (DMEM supplemented with 2% FBS, 50 units/mL penicillin and 50 mg/mL streptomycin). The following day, cells were pretreated with the compounds solubilized in DMSO and diluted in DMEM growth medium to the indicated concentrations and incubated for 1 h at 37°C and 5% CO2. Cells were subsequently infected with SARS-CoV-2 at a MOI of 0.1 in the presence of inhibitors by adding 10 μl of diluted virus to each well. Two days post infection, cells were fixed in 10% neutral buffered formalin for 6 h at 4°C, then removed from the BSL-4 laboratory. The cells were washed in PBS, permeabilized with 1:1 (vol:vol) acetone-methanol solution for 5 min at −20°C, incubated in 0.1 M glycine for 10 min at room temperature, and subsequently incubated in blocking reagent (2% bovine serum albumin, 0.2% Tween 20, 3% glycerin, and 0.05% NaN3 in PBS) for 15 min at room temperature. After each step, the cells were washed three times in PBS. The cells were incubated for 1 h at room temperature with a SARS-CoV-2 nucleoprotein-specific antibody (rabbit polyclonal, 1:2000, Rockland Immunochemicals, 200-401-A50), washed four times in PBS, and incubated with a goat anti-rabbit antibody conjugated with AlexaFluor488 (1:200, Invitrogen) for 1 h at room temperature. 4’,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) was used at 200 ng/mL for nuclei staining. Cells were stored at 4°C in PBS prior to imaging.
iAT2s were seeded on 96-well Transwells or 6.5 mm Transwells (Corning) in CK+DCI media. At 7 days post-seeding, the basolateral media was replaced with dilutions of each drug at the indicated concentrations in CK+DCI media, and drug dilutions in CK+DCI media were added to the apical side of membranes at the same time. After one h, SARS-CoV-2 (MOI 0.04) was added to the apical media and allowed to attach for one h at 37°C and 5% CO2 prior to complete replacement of the apical media containing virus and drug with apical media containing only drug dilutions. At two days post infection, cells were fixed in 10% neutral buffered formalin for 6 h at 4°C and removed from the BSL-4 laboratory. Cells were stained from the apical side as described in the paragraph above. Membranes were then excised from the transwell plate insert using a scalpel and mounted on slides using FluorSafe mounting reagent. Images for both Vero E6 and iAT2 cells were acquired using a Nikon Eclipse Ti2 microscope with Photometrics Prime BSI camera and NIS Elements AR software. Images taken at 4x and 10x magnification, for Vero E6 and iAT2 cells respectively, were used for analysis with the positive cell detection feature of QuPath software (Bankhead et al., 2017). Infection rates were determined by quantifying nuclei and N-positive cells. Staining intensity thresholds were established based on mock-infected and untreated infected samples. Cytotoxicity threshold was set to 50% cell loss compared to DMSO control.
Acknowledgments
We thank team members for expert support. We acknowledge the following sources of funding: NIH F30HL147426 to K.M.A.; CJ Martin Fellowship from the Australian NHMRC to R.B.W.; IM Rosenzweig Award, Pulmonary Fibrosis Foundation to K.D.A.; NIH AG056318, AG064932, AG061706, and AG050471 to B.L.W.; NIH RM1 GM135136 and R21GM127952 and NSF AF1816314 to D. Kozakov; Evergrande MassCPR, C3.ai Digital Transformation Institute Award, and NIH R01HL128172, R01HL095993, R01HL122442, U01HL134745, and U01HL134766 to D.N.K.; CIHR, COVID-19 Action Initiative to B.J.B.; NIH HL007035T32 to E.L.S.; NIH R01AI125453, P01AI120943, and R01AI128364 and Massachusetts Consortium on Pathogen Readiness to J.J.P. and R.D.; NIH U01TR001810, UL1TR001430, R01DK101501, and R01DK117940 to A.A.W.; Fast Grants, Evergrande MassCPR, and NIH R01AI133486, R21AI135912, R21AI137793, and R21AI147285 to E.M.; and NIH 1UL1TR001430, RO1AG064932, and RO1AG061706 to A.E.
We thank Greg Miller and Marianne James (CReM Laboratory and iPSC Core Managers), Mitchell White and Baylee Heiden (technical support Mühlberger lab), and Joseph Corbett (Controls Manager NEIDL). Bulk RNA-seq library preparation, sequencing, and data analysis was performed by the Molecular Biology Core Facility at Dana-Farber Cancer Institute.
Author Contributions
Conceptualization: E.M., D.N.K., A.E.; Experiments: R.M.H., A.J.H., R.K.G., R.B.W., E.L.S., E.B., A.H., U.B., J.H., S.D.; Methodology/Advice: A.A.W., K.D.A., J.H.C., B.C.B., M.S., R.D., M.M., S.W., K.M.A., R.B.W., J.H.; Analysis: A.Y., R.K.G., I.P., B.C.B., E.L.S., A.J.H., D.B., C.V.-M., S.P., D.P., M.D.L., V.P., B.L.W., D. Kozakov, D. Kretov, D.C., R.M.H., E.M., A.J.H., S.W., A.B., C.P.-R.; Validation: A.J.H., E.J.B., A.M.-M., D.C., M.B., P.E.A.A., J.Z., S.O., S.M.L., J.J.P., M.V., A.T.-L., X.V., K.M.A., R.B.W., J.X.-C., J.H., S.F., J.H.K., R.D., R.K.G., B.J.B.; Manuscript: R.K.G., S.P., M.B., I.P., B.C.B., R.B.W., K.M.A., J.H., A.A.W., D.N.K., D.C., J.H.C., S.W., A.J.H., E.L.S., E.M., A.E.; Resources: C.V.-M., B.C.B., I.P., S.P.
Declaration of Interests
B.L.W. declares a position as CSO of Aquinnah Pharmaceuticals. A.E. and D.N.K. declare industry funding from Johnson & Johnson, Merck, and Novartis.
Published: November 19, 2020; corrected online December 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.11.028.
Supplemental Information
Proteomic data and phosphoproteomic data used for analysis, Kinase enrichment analysis based on phosphoproteomic data and RNA-seq data used for analysis.
Prot_1-3-6 Hour: Proteomics abundance data and protein annotation at 1, 3, and 6 h. Normalized data extracted from MaxQuant proteinGroups.txt file, with feature annotation, and differential analysis statistics.
Phospho_1-3-6 Hour: Phosphoproteomic abundance data and protein annotation at 1, 3, and 6 h. Normalized data extracted from MaxQuant PhosphoSites file, with feature annotation, and differential analysis statistics.
Proteomics _24 Hour: Proteomics abundance data and protein annotation at 24 h. Normalized data extracted from MaxQuant proteinGroups.txt file, with feature annotation, and differential analysis statistics.
Phospho_24 Hour: Phosphoproteomic abundance data and protein annotation at 24 h. Phosphoproteomic abundance data and protein annotation at 1, 3, and 6 h. Normalized data extracted from the MaxQuant PhosphoSites file, with feature annotation and differential statistics.
In vitro kinase assay: Phosphoproteomic data corresponding to in vitro kinase assays performed using GSK3 and CNSK2 as kinases and SARS-CoV-2 Nucleoprotein (N) as substrate. Phosphosites on N, upon incubation with either kinase are annotated.
KSEA: Kinase Substrate Enrichment Analysis using KSEAapp R package based on phosphoproteomic data at 1, 3, 6, and 24 h. Top hits filtered and showing relevant Kinase statistics based on motif matching in databases.
KEA2: Kinase Enrichment Analysis 2 (https://www.maayanlab.net/KEA2/) based on phosphoproteomic data at 1, 3, 6, and 24 h.
RNA-seq: RNA-seq data at 24 h for infected and mock iAT2s).
Pathway enrichment results and analysis, and drug-based analysis. Cluster Enrichments: Enrichr-based Reactome pathway enrichments for the clusters of proteomic and phosphoproteomic data in Figures 2 and 4. Clusters based on log2 fold change between mock and infected conditions and all genes within cluster queried using Enrichr tool. Relevant pathways and statistics shown.
iAT2 Enrichments: GSEA-based enrichment results for all time points between infected and mock controls for the iAT2s (Figure 4). GSEA performed using fgsea R package and in-house scripts. Significance, NES, and enriched genes shown for each significant pathway. Pathways filtered for significance (FDR<0.1).
Caco Enrichments, Vero Enrichments, A549 Enrichments: GSEA-based enrichment results for all time points between infected and mock controls for the Caco-2, VeroE6, and A549 cell studies available from public data (Figure 4). GSEA performed using fgsea R package and in-house scripts. Significance, NES, and enriched genes shown for each significant pathway. Pathways filtered for significance (FDR<0.1).
Gene Overlap Studies: Overlap analysis of all genes and differential genes (FDR<0.05 & |log2 fold change|>0.25) across the four cell line studies.
iAT2 Unique Genes Enrichment: Enrichr-based Reactome pathway enrichment for genes differential (FDR<0.05 & |log2 fold change|>0.25) in iAT2s only.
Common Infection Pathways: Pathways that were significantly enriched (FDR < 0.1) in all studies based on GSEA analysis between infected and mock controls using common gene set database.
iAT2 Specific Infection Pathways: Enriched pathways ranked by difference between minimum time point FDR of iAT2 enrichments and the minimum FDR for other studies. A negative number indicates pathways that are most different.
Drug Table: Based on our prediction of drug targets in Figure 5 in the main paper, we annotated confirmed 22 genes as successful targets with their corresponding drugs. We also added drugs that targeted the underlying genes but proved ineffective in hampering SARS-CoV-2.
Candidate Drugs: Candidate drugs that target differential proteins across time points in our dataset.
Curated Viral Suppressors: Curated drugs from the literature that were shown to inhibit viral infection (Bouhaddou et al., 2020; Stukalov et al., 2020).
Curated Unsuccessful Drugs: Curated drugs from the literature that were shown to be unsuccessful in inhibiting viral infection (Bouhaddou et al., 2020; Stukalov et al., 2020
References
- Abo K.M., Ma L., Matte T., Huang J., Alysandratos K.D., Werder R.B., Mithal A., Beermann M.L., Lindstrom-Vautrin J., Mostoslavsky G. Human iPSC-derived alveolar and airway epithelial cells can be cultured at air-liquid interface and express SARS-CoV-2 host factors. bioRxiv. 2020 doi: 10.1101/2020.06.03.132639. [DOI] [Google Scholar]
- Ackermann M., Verleden S.E., Kuehnel M., Haverich A., Welte T., Laenger F., Vanstapel A., Werlein C., Stark H., Tzankov A. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseenko A., Kotelnikov S., Ignatov M., Egbert M., Kholodov Y., Vajda S., Kozakov D. ClusPro LigTBM: Automated Template-based Small Molecule Docking. J. Mol. Biol. 2020;432:3404–3410. doi: 10.1016/j.jmb.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf U., Benoit-Pilven C., Lacroix V., Navratil V., Naffakh N. Advances in Analyzing Virus-Induced Alterations of Host Cell Splicing. Trends Microbiol. 2019;27:268–281. doi: 10.1016/j.tim.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriz G.F., King O.D., Bryant B., Sander C., Roth F.P. Characterizing gene sets with FuncAssociate. Bioinformatics. 2003;19:2502–2504. doi: 10.1093/bioinformatics/btg363. [DOI] [PubMed] [Google Scholar]
- Blaustein M., Pelisch F., Tanos T., Muñoz M.J., Wengier D., Quadrana L., Sanford J.R., Muschietti J.P., Kornblihtt A.R., Cáceres J.F. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 2005;12:1037–1044. doi: 10.1038/nsmb1020. [DOI] [PubMed] [Google Scholar]
- Bojkova D., Klann K., Koch B., Widera M., Krause D., Ciesek S., Cinatl J., Münch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583:469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Correa Marrero M., Polacco B.J., Melnyk J.E., Ulferts S., Kaake R.M. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell. 2020;182:685–712.e19. doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley B.T., Maioli H., Johnston R., Chaudhry I., Fink S.L., Xu H., Najafian B., Deutsch G., Lacy J.M., Williams T. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet. 2020;396:320–332. doi: 10.1016/S0140-6736(20)31305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carsana L., Sonzogni A., Nasr A., Rossi R.S., Pellegrinelli A., Zerbi P., Rech R., Colombo R., Antinori S., Corbellino M. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect. Dis. 2020;20:1135–1140. doi: 10.1016/S1473-3099(20)30434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado P., Rodriguez-Prados J.C., Cosulich S.C., Guichard S., Vanhaesebroeck B., Joel S., Cutillas P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013;6:rs6. doi: 10.1126/scisignal.2003573. [DOI] [PubMed] [Google Scholar]
- Chan W.H., Yu J.S., Yang S.D. PAK2 is cleaved and activated during hyperosmotic shock-induced apoptosis via a caspase-dependent mechanism: evidence for the involvement of oxidative stress. J. Cell. Physiol. 1999;178:397–408. doi: 10.1002/(SICI)1097-4652(199903)178:3<397::AID-JCP14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Chang C.K., Chen C.M., Chiang M.H., Hsu Y.L., Huang T.H. Transient oligomerization of the SARS-CoV N protein--implication for virus ribonucleoprotein packaging. PLoS ONE. 2013;8:e65045. doi: 10.1371/journal.pone.0065045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Xu Z., Zhang T., Lin L., Lu M., Xie C., Yu X. Cep85 Relays Plk1 Activity to Phosphorylated Nek2A for Its Timely Activation in Centrosome Disjunction. iScience. 2019;11:114–133. doi: 10.1016/j.isci.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro I.J., Gil R.S., Ligammari L., Di Giacinto M.L., Vagnarelli P. CDK1 and PLK1 coordinate the disassembly and reassembly of the nuclear envelope in vertebrate mitosis. Oncotarget. 2017;9:7763–7773. doi: 10.18632/oncotarget.23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu R.A., Sass M.M., Johnson J.M., Potts G.K., Choudhary A., Coon J.J., Burkard M.E. Polo-like kinase 4 maintains centriolar satellite integrity by phosphorylation of centrosomal protein 131 (CEP131) J. Biol. Chem. 2019;294:6531–6549. doi: 10.1074/jbc.RA118.004867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E.W., Bandeira N., Sharma V., Perez-Riverol Y., Carver J.J., Kundu D.J., García-Seisdedos D., Jarnuczak A.F., Hewapathirana S., Pullman B.S. The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020;48(D1):D1145–D1152. doi: 10.1093/nar/gkz984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maira G., Salvi M., Arrigoni G., Marin O., Sarno S., Brustolon F., Pinna L.A., Ruzzene M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005;12:668–677. doi: 10.1038/sj.cdd.4401604. [DOI] [PubMed] [Google Scholar]
- Dittmar M., Lee J.S., Whig K., Segrist E., Li M., Jurado K., Samby K., Ramage H., Schultz D., Cherry S. Drug repurposing screens reveal FDA approved drugs active against SARS-Cov-2. bioRxiv. 2020 doi: 10.1101/2020.06.19.161042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrello N.V., Peschiaroli A., Guardavaccaro D., Colburn N.H., Sherman N.E., Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- Du W., Thanos D., Maniatis T. Mechanisms of transcriptional synergism between distinct virus-inducible enhancer elements. Cell. 1993;74:887–898. doi: 10.1016/0092-8674(93)90468-6. [DOI] [PubMed] [Google Scholar]
- Dubois J., Terrier O., Rosa-Calatrava M. Influenza viruses and mRNA splicing: doing more with less. MBio. 2014;5 doi: 10.1128/mBio.00070-14. e00070–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47(D1):D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias J.E., Gygi S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2, Unit 2 9. [DOI] [PubMed] [Google Scholar]
- Evdokimova V., Ruzanov P., Anglesio M.S., Sorokin A.V., Ovchinnikov L.P., Buckley J., Triche T.J., Sonenberg N., Sorensen P.H. Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol. Cell. Biol. 2006;26:277–292. doi: 10.1128/MCB.26.1.277-292.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G., Sharma A., Ramaiah A., Sen C., Kohn D., Gomperts B., Svendsen C.N., Damoiseaux R.D., Arumugaswami V. Antiviral Drug Screen of Kinase inhibitors Identifies Cellular Signaling Pathways Critical for SARS-CoV-2 Replication. bioRxiv. 2020 doi: 10.1101/2020.06.24.150326. [DOI] [Google Scholar]
- Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel R.K., Meyer M., Paczkowska M., Reimand J., Vizeacoumar F., Vizeacoumar F., Lam T.T., Lukong K.E. Global phosphoproteomic analysis identifies SRMS-regulated secondary signaling intermediates. Proteome Sci. 2018;16:16. doi: 10.1186/s12953-018-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz E.M., Ghandi M., Treacy D.J., Wagle N., Garraway L.A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014;74:7079–7089. doi: 10.1158/0008-5472.CAN-14-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., O’Meara M.J., Guo J.Z., Swaney D.L., Tummino T.A., Huttenhain R. A SARS-CoV-2-Human Protein Interaction Map Reveals Targets for Drug Repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes J.M., Grimes K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020;144:63–65. doi: 10.1016/j.yjmcc.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Gui J.F., Lane W.S., Fu X.D. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- Hentges P., Waller H., Reis C.C., Ferreira M.G., Doherty A.J. Cdk1 restrains NHEJ through phosphorylation of XRCC4-like factor Xlf1. Cell Rep. 2014;9:2011–2017. doi: 10.1016/j.celrep.2014.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.J., Okuda K., Edwards C.E., Martinez D.R., Asakura T., Dinnon K.H., 3rd, Kato T., Lee R.E., Yount B.L., Mascenik T.M. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell. 2020;182:429–446.e14. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Hume A.J., Abo K.M., Werder R.B., Villacorta-Martin C., Alysandratos K.-D., Beermann M.L., Simone-Roach C., Lindstrom-Vautrin J., Olejnik J. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell. 2020 doi: 10.1016/j.stem.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley K., Ding J., Villacorta-Martin C., Herriges M.J., Jacob A., Vedaie M., Alysandratos K.D., Sun Y.L., Lin C., Werder R.B. Reconstructed Single-Cell Fate Trajectories Define Lineage Plasticity Windows during Differentiation of Human PSC-Derived Distal Lung Progenitors. Cell Stem Cell. 2020;26:593–608.e8. doi: 10.1016/j.stem.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A., Morley M., Hawkins F., McCauley K.B., Jean J.C., Heins H., Na C.L., Weaver T.E., Vedaie M., Hurley K. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell. 2017;21:472–488.e10. doi: 10.1016/j.stem.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A., Vedaie M., Roberts D.A., Thomas D.C., Villacorta-Martin C., Alysandratos K.D., Hawkins F., Kotton D.N. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat. Protoc. 2019;14:3303–3332. doi: 10.1038/s41596-019-0220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S., Kim T., Iyer V.G., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Karp N.A., Huber W., Sadowski P.G., Charles P.D., Hester S.V., Lilley K.S. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics. 2010;9:1885–1897. doi: 10.1074/mcp.M900628-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann K., Bojkova D., Tascher G., Ciesek S., Münch C., Cinatl J. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell. 2020;80:164–174.e4. doi: 10.1016/j.molcel.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotelnikov S., Alekseenko A., Liu C., Ignatov M., Padhorny D., Brini E., Lukin M., Coutsias E., Dill K.A., Kozakov D. Sampling and refinement protocols for template-based macrocycle docking: 2018 D3R Grand Challenge 4. J. Comput. Aided Mol. Des. 2020;34:179–189. doi: 10.1007/s10822-019-00257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krähling V., Stein D.A., Spiegel M., Weber F., Mühlberger E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J. Virol. 2009;83:2298–2309. doi: 10.1128/JVI.01245-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar L., Futschik M.E. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M., Gouw M., Michael S., Sámano-Sánchez H., Pancsa R., Glavina J., Diakogianni A., Valverde J.A., Bukirova D., Čalyševa J. ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2020;48(D1):D296–D306. doi: 10.1093/nar/gkz1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A., Ma’ayan A. KEA: kinase enrichment analysis. Bioinformatics. 2009;25:684–686. doi: 10.1093/bioinformatics/btp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFemina M.J., Sutherland K.M., Bentley T., Gonzales L.W., Allen L., Chapin C.J., Rokkam D., Sweerus K.A., Dobbs L.G., Ballard P.L., Frank J.A. Claudin-18 deficiency results in alveolar barrier dysfunction and impaired alveologenesis in mice. Am. J. Respir. Cell Mol. Biol. 2014;51:550–558. doi: 10.1165/rcmb.2013-0456OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekmine F., Sassano A., Uddin S., Smith J., Majchrzak B., Brachmann S.M., Hay N., Fish E.N., Platanias L.C. Interferon-gamma engages the p70 S6 kinase to regulate phosphorylation of the 40S S6 ribosomal protein. Exp. Cell Res. 2004;295:173–182. doi: 10.1016/j.yexcr.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Leutert M., Rodríguez-Mias R.A., Fukuda N.K., Villén J. R2-P2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies. Mol. Syst. Biol. 2019;15:e9021. doi: 10.15252/msb.20199021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti S., Sacco F., Calderone A., Perfetto L., Iannuccelli M., Panni S., Santonico E., Palma A., Nardozza A.P., Castagnoli L., Cesareni G. HuPho: the human phosphatase portal. FEBS J. 2013;280:379–387. doi: 10.1111/j.1742-4658.2012.08712.x. [DOI] [PubMed] [Google Scholar]
- Licata L., Lo Surdo P., Iannuccelli M., Palma A., Micarelli E., Perfetto L., Peluso D., Calderone A., Castagnoli L., Cesareni G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2020;48(D1):D504–D510. doi: 10.1093/nar/gkz949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Chen Z., Erdjument-Bromage H., Tempst P., Pandolfi P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Ma-Lauer Y., Carbajo-Lozoya J., Hein M.Y., Müller M.A., Deng W., Lei J., Meyer B., Kusov Y., von Brunn B., Bairad D.R. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA. 2016;113:E5192–E5201. doi: 10.1073/pnas.1603435113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J., Bennett B.D., Parikh A., Feng X.J., McArdle J., Rabitz H.A., Shenk T., Rabinowitz J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylonis I., Giannakouros T. Protein kinase CK2 phosphorylates and activates the SR protein-specific kinase 1. Biochem. Biophys. Res. Commun. 2003;301:650–656. doi: 10.1016/s0006-291x(02)03055-3. [DOI] [PubMed] [Google Scholar]
- Naro C., Sette C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013;2013:151839. doi: 10.1155/2013/151839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayler O., Stamm S., Ullrich A. Characterization and comparison of four serine- and arginine-rich (SR) protein kinases. Biochem. J. 1997;326:693–700. doi: 10.1042/bj3260693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya K., Kataoka N., Hagiwara M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J. Cell Biol. 2011;195:27–40. doi: 10.1083/jcb.201107093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogando N.S., Dalebout T.J., Zevenhoven-Dobbe J.C., Limpens R.W.A.L., van der Meer Y., Caly L., Druce J., de Vries J.J.C., Kikkert M., Bárcena M. SARS-coronavirus-2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020;101:925–940. doi: 10.1099/jgv.0.001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olejnik J., Forero A., Deflubé L.R., Hume A.J., Manhart W.A., Nishida A., Marzi A., Katze M.G., Ebihara H., Rasmussen A.L., Mühlberger E. Ebolaviruses Associated with Differential Pathogenicity Induce Distinct Host Responses in Human Macrophages. J. Virol. 2017;91:91. doi: 10.1128/JVI.00179-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Bartenschlager R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013;2:32–48. doi: 10.5501/wjv.v2.i2.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdikari T.M., Murthy A.C., Ryan V.H., Watters S., Naik M.T., Fawzi N.L. SARS-CoV-2 nucleocapsid protein undergoes liquid-liquid phase separation stimulated by RNA and partitions into phases of human ribonucleoproteins. bioRxiv. 2020 doi: 10.1101/2020.06.09.141101. [DOI] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Isserlin R., Voisin V., Kucera M., Tannus-Lopes C., Rostamianfar A., Wadi L., Meyer M., Wong J., Xu C. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019;14:482–517. doi: 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux P.P., Shahbazian D., Vu H., Holz M.K., Cohen M.S., Taunton J., Sonenberg N., Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D., Sonenberg N. The Akt of translational control. Oncogene. 2005;24:7426–7434. doi: 10.1038/sj.onc.1209098. [DOI] [PubMed] [Google Scholar]
- Saeed M.F., Kolokoltsov A.A., Albrecht T., Davey R.A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010;6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyanarayana A., Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- Saxton R.A., Sabatini D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeman D., Fielding B.C. Coronavirus envelope protein: current knowledge. Virol. J. 2019;16:69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanbeck R., Manfioletti G., Wiśniewski J.R. Architecture of high mobility group protein I-C.DNA complex and its perturbation upon phosphorylation by Cdc2 kinase. J. Biol. Chem. 2000;275:1793–1801. doi: 10.1074/jbc.275.3.1793. [DOI] [PubMed] [Google Scholar]
- Sergushichev A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. 2016 doi: 10.1101/060012. [DOI] [Google Scholar]
- Sgarra R., Maurizio E., Zammitti S., Lo Sardo A., Giancotti V., Manfioletti G. Macroscopic differences in HMGA oncoproteins post-translational modifications: C-terminal phosphorylation of HMGA2 affects its DNA binding properties. J. Proteome Res. 2009;8:2978–2989. doi: 10.1021/pr900087r. [DOI] [PubMed] [Google Scholar]
- Shi C.S., Qi H.Y., Boularan C., Huang N.N., Abu-Asab M., Shelhamer J.H., Kehrl J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014;193:3080–3089. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Limpens R.W.A.L., de Wilde A.H., de Jong A.W.M., Zevenhoven-Dobbe J.C., Maier H.J., Faas F.F.G.A., Koster A.J., Bárcena M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020;18:e3000715. doi: 10.1371/journal.pbio.3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger S.J., Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20:473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stertz S., Reichelt M., Spiegel M., Kuri T., Martínez-Sobrido L., García-Sastre A., Weber F., Kochs G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology. 2007;361:304–315. doi: 10.1016/j.virol.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukalov A., Girault V., Grass V., Bergant V., Karayel O., Urban C., Haas D.A., Huang Y., Oubraham L., Wang A. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. bioRxiv. 2020 doi: 10.1101/2020.06.17.156455. [DOI] [PubMed] [Google Scholar]
- Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., HCA Lung Biological Network SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapial J., Ha K.C.H., Sterne-Weiler T., Gohr A., Braunschweig U., Hermoso-Pulido A., Quesnel-Vallières M., Permanyer J., Sodaei R., Marquez Y. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017;27:1759–1768. doi: 10.1101/gr.220962.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuwen L.A., Geldhof V., Pasut A., Carmeliet P. COVID-19: the vasculature unleashed. Nat. Rev. Immunol. 2020;20:389–391. doi: 10.1038/s41577-020-0343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi Nhu Thao T., Labroussaa F., Ebert N., V’kovski P., Stalder H., Portmann J., Kelly J., Steiner S., Holwerda M., Kratzel A. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature. 2020;582:561–565. doi: 10.1038/s41586-020-2294-9. [DOI] [PubMed] [Google Scholar]
- Thoms M., Buschauer R., Ameismeier M., Koepke L., Denk T., Hirschenberger M., Kratzat H., Hayn M., Mackens-Kiani T., Cheng J. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369:1249–1255. doi: 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toko H., Takahashi H., Kayama Y., Oka T., Minamino T., Okada S., Morimoto S., Zhan D.Y., Terasaki F., Anderson M.E. Ca2+/calmodulin-dependent kinase IIdelta causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation. 2010;122:891–899. doi: 10.1161/CIRCULATIONAHA.109.935296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J., Taylor P., Peterman S.M., Prakash A., Moran M.F. Epidermal growth factor receptor phosphorylation sites Ser991 and Tyr998 are implicated in the regulation of receptor endocytosis and phosphorylations at Ser1039 and Thr1041. Mol. Cell. Proteomics. 2009;8:2131–2144. doi: 10.1074/mcp.M900148-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbag N., Braunstein A., Pagnani A., Huang S.S., Chayes J., Borgs C., Zecchina R., Fraenkel E. Simultaneous reconstruction of multiple signaling pathways via the prize-collecting steiner forest problem. J. Comput. Biol. 2013;20:124–136. doi: 10.1089/cmb.2012.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbag N., Gosline S.J., Kedaigle A., Soltis A.R., Gitter A., Fraenkel E. Network-Based Interpretation of Diverse High-Throughput Datasets through the Omics Integrator Software Package. PLoS Comput. Biol. 2016;12:e1004879. doi: 10.1371/journal.pcbi.1004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- V’kovski P., Gerber M., Kelly J., Pfaender S., Ebert N., Braga Lagache S., Simillion C., Portmann J., Stalder H., Gaschen V. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. eLife. 2019;8:8. doi: 10.7554/eLife.42037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Yang L. Turning the Tide: Natural Products and Natural-Product-Inspired Chemicals as Potential Counters to SARS-CoV-2 Infection. Front. Pharmacol. 2020;11:1013. doi: 10.3389/fphar.2020.01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Codreanu S.G., Wen B., Li K., Chambers M.C., Liebler D.C., Zhang B. Detection of Proteome Diversity Resulted from Alternative Splicing is Limited by Trypsin Cleavage Specificity. Mol. Cell. Proteomics. 2018;17:422–430. doi: 10.1074/mcp.RA117.000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Feng X., Molinolo A.A., Martin D., Vitale-Cross L., Nohata N., Ando M., Wahba A., Amornphimoltham P., Wu X. 4E-BP1 Is a Tumor Suppressor Protein Reactivated by mTOR Inhibition in Head and Neck Cancer. Cancer Res. 2019;79:1438–1450. doi: 10.1158/0008-5472.CAN-18-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh G.I., Price N.T., Bladergroen B.A., Bloomberg G., Proud C.G. Identification of novel phosphorylation sites in the beta-subunit of translation initiation factor eIF-2. Biochem. Biophys. Res. Commun. 1994;201:1279–1288. doi: 10.1006/bbrc.1994.1843. [DOI] [PubMed] [Google Scholar]
- Wichmann D., Sperhake J.P., Lütgehetmann M., Steurer S., Edler C., Heinemann A., Heinrich F., Mushumba H., Kniep I., Schröder A.S. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020;173:268–277. doi: 10.7326/M20-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., Knox C., Guo A.C., Shrivastava S., Hassanali M., Stothard P., Chang Z., Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.H., Yeh S.H., Tsay Y.G., Shieh Y.H., Kao C.L., Chen Y.S., Wang S.H., Kuo T.J., Chen D.S., Chen P.J. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 2009;284:5229–5239. doi: 10.1074/jbc.M805747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C., Sharp J.A., Kawahara M., Davalos A.R., Difilippantonio M.J., Hu Y., Li W., Cao L., Buetow K., Ried T. The XIST noncoding RNA functions independently of BRCA1 in X inactivation. Cell. 2007;128:977–989. doi: 10.1016/j.cell.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Xu L.H., Huang M., Fang S.G., Liu D.X. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase δ. J. Biol. Chem. 2011;286:39546–39559. doi: 10.1074/jbc.M111.242206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K.R., Park S.B., Jung J.W., Seo M.S., Hong I.S., Kim H.S., Seo Y., Kang T.W., Lee J.Y., Kurtz A., Kang K.S. HMGA2 regulates the in vitro aging and proliferation of human umbilical cord blood-derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell Res. (Amst.) 2013;10:156–165. doi: 10.1016/j.scr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., Flodby P., Luo J., Castillo D.R., Liu Y., Yu F.X., McConnell A., Varghese B., Li G., Chimge N.O. Claudin-18-mediated YAP activity regulates lung stem and progenitor cell homeostasis and tumorigenesis. J. Clin. Invest. 2018;128:970–984. doi: 10.1172/JCI90429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proteomic data and phosphoproteomic data used for analysis, Kinase enrichment analysis based on phosphoproteomic data and RNA-seq data used for analysis.
Prot_1-3-6 Hour: Proteomics abundance data and protein annotation at 1, 3, and 6 h. Normalized data extracted from MaxQuant proteinGroups.txt file, with feature annotation, and differential analysis statistics.
Phospho_1-3-6 Hour: Phosphoproteomic abundance data and protein annotation at 1, 3, and 6 h. Normalized data extracted from MaxQuant PhosphoSites file, with feature annotation, and differential analysis statistics.
Proteomics _24 Hour: Proteomics abundance data and protein annotation at 24 h. Normalized data extracted from MaxQuant proteinGroups.txt file, with feature annotation, and differential analysis statistics.
Phospho_24 Hour: Phosphoproteomic abundance data and protein annotation at 24 h. Phosphoproteomic abundance data and protein annotation at 1, 3, and 6 h. Normalized data extracted from the MaxQuant PhosphoSites file, with feature annotation and differential statistics.
In vitro kinase assay: Phosphoproteomic data corresponding to in vitro kinase assays performed using GSK3 and CNSK2 as kinases and SARS-CoV-2 Nucleoprotein (N) as substrate. Phosphosites on N, upon incubation with either kinase are annotated.
KSEA: Kinase Substrate Enrichment Analysis using KSEAapp R package based on phosphoproteomic data at 1, 3, 6, and 24 h. Top hits filtered and showing relevant Kinase statistics based on motif matching in databases.
KEA2: Kinase Enrichment Analysis 2 (https://www.maayanlab.net/KEA2/) based on phosphoproteomic data at 1, 3, 6, and 24 h.
RNA-seq: RNA-seq data at 24 h for infected and mock iAT2s).
Pathway enrichment results and analysis, and drug-based analysis. Cluster Enrichments: Enrichr-based Reactome pathway enrichments for the clusters of proteomic and phosphoproteomic data in Figures 2 and 4. Clusters based on log2 fold change between mock and infected conditions and all genes within cluster queried using Enrichr tool. Relevant pathways and statistics shown.
iAT2 Enrichments: GSEA-based enrichment results for all time points between infected and mock controls for the iAT2s (Figure 4). GSEA performed using fgsea R package and in-house scripts. Significance, NES, and enriched genes shown for each significant pathway. Pathways filtered for significance (FDR<0.1).
Caco Enrichments, Vero Enrichments, A549 Enrichments: GSEA-based enrichment results for all time points between infected and mock controls for the Caco-2, VeroE6, and A549 cell studies available from public data (Figure 4). GSEA performed using fgsea R package and in-house scripts. Significance, NES, and enriched genes shown for each significant pathway. Pathways filtered for significance (FDR<0.1).
Gene Overlap Studies: Overlap analysis of all genes and differential genes (FDR<0.05 & |log2 fold change|>0.25) across the four cell line studies.
iAT2 Unique Genes Enrichment: Enrichr-based Reactome pathway enrichment for genes differential (FDR<0.05 & |log2 fold change|>0.25) in iAT2s only.
Common Infection Pathways: Pathways that were significantly enriched (FDR < 0.1) in all studies based on GSEA analysis between infected and mock controls using common gene set database.
iAT2 Specific Infection Pathways: Enriched pathways ranked by difference between minimum time point FDR of iAT2 enrichments and the minimum FDR for other studies. A negative number indicates pathways that are most different.
Drug Table: Based on our prediction of drug targets in Figure 5 in the main paper, we annotated confirmed 22 genes as successful targets with their corresponding drugs. We also added drugs that targeted the underlying genes but proved ineffective in hampering SARS-CoV-2.
Candidate Drugs: Candidate drugs that target differential proteins across time points in our dataset.
Curated Viral Suppressors: Curated drugs from the literature that were shown to inhibit viral infection (Bouhaddou et al., 2020; Stukalov et al., 2020).
Curated Unsuccessful Drugs: Curated drugs from the literature that were shown to be unsuccessful in inhibiting viral infection (Bouhaddou et al., 2020; Stukalov et al., 2020