

Monophosphate prodrug analogs of 2′-deoxy-2′-fluoro-2′-C-methylguanosine have been reported as potent inhibitors of hepatitis C virus (HCV) RNA-dependent RNA polymerase. These prodrugs also display potent anti-dengue virus activities in cellular assays although their prodrug moieties were designed to produce high levels of triphosphate in the liver. Since peripheral blood mononuclear cells (PBMCs) are among the major targets of dengue virus, different prodrug moieties were designed to effectively deliver 2′-deoxy-2′-fluoro-2′-C-methylguanosine monophosphate prodrugs and their corresponding triphosphates into PBMCs after oral administration.
KEYWORDS: dengue, nucleotide analog, monophosphate prodrug, cyclic phosphoramidate, nucleoside triphosphate, polymerase inhibitor
ABSTRACT
Monophosphate prodrug analogs of 2′-deoxy-2′-fluoro-2′-C-methylguanosine have been reported as potent inhibitors of hepatitis C virus (HCV) RNA-dependent RNA polymerase. These prodrugs also display potent anti-dengue virus activities in cellular assays although their prodrug moieties were designed to produce high levels of triphosphate in the liver. Since peripheral blood mononuclear cells (PBMCs) are among the major targets of dengue virus, different prodrug moieties were designed to effectively deliver 2′-deoxy-2′-fluoro-2′-C-methylguanosine monophosphate prodrugs and their corresponding triphosphates into PBMCs after oral administration. We identified a cyclic phosphoramidate, prodrug 17, demonstrating well-balanced anti-dengue virus cellular activity and in vitro stability profiles. We further determined the PBMC concentration of active triphosphate needed to inhibit virus replication by 50% (TP50). Compound 17 was assessed in an AG129 mouse model and demonstrated 1.6- and 2.2-log viremia reductions at 100 and 300 mg/kg twice a day (BID), respectively. At 100 mg/kg BID, the terminal triphosphate concentration in PBMCs exceeded the TP50 value, demonstrating TP50 as the target exposure for efficacy. In dogs, oral administration of compound 17 resulted in high PBMC triphosphate levels, exceeding the TP50 at 10 mg/kg. Unfortunately, 2-week dog toxicity studies at 30, 100, and 300 mg/kg/day showed that “no observed adverse effect level” (NOAEL) could not be achieved due to pulmonary inflammation and hemorrhage. The preclinical safety results suspended further development of compound 17. Nevertheless, present work has proven the concept that an efficacious monophosphate nucleoside prodrug could be developed for the potential treatment of dengue virus infection.
INTRODUCTION
The mosquito-borne dengue virus is endemic to tropical and subtropical regions throughout the world, making dengue fever the most important mosquito-borne viral disease afflicting humans. Its global distribution is comparable to that of malaria, with an estimated 2.5 billion people at risk for epidemic transmission (1). There have been steady increases in countries affected and incidence since the 1950s, and recent estimates suggest annual rates of 390 million cases accompanied by 20,000 deaths (2).
Dengue viruses (DENVs) can be further classified into four different serotypes (DENV-1 to -4), all of which can lead to disease symptoms with various degrees of severity. Secondary infection by a different serotype may increase the risk of severe dengue diseases (3). While diagnosis of dengue virus infection can be rapid and simple, distinguishing the serotype requires additional instrumentation, usually in a laboratory setting. Thus, the ideal treatment for dengue fever should possess pan-serotype activities. Although there is a dengue vaccine available, it is approved in only a few countries for patients 9 to 45 years old with a history of infection (4). No antiviral is currently available for the treatment of dengue.
DENV is an enveloped, positive-strand RNA virus belonging to the Flaviviridae family and the genus Flavivirus. Medically important viruses in this class include yellow fever virus (YFV), Japanese encephalitis virus (JEV), West Nile virus (WNV), and Zika virus (ZIKV). The dengue virus genome encodes three structural (C-prM-E) and seven nonstructural (NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5) proteins. The nonstructural protein NS5 contains both methyltransferase and RNA-dependent RNA polymerase (RdRp) activities. The RdRp is a virus-specific enzyme which catalyzes the replication of viral RNA from its own complementary template. It is essential for viral replication and an attractive target for therapeutic intervention (5).
Nucleoside/nucleotide analogs are a highly successful compound class of antivirals, as exemplified in the treatment of human immunodeficiency virus (HIV), herpes simplex virus (HSV), hepatitis B virus (HBV), and hepatitis C virus (HCV) (6). These inhibitors are converted to their active nucleoside triphosphate forms by host cell machinery and inhibit the synthesis of viral RNAs or DNAs by acting as chain terminators or substrate mimics (7). Because the modified nucleoside triphosphates must be recognized by the highly conserved active site of DENV polymerase, they have a high likelihood of pan-DENV serotype activity and of providing a high barrier to drug resistance (8). These features make them very attractive for dengue drug development (9). We previously reported an adenosine-based nucleoside (NITD-008, compound 1; 7-deaza-2′-C-ethynyl-adenosine) (Fig. 1A), which potently inhibited DENV both in vitro and in vivo. However, the development of NITD-008 was terminated due to an insufficient safety profile (10). 4′-Azido-cytidine (R-1479, compound 2) and its ester prodrug, balapiravir (compound 3) (Fig. 1A), were originally developed for the treatment of HCV, but their development was terminated due to hematologic adverse events such as lymphopenia (11). Azido-cytidine (compound 2) was weakly active against dengue virus infection (12), and balapiravir (compound 3) failed to reduce viral load in dengue patients (13). Uridine- and guanosine-based prodrugs are effective against dengue virus in vitro although in vivo activity has yet to be demonstrated (14, 15).
FIG 1.

Structures and in vitro biological activities of selected dengue antiviral compounds. (A) Structures and in vitro biological profile of NITD-008 (1), R-1479 (2), and balapiravir (3). (B) Structures and in vitro biological profile of PSI-352938 (4), PSI-353661 (5), and their corresponding triphosphate 6 and nucleoside 7 metabolites. (C) Synthesis of cyclic phosphoramidate prodrugs of 6-O-alkyl-2′-deoxy-2′-fluoro-2′-C-methylguanosine. Reactions conditions are as follows: (i) t-BuMgCl in tetrahydrofuran; (ii) t-BuOK in DMSO; (iii) separation by preparative reverse-phase HPLC.
It is not uncommon that many nucleoside analogs suffer the lack of biological activities in cellular assays due to poor intracellular conversion to their triphosphates. In particular, conversion of nucleoside analogs into their nucleotide or nucleoside monophosphates is often rate limiting or nonproductive (16). On the other hand, the unprotected monophosphate species are poor drug candidates as they have inadequate cellular permeability due to the inability of negatively charged phosphates to cross the cell membrane. To circumvent these problems, the monophosphate prodrug approach has been developed to deliver the nucleoside monophosphate directly into target cells (17). This prodrug approach has proven to be effective in improving the therapeutic potential of antiviral and anticancer nucleosides (18). For instance, the uridine-based monophosphate prodrug sofosbuvir is a key component in a number of HCV combination therapies (19). The excellent efficacy of sofosbuvir is due to the efficient delivery of the nucleoside triphosphate into the target organ, the liver (20).
Similarly, using nucleotide prodrugs to deliver a high level of active triphosphates into peripheral blood mononuclear cells (PBMCs) has been successfully demonstrated in tenofovir disoproxil fumarate and tenofovir alafenamide for HIV (21). The prodrug tenofovir alafenamide was transiently present in plasma (half-life [t1/2] of ∼30 min) when dosed orally to dogs. The exposure was sufficient to drive a high and sustained level of the active metabolite in PBMCs (21). However, this acyclic ribose modification has not worked for any RNA viruses, and the strategy could not be generalized to deliver compounds systemically including to PBMCs. GS-5734 (also known as remdesivir) (22) was initially developed for Ebola virus but was repurposed for the treatment of SARS-CoV-2 and is administered intravenously (i.v.).
In 2010, Pharmasset (acquired by Gilead in 2012) reported two nucleotide prodrugs of 2′-deoxy-2′-fluoro-2′-C-methylguanosine, PSI-352938 (compound 4) (23, 24) and PSI-353661 (compound 5) (25), as potent inhibitors of HCV replication (Fig. 1B). Their common guanosine-based nucleoside triphosphate 6 was reported to be a potent inhibitor of HCV NS5B polymerase, with a 50% inhibitory concentration (IC50) value of 5.94 μM. We evaluated these prodrugs and the active triphosphate in our PBMC dengue virus plaque and dengue virus RdRp enzyme assays, respectively. We subsequently embarked on the optimization of the prodrug moieties to deliver the active triphosphate 6 into PBMCs, one of the major dengue virus replication sites (26).
Here, we report our research leading to a cyclic phosphoramidate prodrug of 2′-deoxy-2′-fluoro-2′-C-methylguanosine for the treatment of dengue starting from the liver-targeting prodrugs. By design, all prodrugs had amino acid ester linkage which could serve as a target for ubiquitous esterases in the target cells. The optimized prodrug 17 resulted in high triphosphate loading in PBMCs after oral administration in dogs. In addition, compound 17 demonstrated oral efficacies at 100 mg/kg twice a day (BID) in the dengue mouse model with high triphosphate concentrations in PBMCs, while the liver-targeting prodrug 5 failed to reduce viremia at an even higher dose (25). Based on the correlation between the intracellular triphosphate concentration needed to inhibit 50% of virus replication (TP50) and the viremia reduction observed in in vivo efficacy studies, we defined the minimum efficacious dose for a nucleoside/nucleotide prodrug. Finally, we also report the preclinical development and safety assessment of compound 17.
RESULTS
Chemistry and structure-activity/-property relationship (SAR/SPR).
Figure 1A illustrates adenosine-based (compound 1) and cytidine-based (compound 2) nucleoside analogs active against dengue virus. We employed guanosine-based nucleoside analog 7 (2′-deoxy-2′-fluoro-2′-C-methylguanosine) and investigated a suitable prodrug moiety to effectively deliver the monophosphate into PBMCs. As shown in Fig. 1B, compound 7 is not active, but the linear prodrug 5 had excellent activity, suggesting that the conversion of compound 7 to the corresponding monophosphate is the rate-limiting step. To confirm activity against dengue virus RdRp, we performed an in vitro dengue virus RdRp assay with the active triphosphate form. The resulting IC50 is 1.1 μM, confirming our hypothesis.
From various types of nucleotide prodrugs, we focused our attention on 3′,5′-cyclic phosphoramidates (27). To access the cyclic phosphoramidate prodrugs, the nucleoside starting material, 6-O-alkyl-2′-deoxy-2′-fluoro-2′-C-methylguanosine 8, was prepared according to the literature (28). The guanosine analog 8 was reacted with pentafluorophenyl ester agent 9 in the presence of tert-butylmagnesium chloride (tert-BuMgCl) as a base to yield the linear phosphoramidate product 10 as a mixture of diastereomers. The cyclization step was carried out by treatment of compound 10 with tert-butoxide (t-BuOK) in dimethyl sulfoxide (DMSO) to afford a diastereomeric mixture of cyclic product 11, which was separated by reverse-phase high-performance liquid chromatography (RP-HPLC) to yield each single phosphorous stereoisomer, Sp or Rp (Fig. 1C). The phosphorous stereochemistry of one of the cyclic phosphoramidates, compound 17, was assigned an Rp configuration as determined by single-crystal X-ray analysis (Fig. 2). The X-ray structure of compound 17 indicates that the phosphoramidate moiety is cis oriented to the guanine base through H bond formation between the carbonyl and 7-NH2. In the 31P nuclear magnetic resonance (NMR) spectroscopy, the phosphorous peak of Rp isomer 17, rather than the corresponding Sp isomer, appeared in the upper field. This observation was applied to assign the phosphorous stereochemistry for the rest of the analogs.
FIG 2.
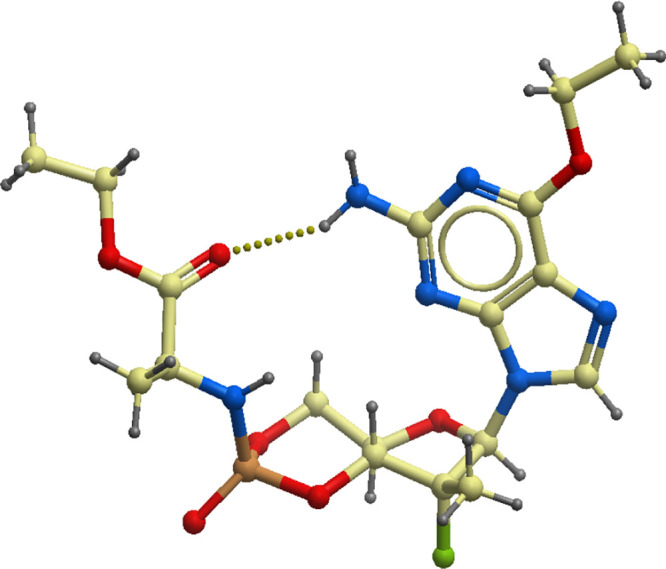
Single X-ray crystal structure of compound 17, represented as ball and stick model. The oxygen, nitrogen, fluorine, and phosphorous atoms are colored in red, blue, green, and orange, respectively.
Over 150 cyclic phosphoramidates were synthesized with variations in the ester, amino acid, phosphorous stereoisomer, and C-6 substitutions on the guanine base. As highlighted in Table 1, the prodrugs were assessed by cellular anti-dengue virus activity, plasma stability, liver fraction S9 stability, and intestinal fraction S9 stability to select the best prodrug with a balanced profile. Most of the prodrugs had a half-life of over 120 min in plasma and in the intestinal S9 fraction in higher species (dogs and human). The cyclic prodrug 12 had the same amino acid moiety and phosphorus stereochemistry (Sp) as compound 5 but showed a 2-fold-longer half-life than the linear prodrug 5 in the human liver S9 fraction. The Rp isomer 13 was much less potent, with a 2-fold-longer liver S9 fraction half-life than the Sp isomer 12. The corresponding (R)-alanine analogs 14 and 15 led to reduced anti-dengue virus activity. The glycine analog 16 had similar potency and stability profiles as the alanine prodrug 13. The ethyl ester 17 showed 3-fold improvement in potency while maintaining similar stability in the liver S9 fraction. The methyl ester 18 had a good stability profile but was less potent than compound 17. The more lipophilic 6-O-iPr analog 19 did not change levels of anti-dengue virus activity and in vitro stability. Based on balancing antiviral activities and in vitro stability profiles, alanine-based prodrugs with different combinations of esters and stereoisomers (compounds 12, 14, 17, and 18) were selected for further characterization.
TABLE 1.
Anti-dengue virus activities and in vitro stability profiles of cyclic phosphoramidate prodrugs of 6-O-alkyl-2′-deoxy-2′-fluoro-2′-C-methylguanosinea
| Compound | R1b | R2c | R3 | Sp or Rp | PBMC EC50 (μM)d |
t1/2 (min) in plasma in: |
t1/2 (min) in liver S9 fraction in: |
t1/2 (min) in intestinal S9 fraction in: |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | Dog | Rat | Human | Dog | Human | Dog | ||||||
| 4e | Rp | >25 | >120 | NDf | >120 | >120 | ND | ND | ND | |||
| 5e | Sp | 0.17 | >120 | >120 | <5 | 18 | 51 | >120 | 72 | |||
| 12 | i-Pr | Me (l-Ala) | Et | Sp | 0.072 | >120 | >120 | <5 | 36 | 56 | >120 | >120 |
| 13 | i-Pr | Me (l-Ala) | Et | Rp | 0.67 | >120 | >120 | <5 | 63 | 110 | >120 | >120 |
| 14 | i-Pr | Me (d-Ala) | Et | Sp | 0.37 | >120 | >120 | 6 | >120 | >120 | >120 | >120 |
| 15 | i-Pr | Me (d-Ala) | Et | Rp | 1.1 | >120 | >120 | 6 | 105 | 73 | ND | ND |
| 16 | i-Pr | H (Gly) | Et | Rp | 0.33 | >120 | >120 | <5 | 58 | 91 | ND | ND |
| 17 | Et | Me (l-Ala) | Et | Rp | 0.23 | >120 | >120 | <5 | 76 | 116 | >120 | >120 |
| 18 | Me | Me (l-Ala) | Et | Rp | 0.46 | >120 | >120 | <5 | >120 | >120 | >120 | >120 |
| 19 | Me | Me (l-Ala) | i-Pr | Rp | 0.43 | 113 | >120 | <5 | >120 | >120 | ND | ND |
| 20 | Et | i-Pr (d-Val) | Et | Sp | 0.23 | >120 | >120 | 66 | 82 | 61 | >120 | >120 |
Cyclic phosphoramidate prodrugs of 2′-deoxy-2′-fluoro-2′-C-methylguanosine show correlations between triphosphate levels and antiviral activities in PBMCs.
Selected cyclic phosphoramidate prodrugs 12, 14, 17, and 18 as well as the linear phosphoramidate PSI-353661 (compound 5) were assessed for their levels of triphosphate conversion in vitro. Compounds were incubated in human PBMCs at 10 μM for 24 h. At least two biological replicates were conducted for each compound (three technical replicates for each biological replicate). A trend of linear correlation was observed between triphosphate levels and potencies in human PBMCs (Fig. 3), and the most potent compound, 12, showed the highest triphosphate level in vitro.
FIG 3.
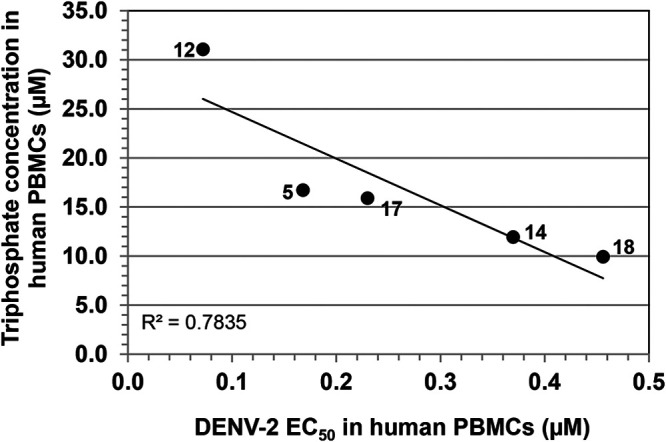
Correlation between triphosphate levels versus potencies in human PBMCs. For the PBMC triphosphate level, different prodrugs (compounds 12, 5, 17, 14, and 18) were incubated in human PBMCs at 10 μM for 24 h, and then the intracellular triphosphate concentration was measured by LC-MS/MS. Data were obtained from at least two biological replicates. The DENV-2 EC50 was obtained from the human PBMCs plaque assay. See Materials and Methods for further details.
Cyclic phosphoramidate prodrugs of 2′-deoxy-2′-fluoro-2′-C-methylguanosine show different PK profiles in dogs.
The cyclic phosphoramidate prodrugs showed low plasma stability in rodents but high plasma and liver S9 fraction stability in dogs and humans (Table 1). Thus, dogs were chosen as the preclinical species relevant for the pharmacokinetic (PK) assessment of the selected prodrugs.
Compounds 12, 14, 17, and 18 were dosed i.v. (0.5 mg/kg) and orally (p.o.) (3 mg/kg) in dogs to determine the pharmacokinetic parameters of the intact prodrug and metabolite 7 in plasma (Table 2) as well as the triphosphate 6 in PBMCs (Table 2 and Fig. 4). Compound 17 showed the highest intracellular triphosphate level in PBMCs after oral dosing; thus, it was selected for further characterization.
TABLE 2.
Pharmacokinetic parameters of prodrugs 12, 14, 17, and 18
| Drug and routea | Parameterb | Value for the prodrugc
|
|||
|---|---|---|---|---|---|
| 12 | 14 | 17 | 18 | ||
| Intact prodrug in plasma | |||||
| i.v. | Vss (l/kg) | 0.4 | ND | 0.5 | 0.5 |
| ER (%) | >100 | 67 | 45 | ||
| t1/2 (h) | 0.1 | 0.3 | 0.3 | ||
| p.o. | Cmax (μM) | 1.0 | 0.8 | 2.5 | 1.8 |
| Tmax (h) | 0.08 | 0.25 | 0.08 | 0.25 | |
| AUClast (μM⋅h) | 0.3 | 0.5 | 1.6 | 1.3 | |
| F (%) | 25 | ND | 44 | 24 | |
| Nucleoside metabolite 7 in plasma | |||||
| i.v. | t1/2 (h) | 4.0 | 4.0 | 3.4 | 3.9 |
| p.o. | Cmax (μM) | 0.8 | 0.8 | 0.5 | 1.3 |
| Tmax (h) | 0.5 | 0.5 | 2 | 1 | |
| AUC0–24 (μM⋅h) | 4.1 | 2.4 | 4.5 | 6.8 | |
| Triphosphate metabolite 6 in PBMCs | |||||
| p.o. | Cmax | 0.7 | 0.9 | 2.7 | 0.7 |
| Tmax | 1.0 | 16 | 11 | 4.7 | |
| AUC0–24 | 8.5 | 13.0 | 43.5 | 12.3 | |
Beagle dogs (n = 3) were dosed i.v. at 0.5 mg/kg and p.o. at 3 mg/kg.
Vss, volume of distribution at steady state; ER, hepatic extraction ratio, calculated from the i.v. clearance using a dog liver blood flow of 42 ml/min/kg; t1/2, elimination half-life; Cmax, maximum concentration; Tmax, time to reach maximum concentration; AUClast, area under the concentration-time curve (from t = 0 until the last measurable concentration); AUC0–24, area under the concentration-time curve for t = 0 to 24 h; F, bioavailability.
ND, not determined.
FIG 4.

Pharmacokinetic profiles of triphosphate 6 in PBMCs. Selected prodrugs (compounds 12, 14, 17, and 18) were dosed orally (3 mg/kg) to Beagle dogs, and the PBMC triphosphate concentration from each prodrug was measured by LC-MS/MS. Error bars represent standard deviations (n = 3 dogs).
Compound 17 shows pan-serotype and good antiviral activities in multiple cell lines.
The activities of compound 17 against all four DENV serotypes were examined in primary human PBMCs and in cell lines other than PBMCs. Compound 17 was active against all DENV serotypes, as shown in Table 3. It also showed good activity in multiple cell lines, which may be important for in vivo efficacy as DENV infection shows a broad tissue tropism (29).
TABLE 3.
In vitro activities and cytotoxicities of compound 17 across multiple serotypes and cell lines
| Cell type | Assay type | Parameter | Activity (μM) |
|---|---|---|---|
| PBMC | DENV-1 (48 h) | EC50 | 0.18 ± 0.06 |
| DENV-2 (48 h) | EC50 | 0.23 ± 0.04 | |
| DENV-3 (48 h) | EC50 | 0.36 ± 0.33 | |
| DENV-4 (48 h) | EC50 | 0.37 ± 0.14 | |
| THP-1 | DENV-2 | EC50 | 0.46 ± 0.20 |
| KU812 | DENV-2 high-content imaging | EC50 | 1.41 |
| K562 | DENV-2 | EC50 | 2.79 ± 0.22 |
| Cytotoxicity | CC50 | >100 | |
| 293T | DENV-2 | EC50 | 3.40 |
| Huh-7 | DENV-2 replicon expressing luciferase | EC50 | 1.73 ± 1.06 |
| HepG2 | Cytotoxicity | CC50 | >100 |
| MT-4 | Cytotoxicity | CC50 | >100 |
Compound 17 is converted to the active triphosphate 6 in PBMCs across multiple species.
For preclinical development, various animal models are typically used to assess the pharmacokinetics, efficacy, and safety of a compound. To assess the relevant species for further characterization, the triphosphate conversion of compound 17 was assessed in vitro. As shown in Table 4, the prodrug was converted to different levels of triphosphate in the PBMCs of all the species relevant for safety and efficacy assessment (mouse, rat, dog, monkey, and human).
TABLE 4.
Conversion of prodrug 17 to triphosphate in the PBMCs of multiple species
| Compound 17 concn (μM)a | Triphosphate concn (μM) in PBMCs of:b
|
||||
|---|---|---|---|---|---|
| Mouse | Rat | Dog | Monkey | Human | |
| 10 | 8.5 ± 2.5 | ND | 2.8 ± 0.4 | 39.2 ± 14.5 | 15.9 ± 10.6 |
| 100 | 53.4 ± 8.4 | 8.8 ± 0.5 | 13.8 ± 1.8 | 22.6 ± 6.4 | 59.7 ± 19.4 |
Compound was incubated in PBMCs at 10 and 100 μM for 24 h, and then the triphosphate concentration was measured by LC-MS/MS.
Data are means ± standard deviations (n = 3 biological replicates). ND, not determined.
TP50 and determination of exposure target for efficacy.
Although the AG129 viremia mouse model was considered a suitable model for testing anti-dengue virus efficacy (30), it was very challenging to use this animal model due to high esterase activity in plasma, resulting in in vivo metabolism and thus little exposure of the intact prodrug. We measured the active triphosphate 6 in PBMCs as potency is driven by the intracellular triphosphate level (Fig. 3). For pharmacokinetic/pharmacodynamic (PK/PD) correlation, we determined the TP50, which is the intracellular triphosphate concentration that inhibits virus replication by 50% (or the amount of triphosphate generated in cells at the 50% effective concentration [EC50] of the prodrug). The TP50 becomes the exposure target in PBMCs for in vivo efficacy.
Due to the sensitivity limitation of the analytical method, it was not possible to measure the PBMC triphosphate concentration at the EC50 of the prodrug. Thus, we incubated human PBMCs for 24 h with higher prodrug concentrations (3, 10, 30, and 100 μM) and observed the intracellular triphosphate levels, which followed Michaelis-Menten enzyme kinetics (Fig. 5), as has been reported for other triphosphates (12). Saturation was observed at the higher prodrug concentration.
FIG 5.
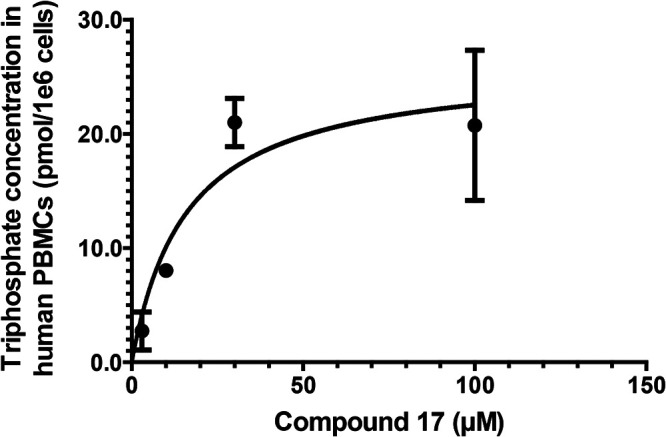
TP50 determination. Compound 17 (3, 10, 30, and 100 μM) was incubated in human PBMCs for 24 h, and the intracellular triphosphate concentration was measured. A Michaelis-Menten equation was used for curve fitting. The TP50 was calculated from extrapolating the DENV-2 EC50 of compound 17 to the triphosphate concentration. See Materials and Methods for more details. Experiments were conducted in three biological replicates. The graph represents one of the three biological experiments (error bars represent standard deviations from three technical replicates in each experiment). The average TP50 is 0.22 ± 0.12 pmol/1e6 cells (0.78 ± 0.42 μM) (means ± standard deviations from 3 biological replicates).
The TP50 experiments were conducted with compound 17 in three biological replicates (three technical replicates for each biological replicate). From each biological replicate, the Michaelis-Menten equation was used to fit the curve, as described in Materials and Methods (see “TP50 determination”). Using the same batch of PBMCs, the EC50 was also determined as described in Materials and Methods (see “In vitro antiviral assays”). The TP50 value was extrapolated from the EC50 of the prodrug. The average TP50 value from all the biological replicates was 0.78 ± 0.43 μM.
Compound 17 shows in vivo efficacy in a dengue mouse model.
Compound 17 (given p.o. at 10, 30, 100, and 300 mg/kg BID for 3 days) was assessed in the AG129 viremia mouse model. The infection of mice with DENV-2 (strain TSV01) leads to viremia which peaks on day 3 postinfection (30). A dosing scheme with the blood-sampling schedule is depicted in Fig. 6A.
FIG 6.

Efficacy study and PBMC triphosphate analysis in the dengue viremia mouse model. (A) Dosing scheme of efficacy study in AG129 mice. Compound 17 was dosed p.o. immediately after infection at 10, 30, 100, and 300 mg/kg BID for 3 days (total doses of 20, 60, 200, and 600 mg/kg/day). Each dose group contained 6 mice. Plasma was sampled at 1, 3, 6, 24, 48, 50, 52, 55, and 72 h after the first dose for intact prodrug and nucleoside 7 analysis. A plasma sample was also taken at 72 h after the first dose (terminal sampling) for viremia readout. PBMCs were collected during the terminal time points for triphosphate analysis. (B) Compound 17 shows efficacy in AG129 mice. Viremia readout from each mouse was done on day 3 (at terminal time points) by plaque assay. Error bars represent standard deviations (n = 6 mice). Compound 17 reduced viremia by 3-, 4-, 28- and 54-fold at doses of 10, 30, 100, and 300 mg/kg/day BID, respectively. The viremia reductions at doses of 100 and 300 mg/kg BID are significant (P < 0.0001). The difference in the viremia reduction levels between the 30- and 100- or 300-mg/kg BID groups are also significant (P < 0.01 or P < 0.0001, respectively). *, P < 0.01; ***, P < 0.0001. (C) PBMC triphosphate concentration from 30- and 100-mg/kg BID groups. Blood from 6 mice was pooled at the terminal time point (72 h after the first dose) to collect PBMCs. The terminal triphosphate concentrations were 0.39 and 1.43 μM for the 30- and 100-mg/kg BID groups, respectively. The terminal triphosphate level from the 100-mg/kg BID group exceeded TP50.
Nucleoside metabolite 7, but not the intact prodrug, was detectable in plasma. This is expected as the prodrug half-life in mouse plasma is likely less than 5 min, similar to that in rat plasma (Table 1). Triphosphate 6 was not detected in plasma, as expected, as the conversion of the prodrug to the active triphosphate happens intracellularly. Table S1 in the supplemental material shows the pharmacokinetic parameters obtained for metabolite 7 and a trend of dose proportionality.
Efficacy was observed at 100- and 300-mg/kg BID doses as the viremia was significantly reduced by 28- and 54-fold (or 1.6- and 2.2 logs), respectively. The 10- and 30-mg/kg BID doses were not efficacious as the viremia reduction was only 3- and 4-fold, respectively, and was not significant (Fig. 6B).
The PBMC triphosphate level was quantified from a pool of 6 mice from the 30- and 100-mg/kg BID groups. At 72 h after the first dose (terminal sampling, the same time points as the viremia readout), the intracellular triphosphate levels of the 30- and 100-mg/kg BID groups were 0.39 and 1.43 μM, respectively (Fig. 6C). Corresponding to the observed efficacy, the terminal triphosphate concentration exceeded the TP50 (0.78 μM) for the 100-mg/kg BID group but not for the 30-mg/kg BID group.
Dose escalation study for preclinical development.
Compound 17 underwent crystal form selection and formulation optimization to obtain the highest oral bioavailability in dogs (Table S2). Using the optimized solid dispersion formulation, the PBMC triphosphate levels and dose proportionality were reassessed in dogs (p.o. at 10, 30, 100, and 300 mg/kg). A dose proportionality was observed for the intact prodrug and metabolite 7 in plasma (Fig. 7A and B and Table 5). The intact prodrug had a short half-life (<1 h), while metabolite 7 had a long half-life (9 to 13 h) and constituted the major metabolite in dog plasma. Other phosphoramidate intermediates were also detected in plasma but at lower levels and with much shorter half-lives (data not shown).
FIG 7.

Pharmacokinetic profiles of the intact prodrug (A), major metabolite nucleoside 7 in plasma (B), and triphosphate metabolite 6 in PBMCs (C) upon single rising doses of compound 17 in Beagle dogs. Error bars represent standard deviations (n = 3 dogs). The triphosphate concentration in PBMCs was determined only from the 10- and 30-mg/kg groups. At 10 mg/kg, the PBMC triphosphate level exceeded TP50.
TABLE 5.
Pharmacokinetic parameters of compound 17 dosed orally in solid dispersion formulation to Beagle dogsa
| Parameterb | Value for the parameter at the indicated dose (mg/kg) of:c
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intact prodrug |
Nucleoside 7 |
Triphosphate 6 |
||||||||
| 10 | 30 | 100 | 300 | 10 | 30 | 100 | 300 | 10 | 30 | |
| Cmax (μM) | 1.9 | 4.6 | 17.3 | 24.9 | 0.5 | 1.3 | 3.4 | 7.5 | 13.1 | 22.3 |
| Tmax (h) | 0.5 | 0.5 | 0.7 | 0.7 | 2.3 | 3.0 | 3.0 | 3.0 | 32d | 9.3d |
| AUClast (μM⋅h) | 1.9 | 5.6 | 33.6 | 72.6 | 7.2 | 17.0 | 46.4 | 130.6 | 957.6 | 1879.9 |
| t1/2 (h) | 0.4 | 0.7 | 1.1 | 4.7 | 8.9 | 26.9 | 8.6 | 31.1 | 86.6 | 86.6 |
Compound 17 was dosed orally at 10, 30, 100, and 300 mg/kg in solid dispersion formulation to Beagle dogs (n = 3).
Cmax, maximum concentration; Tmax, time to reach maximum concentration; AUClast, area under the concentration-time curve (t = 0 until the last measurable concentration; for intact prodrug and nucleoside, t = 0 up to 24 h [Fig. 7A and B]; for triphosphate, t = 0 to 168 hours); t1/2, half-life.
Intact prodrug and major metabolite 7 were measured in plasma from all dose groups. Triphosphate 6 was measured in PBMCs only from the 10- and 30-mg/kg dose groups.
High interanimal variability observed (Fig. 7C).
Upon administration of a single oral dose of compound 17, prolonged exposure of triphosphate 6 in PBMCs was observed. The triphosphate half-life was 3.5 days in dogs. A trend of dose proportionality was observed in spite of the high variability of the triphosphate levels (Fig. 7C and Table 5).
Compound 17 in vitro and in vivo safety assessment.
Compound 17 was assessed in multiple cell lines (HepG2, K562, and MT-4) (31) and found to be clean (Table 3), as well as in various in vitro biochemical assays, including the mini-Ames test for genotoxicity, assays of the hERG (human ether-a-go-go related gene) channel for cardiovascular toxicity and CYP450 inhibition for drug-drug interactions, a micronucleus assay for mutagenicity, and determination of various receptor, ion channel, and kinase profiles. The compound did not show significant inhibition in any of these assays. Of note, PSI-353661 (compound 5) had also been assessed in various cytotoxicity assays, including in HepG2, Huh-7, and BxPC3 cells, and the compound demonstrated a CC50 of ≥80 μM (32), suggesting low in vitro cytotoxic liability for this compound series.
With no observed in vitro toxicity, compound 17 was progressed to 2-week rat and dog toxicology studies. Compound 17 was administered by oral gavage at 100, 300, and 1,000 mg/kg/day in rats and at 30, 100, and 300 mg/kg/day in dogs. The compound was tolerated in rats when given at doses up to 1,000 mg/kg/day for 14 consecutive days. Unfortunately, compound 17 was poorly tolerated in dogs. On days 7 to 9, significant findings in the lung (inflammation and hemorrhage) led to a severe decline in canine health, moribund animals, and necessary termination of some animals from the highest dose group. The findings were dose related, and mild pulmonary inflammation and hemorrhage were already observed in one of the six dogs in the 30-mg/kg/day group. The no observed adverse effect level (NOAEL) was not achieved in dogs. Table S3 shows the triphosphate levels in dog PBMCs obtained on day 1 and day 14.
DISCUSSION
Our objective is to develop an oral dengue drug that is efficacious and safe. Nucleoside analogs offer several advantages as dengue drug candidates as they target an essential virus-specific enzyme, RdRp (5), and have pan-serotypic activity and a high resistance barrier (8). As PBMCs are among the major viral replication sites (26), the active triphosphate concentration in PBMCs was used as a pharmacological marker for efficacy in addition to the EC50 value generated in PBMCs. The same measurement of triphosphate concentration in the target organs has also been used as a pharmacological marker for HIV (33). Since there are several steps for nucleoside analogs to become active in the cell, it is essential to set up the in vitro assay with conditions that are as similar to those in the target organ as possible.
The use of 4G2 antibody-enhanced PBMC infection warrants special explanation. We tried several cell types and were surprised that the variation is much greater for nucleoside analogs than for other antiviral compounds. For instance, PSI-352938 (compound 4) (Fig. 1B) has submicromolar activity in Huh-7 cells but is inactive in PBMCs. The discrepancy is due to initial CYP3A4-mediated dealkylation of the cyclic phosphate moiety (34). This prompted us to carefully select the cell types which best mimic the pathogenesis of dengue virus. We finally settled for PBMCs as they are among the target cells for dengue virus. To improve the efficiency of the infection, we used antibody enhancement using 4G2 antibody and detection by plaque assay.
Several successful examples of nucleoside antivirals have been developed for HIV, HSV, HBV, and HCV therapeutic areas (6). For dengue virus, several different nucleosides have been studied, i.e., NITD-008 and balapiravir. NITD-008 demonstrated good oral efficacy in the dengue mouse model, but it was unable to progress to human clinical trials due to an insufficient safety profile (10). Balapiravir was repurposed from HCV in a phase II dengue clinical trial, but failed to reduce viral load in the patients (13). Although the reasons remain to be fully understood, one potential reason could be that the activation of PBMCs during dengue virus infection led to reduction in the potency of balapiravir, as described previously (12). We chose our starting point to be 2′-deoxy-2′-fluoro-2′-C-methylguanosine as its active triphosphate showed potent inhibition for dengue virus RdRp with an IC50 value of 1.1 μM. PSI-352938 (compound 4) and PSI-353661 (compound 5) also displayed inhibition of DENV in Huh-7 cells containing dengue virus replicon, with EC50 values of 0.76 μM and 0.044 μM, respectively (Fig. 1B). PSI-352938 passed preclinical safety assessments and was well tolerated at doses of up to 1,600 mg once daily in a phase I study (20). In a later phase II study, this compound caused liver function abnormalities after 13 weeks of dosing (35). Similarly, PSI-353661 was not ideal against dengue virus as it had a short half-life in the liver S9 fraction (<20 min) and was designed for targeting the liver, not for systemic distribution, as required.
We suspected that the triphosphate metabolite could be the reason for liver toxicity and hypothesized that modification of the prodrug moiety could change the compound distribution and thus reduce liver toxicity. Our prodrugs are specifically designed to maximize the stability in gastrointestinal and liver tissue and in systemic circulation before penetrating into PBMCs. Once inside PBMCs, the intracellular enzymes would unmask the prodrug moieties easily, allowing further metabolism to the active triphosphate. We used in vitro stability in liver and intestinal S9 fractions and plasma stability, together with cellular anti-dengue virus activity in PBMCs, to find the most balanced compound. From various types of nucleotide prodrugs, we became interested in the 3′,5′-cyclic phosphoramidates (27). This particular prodrug could offer additional advantages compared to those of the linear phosphoramidates like PSI-353661 (compound 5) as they masked the 3′-free OH and reduced a degree of rotational freedom, potentially allowing for improved cell entry and prolonged metabolic stability in the liver. In addition, 3′,5′-cyclization eliminated the release of toxic aromatic alcohols like phenol and naphtol. Over 150 cyclic phosphoramidates were synthesized, with variations in the ester, amino acid, phosphorous stereoisomer, and C-6 substitution on the guanine base. Based on balanced in vitro dengue virus and stability profiles (Table 1), we selected alanine prodrugs with different combinations of esters and stereoisomers (compounds 12, 14, 17, and 18) for further characterization. We directly monitored the concentration of the active triphosphate 6 in PBMCs and observed a trend of linear correlation between potencies and PBMC triphosphate levels (Fig. 3). This correlation is expected as the triphosphate is the pharmacologically active form inhibiting viral replication through termination of RNA chain synthesis (7, 10).
In vivo pharmacokinetic profiling for the selected prodrugs 12, 14, 17, and 18 was performed in dogs with i.v. administration at 0.5 mg/kg and p.o. at 3 mg/kg to establish plasma clearance, oral bioavailability, and triphosphate loading in PBMCs over 24 h (Table 2; Fig. 4). Prodrug 12 had the least stability (the highest clearance) both in vitro in the liver S9 fraction and in vivo. Compound 17 showed the highest level of triphosphate in PBMCs (about 3-fold), albeit with high interanimal variabilities due to the multiple enzymes required for the conversion. It has been reported that small modifications of phosphoramidate prodrugs could change the triphosphate conversion dramatically (36). Overall, compound 17 becomes our lead compound to progress for further characterization as it achieved both good pharmacokinetic profiles (Tables 1 and 2) and in vitro potencies across different serotypes and cell lines (Table 3). To assess the relevance of various animal models for pharmacokinetics, efficacy, and safety evaluation for further preclinical development, we tested compound 17 for triphosphate conversion in PBMCs of multiple species (Table 4). In fact, species difference in triphosphate conversion has been well documented (34, 36–38). We demonstrated that compound 17 was converted to the active triphosphate in the PBMCs of all the relevant species, including mice and monkeys, which enable compound assessment in the two different dengue virus infection animal models if needed (30, 39).
To develop pharmacokinetic/pharmacodynamic (PK/PD) correlation and determine the minimum efficacious dose of prodrug 17, we determined the TP50 (Fig. 5). TP50 is the intracellular triphosphate concentration at which 50% of the viral replication is inhibited (12). Next, we wanted to assess if the level of triphosphate can be translated to efficacy in vivo using a dengue viremia mouse model (30). Due to the abundant carboxylesterases in the plasma of rodent species (40), oral doses of 100 and 300 mg/kg BID for 3 days were needed to achieve at least a 1-log viremia reduction (1.6- and 2.2-log viremia reductions, respectively), while the efficacy was not observed at 10 and 30 mg/kg (Fig. 6). The PBMC triphosphate concentration on day 3 reached TP50 (0.78 μM) for the 100-mg/kg BID group (1.43 μM) but not for the 30-mg/kg BID group (0.39 μM). Based on these in vitro and in vivo results, we defined here for the first time the minimum efficacious dose for a nucleos(t)ide prodrug as the dose that is required to maintain a triphosphate concentration in PBMCs above the TP50. By applying this principle to dog species, whose plasma stability is close to that of human, compound 17 reached the TP50 already at 10 mg/kg (Fig. 7C).
Once we were able to estimate the minimum efficacious dose in dogs, the compound was prepared for preclinical toxicology evaluation in vivo. A solid dispersion formulation was developed to improve the oral bioavailability and enable compound safety assessment at high doses. Single-dose pharmacokinetic studies were conducted in dogs to confirm the PBMC triphosphate level and assess the dose proportionality. Compound 17 showed a trend of dose proportionality for the intact prodrug and free nucleoside metabolite in plasma as well as for the triphosphate level in PBMCs. The level of triphosphate was sustained at a level above the TP50 for at least 1 week after a single dose of 10 mg/kg (half-life of 3.5 days), making the compound possible for a single-dose cure (Fig. 7; Table 5). The long intracellular half-life has been reported for other nucleoside triphosphates in vivo (19) as well as in vitro in lymphocyte- or monocyte-derived cells (41, 42). We also observed it when we conducted PBMC studies with a close analog of compound 17 (see Fig. S1 in the supplemental material).
Compound 17 was further assessed for safety in 2-week rat and dog toxicology studies. Although the half-life of compound 17 in rat plasma is <5 min, the triphosphate could still be detected in PBMCs upon multiple oral doses. Compound 17 was tolerated in rats but not in dogs. At 100 and 300 mg/kg/day, clinical signs accompanied by weight loss were already observed on day 7, causing early termination for these two groups. Liver was not the target organ for this compound, unlike PSI-352938. This indicates that our prodrug moiety has changed the compound distribution. However, tubular degeneration of the kidneys, lung inflammation, and hemorrhage were observed, among other findings. The pathology findings in dogs were dose related. NOAEL (no observed adverse effect level) was not achieved in this study. Due to the severity and only partial reversibility of the adverse findings, further development of compound 17 was not pursued.
Although compound 17 did not progress to the clinical stage, the orally bioavailable prodrug is pioneering the field. A phosphoramidate prodrug has never been shown to be suitable for oral administration as phosphoramidates usually break down in the liver. Although the tenofovir-based prodrugs are orally bioavailable, they are significantly different as this type of modification is unlikely to work in RNA viruses. Another example of a phosphoramidate prodrug is remdesivir. It was initially developed for Ebola virus but later repurposed for the treatment of SARS-CoV-2. It has to be delivered intravenously as the drug will be extensively metabolized if given orally. This severely limits the accessibility of the drug as it has to be administered in a hospital setting. We demonstrated that it is possible to have an orally available phosphoramidate drug by modifying the prodrug moiety. We postulate that this concept can be applied to remdesivir. By changing the linear prodrug portion to a cyclic one, we believe that it can make remdesivir orally bioavailable and broaden its application outside a hospital setting.
Taking these results together, we have shown the potential of monophosphate prodrugs for dengue virus infection and demonstrated a suitable prodrug moiety to effectively deliver the monophosphate into PBMCs upon oral dosing. We have addressed the efficacy of monophosphate prodrugs by demonstrating a proof of concept in a mouse model. We established the TP50 (the intracellular triphosphate concentration at which 50% of the virus replication is inhibited) for a guanosine analog as an exposure target and defined the minimum efficacious dose as the one that is required to maintain a triphosphate concentration in PBMCs above the TP50. This concept could be universally applied and will be useful to evaluate the efficacy of any dengue virus nucleos(t)ide monophosphate prodrug.
MATERIALS AND METHODS
Materials.
All nucleoside/nucleotide compounds were synthesized at the Novartis Institute for Tropical Diseases (NITD). The solid dispersion batch of compound 17 consisted of 20% (wt/wt) active ingredient, 40% (wt/wt) hypromellose acetyl succinate (HPMC-ASLF; Shin-Etsu Chemical, Tokyo, Japan), 35% (wt/wt), hypromellose (HPMC-E3; Shin-Etsu Chemical, Tokyo, Japan), and 5% (wt/wt) sodium lauryl sulfate (SLS; Sigma-Aldrich, St. Louis, MO). 8-Bromoadenosine 5′-triphosphate (Br-ATP) and hexylamine were from Sigma-Aldrich. Acetonitrile (liquid chromatography mass spectrometry [LC-MS] grade) was from Merck (Darmstadt, Germany). All other solvents, reagents, and chemicals were either of molecular biology grade or of the highest chemical grade available from Sigma-Aldrich or Thermo Fisher Scientific (Waltham, MA) unless otherwise mentioned.
Pooled liver S9 fraction (Gentest, Corning, NY) and intestinal S9 fraction (XenoTech, Kansas City, KS) were from mixed genders for human and from males for all other species. Plasma matrix for in vitro stability assays were from mixed genders and obtained from Seralab (West Sussex, UK). Cryopreserved human PBMCs (individual donors) were purchased from AllCells (Alameda, CA) or ReachBio (Seattle, WA). Written consent from the donors was available for all samples. All experiments involving human matrices were approved by the Institutional Review Board of Novartis prior to the start of the experiments. PBMCs from other species (pooled) were from 3H Biomedical (Uppsala, Sweden). C6/36, THP-1, KU812, K562, and 293T cells were from American Type Culture Collection (ATCC, Manassas, VA).
Vacutainer cell preparation tubes (CPTs) (with sodium citrate; 4-ml draw capacity) were from BD Biosciences (Franklin Lakes, NJ). The collagen I-coated plates were from Thermo Fisher Scientific. HEPES buffer, RPMI 1640 medium, and penicillin-streptomycin were from Life Technologies (Carlsbad, CA). PhosSTOP (phosphatase inhibitor) and protease inhibitor cocktail (cOmplete) tablets were from Roche Applied Science (Penzberg, Germany).
Stability in plasma, liver, and intestinal S9 fraction.
Stability assays in plasma, liver, and the intestinal S9 fraction were performed as previously described (43, 44). Briefly, the metabolic reaction was initiated by addition of compounds to the matrices. Incubation was conducted in a shaking water bath (37°C), and time points were sequentially taken. Samples were quenched with ice-cold acetonitrile and analyzed by LC with tandem MS (LC-MS/MS). The half-life (t1/2) was calculated from the rate of compound depletion.
In vitro antiviral assays.
Antiviral assays in PBMCs were performed by plaque assay as previously described (12). The EC50 was calculated by Prism (GraphPad Software, La Jolla, CA) using the equation for a sigmoidal dose-response (variable slope) curve.
The Huh-7 cell dengue virus replicon assay was previously described (14). The cells harbor stable subgenomic dengue virus RNA capable of replicating in the cells without the structural proteins that are necessary for formation of mature virions. For the 293T cell dengue virus plaque assay, 3 × 104 cells were seeded in a 96-well collagen I-coated plate in 100 μl of medium (Dulbecco’s modified Eagle’s medium [DMEM] supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin) 1 day prior to infection. The medium was removed, and the cells were infected with the virus-antibody complex at a multiplicity of infection (MOI) of 3 in the same medium without serum for 1 h at 37°C. The medium was then removed and replaced with DMEM supplemented with 2% fetal bovine serum and 1% penicillin-streptomycin for another 2 days. The EC50 was calculated using a plaque assay as described previously. THP-1 and KU812 assays were performed as described previously (45). A K562 assay was performed similarly at an MOI of 1. A cytotoxicity assay was also performed as previously described (15, 31).
An RdRp assay was conducted as previously described (12). Briefly, a 244-nucleotide RNA with the sequence 5′-(TCAG)20(TCCAAG)14(TCAG)20-3′ was used as a template (46). Compounds with various concentrations were mixed with the RNA template (100 nM), dengue virus RdRp (100 nM), 0.5 μM BBT-GTP (where BBT is 2'-[2-benzothiazoyl]-6'-hydroxybenzothiazole), and 2 μM ATP, CTP, and UTP in the buffer containing 50 mM Tris HCl, pH 7.5, 10 mM KCl, 0.5 mM MnCl2, and 0.01% Triton X-100 for 120 min. The amount of substrate produced and IC50 were determined as described previously (47).
In vitro triphosphate conversion studies in PBMCs.
Cryopreserved PBMCs were thawed and incubated in RPMI medium containing 2% fetal bovine serum, 1% penicillin-streptomycin, and a designated concentration of the prodrug. After the intracellular conversion into the corresponding triphosphate reached steady state (24 h at 37°C in the incubator), the cells were spun down for 10 min (at ambient temperature, 135 × g) and washed with cold 0.9% NaCl solution in 1 mM HEPES. The wash buffer was carefully removed with a micropipette, and PBMC lysis was carried out before triphosphate measurement by LC-MS/MS.
TP50 determination.
The TP50 was derived using Michaelis-Menten kinetics with increasing prodrug concentrations in human PBMCs as previously described (12). Briefly, cryopreserved human PBMCs were incubated with compound 17 (3, 10, 30, and 100 μM) for 24 h. The cells were then lysed, and the PBMC triphosphate level was analyzed by LC-MS/MS. The intracellular triphosphate concentrations were plotted against the prodrug concentrations used for the incubation. A Michaelis-Menten equation of Y = A[X]/B + [X] was used to fit the curve, where [Y] is the triphosphate concentration, A is the calculated maximum triphosphate concentration extrapolated from the graph, [X] is the prodrug concentration, and B is the calculated prodrug concentration at which its triphosphate reached half of its maximal value. The TP50 value [Y] was extrapolated when the prodrug concentration [X] was equal to its EC50 in PBMCs.
PBMCs lysis.
Cell lysis buffer containing 50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1% IGEPAL CA-630, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1× protease inhibitor stock solution (Roche), and 1× PhosSTOP tablet was freshly made and used within 2 h.
PBMCs were lysed by addition of cell lysis buffer at 10 million cells/ml (or as otherwise mentioned) and incubated at room temperature for 10 min. The cell debris was then spun down at 15,800 × g for 20 min (4°C). The lysate was transferred into new tubes, snap-frozen in liquid nitrogen, and stored at −80°C until used for triphosphate LC-MS/MS analysis.
PBMC isolation and lysis in in vivo studies.
To increase the sensitivity of triphosphate detection, PBMCs were collected and lysed at a concentration of 30 million cells/ml from each dog or a pool from AG129 mice. To isolate PBMCs from animals, blood was drawn to Vacutainer CPTs at the indicated time points in the figures and immediately processed. The CPTs were centrifuged at ambient temperature for 20 min (1,500 × g). A whitish layer (PBMCs) under the plasma layer was pipetted into a 15-ml conical centrifuge tube. Phosphate-buffered saline (PBS; 10 ml) was added, and the mixture was centrifuged at ambient temperature for another 10 min (700 × g). The plasma supernatant was then carefully aspirated using a vacuum pump while ensuring that the PBMC pellet remained at the bottom of the tube. The pellet was then resuspended with PBS (1 ml) and vortexed at the lowest setting. The suspension was transferred to an Eppendorf tube and lysed. A 5-fold-larger amount of protease inhibitor was used for lysing mouse PBMCs to ensure the stability of the triphosphate. The samples (cell lysate) were stored at −80°C for triphosphate LC-MS/MS analysis.
Bioanalysis by LC-MS/MS.
Analyses were performed with reverse-phase liquid chromatography (LC) coupled to tandem mass spectrometry (MS/MS). Electrospray ionization (ESI) was used in positive mode for intact prodrug and nucleoside 7 and in negative mode for triphosphate 6. Analysis was most challenging in the rodent matrix due to the ex vivo instability of intact prodrug in plasma and triphosphate in cell lysate.
To stabilize the intact prodrug in plasma, an inhibitor cocktail of NaF and citric acid (20 mM NaF and 40 mM citric acid final concentration) was added to the blood collection tubes. Plasma was obtained by centrifuging the blood for 5 min (4°C) at 10,000 × g. A 175-μl extraction solution mixture (acetonitrile-methanol-acetic acid, 90:10:0.2 [vol/vol]) containing a generic internal standard (warfarin) was added to 25 μl of plasma. The sample plates were shaken and then centrifuged for 10 min (4°C, 2,884 × g). The supernatant was collected, and 5 μl was injected to the LC-MS/MS system. The mobile phase was 20 mM ammonium acetate plus 1% acetic acid (A) or 1% acetic acid in 100% acetonitrile (B). Prodrug and metabolite separation was performed on a Hydro-RP column (100 by 3 mm, 2.5-μm particle size; Phenomenex, Torrance, CA) at 600 μl/min with the following gradient: 0% to 80% B for 2 min and 80% B for another 1.5 min. MS/MS detection was performed with a 4000 QTRAP instrument (Sciex, Framingham, MA). Multiple reaction monitoring (MRM) transitions of m/z 489.4/310.2 and m/z 300.3/152.1 were used to detect prodrug 17 and metabolite 7, respectively. Calibration and quality control (QC) samples used a matched matrix (e.g., naive AG129 mouse plasma), and the lower limit of quantification (LLOQ) obtained was 15.75 nM for both analytes.
To stabilize the triphosphate in PBMC lysates extracted from mice, a larger amount of protease inhibitor was used, as described above for PBMC isolation in in vivo studies. To retain the very polar triphosphate on a reverse-phase LC column, hexylamine was used for ion pairing.
An equal volume of acetonitrile-water mixture (1:1) containing an internal standard (Br-ATP) was added to the PBMC lysate. The mixture was vortexed and centrifuged (1,431 × g at ambient temperature) for 10 min. The supernatant (5 μl) was then injected to the LC-MS/MS system (4000 QTRAP; Sciex, Framingham, MA). Ion pair chromatography was used for separation on a Gemini-NX column (5 by 2 mm, 3-μm pore size; Phenomenex, Torrance, CA). Mobile phases A and B were water and an acetonitrile mixture, respectively, containing 5 mM ammonium acetate and 5 mM hexylamine, buffered at pH 8.5. Gradient elution (700 μl/min) was achieved with 5% to 60% mobile phase B within 3.5 min. Detection was in negative mode with MRM transitions of m/z 538.2/159.0 and m/z 585.9/159.0 for the triphosphate 6 and Br-ATP, respectively. A matched matrix (e.g., mouse PBMC lysate) with cell concentrations similar to those of the samples was used for calibration and QC. The LLOQ was 0.065 pmol/3 million cells. The triphosphate concentration per cell volume (in μM) was calculated using the corpuscular volume of 283 fl for PBMCs (48).
In vivo pharmacokinetic and toxicology studies.
All Novartis animal studies were approved by the institutional review boards of the different sites where the experiments were carried out and by the respective authorities.
The pharmacokinetic studies of compounds 12, 14, 17, and 18 were conducted in Beagle dogs by intravenous administration (i.v., 0.5 mg/kg) or oral gavage (p.o., 3 mg/kg). A solution formulation containing 20% polyethylene glycol (PEG) 300, 5% Solutol HS-15, and 5% dextran in water was used for this study (Fig. 4; Table 2). Blood samples (3.5 ml; anticoagulant, K2EDTA) were collected at 0.08, 0.25, 0.5, 1, 2, 4, 8, and 24 h postdosing.
The rising dose and 2-week toxicology studies of compound 17 (Fig. 7; Table 5) were performed in Wistar Han rats and Beagle dogs at Charles River Laboratories Preclinical Services Montreal (Sherbrooke, Canada) under the sponsorship of Novartis with solid dispersion formulation (see Materials and Methods section) and doses as described in Results.
The noncompartmental pharmacokinetic parameters from various studies were calculated using either Watson LIMS (Thermo Fisher Scientific, Waltham, MA) or WinNonlin, version 5.01 (Pharsight Corporation, Mountain View, CA).
In vivo efficacy studies.
AG129 mice (lacking alpha/beta interferon [IFN-α/β] and IFN-γ receptors [30]) were obtained from Biological Resource Center (BRC), Singapore. Male and female AG129 mice aged 8 to 14 weeks (weighing 20 to 30 g) were used. Infection with DENV-2 (strain TSV01) was given intraperitoneally (500 μl, 1.4 × 107 PFU/ml). Solid dispersion of compound 17 was dosed p.o. immediately after infection. The doses were 10, 30, 100, and 300 mg/kg BID for 3 consecutive days. Plasma samples were collected on days 1 and 3 postinfection for intact prodrug and nucleoside 7 analysis (Fig. 6A, dosing scheme). Pooled blood samples of 6 mice were collected at 72 h after the first dose (terminal sampling) for PBMC triphosphate analysis. Stabilization of the prodrug and triphosphate in the matrices was as described in the LC-MS/MS section above. Plasma was also obtained from the terminal time point of each mouse for viremia readout by plaque assay.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kwan Leung, David Beer, Paul Smith, Lv Liao, Margaret Weaver, and Thierry Diagana for their helpful discussions on this project. Peter Wipfli, Ying-Bo Chen, Meng Hui Lim, and Mahesh Nanjundappa are kindly acknowledged for their analytical and pharmacokinetic work. We also appreciate Kah Fei Wan and his team for generating high-throughput antiviral activities. We are grateful to Caroline Rynn and her team for generating stability data in plasma, liver, and the intestinal S9 fraction. Ina Dix and Trixie Wagner are kindly acknowledged for their X-ray analysis of compound 17.
All authors were employees of Novartis at the time the work described in the manuscript was performed.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Whitehorn J, Simmons CP. 2011. The pathogenesis of dengue. Vaccine 29:7221–7228. doi: 10.1016/j.vaccine.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 2.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guzman MG, Alvarez M, Halstead SB. 2013. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: an historical perspective and role of antibody-dependent enhancement of infection. Arch Virol 158:1445–1459. doi: 10.1007/s00705-013-1645-3. [DOI] [PubMed] [Google Scholar]
- 4.Capeding MR, Tran NH, Hadinegoro SR, Ismail HI, Chotpitayasunondh T, Chua MN, Luong CQ, Rusmil K, Wirawan DN, Nallusamy R, Pitisuttithum P, Thisyakorn U, Yoon IK, van der Vliet D, Langevin E, Laot T, Hutagalung Y, Frago C, Boaz M, Wartel TA, Tornieporth NG, Saville M, Bouckenooghe A, CYD14 Study Group. 2014. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet 384:1358–1365. doi: 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- 5.Lim SP, Noble CG, Shi PY. 2015. The dengue virus NS5 protein as a target for drug discovery. Antiviral Res 119:57–67. doi: 10.1016/j.antiviral.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Jordheim LP, Durantel D, Zoulim F, Dumontet C. 2013. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov 12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 7.Chen YL, Yin Z, Duraiswamy J, Schul W, Lim CC, Liu B, Xu HY, Qing M, Yip A, Wang G, Chan WL, Tan HP, Lo M, Liung S, Kondreddi RR, Rao R, Gu H, He H, Keller TH, Shi PY. 2010. Inhibition of dengue virus RNA synthesis by an adenosine nucleoside. Antimicrob Agents Chemother 54:2932–2939. doi: 10.1128/AAC.00140-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delang L, Vliegen I, Froeyen M, Neyts J. 2011. Comparative study of the genetic barriers and pathways towards resistance of selective inhibitors of hepatitis C virus replication. Antimicrob Agents Chemother 55:4103–4113. doi: 10.1128/AAC.00294-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B, Yokokawa F, Nilar S, Smith P, Beer D, Lescar J, Shi PY. 2013. Ten years of dengue drug discovery: progress and prospects. Antiviral Res 100:500–519. doi: 10.1016/j.antiviral.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 10.Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J, Kondreddi RR, Niyomrattanakit P, Lakshminarayana SB, Goh A, Xu HY, Liu W, Liu B, Lim JY, Ng CY, Qing M, Lim CC, Yip A, Wang G, Chan WL, Tan HP, Lin K, Zhang B, Zou G, Bernard KA, Garrett C, Beltz K, Dong M, Weaver M, He H, Pichota A, Dartois V, Keller TH, Shi PY. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A 106:20435–20439. doi: 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klumpp K, Smith DB. 2011. Antiviral drugs: from basic discovery through clinical trials. John Wiley & Sons, Hoboken, NJ. [Google Scholar]
- 12.Chen YL, Abdul Ghafar N, Karuna R, Fu Y, Lim SP, Schul W, Gu F, Herve M, Yokohama F, Wang G, Cerny D, Fink K, Blasco F, Shi PY. 2014. Activation of peripheral blood mononuclear cells by dengue virus infection depotentiates balapiravir. J Virol 88:1740–1747. doi: 10.1128/JVI.02841-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen NM, Tran CNB, Phung LK, Duong KTH, Huynh H. l A, Farrar J, Nguyen QTH, Tran HT, Nguyen CVV, Merson L, Hoang LT, Hibberd ML, Aw PPK, Wilm A, Nagarajan N, Nguyen DT, Pham MP, Nguyen TT, Javanbakht H, Klumpp K, Hammond J, Petric R, Wolbers M, Nguyen CT, Simmons CP. 2013. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis 207:1442–1450. doi: 10.1093/infdis/jis470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeo KL, Chen YL, Xu HY, Dong H, Wang QY, Yokokawa F, Shi PY. 2015. Synergistic suppression of dengue virus replication using a combination of nucleoside analogs and nucleoside synthesis inhibitors. Antimicrob Agents Chemother 59:2086–2093. doi: 10.1128/AAC.04779-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang G, Lim SP, Chen YL, Hunziker J, Rao R, Gu F, Seh CC, Ghafar NA, Xu H, Chan K, Lin X, Saunders OL, Fenaux M, Zhong W, Shi PY, Yokokawa F. 2018. Structure-activity relationship of uridine-based nucleoside phosphoramidate prodrugs for inhibition of dengue virus RNA-dependent RNA polymerase. Bioorg Med Chem Lett 28:2324–2327. doi: 10.1016/j.bmcl.2018.04.069. [DOI] [PubMed] [Google Scholar]
- 16.Stein DS, Moore KH. 2001. Phosphorylation of nucleoside analog antiretrovirals: a review for clinicians. Pharmacotherapy 21:11–34. doi: 10.1592/phco.21.1.11.34439. [DOI] [PubMed] [Google Scholar]
- 17.McGuigan C, Harris SA, Daluge SM, Gudmundsson KS, McLean EW, Burnette TC, Marr H, Hazen R, Condreay LD, Johnson L, De Clercq E, Balzarini J. 2005. Application of phosphoramidate pronucleotide technology to abacavir leads to a significant enhancement of antiviral potency. J Med Chem 48:3504–3515. doi: 10.1021/jm0491400. [DOI] [PubMed] [Google Scholar]
- 18.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. 2014. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem Rev 114:9154–9218. doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a beta-d-2'-deoxy-2'-alpha-fluoro-2'-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 53:7202–7218. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 20.Sofia MJ. 2013. Nucleotide prodrugs for the treatment of HCV infection. Adv Pharmacol 67:39–73. doi: 10.1016/B978-0-12-405880-4.00002-0. [DOI] [PubMed] [Google Scholar]
- 21.Ray AS, Fordyce MW, Hitchcock MJ. 2016. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of Human Immunodeficiency virus. Antiviral Res 125:63–70. doi: 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Siegel D, Hui HC, Doerffler E, Clarke MO, Chun K, Zhang L, Neville S, Carra E, Lew W, Ross B, Wang Q, Wolfe L, Jordan R, Soloveva V, Knox J, Perry J, Perron M, Stray KM, Barauskas O, Feng JY, Xu Y, Lee G, Rheingold AL, Ray AS, Bannister R, Strickley R, Swaminathan S, Lee WA, Bavari S, Cihlar T, Lo MK, Warren TK, Mackman RL. 2017. Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo[2,1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) for the treatment of Ebola and emerging viruses. J Med Chem 60:1648–1661. [DOI] [PubMed] [Google Scholar]
- 23.Lam AM, Espiritu C, Murakami E, Zennou V, Bansal S, Micolochick Steuer HM, Niu C, Keilman M, Bao H, Bourne N, Veselenak RL, Reddy PG, Chang W, Du J, Nagarathnam D, Sofia MJ, Otto MJ, Furman PA. 2011. Inhibition of hepatitis C virus replicon RNA synthesis by PSI-352938, a cyclic phosphate prodrug of beta-d-2'-deoxy-2'-alpha-fluoro-2'-beta-C-methylguanosine. Antimicrob Agents Chemother 55:2566–2575. doi: 10.1128/AAC.00032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy PG, Bao D, Chang W, Chun BK, Du J, Nagarathnam D, Rachakonda S, Ross BS, Zhang HR, Bansal S, Espiritu CL, Keilman M, Lam AM, Niu C, Steuer HM, Furman PA, Otto MJ, Sofia MJ. 2010. 2'-Deoxy-2'-alpha-fluoro-2'-beta-C-methyl 3',5'-cyclic phosphate nucleotide prodrug analogs as inhibitors of HCV NS5B polymerase: discovery of PSI-352938. Bioorg Med Chem Lett 20:7376–7380. doi: 10.1016/j.bmcl.2010.10.035. [DOI] [PubMed] [Google Scholar]
- 25.Chang W, Bao D, Chun BK, Naduthambi D, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Zhang HR, Bansal S, Espiritu CL, Keilman M, Lam AM, Niu C, Steuer HM, Furman PA, Otto MJ, Sofia MJ. 2011. Discovery of PSI-353661, a novel purine nucleotide prodrug for the treatment of HCV infection. ACS Med Chem Lett 2:130–135. doi: 10.1021/ml100209f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fink K, Ng C, Nkenfou C, Vasudevan SG, van Rooijen N, Schul W. 2009. Depletion of macrophages in mice results in higher dengue virus titers and highlights the role of macrophages for virus control. Eur J Immunol 39:2809–2821. doi: 10.1002/eji.200939389. [DOI] [PubMed] [Google Scholar]
- 27.Meppen M, Pacini B, Bazzo R, Koch U, Leone JF, Koeplinger KA, Rowley M, Altamura S, Di Marco A, Fiore F, Giuliano C, Gonzalez-Paz O, Laufer R, Pucci V, Narjes F, Gardelli C. 2009. Cyclic phosphoramidates as prodrugs of 2'-C-methylcytidine. Eur J Med Chem 44:3765–3770. doi: 10.1016/j.ejmech.2009.04.043. [DOI] [PubMed] [Google Scholar]
- 28.Reddy PG, Chun BK, Zhang HR, Rachakonda S, Ross BS, Sofia MJ. 2011. Stereoselective synthesis of PSI-352938: a beta-d-2'-deoxy-2'-alpha-fluoro-2'-beta-C-methyl-3',5'-cyclic phosphate nucleotide prodrug for the treatment of HCV. J Org Chem 76:3782–3790. doi: 10.1021/jo200060f. [DOI] [PubMed] [Google Scholar]
- 29.Martina BE, Koraka P, Osterhaus AD. 2009. Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev 22:564–581. doi: 10.1128/CMR.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schul W, Liu W, Xu HY, Flamand M, Vasudevan SG. 2007. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J Infect Dis 195:665–674. doi: 10.1086/511310. [DOI] [PubMed] [Google Scholar]
- 31.Fenaux M, Lin X, Yokokawa F, Sweeney Z, Saunders O, Xie L, Lim SP, Uteng M, Uehara K, Warne R, Gang W, Jones C, Yendluri S, Gu H, Mansfield K, Boisclair J, Heimbach T, Catoire A, Bracken K, Weaver M, Moser H, Zhong W. 2016. Antiviral nucleotide incorporation by recombinant human mitochondrial RNA polymerase is predictive of increased in vivo mitochondrial toxicity risk. Antimicrob Agents Chemother 60:7077–7085. doi: 10.1128/AAC.01253-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furman PA, Murakami E, Niu C, Lam AM, Espiritu C, Bansal S, Bao H, Tolstykh T, Micolochick Steuer H, Keilman M, Zennou V, Bourne N, Veselenak RL, Chang W, Ross BS, Du J, Otto MJ, Sofia MJ. 2011. Activity and the metabolic activation pathway of the potent and selective hepatitis C virus pronucleotide inhibitor PSI-353661. Antiviral Res 91:120–132. doi: 10.1016/j.antiviral.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bazzoli C, Jullien V, Le Tiec C, Rey E, Mentre F, Taburet AM. 2010. Intracellular pharmacokinetics of antiretroviral drugs in HIV-infected patients, and their correlation with drug action. Clin Pharmacokinet 49:17–45. doi: 10.2165/11318110-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 34.Niu C, Tolstykh T, Bao H, Park Y, Babusis D, Lam AM, Bansal S, Du J, Chang W, Reddy PG, Zhang HR, Woolley J, Wang LQ, Chao PB, Ray AS, Otto MJ, Sofia MJ, Furman PA, Murakami E. 2012. Metabolic activation of the anti-hepatitis C virus nucleotide prodrug PSI-352938. Antimicrob Agents Chemother 56:3767–3775. doi: 10.1128/AAC.00530-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gentile I, Buonomo AR, Zappulo E, Borgia G. 2015. Discontinued drugs in 2012 – 2013: hepatitis C virus infection. Expert Opin Invest Drugs 24:239–251. doi: 10.1517/13543784.2015.982274. [DOI] [PubMed] [Google Scholar]
- 36.Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA. 2010. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem 285:34337–34347. doi: 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birkus G, Wang R, Liu X, Kutty N, MacArthur H, Cihlar T, Gibbs C, Swaminathan S, Lee W, McDermott M. 2007. Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob Agents Chemother 51:543–550. doi: 10.1128/AAC.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami E, Wang T, Babusis D, Lepist EI, Sauer D, Park Y, Vela JE, Shih R, Birkus G, Stefanidis D, Kim CU, Cho A, Ray AS. 2014. Metabolism and pharmacokinetics of the anti-hepatitis C virus nucleotide prodrug GS-6620. Antimicrob Agents Chemother 58:1943–1951. doi: 10.1128/AAC.02350-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onlamoon N, Noisakran S, Hsiao HM, Duncan A, Villinger F, Ansari AA, Perng GC. 2010. Dengue virus-induced hemorrhage in a nonhuman primate model. Blood 115:1823–1834. doi: 10.1182/blood-2009-09-242990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bahar FG, Ohura K, Ogihara T, Imai T. 2012. Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci 101:3979–3988. doi: 10.1002/jps.23258. [DOI] [PubMed] [Google Scholar]
- 41.Ray AS, Vela JE, Boojamra CG, Zhang L, Hui H, Callebaut C, Stray K, Lin KY, Gao Y, Mackman RL, Cihlar T. 2008. Intracellular metabolism of the nucleotide prodrug GS-9131, a potent anti-human immunodeficiency virus agent. Antimicrob Agents Chemother 52:648–654. doi: 10.1128/AAC.01209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, Siegel D, Perron M, Bannister R, Hui HC, Larson N, Strickley R, Wells J, Stuthman KS, Van Tongeren SA, Garza NL, Donnelly G, Shurtleff AC, Retterer CJ, Gharaibeh D, Zamani R, Kenny T, Eaton BP, Grimes E, Welch LS, Gomba L, Wilhelmsen CL, Nichols DK, Nuss JE, Nagle ER, Kugelman JR, Palacios G, Doerffler E, Neville S, Carra E, Clarke MO, Zhang L, Lew W, Ross B, Wang Q, Chun K, Wolfe L, Babusis D, Park Y, Stray KM, Trancheva I, Feng JY, Barauskas O, Xu Y, Wong P, et al. 2016. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531:381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di L, Kerns EH, Hong Y, Chen H. 2005. Development and application of high throughput plasma stability assay for drug discovery. Int J Pharm 297:110–119. doi: 10.1016/j.ijpharm.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 44.Plant N. 2004. Strategies for using in vitro screens in drug metabolism. Drug Discov Today 9:328–336. doi: 10.1016/s1359-6446(03)03019-8. [DOI] [PubMed] [Google Scholar]
- 45.Fu Y, Chen YL, Herve M, Gu F, Shi PY, Blasco F. 2014. Development of a FACS-based assay for evaluating antiviral potency of compound in dengue infected peripheral blood mononuclear cells. J Virol Methods 196:18–24. doi: 10.1016/j.jviromet.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 46.Hung M, Gibbs CS, Tsiang M. 2002. Biochemical characterization of rhinovirus RNA-dependent RNA polymerase. Antiviral Res 56:99–114. doi: 10.1016/s0166-3542(02)00101-8. [DOI] [PubMed] [Google Scholar]
- 47.Niyomrattanakit P, Abas SN, Lim CC, Beer D, Shi PY, Chen YL. 2011. A fluorescence-based alkaline phosphatase-coupled polymerase assay for identification of inhibitors of dengue virus RNA-dependent RNA polymerase. J Biomol Screen 16:201–210. doi: 10.1177/1087057110389323. [DOI] [PubMed] [Google Scholar]
- 48.Simiele M, D'Avolio A, Baietto L, Siccardi M, Sciandra M, Agati S, Cusato J, Bonora S, Di Perri G. 2011. Evaluation of the mean corpuscular volume of peripheral blood mononuclear cells of HIV patients by a coulter counter to determine intracellular drug concentrations. Antimicrob Agents Chemother 55:2976–2978. doi: 10.1128/AAC.01236-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.