

Abstract
Straightforward methods for detecting adenosine-to-inosine (A-to-I) RNA editing are key to better understanding its regulation, function, and connection with disease. We address this need by developing a novel reagent, N-(4-ethynylphenyl)acrylamide (EPhAA), and illustrating its ability to selectively label inosine in RNA. EPhAA is synthesized in a single step, reacts rapidly with inosine, and is “click”-compatible, enabling flexible attachment of fluorescent probes at editing sites. We first validate EPhAA reactivity and selectivity for inosine in both ribonucleosides and RNA substrates, and then apply our approach to directly monitor in vitro A-to-I RNA editing activity using recombinant ADAR enzymes. This method improves upon existing inosine chemical labeling techniques and provides a cost-effective, rapid, and non-radioactive approach for detecting inosine formation in RNA. We envision this method will improve study of A-to-I editing and enable better characterization of RNA modification patterns in different settings.
Keywords: RNA, A-to-I editing, ADAR, click-chemistry, bioconjugation, RNA chemistry
Graphical Abstract
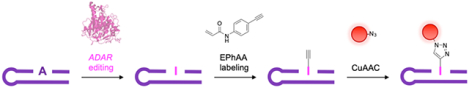
Detecting Adenosine-to-Inosine (A-to-I) RNA editing is vital to improving our understanding of its regulation and function in humans. Here we develop a new “click”-compatible reagent, N-(4-ethynylphenyl)acrylamide (EPhAA), to selectively functionalize inosine in RNA. EPhAA offers a cost-effective and rapid approach for detecting enzymatic A-to-I RNA editing activity in vitro.
RNA is chemically modified by a number of enzymes after transcription, in turn influencing RNA stability, localization and activity within the cell. Adenosine-to-inosine (A-to-I) RNA editing is one of the most widespread modifications, and is performed by adenosine deaminases acting on RNA (ADARs) (Scheme 1a).1 Adenosine deamination changes the molecular structure and hydrogen bonding pattern of the nucleobase, and resulting inosines instead base pair with cytidine to effectively recode these sites as guanosine. Editing sites within protein-coding mRNAs directly alter amino acid sequences and produce different protein isoforms. Non-coding RNAs also undergo extensive editing, including microRNAs and small-interfering RNAs, significantly altering their biosynthesis, localization, and gene regulation properties.2–3 A-to-I editing is essential for a number of biological processes including tissue development,4–5 neurological function,6 and immune system activation.7 Dysfunctional editing is also directly linked with autoimmune diseases,8–9 neurological disorders,10 and several types of cancer.11–12
Scheme 1.
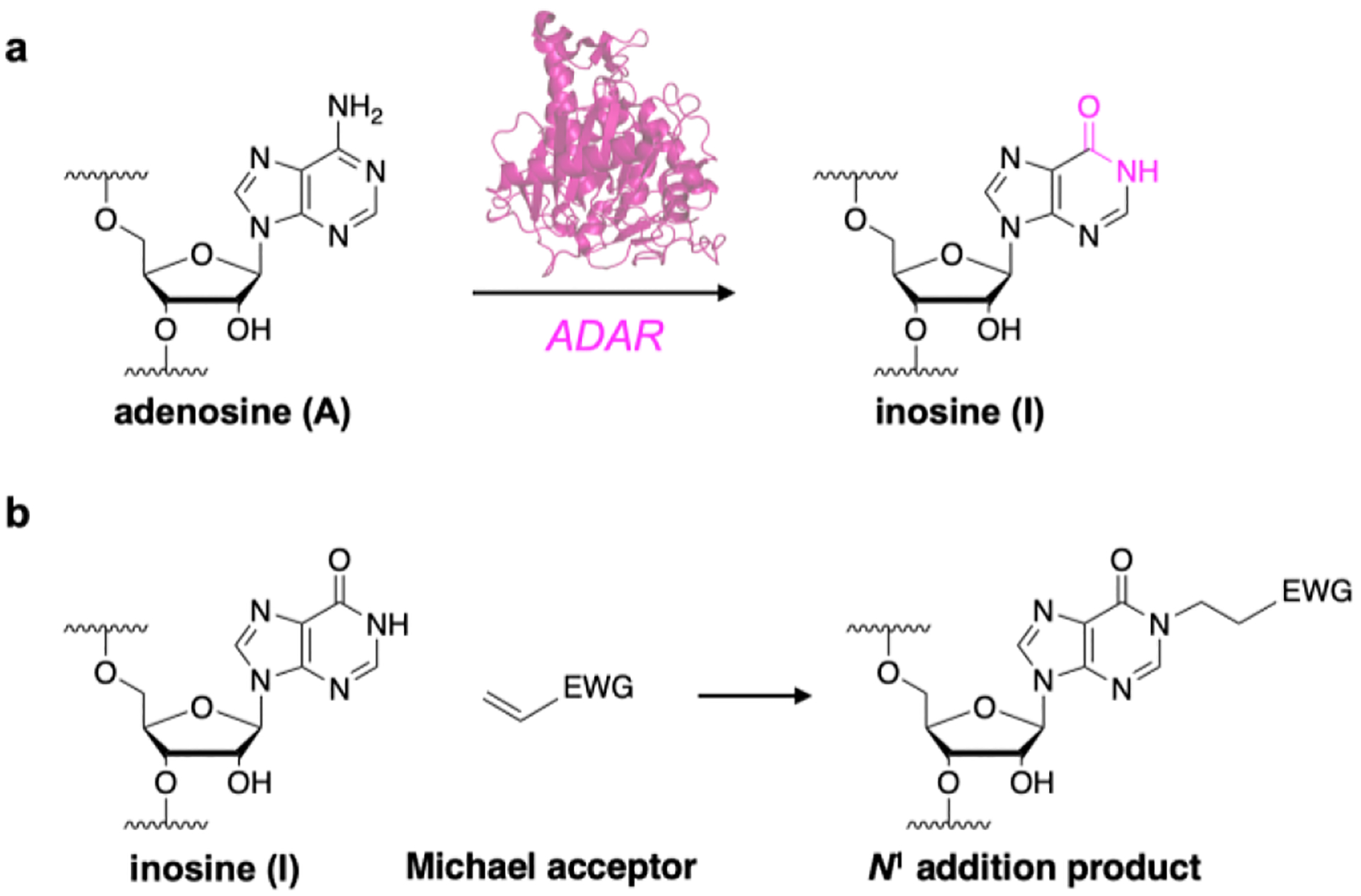
Formation and detection of inosine in RNA. a) A-to-I editing is catalyzed by adenosine deaminases acting on RNA (ADAR, pink). b) Inosine nucleobases can be detected by reacting with Michael acceptors to yield N1 addition products.
Despite this importance, our overall understanding of A-to-I editing regulation is limited. In particular, while many sites have been identified (> 5 million),13–14 it is unclear why certain sites are edited at higher frequency than others and what precise function they each serve.15 Efforts to map A-to-I locations and ADAR binding sites have revealed that editing patterns are highly complex and variable in humans,7, 16–18 and the precise mechanisms by which ADAR enzymes bind to and edit specific RNA sequences remain unclear. This gap is also significant for therapeutic site-directed RNA editing strategies,19 as both the design and precise implementation of this machinery is reliant on a thorough understanding of ADAR regulation.
Detecting inosine formation in RNA is of central importance for characterizing editing mechanisms. While high-throughput RNA sequencing (RNA-seq) is commonly employed for large scale detection and mapping of A-to-I sites,20 this method is also costly, prone to random sampling errors, and requires complex bioinformatic analyses.21–22 Alternatively, model reactions using ADAR enzymes with small RNA substrates (~20–50 nt) have yielded substantial insights into how certain RNA sequences and structural motifs are recognized and edited.17, 23–26 Although A-to-I sites are “visible” as A-G transitions in Sanger sequencing,27–28 these methods require relatively large RNA substrates (>300–400 nt), and are incompatible with the small RNA strands that are ideal for these experiments. To detect inosine in smaller substrates, adenosines within chimeric RNA strands are often internally radiolabeled with 32P. After ADAR editing, RNA substrates are then digested with nuclease P1 and A-to-I nucleotide changes are detected with autoradiographic thin layer chromatography.23, 25–26 While this method is effective, it is also time-consuming to construct each RNA substrate, and assays using these radioactive materials require specialized training, instrumentation, and waste disposal protocols. Alternatively, deamination can be detected by incorporating alkyne-modified or fluorogenic thiolated-adenosine analogues into RNA substrates,29–30 but these approaches can introduce structural alterations into RNA targets that impact ADAR targeting, and they require lengthy phosphoramidite monomer synthesis.
Direct chemical detection of A-to-I editing would circumvent these assay limitations, and inosine has been shown to react with Michael acceptors to yield N1 addition products (scheme 1b).31 In particular, a recent approach utilized acrylonitrile to alkylate inosines for reverse-transcription termination sequencing. Termed “inosine chemical erasing sequencing” (ICE-seq), this technique improved the accuracy of detecting A-to-I sites using Sanger sequencing, but also suffered from significant limitations in sensitivity, and requires matched DNA and RNA samples for each assay.32 Subsequent work derivatized acrylonitrile for use in “clickable”-biotinylation and enrichment of A-to-I edited transcripts.33 While acrylonitrile is a promising scaffold for chemical detection of inosine, derivatizing these reagents is difficult and requires several synthetic and purification steps. Alternatively, we recently reported an acrylamidofluorescein reagent that enables fluorescent detection and enrichment of inosine in RNA.34 While our initial study demonstrated feasibility, acrylamidofluorescein also displayed poor solubility and was restricted to fluorescein addition. However, acrylamide scaffolds are simple to modify, and we were interested in elaborating upon this architecture to develop an improved and more generalizable inosine probe. Toward this goal, we first screened potential acrylamide scaffolds for inosine reactivity using our previously established reaction conditions (50:50 EtOH:1M triethylammonium acetate pH 8.6, 70 °C) (Fig. 1). Acrylamide and N-phenylacrylamide were both highly reactive toward inosine, whereas alkylacrylamide scaffolds (mPEG acrylamide and N-hydroxyethylacrylamide) gave little to no product formation (Figs. 1b, S1). Interestingly, N-phenylacrylamide is structurally similar to acrylamidofluorescein (Fig. 1c), and it is likely this moiety exhibits sufficient electron-withdrawing properties consistent in other Michael acceptors.
Figure 1.
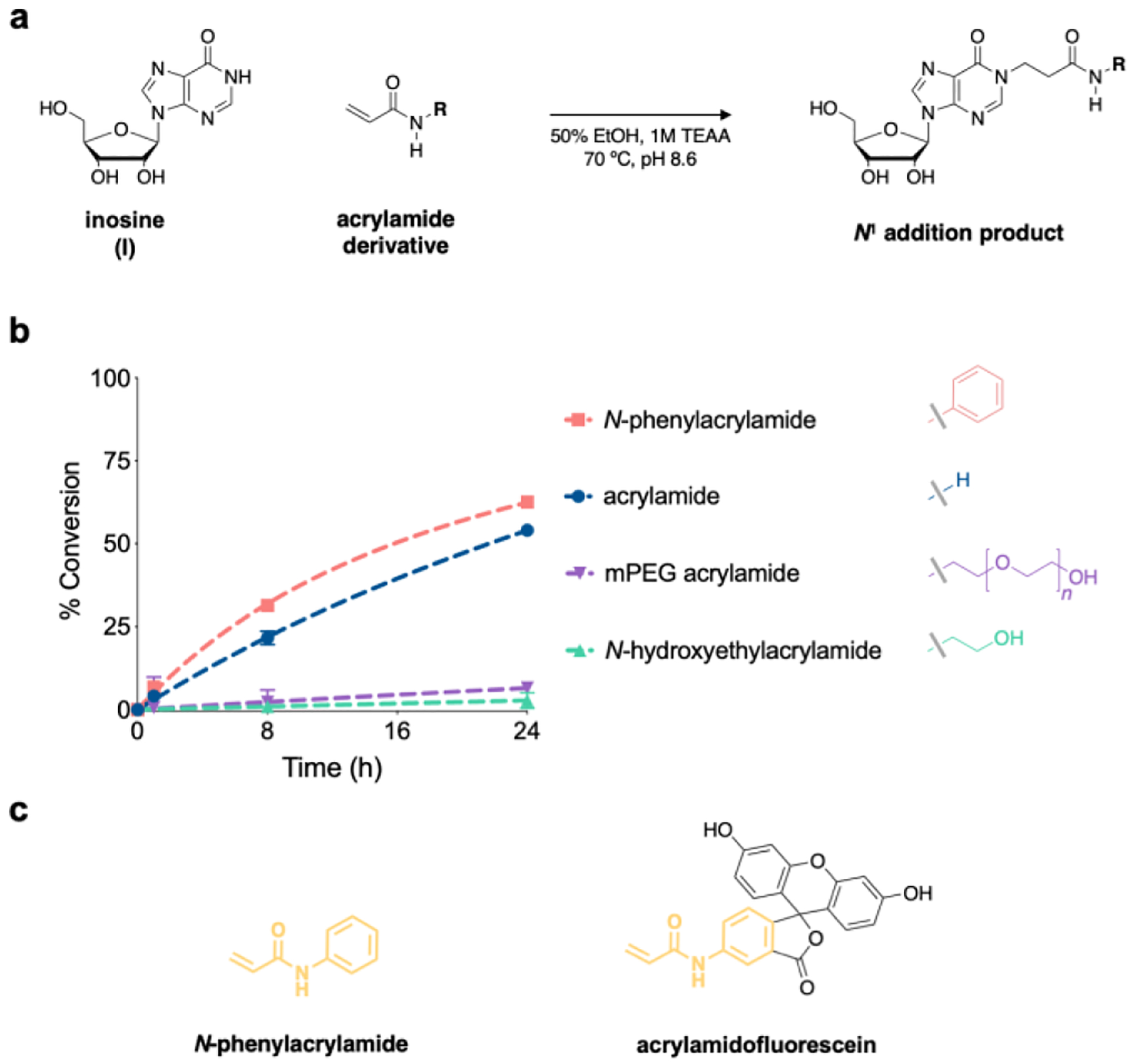
a) Acrylamide derivatives were evaluated for reactivity with inosine by b) monitoring product formation via HPLC. Values represent mean with S.D. error bars (n = 3). c) Structural similarities (yellow) between N-phenylacrylamide and acrylamidofluorescein.
We next sought to derivatize N-phenylacrylamide to enable secondary functionalization with fluorescent probes using copper-catalyzed azide-alkyne cycloaddition (CuAAC), or “click” chemistry. We quickly identified 4-ethynylaniline as a commercially available, alkyne-functionalized intermediate, enabling us to employ a one-step coupling with acrylic acid to yield N-(4-ethynylphenyl)acrylamide (EPhAA, scheme 2). After verifying product identity (Figs. S3–S5), we tested EPhAA for reactivity with inosine and confirmed appearance of the expected addition product N1-ethynylphenylamidoethylinosine (EPhAE1I) by HPLC and ESI-MS analysis (Figs 2a–b, S6a, S7). Deprotonation of N1 on inosine is known to mechanistically drive this reaction,31 and so to further confirm that EpHAA undergoes addition at this position, we tested our labeling reaction across different pH values (Fig. 2c). As expected, we observed a steep increase in reaction rates consistent with the known pKa value of inosine N1 (~8.7).35 In assembling reaction mixtures, we also noted improved solubility of EPhAA compared to acrylamidofluorescein, and we were able to double our normal working concentrations to ~500 mM. As expected, this resulted in more rapid overall reaction kinetics, and when compared to our previous reagent, we observed a ~2–3-fold increase in conversion percentages at similar reaction times (Fig. 2d).34 Next, we incubated EPhAA with the remaining ribonucleosides uridine (U), guanosine (G), adenosine (A), and cytidine (C) (Figs. 2d, S6). In this test, we observed robust selectivity towards I, and while U and G have similar acidic nitrogens that can be labeled with Michael acceptors,36–37 N1 on inosine displays much higher nucleophilicity and reactivity with these reagents,31 and off-target labeling was only observed at extended reaction times. Acrylonitrile and acrylamide reagents are also known to react with pseudouridine (Ψ),31 and we determined that our reagent exhibited similar reactivity characteristics, as we observed the expected N1 addition product (Figure S6f, S7, S9). While this off-target reactivity may seem problematic, the primary application we envision for our EPhAA reagent is detecting inosine in model RNA strands to monitor ADAR activity, and Ψ can be omitted from these substrates. Additionally, if assays necessitate the use of cellular RNA or require Ψ content, existing carbodiimide reagents can be employed to selectively block and deplete Ψ sites.38–39 Lastly, we assessed general EPhAA stability by incubating the reagent in the absence of ribonucleoside, and while we observed some degradation of the reagent in our labeling conditions, this effect was minimal and only seen in extended reaction times (Fig. S6g).
Scheme 2.
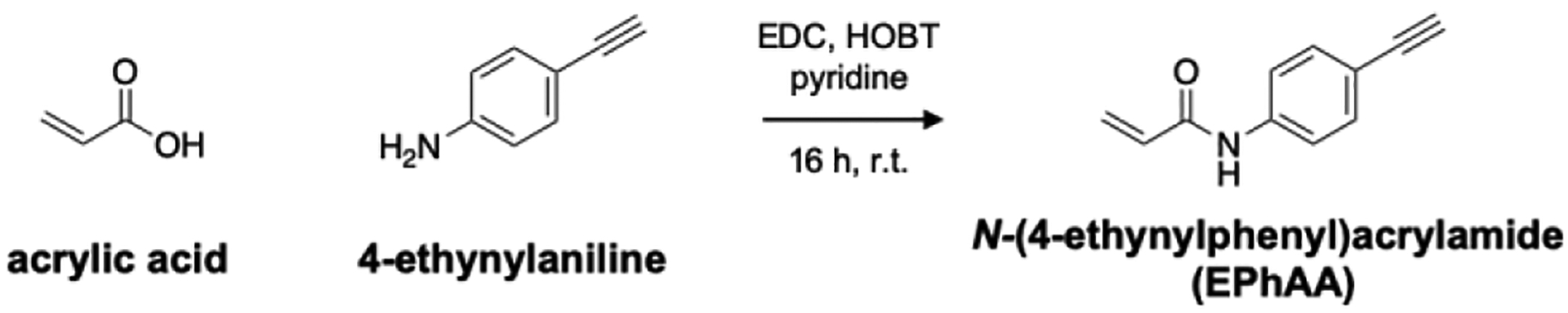
One-step synthesis of EPhAA.
Figure 2.
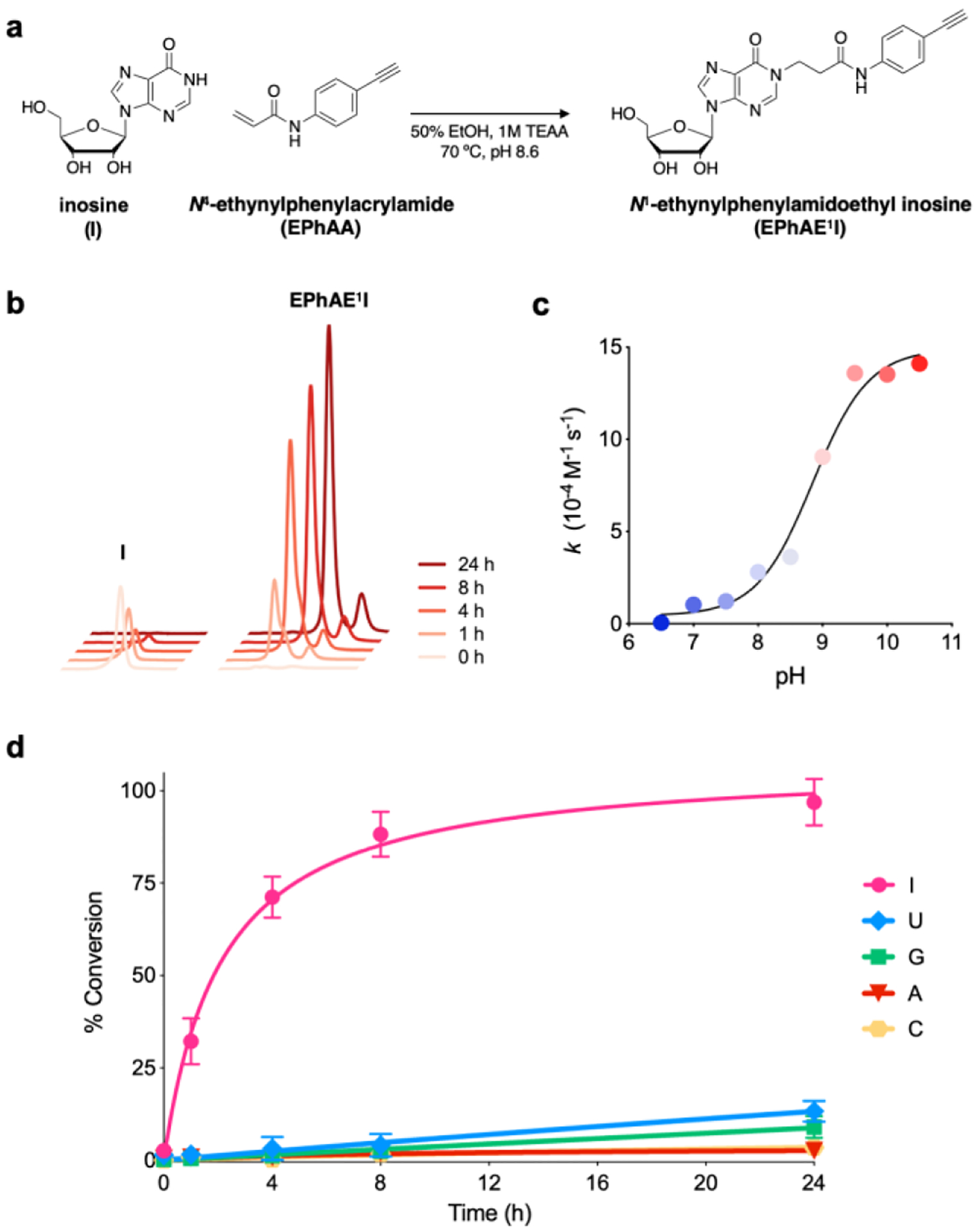
Reactivity assessment of EPhAA with ribonucleosides. a) Reaction scheme of EPhAA with inosine. b) Representative HPLC traces depicting formation of EPhAE1I over 24 hours. c) Dependence of pH on reaction rate constants for EPhAA addition on inosine. d) EPhAA reactivity with each of the major ribonucleosides over 24 hours. Values represent mean with S.D. error bars (n = 3).
With our validated reagent in hand and given our ultimate goal of detecting ADAR-mediated A-to-I editing, we next sought to label inosine in RNA oligonucleotides. ADARs commonly deaminate double-stranded RNA substrates at A:C mismatches, “flipping out” the target A into the active site to leave behind an “orphan C” base.1–2, 40 To mimic this, we synthesized a target strand inspired by an mRNA hairpin (HER1) that undergoes editing by human ADAR1 (hADAR1) at a defined site (Fig. 3a).23 To detect inosine, we planned to use CuAAC to install a fluorophore after EPhAA labeling. We first wanted to verify that our click-labeling conditions were optimal, so we subjected an alkyne-functionalized DNA oligonucleotide to a standard CuAAC protocol with a picolyl azide-functionalized Cyanine5 fluorophore (Cy5-N3).41 As shown in Fig. S10, we observed complete labeling of the alkyne-modified strand, indicating these conditions would be compatible with our workflow. Given our previous data showing background labeling of U and G nucleotides at very long reaction times (Fig. 2d), we were next interested in optimizing EPhAA labeling time to minimize off-target attachment. To test this, we reacted HER1 RNA A and I substrates with EPhAA for increasing amounts of time in independent duplicate trials followed by CuAAC labeling (Fig. S11). In RNA I samples, we expectedly saw a rapid appearance of a major product band, indicative of inosine fluorescent chemical labeling. RNA A did not produce significant signal, but did exhibit a “smear” in longer reaction times, likely indicating a mixture of products resulting from off-target labeling at U and G residues. This distribution was also observed in RNA I reactions with extended labeling times, further corroborating this hypothesis. We also stained all RNA species in gels using SYBR gold to assess overall labeling efficiency (Fig. S11). Although we did not achieve full conversion of the RNA I strand, we identified 6 hours as an optimal EPhAA reaction time to achieve robust selectivity (~60-fold I vs A labeling, Fig. S11). In particular, when compared to our previous acrylamidofluorescein reagent, we achieved significantly better selectivity (~60-fold vs ~8-fold) and with much shorter reaction times (6 h vs 24 h).34 Lastly, given our ultimate goal of detecting RNA editing, we were also interested in assessing the linearity of our method for measuring different A-to-I editing “rates.” To test this, we performed a series of duplicate labeling reactions using varying ratios of A and I substrate while keeping the total amount of RNA constant. As shown in Fig. 3b–c, inosine content was highly proportional to fluorescent intensity and we observed linearity between these variables (R2 = 0.95, r = 0.98), providing additional confidence that our method could accurately measure A-to-I editing activity.
Figure 3.
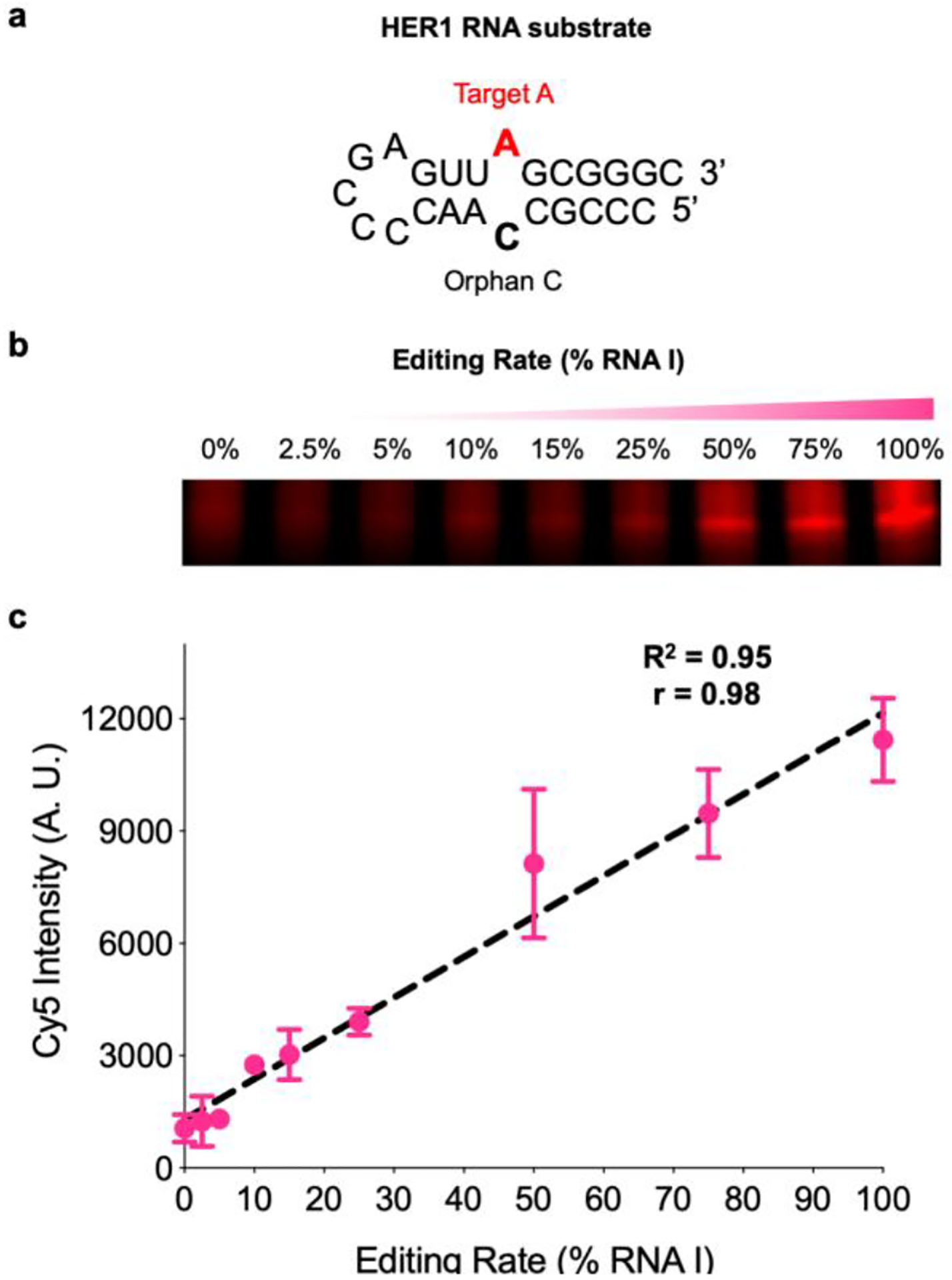
Validating chemical detection of inosine in RNA. a) Sequence and structure of the model HER1 RNA hairpin substrate with target A (red) and the orphan C base (black). b) EPhAA labelling and PAGE analysis of different simulated RNA editing rates and c) densitometric quantification of signal (arbitrary units, A.U.) across reactions. Values represent mean with S.D. error bars (n = 2).
Finally, we wanted to directly illustrate the utility of our method for detecting ADAR-mediated A-to-I editing. Given that HER1 is selectively recognized and edited by hADAR1, we first expressed and purified recombinant deaminase domains from this enzyme. Additionally, we prepared a mutant hADAR1 enzyme (E1008Q) which displays increased catalytic activity and speed, likely by providing enhanced stability of the orphan C nucleobase (Fig. 4a).23, 40 We envisioned that these enzyme variants would be a suitable test of our labeling method and further validate this approach for detecting catalytic deamination differences arising from biochemical variations in ADAR enzymes. As shown in Figs. 4b–d, we performed duplicate in vitro deamination experiments on our HER1 RNA A substrate with both enzymes, and we were able to fluorescently detect A-to-I conversion and robustly distinguish activity between wild type ADAR and the hyperactive E1008Q mutant. In addition to plotting overall editing activity, we also estimated initial velocities (vi) for both enzymes and observed ~14-fold increase in turnover speed for the E1008Q mutant (Fig. S12), which is in close agreement with previous activity comparisons of these hADAR1 isoforms.23
Figure 4.
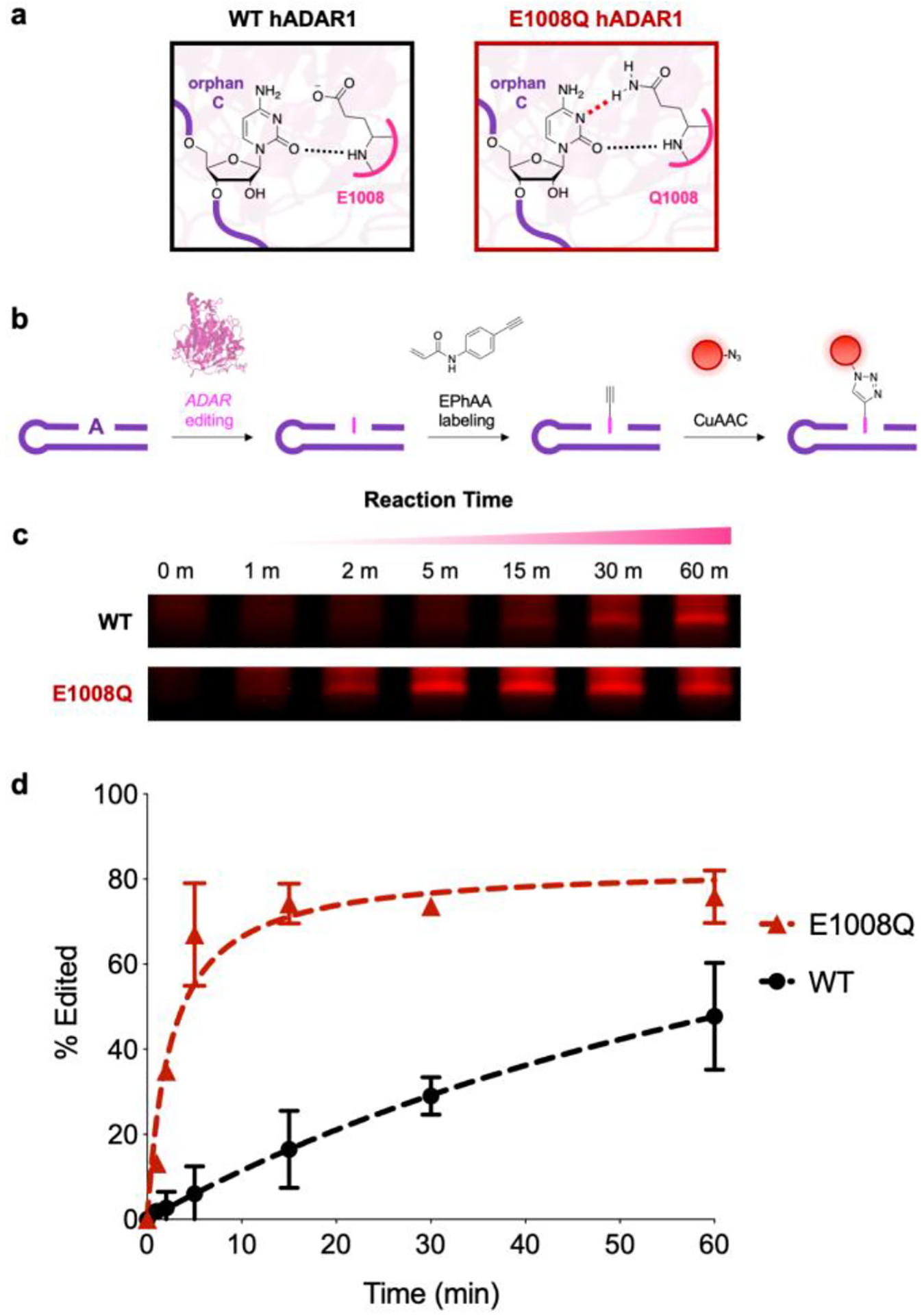
Chemical detection of ADAR1-mediated A-to-I RNA editing. a) ADAR1 amino acids interact with the orphan C base, with the E1008Q point mutation providing increased stability and overall catalytic efficiency. b) Overall workflow for detecting A-to-I editing with EPhAA labeling and CuAAC. c) EPhAA labelling and PAGE analysis of in vitro A-to-I RNA editing reactions. d) Densitometric quantification of signal across different RNA editing reactions using wild type (WT, black) and E1008Q mutant (red) ADAR enzymes. Values represent mean with S.D. error bars (n = 2).
A-to-I RNA editing is a widespread post-transcriptional modification that is essential for a variety of cellular processes, and aberrant RNA editing is directly linked to a number of diseases. Despite progress in characterizing A-to-I editing regulation and dynamics, significant gaps remain in our understanding of why certain sites are edited more than others, and what functional roles these editing events play. Simple and straightforward methods for detecting inosine formation in RNA and measuring ADAR activity are integral to addressing these knowledge gaps. In this work, we show the development and validation of a novel reagent, N-(4-ethynylphenyl)acrylamide (EPhAA), as an economical and rapid chemical labeling method for assaying A-to-I RNA editing in vitro. This reagent is simple to synthesize, improves upon existing labeling approaches, and robustly detects inosine in RNA. We envision this method will be a valuable tool to complement existing techniques for characterizing ADAR mechanisms and deciphering A-to-I RNA editing signatures in a variety of contexts. In particular, we view EPhAA labeling as a cost-effective and rapid method to elucidate the effects of RNA sequence and structure on ADAR editing activity in vitro, better assess the biochemical impact of disease-relevant ADAR mutations on pathological A-to-I editing, and accurately measure the activity of engineered recombinant enzymes for site-directed RNA editing.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01GM116991 to J.M.H.). P.A.B acknowledges support from the National Institutes of Health in the form of R01GM061115. The authors also thank Tewoderos Ayele and Travis Loya for helpful discussions and advice.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- (1).Bass BL Annu. Rev. Biochem 2002, 71, 817–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nishikura K Nat. Rev. Mol. Cell. Biol 2016, 17 (2), 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kawahara Y; Zinshteyn B; Sethupathy P; Iizasa H; Hatzigeorgiou AG; Nishikura K Science 2007, 315 (5815), 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wahlstedt H; Daniel C; Ensterö M; Öhman M Genome Res. 2009, 19 (6), 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shtrichman R; Germanguz I; Mandel R; Ziskind A; Nahor I; Safran M; Osenberg S; Sherf O; Rechavi G; Itskovitz-Eldor J PLoS One 2012, 7 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hwang T; Park C-K; Leung AK; Gao Y; Hyde TM; Kleinman JE; Rajpurohit A; Tao R; Shin JH; Weinberger DR Nat. Neurosci 2016, 19 (8), 1093. [DOI] [PubMed] [Google Scholar]
- (7).Tan MH; Li Q; Shanmugam R; Piskol R; Kohler J; Young AN; Liu KI; Zhang R; Ramaswami G; Ariyoshi K Nature 2017, 550 (7675), 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Roth SH; Danan-Gotthold M; Ben-Izhak M; Rechavi G; Cohen CJ; Louzoun Y; Levanon EY Cell Rep. 2018, 23 (1), 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vlachogiannis NI; Gatsiou A; Silvestris DA; Stamatelopoulos K; Tektonidou MG; Gallo A; Sfikakis PP; Stellos KJ Autoimmun. 2020, 106, 102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tran SS; Jun H-I; Bahn JH; Azghadi A; Ramaswami G; Van Nostrand EL; Nguyen TB; Hsiao Y-HE; Lee C; Pratt GA Nature Neurosci. 2019, 22 (1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Maas S; Kawahara Y; Tamburro KM; Nishikura K RNA Biol. 2006, 3 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Han L; Diao L; Yu S; Xu X; Li J; Zhang R; Yang Y; Werner HMJ; Eterovic AK; Yuan Y; Li J; Nair N; Minelli R; Tsang YH; Cheung LWT; Jeong KJ; Roszik J; Ju Z; Woodman SE; Lu Y; Scott KL; Li JB; Mills GB; Liang H Cancer Cell 2015, 28 (4), 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Picardi E; D’Erchia AM; Lo Giudice C; Pesole G Nucleic Acids Res. 2016, 45 (D1), D750–D757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ramaswami G; Li JB Nucleic Acids Res. 2013, 42 (D1), D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chalk AM; Taylor S; Heraud-Farlow JE; Walkley CR Genome Biol. 2019, 20 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Song Y; Yang W; Fu Q; Wu L; Zhao X; Zhang Y; Zhang R Nat. Struct.Mol. Biol 2020, 27, 351–362. [DOI] [PubMed] [Google Scholar]
- (17).Wong SK; Sato S; Lazinski DW RNA 2001, 7 (6), 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Deffit SN; Hundley HA Wiley Interdiscip. Rev. RNA 2016, 7 (1), 113–127. [DOI] [PubMed] [Google Scholar]
- (19).Montiel-Gonzalez MF; Quiroz JFD; Rosenthal JJ Methods 2019, 156, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Oakes E; Vadlamani P; Hundley HA, Methods for the Detection of Adenosine-to-Inosine Editing Events in Cellular RNA. Methods Mol. Biol 2017, 1648, 103–127. [DOI] [PubMed] [Google Scholar]
- (21).Pinto Y; Levanon EY Methods 2019, 156, 25–31. [DOI] [PubMed] [Google Scholar]
- (22).Ouyang Z; Liu F; Zhao C; Ren C; An G; Mei C; Bo X; Shu W Sci. Rep 2018, 8 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang Y; Park S; Beal PA Biochemistry 2018, 57 (10), 1640–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Herbert A; Rich A Proc. Natl. Acad. Sci 2001, 98 (21), 12132–12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Phelps KJ; Tran K; Eifler T; Erickson AI; Fisher AJ; Beal PA Nucleic Acids Res. 2015, 43 (2), 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Stephens OM; Yi-Brunozzi HY; Beal PA Biochemistry 2000, 39 (40), 12243–12251. [DOI] [PubMed] [Google Scholar]
- (27).Sanger F; Nicklen S; Coulson AR Proc. Natl. Acad. Sci 1977, 74 (12), 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Burns CM; Chu H; Rueter SM; Hutchinson LK; Canton H; Sanders-Bush E; Emeson RB Nature 1997, 387 (6630), 303–308. [DOI] [PubMed] [Google Scholar]
- (29).Mizrahi RA; Shin D; Sinkeldam RW; Phelps KJ; Fin A; Tantillo DJ; Tor Y; Beal PA Angew. Chem 2015, 54 (30), 8713–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Phelps KJ; Ibarra-Soza JM; Tran K; Fisher AJ; Beal PA ACS Chem. Biol 2014, 9 (8), 1780–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yoshida M; Ukita T Biochim. Biophys. Acta- Nuc. Acids and Prot. Syn 1968, 157 (3), 455–465. [PubMed] [Google Scholar]
- (32).Sakurai M; Yano T; Kawabata H; Ueda H; Suzuki T Nat. Chem. Biol 2010, 6 (10), 733–40. [DOI] [PubMed] [Google Scholar]
- (33).Li Y; Göhl M; Ke K; Vanderwal CD; Spitale RC Org. Lett 2019, 21(19), 7948–7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Knutson SD; Ayele TM; Heemstra JM Bioconjug. Chem 2018, 29 (9), 2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fox JJ; Wempen I; Hampton A; Doerr IL J. Am. Chem. Soc 1958, 80 (7), 1669–1675. [Google Scholar]
- (36).Boncel S; Gondela A; Walczak K Synthesis 2010, 2010 (10), 1573–1589. [Google Scholar]
- (37).Manso JA; Camacho IFC; Calle E; Casado J Org. Biomol. Chem 2011, 9 (18), 6226–6233. [DOI] [PubMed] [Google Scholar]
- (38).Carlile TM; Rojas-Duran MF; Zinshteyn B; Shin H; Bartoli KM; Gilbert WV Nature 2014, 515 (7525), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Li X; Zhu P; Ma S; Song J; Bai J; Sun F; Yi C Nat. Chem. Biol 2015, 11 (8), 592. [DOI] [PubMed] [Google Scholar]
- (40).Kuttan A; Bass BL Proc. Natl. Acad. Sci 2012, 109 (48), E3295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Uttamapinant C; Tangpeerachaikul A; Grecian S; Clarke S; Singh U; Slade P; Gee KR; Ting AY Angew. Chem 2012, 51 (24), 5852–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.