

CONSPECTUS
Since its discovery, vancomycin has been used in clinic for >60 years. Because of their durability, vancomycin and related glycopeptides serve as the antibiotics of last resort for the treatment of protracted bacterial infections of resistant Gram-positive pathogens, including methicillin-resistant S. aureus (MRSA) and multidrug-resistant (MDR) S. pneumoniae. After 30 years of use, vancomycin resistance was first observed and is now widespread in enterococci, and more recently in S. aureus. The widespread prevalence of vancomycin-resistant enterococci (VRE) and the emergence of vancomycin-resistant S. aureus (VRSA) represent a call to focus on the challenge of resistance, highlight the need for new therapeutics, and provide the inspiration for the design of more durable antibiotics less prone to bacterial resistance than even vancomycin.
Herein, we summarize progress on efforts to overcome vancomycin resistance, first addressing recovery of its original durable mechanism of action and then introducing additional independent mechanisms of action intended to increase the potency and durability beyond that of vancomycin itself. The knowledge of the origin of vancomycin resistance and an understanding of the molecular basis of the binding affinity loss between vancomycin and the altered target ligand D-Ala-D-Lac provided the basis for the subtle and rational redesign of the vancomycin binding pocket to remove the destabilizing lone pair repulsion or reintroduce a lost H-bond, while not impeding binding to the unaltered ligand D-Ala-D-Ala. Preparation of the modified glycopeptide core structure was conducted by total synthesis, providing binding pocket-modified vancomycin aglycons with dual D-Ala-D-Ala/D-Lac binding properties that directly address the intrinsic mechanism of resistance to vancomycin. Fully glycosylated pocket-modified vancomycin analogues were generated through a subsequent two-step enzymatic glycosylation, providing a starting point for peripheral modifications used to introduce additional mechanisms of action. A well-established vancosamine N-(4-chlorobiphenyl)methyl (CBP) modification as well as newly discovered C-terminus trimethylammonium cation (C1) or guanidine modifications were introduced, providing two additional synergistic mechanisms of action independent of D-Ala-D-Ala/D-Lac binding. The CBP modification provides an additional stage for inhibition of cell wall synthesis that results from direct competitive inhibition of transglycosylase, whereas the C1/guanidine modification induces bacteria cell permeablization. The synergistic behavior of the three independent mechanisms of action combined in a single molecule provides ultrapotent antibiotics (MIC 0.01–0.005 μg/mL against VanA VRE). Beyond the remarkable antimicrobial activity, the multiple mechanisms of action suppress the rate at which resistance may be selected, where any single mechanism of action is protected by the action of others. The results detailed herein that rationally targeted durable vancomycin-derived antibiotics have generated compounds with a “resistance against resistance”, provided new candidate antibiotics, and may serve as a generalizable strategy to combat antibacterial resistance.
Graphical Abstract
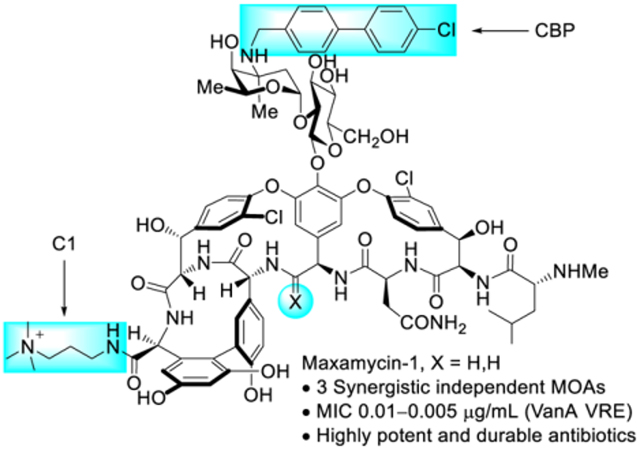
1. INTRODUCTION
The extensive use of antibiotics in clinical treatments of microbial infections has resulted in widespread antibiotic resistance that is now an urgent threat worldwide. A 2019 CDC report on antimicrobial resistance found that at least 2.8 million people were infected by antibiotic-resistant pathogens, causing at least 35,000 deaths/year in the US alone.5 Moreover, the ongoing accumulation of multiple resistance mechanisms in a single pathogen results in multidrug resistance,6 introducing further challenges in the treatment of such infections. Those caused by both methicillin-resistant S. aureus (MRSA) and multidrug-resistant S. pneumoniae are regarded as serious threats, constituting half of these total infection numbers.5
Since the disclosure (1956) and clinical introduction (1958) of vancomycin (1, Figure 1) as the first member of glycopeptide antibiotics,7,8 the clinical importance of this family of natural products has only increased in the treatment of infections caused by Gram-positive pathogens, including MRSA and S. pneumoniae.9–13 The target of vancomycin and related glycopeptide antibiotics is the D-Ala-D-Ala moiety at the C-terminus of peptidoglycan biosynthetic precursors, including Lipid II, where their binding and sequestration of the substrate for transpeptidase inhibits peptidoglycan cross-linking and cell wall maturation.14–16 This robust mechanism of action has allowed vancomycin to largely avoid resistance development. The target is neither a protein or nucleic acid that may be altered by single genetic mutations, but rather is the substrate of an essential and highly conserved enzyme-catalyzed reaction in the maturation of the cell wall. The class acts at the outside periphery of the cell wall, avoiding other common mechanisms of resistance that include cytosolic metabolic deactivation, efflux, or blocked cellular entry. Consequently, vancomycin resistance emerged only 30 years after clinical introduction, much slower than other antibiotic classes.17–19 The resistance was initially observed and is now widespread in enterococci (VRE),20 and recently emerged in S. aureus (VRSA).21 This resistance was not evolved by pathogenic bacteria, but arose by acquisition of the protection genes (vanRSHAX) found in nonpathogenic vancomycin-producing organisms.15,22–25 The basis of this resistance entails an intricate inducible late-stage remodeling of the peptidoglycan precursor C-terminus from D-Ala-D-Ala to D-Ala-D-Lac, resulting in a loss in binding affinity between vancomycin and its target (1000-fold) and producing a corresponding loss in antimicrobial activity (1000-fold resistance).26,27
Figure 1.
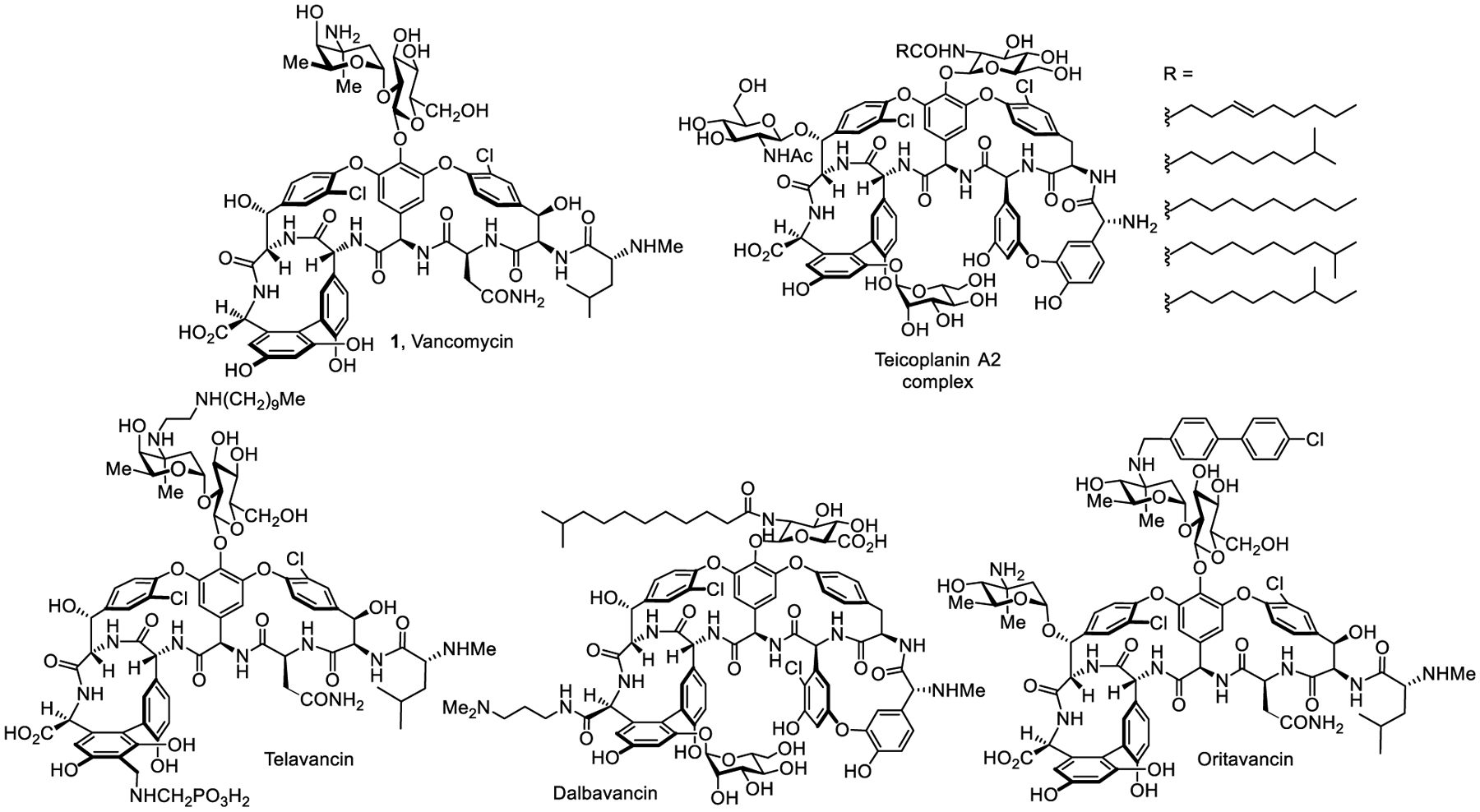
Vancomycin, teicoplanin, and semi-synthetic glycopeptide antibiotics.
As the antibiotic of last resort for protracted Gram-positive bacterial infections, the widespread prevalence of VRE and the emergence of VRSA represent urgent threats and a need for new therapeutics. Extensive studies with semi-synthetic derivatives of the natural products have been conducted and some exhibit enhanced antimicrobial potency,28–30 introduce additional mechanisms of action,31–34 or improve pharmacokinetic (PK) properties.35 As a result, three second-generation glycopeptide antibiotics, telavancin,36 dalbavancin37 and oritavancin,38 have been approved for clinical use although their paths to approval were anything but straightforward (Figure 1).
We focused on the rational redesign of the vancomycin binding pocket for dual D-Ala-D-Ala/D-Lac binding to overcome the molecular basis of resistance, building on our total syntheses of the glycopeptide antibiotic aglycons that now include vancomycin,39–41 teicoplanin,42,43 ristocetin,44 and chloropeptins45,46. Expectations are that this alone might extend its clinical lifetime for another half century. In addition, we wished to explore whether peripheral modifications of such pocket-modified vancomycins could introduce additional mechanisms of action independent of D-Ala-D-Ala/D-Lac binding, providing potent and even more durable antibiotics. The question was whether we could rationally design antibiotics that are even less prone to resistance than vancomycin itself. It is possible such compounds that act by up to three synergistic mechanisms of action as detailed herein, only one of which is dependent on D-Ala-D-Ala/D-Lac binding, might provide extraordinarily potent antibiotics and display clinical lifetimes measured not in decades or even the half a century of vancomycin, but perhaps could remain effective for centuries. In this account, we present our progress on this redesign of vancomycin to generate such “supernatural” products.47,48
2. Vancomycin binding pocket redesign: overcoming resistance
2.1. Delineating details of the molecular basis of resistance
Based on the attribution of resistance to the decreased binding of vancomycin with its altered target (D-Ala-D-Lac vs D-Ala-D-Ala),26,27 we first addressed the origin of the affinity loss.49 Created by the rigid macrocyclic backbone, the vancomycin binding pocket interacts with D-Ala-D-Ala through a series of hydrophobic interactions and an array of five H-bonds. The single-atom alteration in the target ligand (NH→O) not only results in the loss of one H-bond, but also introduces a destabilizing lone-pair repulsion. The contribution of each feature to the 1000-fold binding affinity loss was established by binding constant measurements of vancomycin with model ligands 2–4, where a methylene group was incorporated in 3 in place of the amide NH in 2 (D-Ala-D-Ala) and ester O in 4 (D-Ala-D-Lac).49 The loss in affinity was thereby partitioned into a major impact (100-fold, 2.6 kcal/mol) derived from introduction of the destabilizing lone-pair repulsion of the ester oxygen and a smaller effect (10-fold, 1.5 kcal/mol) derived from loss of an amide H-bond (Figure 2), providing the basis for our vancomycin binding pocket redesign. Thus, removal of the lone-pair repulsive interaction would provide a vancomycin analogue that recovers most of the binding affinity loss with D-Ala-D-Lac, while reinstallation of a reversed H-bond would provide complete recovery. Complicating the design and because even resistant organisms enlist D-Ala-D-Ala peptidoglycan precursors until challenged by a glycopeptide antibiotic, this requires enhancement of D-Ala-D-Lac binding while maintaining D-Ala-D-Ala binding. This also ensures the antibiotic remains active against vancomycin-sensitive bacteria as well as reinstating activity against vancomycin-resistant organisms.
Figure 2.
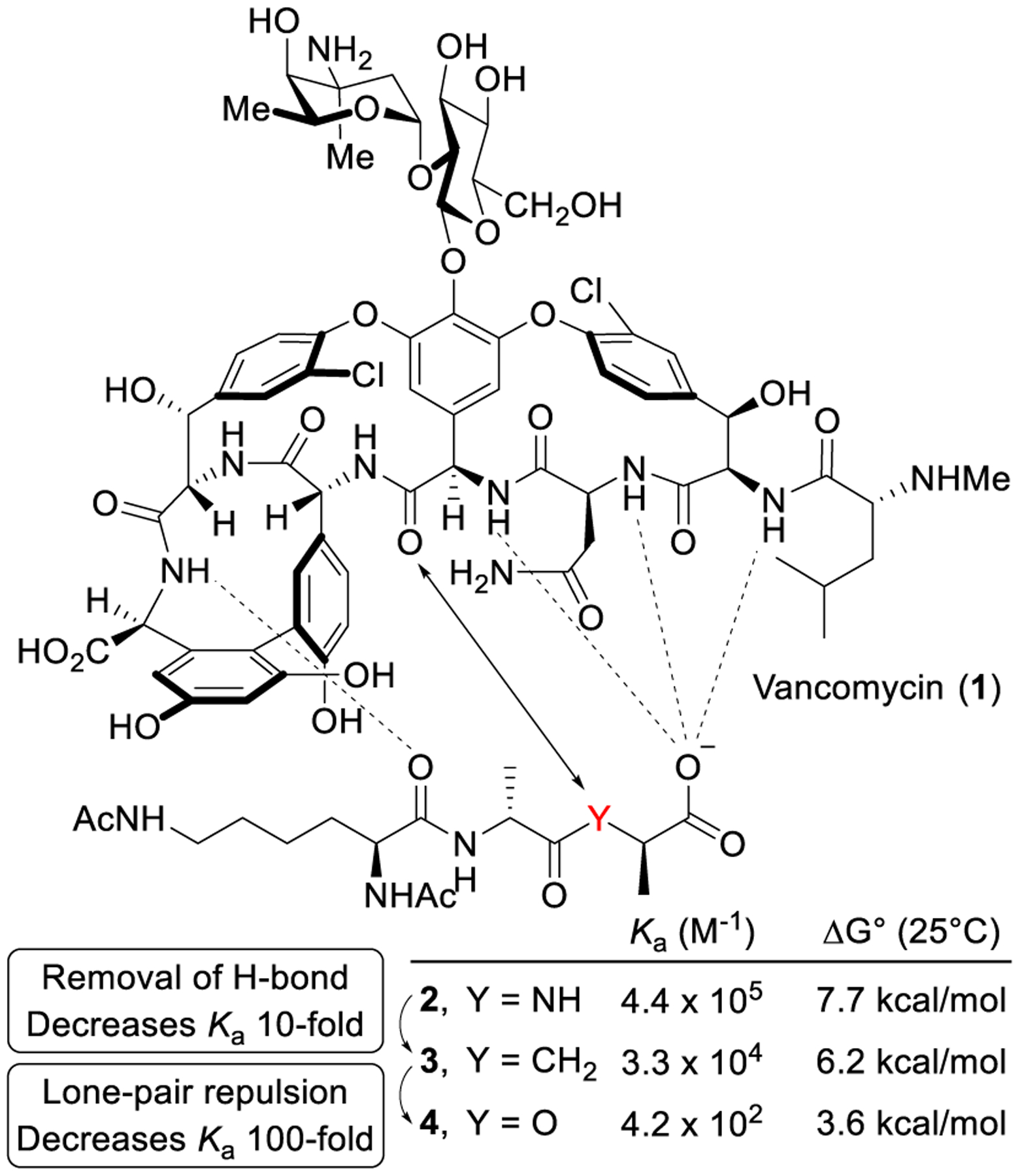
Interaction of vancomycin with ligands 2–4.
2.2. Rational design of binding pocket-modified vancomycins
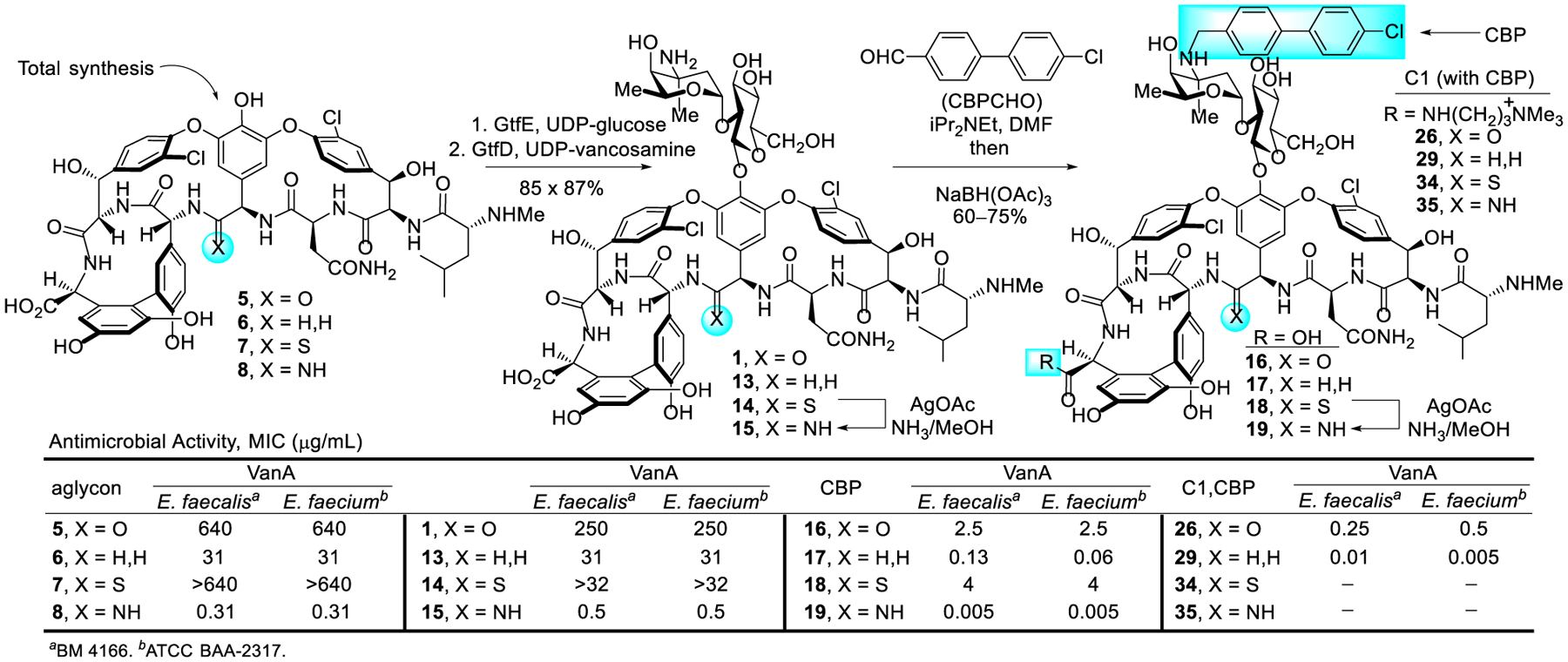
The effect of a single heavy-atom change in the peptidoglycan ligand suggested the introduction of a compensating single atom modifications at the vancomycin residue 4 amide (D ring) could improve binding with D-Ala-D-Lac by removing the destabilizing lone-pair repulsive interaction (100-fold) and/or reintroducing a H-bond (additional 10-fold). Two such modifications were targeted that first replaced the amide carbonyl with a methylene group to recover the majority of binding affinity lost with D-Ala-D-Lac, and subsequently with an amidine to fully restore the binding.1 In the absence of semi-synthetic methods to selectively target the key amide, our total syntheses of the glycopeptide antibiotics39–46 laid the foundation for preparation of the modified glycopeptide core structures (Figure 3). [Ψ[CH2NH]Tpg4]vancomycin aglycon (6) was synthesized by following our strategy for the synthesis of vancomycin,50 introducing the modified aminomethylene group through a reductive amination prior to construction of the CD ring system. The preparation of [Ψ[C(=NH)NH]Tpg4]vancomycin aglycon (8) enlisted a divergent synthesis strategy,51,52 relying on the thioamide derivative [Ψ[C(=S)NH]Tpg4]vancomycin aglycon (7). A selective thionation of the macrocyclic CD ring system with Lawesson’s reagent provided the thioamide used in the synthesis of the thioamide vancomycin aglycon 7. A newly invented AgOAc-promoted late-stage amination of the fully deprotected substrate provided 8 and a series of analogues,53 which has been reviewed elsewhere.3
Figure 3.
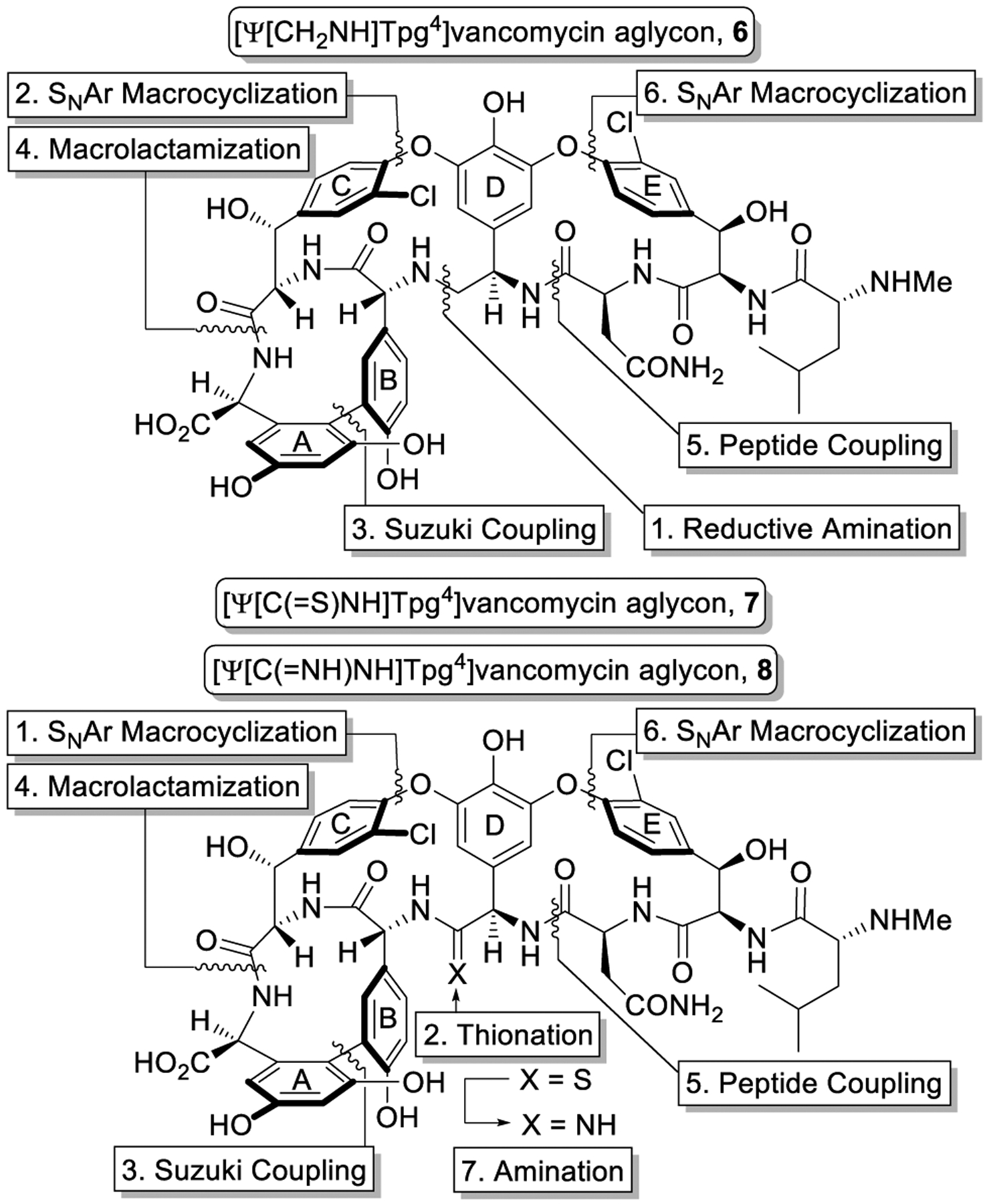
Binding pocket redesign and synthetic strategy.
Significant (with 6) or more remarkable (with 8) improvements in binding affinity for D-Ala-D-Lac were observed with the pocket-modified vancomycin aglycons, and both exhibited the dual ligand binding characteristics (Figure 4). In turn, these increases were expressed in their activity against a vancomycin-resistant E. faecalis (VanA VRE) strain. Whereas 6 shows a 40-fold increase in binding affinity with D-Ala-D-Lac and a near equivalent improvement in MIC compared with vancomycin aglycon, 8 completely restored the activity against vancomycin-resistant organisms with sub pg/mL potency, displaying a further >10-fold increase in binding affinity with D-Ala-D-Lac compared with 6 and 600-fold increase compared with vancomycin aglycon. Although introduction of a reversed H-bond may be invoked to explain this latter result, it is more accurately attributable to introduction of a now stabilizing electrostatic interaction that results from binding of 8 as the protonated amidine.49,51–53
Figure 4.
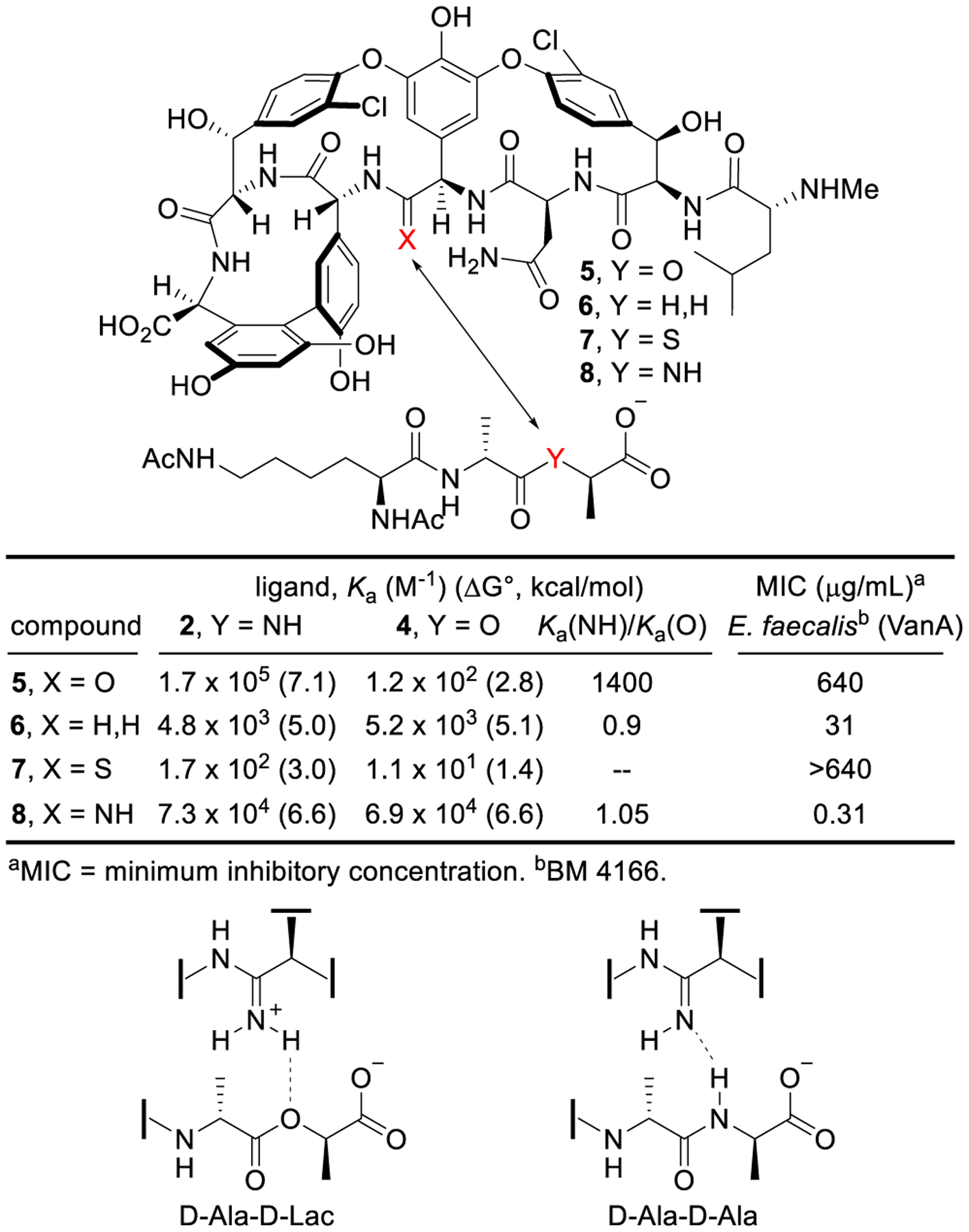
Ligand binding affinity and antimicrobial activity of pocket-modified vancomycin aglycons and dual binding behavior of amidine 8.
Importantly, 6 and 8 still bind D-Ala-D-Ala and remain active against vancomycin-sensitive organisms. As expected, the removal of one H-bond for binding D-Ala-D-Ala with 6 resulted in a 35-fold decrease in binding affinity and a corresponding lower antimicrobial activity than vancomycin for sensitive organisms, but it does exhibit the desired dual D-Ala-D-Ala/D-Lac binding. The behavior of 8 was more remarkable and not as easily predicted. Its binding with D-Ala-D-Ala exhibits only a 2-fold decrease compared with vancomycin aglycon, indicating that the amidine also effectively serves as a H-bond acceptor when binding D-Ala-D-Ala. This is achieved by binding to D-Ala-D-Ala as a free base amidine, whereas its binding to D-Ala-D-Lac utilizes the protonated amidine. Together, 6 and 8 represent a class of synthetic vancomycin analogues that exhibit dual D-Ala-D-Ala/D-Lac binding, overcoming the mechanism of resistance to vancomycin while remaining active against vancomycin-sensitive bacteria.
3. Carbohydrate introduction: the challenge and solution
Although the vancomycin disaccharide does not impact ligand binding or in vitro antimicrobial activity, it does improve the hydrophilicity, impacts in vivo efficacy, and affects PK and distribution properties. The disaccharide may also introduce an additional, albeit weak, mechanism of action through indirect inhibition of transglycosylase.6 Subsequent to the successful pocket modifications that address the mechanism of resistance, we turned to the carbohydrate installation. This not only presented an opportunity to complete an improved total synthesis of vancomycin54 but also provide access to fully glycosylated pocket-modified analogues.
An enzymatic approach was adopted for the introduction of the disaccharide, providing a two-step glycosylation of the vancomycin aglycon and analogues on a preparative scale.54 With stably overexpressed recombinant enzymes and minor modifications to the Walsh and Wong reaction conditions,55,56 the carbohydrates are sequentially added to the aglycons (5–7) with commercial or synthetic glycosyl donors (UDP-glucose/GtfE and UDP-vancosamine/GtfD) to provide the pseudoaglycons (9–11) and subsequently vancomycin and its analogues [Ψ[CH2NH]Tpg4]vancomycin (13) and [Ψ[C(=S)NH]Tpg4]vancomycin (14) in superb yields (Scheme 1). Although the direct glycosylation of [Ψ[C(=NH)NH]Tpg4]vancomycin aglycon (8) to give pseudoaglycon 12 was not successful, 15 was obtained directly in a single step from thioamide 14 with our newly developed AgOAc-mediated amination reaction53 capable of conduct on a fully deprotected and carbohydrate-bearing vancomycin analogue. The introduction of the disaccharide onto 6 and 7 represents a beautiful demonstration of the enzymatic glycosylations for functionalization of vancomycin analogues that contain deep-seated structural modifications (Scheme 1).2,57
Scheme 1.
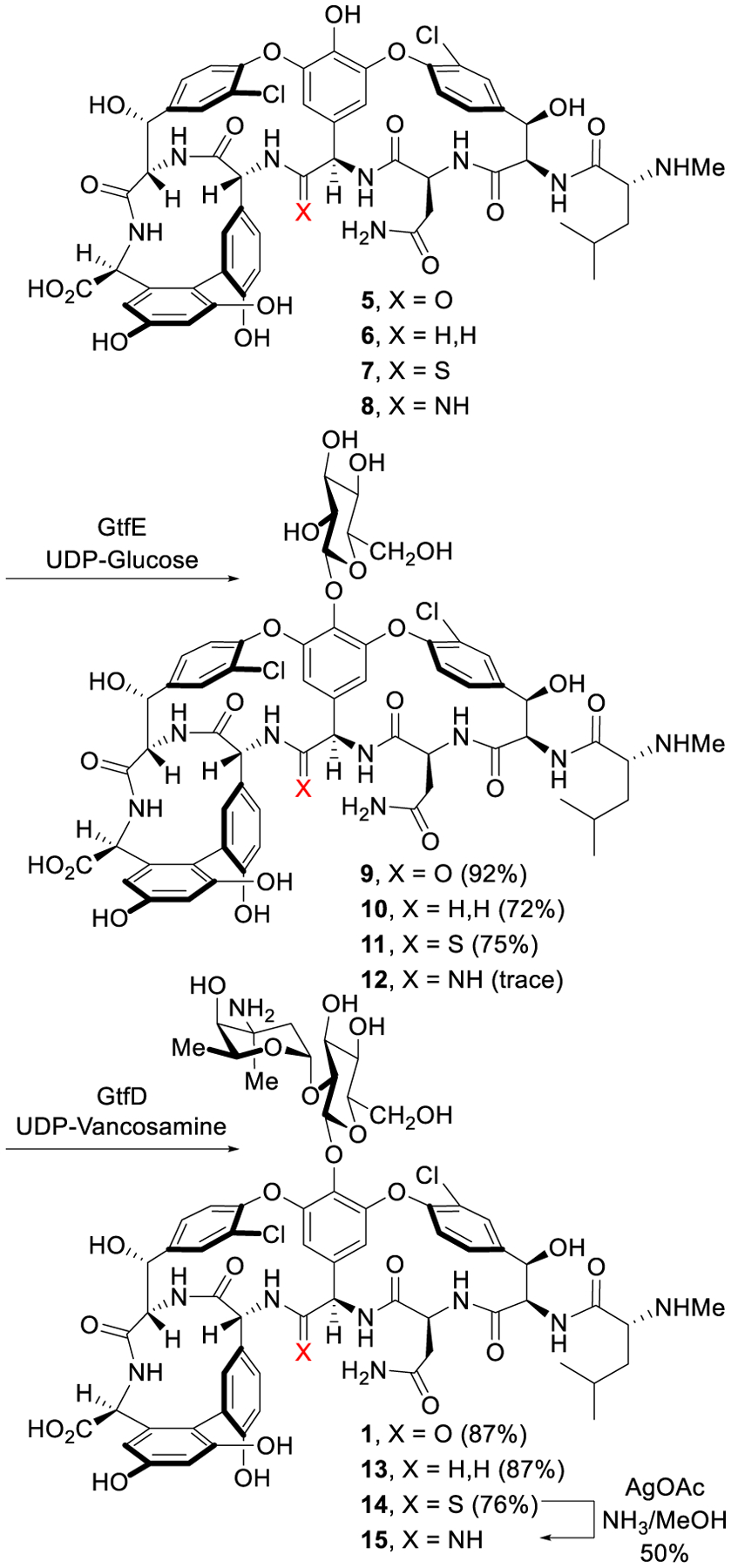
Enzymatic glycosylation of vancomycin aglycon and pocket-modified analogues.
4. Peripheral modifications: added synergistic mechanisms of action improve potency and durability
4.1. Peripheral CBP modification
Peripheral modifications of the glycopeptide natural products have generated semi-synthetic antibiotics with improved potency against vancomycin-sensitive and vancomycin-resistant organisms, introduced additional mechanisms of action independent of D-Ala-D-Ala/D-Lac binding, and improved PK/ADME properties. The vancosamine N-(4-chlorobiphenyl)methyl (CBP) modification represents an important example of such modifications. First reported by Nagarajan,58,59 this modification restored antimicrobial activity against vancomycin-resistant organisms and improved the activity against vancomycin-sensitive organisms. As demonstrated first by investigators at Lilly with cell free extracts60 and later by Kahne and Walker with pure enzyme,61,62 CBP-vancomycin and related compounds were shown to competitively inhibit transglycosylase independent of D-Ala-D-Ala binding, providing a newly added mechanism of action for cell wall synthesis inhibition.
Central to our goal of not just reinstating the activity of vancomycin but rather to exceed its properties, such peripheral modifications were examined with the pocket-modified vancomycin analogues (Figure 5). By modifying conditions disclosed for CBP-vancomycin, a CBP group was installed on unprotected 13 and 14 by reductive amination without competitive reaction of the N-terminus/residue 4 secondary amines or thioamide reduction, providing CBP-[Ψ[CH2NH]Tpg4]vancomycin (17) and CBP-[Ψ[C(=S)NH]Tpg4]vancomycin (18). Direct AgOAc-mediated amination of 18 provided CBP-[Ψ[C(=NH)NH]Tpg4]vancomycin (19).2,57
Figure 5.
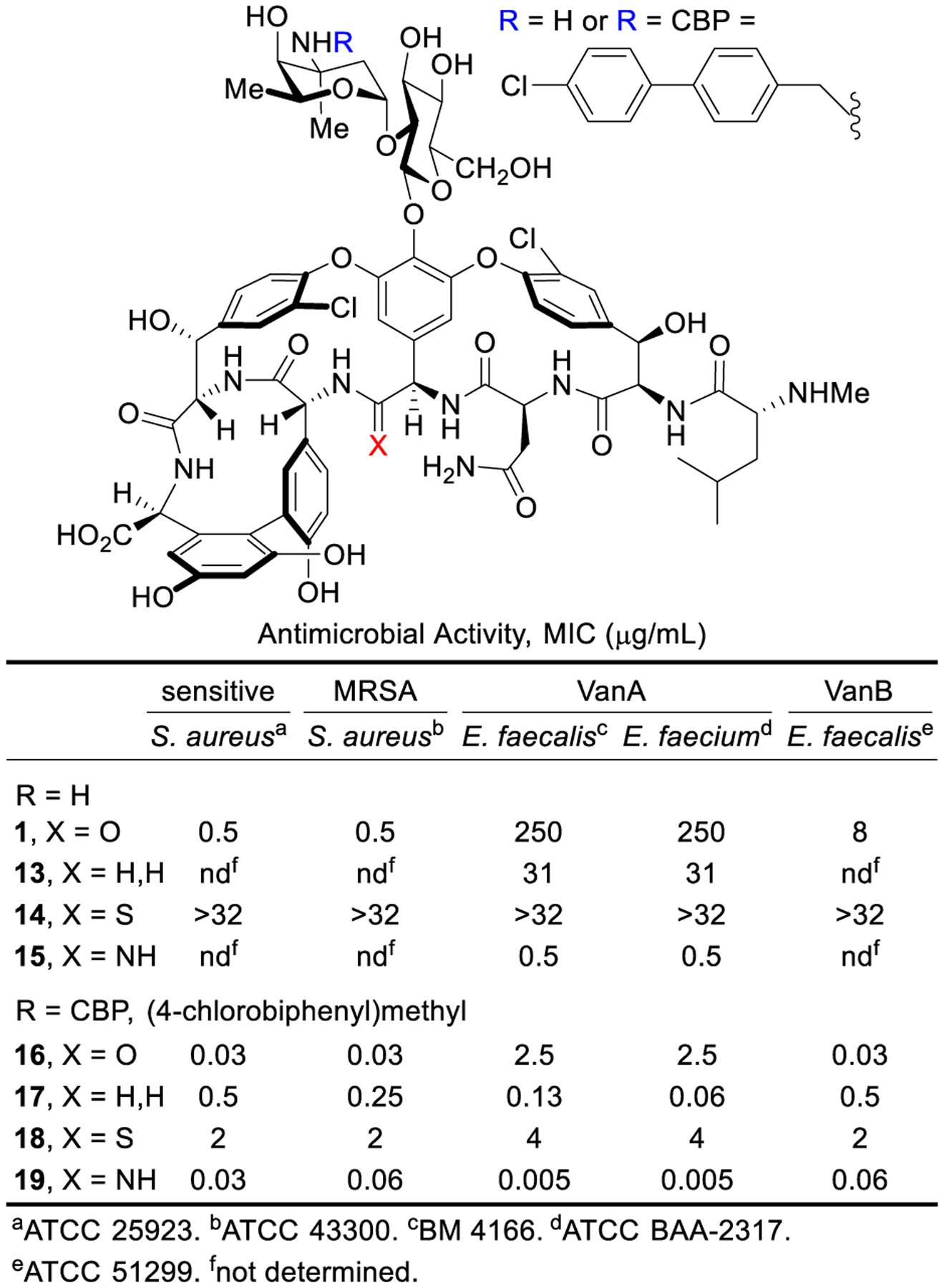
Activity of vancomycin, binding-pocket analogues 13–15, and their CBP derivatives 16–19.
The activity of the pocket-modified vancomycin analogues and their CBP derivatives against Gram-positive bacteria is remarkable (Figure 5). Whereas the pocket-modified analogues (13 and 15) expectedly display activity essentially identical to their aglycons, the attachment of the CBP group (17 and 19) improved the potency as much as 100-fold, now exhibiting superb activity against both vancomycin-sensitive and vancomycin-resistant organisms (MIC 0.13–0.005 μg/mL). The potency of 19 is stunning, being >104-fold more active than vancomycin against VanA VRE and 100-fold more potent than vancomycin against sensitive organisms. Subtle in these studies is the demonstration that CBP-[Ψ[C(=S)NH]Tpg4]vancomycin (18), which is incapable of binding D-Ala-D-Ala/D-Lac, exhibits good antimicrobial activity against both sensitive and resistant organisms derived only from direct competitive transglycosylase inhibition. As a result of the combined binding pocket and CBP modifications, 17 and 19 represent analogues that possess two independent synergistic mechanisms of action effective against both vancomycin-sensitive and vancomycin-resistant organisms, only one of which requires the dual D-Ala-D-Ala/D-Lac binding.
4.2. Introduction of a C-terminus C1 modification
A second modification examined was introduction of a protonated tertiary amine or trialkylammonium cation on the C-terminus. The C-terminus attachment of a tertiary amine in dalbavancin improved its antimicrobial potency against S. aureus.63 More recently, the introduction of C-terminus long chain quaternary ammonium salts for membrane anchoring provided compounds with enhanced activity against vancomycin-resistant organisms, possessing an additional mechanism of action involving disruption of the cell membrane.32,34
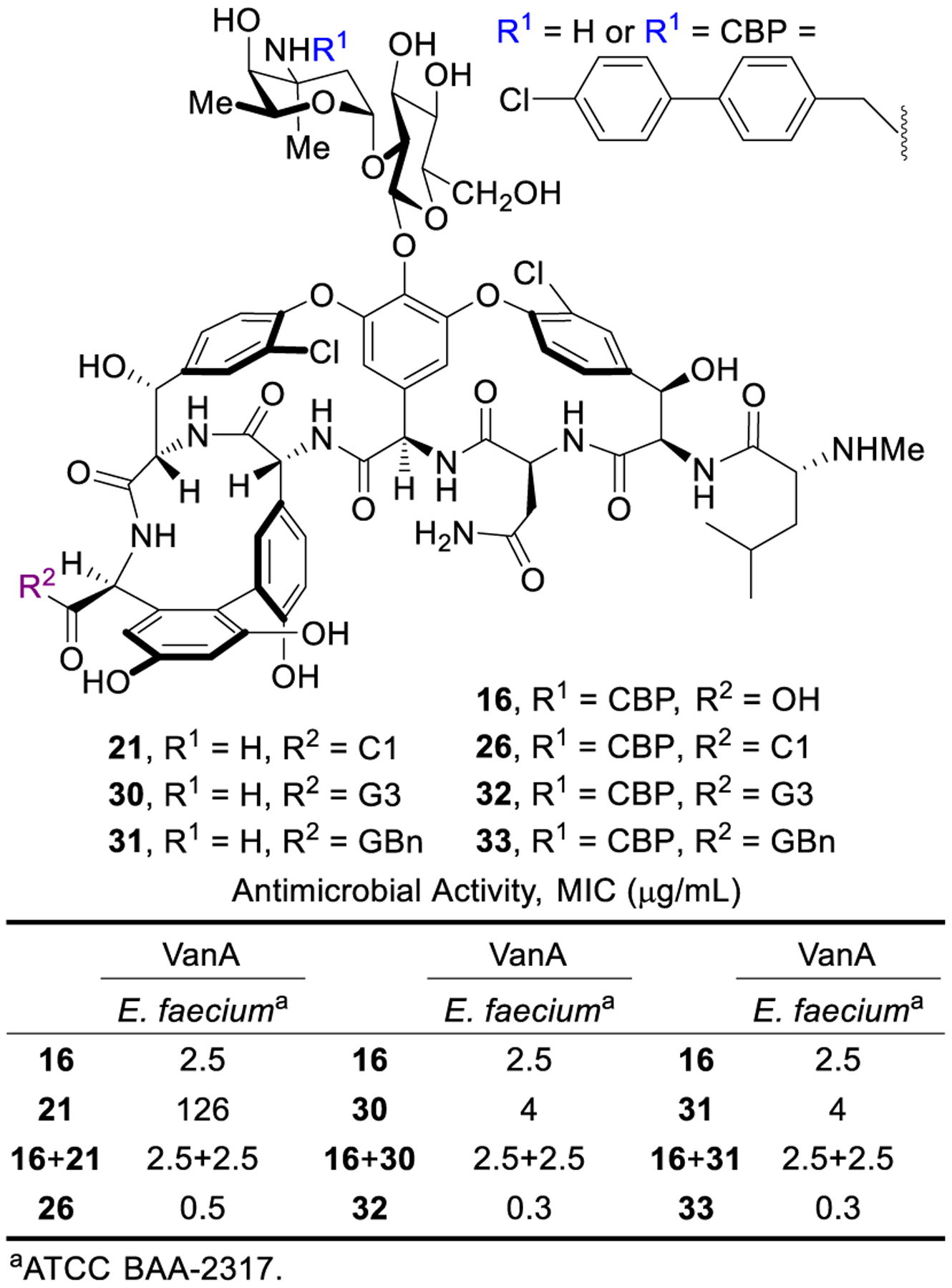
A variety of trialkylammonium salts were attached to the C-terminus carboxylic acid of vancomycin and analogues 13, 16 and 17 to establish whether such modifications are effective with compounds that incorporate binding pocket and/or CBP modifications.4 The modest dual ligand binding affinity and antimicrobial potency of pocket-modified analogue 13 permitted the most accurate assessment of the impact of sequential peripheral modifications. Although the C14-substituted ammonium salt modification on vancomycin (C14-vancomycin, 23) was the most potent derivative with a 125-fold improved activity against VanA VRE (Figure 6), no improvement was observed when this modification was attached to CBP-vancomycin (28 vs 16/23). Instead, only the simple trimethylammonium cation (C1) not previously described displayed an improvement (26, 5 to 10-fold) in activity against VanA VRE. The attachment of C1 to 17 provided a similar improvement in activity (10-fold), generating C1-CBP-[Ψ[CH2NH]Tpg4]vancomycin (29) with outstanding antimicrobial activity against vancomycin-resistant organisms (MIC 0.01–0.005 pg/mL) (Figure 6) and being nearly 105-fold more potent than vancomycin. It represents the first member of this special class of analogues that we have come to refer to as Maxamycins (Maxamycin-1).64
Figure 6.
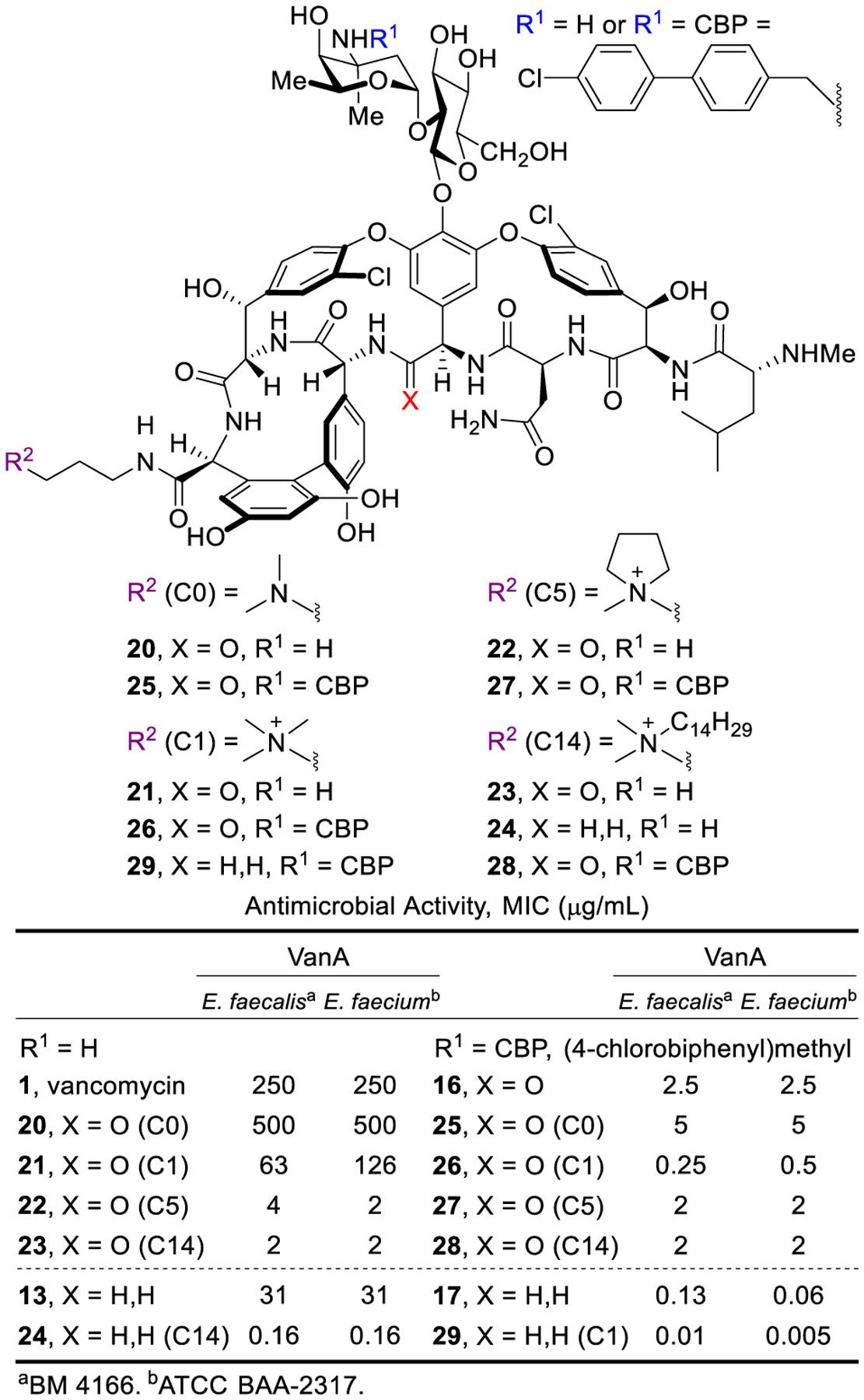
Activity of CBP and C-terminus modified vancomycin analogues against vancomycin-resistant organisms.
Studies of the C1 modification revealed that it induces bacteria cell permeabilization, but not membrane depolarization or disruption like the C14 modification. Moreover, this functional activity was the only feature that correlated with its impact on antimicrobial activity of the analogues examined. While this induction of permeabilization was modest with 21, it was more significant when C1 was attached to CBP-vancomycin (26) and the pocket-modified vancomycin analogue 29 (Figure 7). Together, the results indicate that an additional independent mechanism of action was introduced by attachment of C1 that is synergistic with that derived from D-Ala-D-Ala/D-Lac binding (transpeptidase inhibition) and direct transglycosylase inhibition (CBP).
Figure 7.
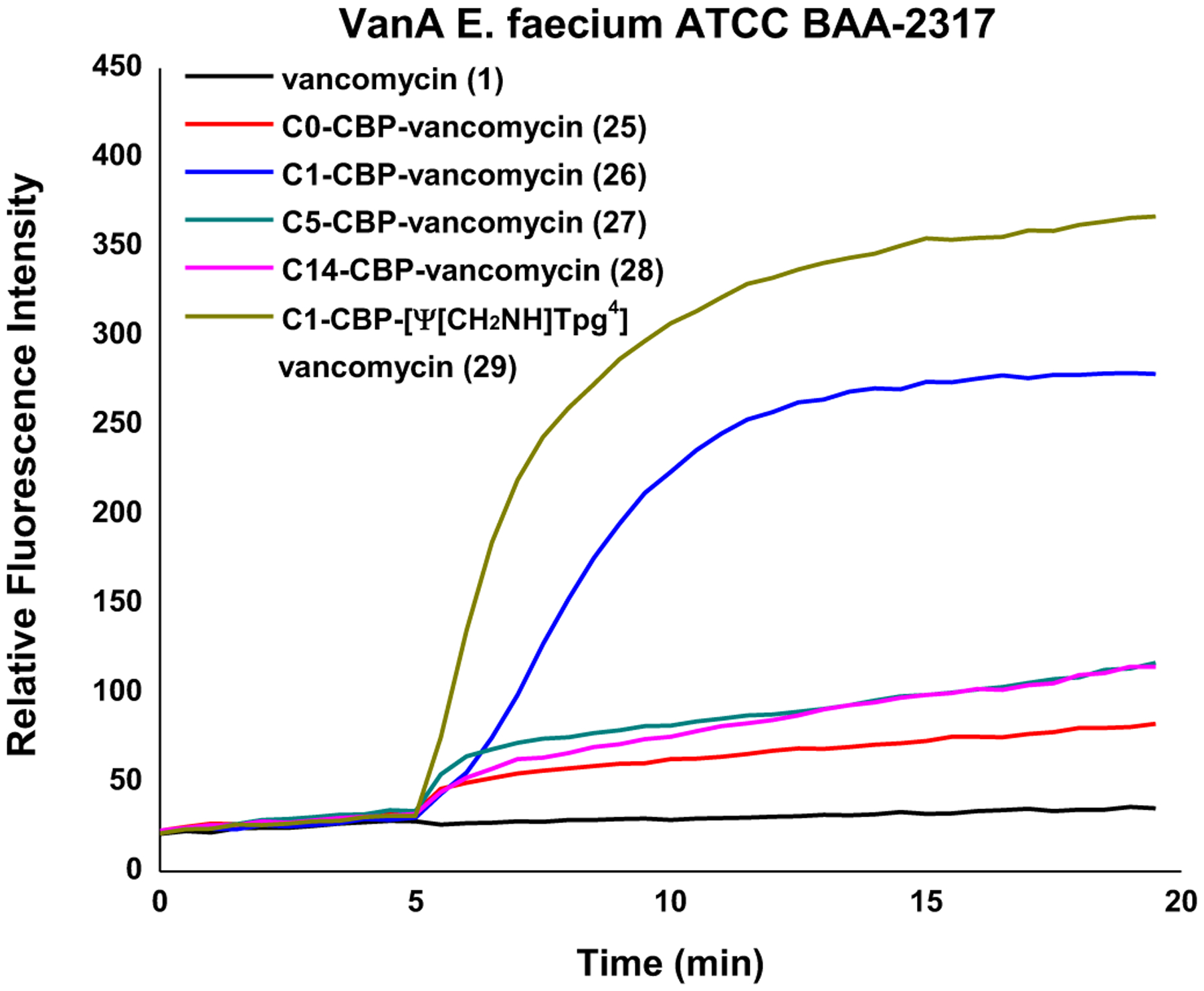
Bacterial cell permeability induced by 1 and 25–29 (10 μM added at 5 min) in VanA VRE.
In addition to the remarkable improvement in activity (>104-fold) relative to vancomycin, the analogues overcome vancomycin resistance. Among the modifications introduced in 29, the binding-pocket modification of vancomycin allows dual D-Ala-D-Ala/D-Lac binding that directly addresses the intrinsic mechanism of vancomycin resistance. A second synergistic mechanism of action was introduced with the CBP modification, leading to direct competitive inhibition of transglycosylase and cell wall synthesis independent of D-Ala-D-Ala/D-Lac binding. The C-terminus trimethylammonium cation introduced a third synergistic mechanism of action, induction of bacteria cell permeabilization independent of D-Ala-D-Ala/D-Lac binding or transglycosylase inhibition. Because all three mechanisms impact cell wall synthesis or integrity, they act synergistically with each enhancing the antimicrobial potency. Moreover, the incorporation of multiple independent mechanisms of action within a single molecule impacts the rate at which resistance would emerge, the crucial question we were focused on. This durability, or “resistance to resistance”, was assessed with an extended 50-day serial exposure of stringent vancomycin-resistant bacterial strains (VanA VRE) to the antibiotics. Alongside this study are results derived from two compounds discussed later in Section 4.3 (30 and 32) that further improve the C-terminus activity (potency, durability, and efficacy). While resistance was observed to arise against 16 and 23 that possess single mechanisms of action, much more muted changes in potency were observed for 17 and 26, outperforming the current front-line single-target therapies daptomycin, linezolid, and tigecycline. The activity of 29, expressing three independent mechanisms of action, remained unchanged after 25 days and exhibited a mere 2 to 4-fold loss in potency after the extended 50 days.This represents not resistance but a thickening of the cell wall and increased number of target sites (Figure 8). The magnitude of the MIC changes for the analogues acting by multiple mechanisms was sufficiently small to indicate that full loss of any one of the contributing mechanisms was not observed, being protected by the presence of the others. Combined, these results represent remarkable achievements derived by introduction of additional synergistic mechanisms of action, overcoming the intrinsic resistance, providing remarkable improvements in potency, and mitigating of the rate of resistance development. Both the potency and the durability of the activity to such challenges were found to follow predictable trends (three > two > one mechanism of action).
Figure 8.
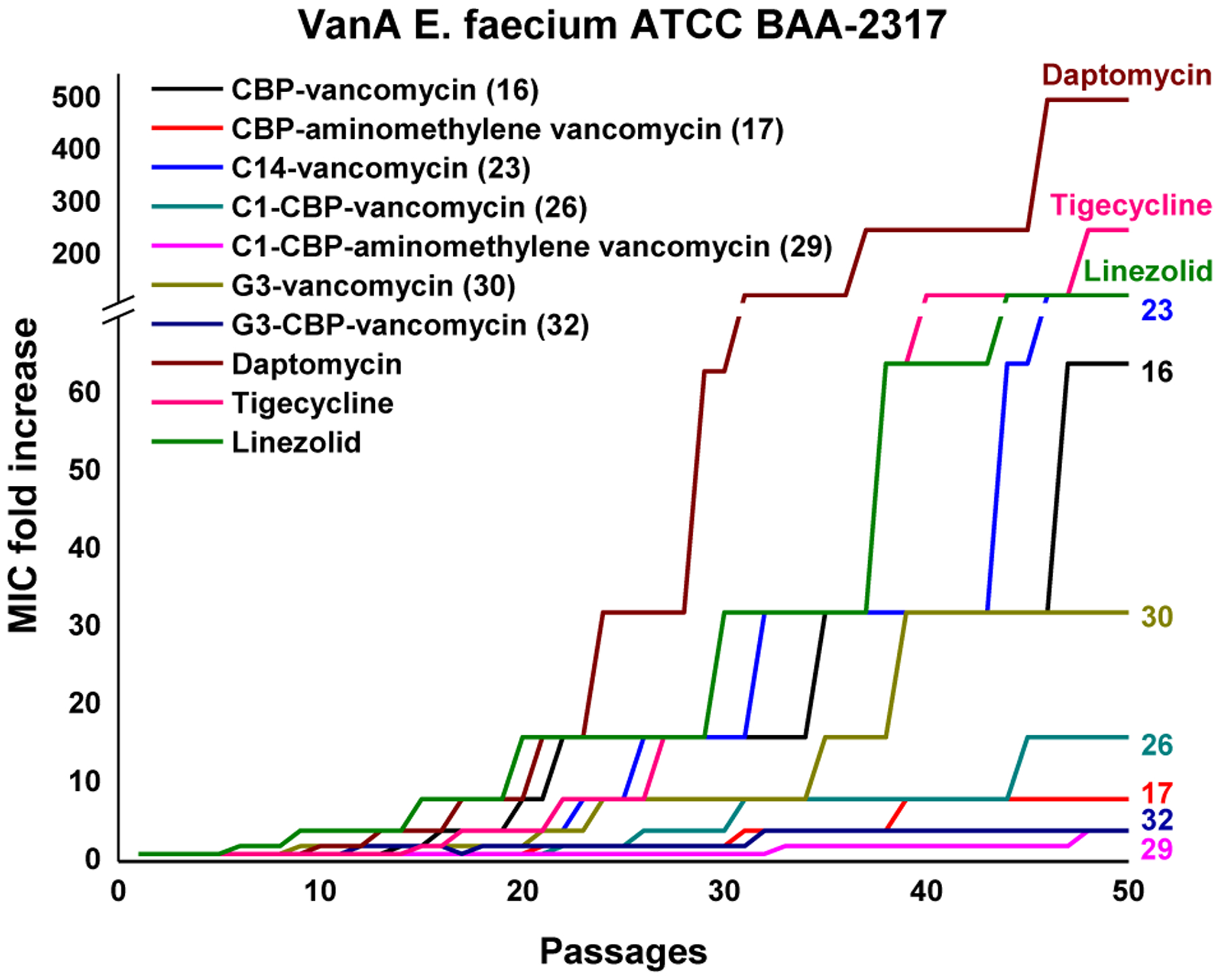
Resistance acquisition on serial passaging of VanA VRE in presence of 0.5xMIC levels of compound.
Derivatives that bear a trimethylammonium cation at the N-terminus or on the A-ring were prepared from vancomycin or CBP-vancomycin in studies that established the C1 incorporation is sensitive to the modification site.65,66 While the activity of vancomycin or CBP-vancomycin remained unchanged or decreased with a N-terminus trimethylammonium cation modification, the A-ring modification led only to a subtle improvement in activity (2 to 4-fold) less significant than the C-terminus derivatives. As expected, the ability of these analogues to induce bacteria cell permeabilization corresponded with their antimicrobial activity, following the same trend (C-terminus > A-ring > N-terminus modification). These results indicate that the effect of trimethylammonium cation is not only structure specific (C1 vs other trialkylammonium salts), but also site specific (C-terminus vs N-terminus or A-ring), implying the impact is derived from interaction with a specific binding partner.
Because of the nature of the C1 modification, studies were conducted with 26 to establish its potential impact. These revealed that the trimethylammonium cation does not introduce new liabilities in common pharmacological properties, established that it is well tolerated in mice, and that it even imparts PK improvements over vancomycin and CBP-vancomycin.67 Finally, 26 was shown to exhibit in vivo efficacy against a challenging vancomycin-resistant MRSA strain (VanA VRSA, VRS2) that is representative of the pathogens all fear will emerge in the general population. Without dosing optimization, 26 provided a nearly 3-log10 reduction in bacteria count (Figure 9).67 The results highlight that the improvement in antimicrobial potency by the added mechanism of action also translated into improved in vivo behavior, laying the foundation for studies to be conducted with peripherally modified vancomycin analogues that contain binding-pocket modifications.
Figure 9.
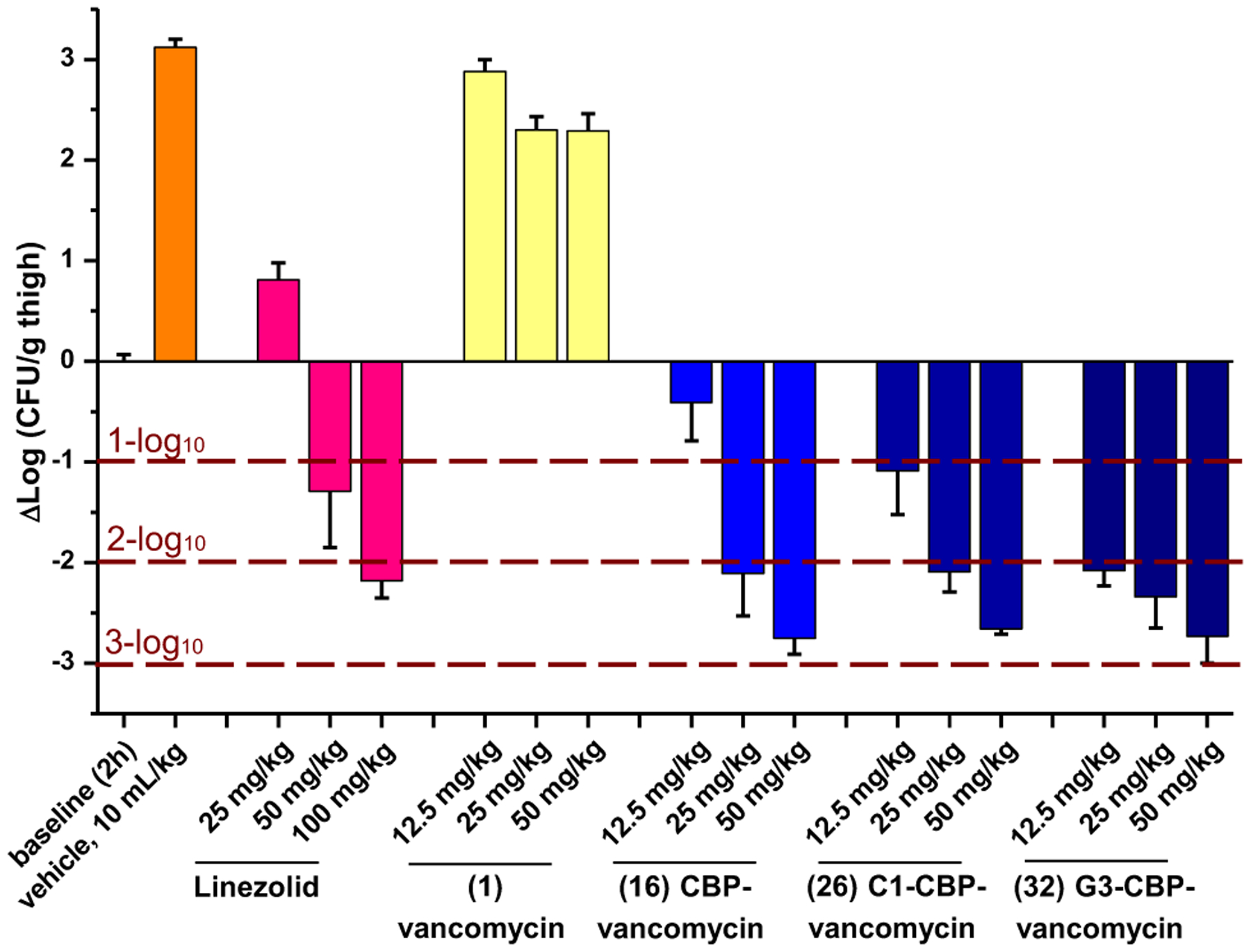
Efficacy of vancomycin, CBP-vancomycin (16), C1-CBP-vancomycin (26), G3-CBP-vancomycin (32) and linezolid against the multidrug-resistant vancomycin-resistant S. aureus (VRSA) strain VRS-2 in a mouse thigh infection model (n=5/dose). Dose-dependent bactericidal effect (relative to 2 h baseline): 32 > 26 ≥ 16 > linezolid > 1.
4.3. Improvements through introduction of C-terminus guanidine
Improvements in both antimicrobial potency and durability against resistant organisms by the C-terminus trimethylammonium cation led us examine alternative modifications. Compared to a tertiary amine (pKa = 10.6), a protonated guanidine (pKa = 13.2) could act as a more persistent positive charge under physiological conditions. In contrast to a trimethylammonium cation, a guanidine could also serve as a multiple H-bond donor with an increased affinity for anionic groups (carboxylate, phosphate) of potential binding partners in the bacteria cell envelope.
A series of C-terminus guanidine modifications on vancomycin and CBP-vancomycin was introduced in a single step amide coupling reaction with guanidine-containing amines without protection/deprotection steps.68 The guanidine modifications provided up to 64-fold improvements in antimicrobial activity over vancomycin (30 and 31) and up to a further 8-fold improvement to CBP-vancomycin (32 and 33) (Figure 10). Whereas the linker length and rigidity did not influence the improved activity within the series examined, the presence of the guanidine as well as the net positive charge were essential. G3-CBP-vancomycin (32) and GBn-CBP-vancomycin (33) emerged as representative of the most effective compounds in the series. Moreover, the derivatives (30, 32, 33) were found to express the additional mechanism of action observed with the C1 analogues with comparable induced bacterial cell permeabilization (Figure 11).
Figure 10.
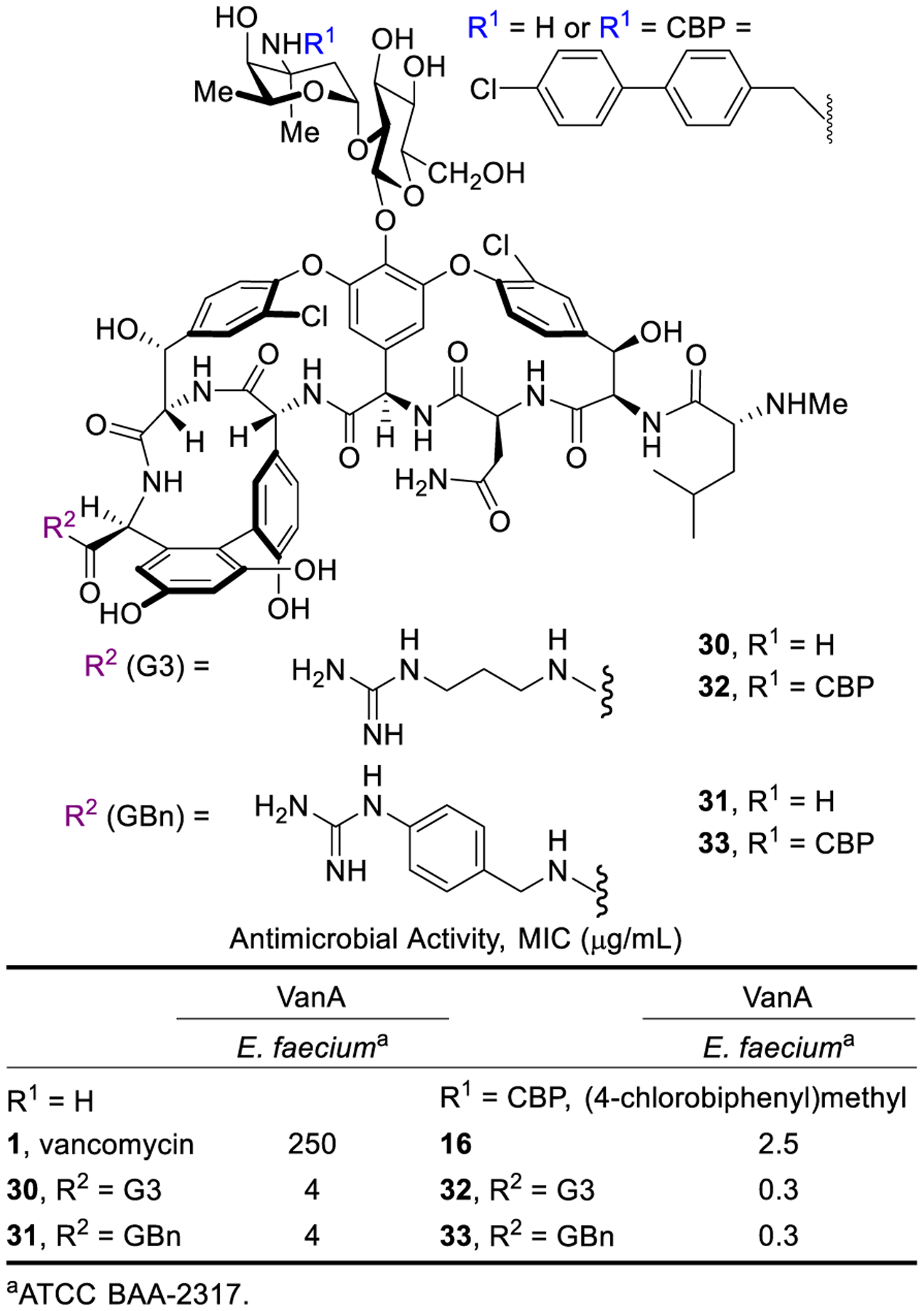
Activity of C-terminus guanidine-modified vancomycin and CBP-vancomycin against vancomycin-resistant strains.
Figure 11.
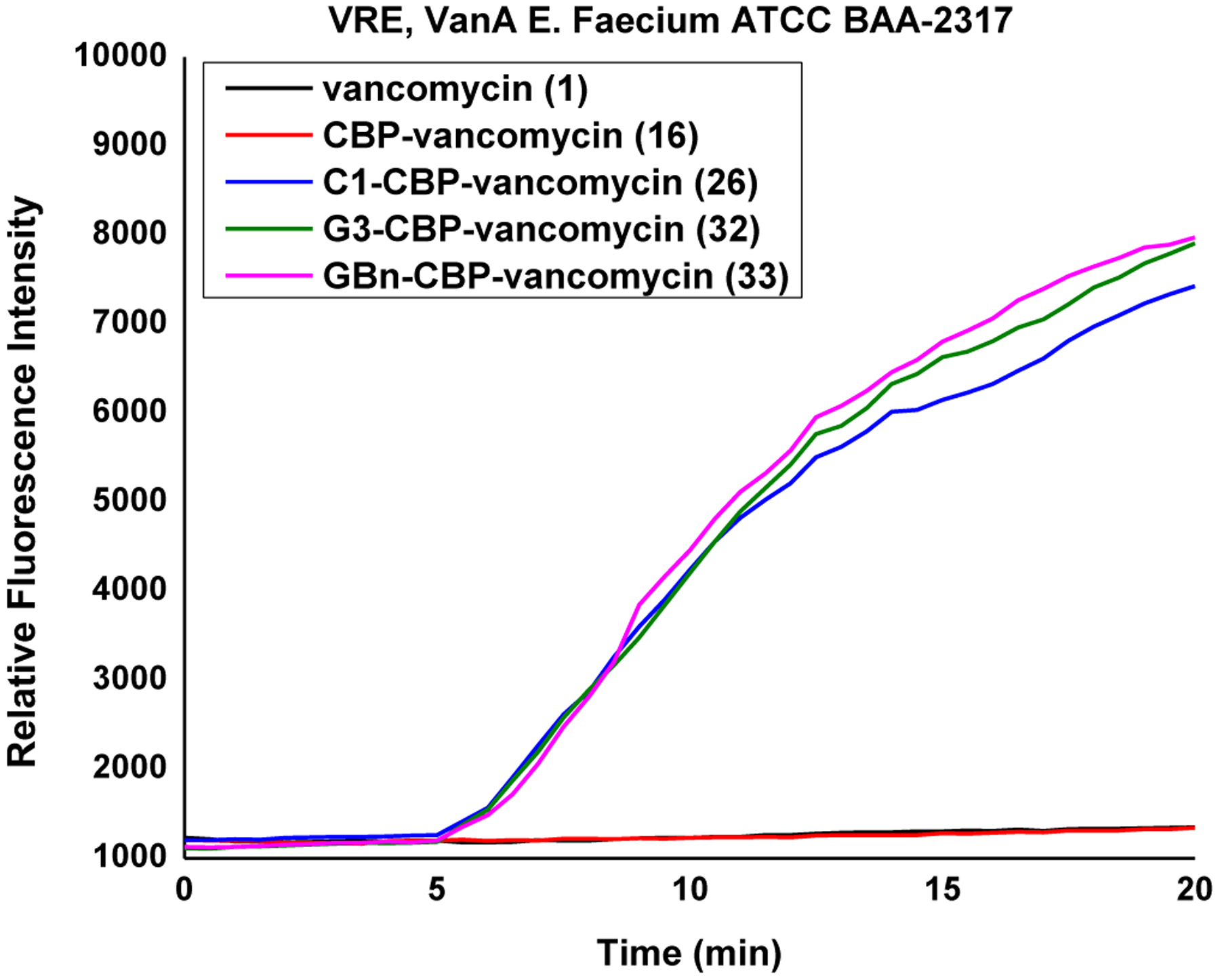
Bacterial cell permeability induced by 1, 16, 26, 32 and 33 (10 μM added at 5 min) in VanA VRE.
As with C1-CBP-vancomycin (26), derivatives bearing two peripheral modifications exhibited a substantial decrease in the rate of resistance development. This behavior was even more pronounced with G3-CBP-vancomycin (32, Figure 8).68 While the two VanA VRE strains examined rapidly developed resistance to 16 (CBP-vancomycin) and 30 (G3-vancomycin) with single modifications, only a minor decrease in activity was observed with 32 (G3-CBP-vancomycin, 4-fold) at the end of the 50 day assessment (Figure 8). This improvement in the durability not only confirmed introduction of an independent mechanism of action, but can be attributed to the more robust activity of the G3 versus C1 modification. Notably, this is observed in VanA VRE strains resistant to vancomycin and with derivatives that lack a pocket modification, expressing two mechanisms of action that act independent of D-Ala-D-Ala/D-Lac binding.
Finally, the activity of a 1:1 mixture of CBP-vancomycin and C-terminus modified vancomycins that contain C1, G3 or GBn (21, 30 and 31) was determined to establish whether the improved activity requires the two modifications to be located on the same glycopeptide molecule (Figure 12). Unlike the synergistic activity observed with 26, 32 and 33, the equimolar mixtures of the singly-modified vancomycins displayed simple additive effects and activity only at the level of the most potent compound in the mixture (CBP-vancomycin, 16).68 The improved activity only observed when both peripheral modifications are found in a single molecule highlights the synergistic nature of the two peripheral modifications.
Figure 12.
Activity of equimolar mixtures of CBP-vancomycin and C-terminus vancomycin analogues (21, 30, 31) against VanA VRE presented alongside that of 26, 32, 33.
The G3 modification in 32 did not introduce liabilities in common pharmacological properties and exhibited even better in vivo efficacy against a feared MDR VRSA strain than 26 (Figure 9).68 Together, the studies show that peripheral modifications to vancomycin and its binding-pocket analogues provide enhanced in vitro and in vivo antimicrobial potency and efficacy, additional synergistic mechanisms of action independent of D-Ala-D-Ala/D-Lac binding, and improved durability against the emergence of antimicrobial resistance. The analogues generated (26, 29, 32) represent a class of rationally designed glycopeptide antibiotics with multiple synergistic mechanisms of action against vancomycin-sensitive and vancomycin-resistant organisms. For vancomycin-sensitive organisms, all three compounds effectively express their activity through three independent mechanisms of action. For vancomycin-resistant organisms, 26 and 32 act by two mechanisms of action derived from the peripheral modifications, whereas 29 acts by three independent mechanisms of action, only one of which relies on D-Ala-D-Ala/D-Lac binding.
4.4. Insights into mechanism of action introduced by C-terminus modifications
The structure and site specificity of the trimethylammonium cation and guanidinium modifications as well as the synergistic activity they dispaly imply interaction with specific cell envelope components. Our efforts have suggested teichoic acids may be a binding partner for the modifications. Teichoic acids are polyanionic alditol phosphate polymers in the cell envelope contributing to its stability/rigidity, cation homeostasis/transport, and sequestration/regulation of autolysins.69–73 Added exogeneous lipoteichoic acid (LTA) reduced the antimicrobial activity of such compounds (Figure 13) to the level expressed by CBP-vancomycin, rescued their bacteria cell growth inhibition, and blocked their induced bacteria cell permeabilization.67,68 These studies suggest a direct interaction of the C-terminus persistent positive charge present in 26, 32 and 33 with teichoic acid is likely contributing to their bacterial cell permeabilization properties and associated antimicrobial activity.
Figure 13.
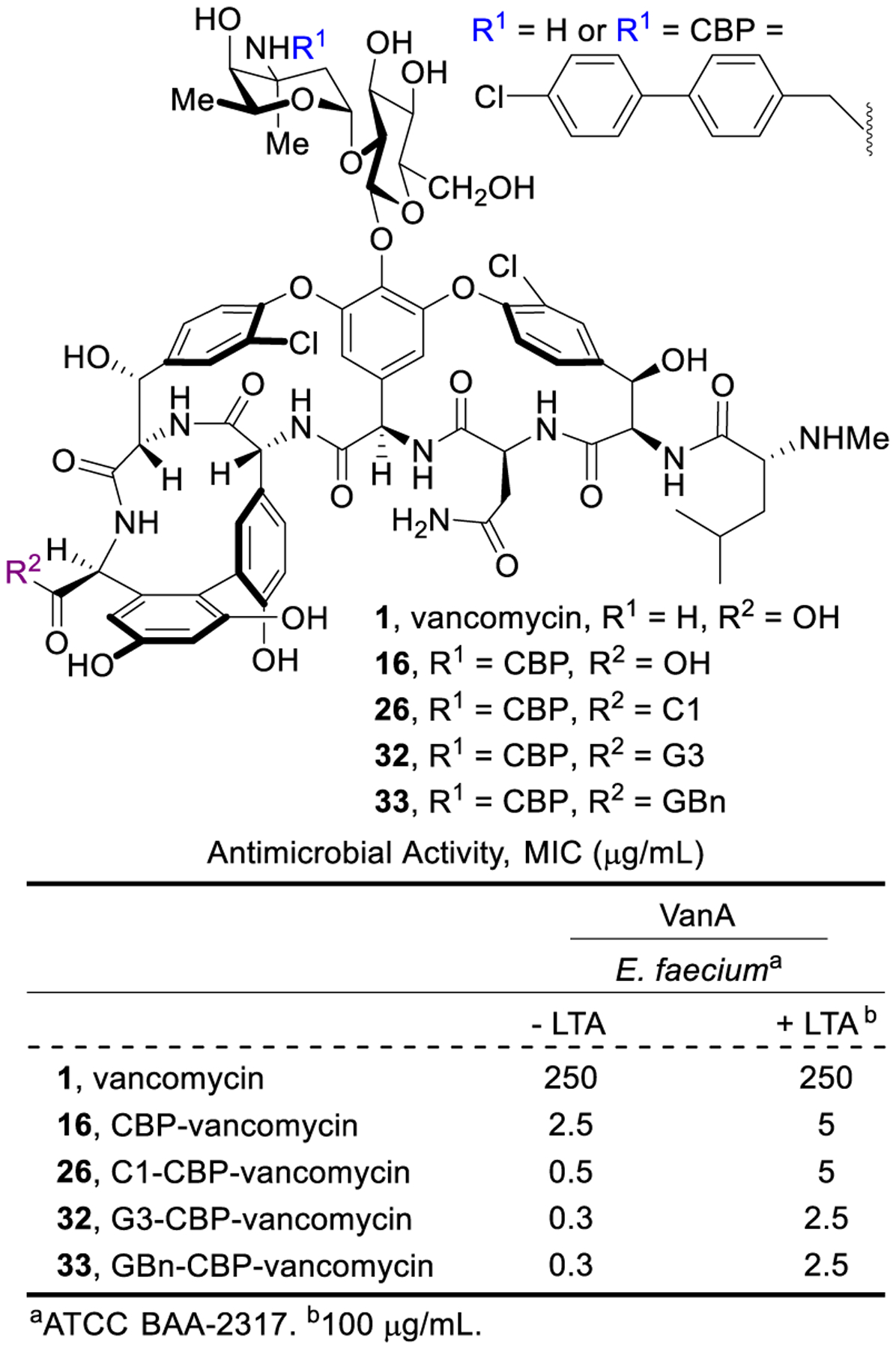
Impact of added lipoteichoic acid (LTA) on the activity of vancomycin and 16, 26, 32 and 33.
4.5. A bonus: improved PK properties with combined peripheral modifications
The tolerability and pharmacokinetic properties were established for C1-CBP-vancomycin (26) and G3-CBP-vancomycin (32).67,68 Maximum tolerated doses (MTD, iv) in mice were found to be 300 mg/kg for vancomycin (1), 75 mg/kg for CBP-vancomycin (16), and ≥50 mg/kg for 26 and 32. Although a small decrease in tolerability was observed with the introduction of the CBP peripheral modification, the C-terminus modifications did not significantly further impact this behavior. The larger progressive improvements in the antimicrobial activity of CBP-vancomycin (100-fold) and C1-CBP-vancomycin/G3-CBP-vancomycin (1000-fold) make the small differences in the MTDs relative to vancomycin even more impressive. Importantly, 26 and 32 improved the short terminal half-life (t1/2), low exposure (AUC) and rapid clearance (CL) of vancomycin and mitigated the poor dose proportionality and extended terminal half-life (t1/2) of 16 that makes clinical administration of the oritavancin challenging.74 The improved half-life of 26 and 32 relative to vancomycin indicate that both trimethylammonium cation and guanidine are not subject to rapid metabolism. Together, the improvements in PK properties and lack of other assessed liabilities67,68 displayed by the combined peripheral modifications represent a welcomed bonus derived from the design elements (Table 1).
Table 1.
PK properties of 1, 16, 26and 32.a
| Parameter | vancomycin (1) | CBP-vancomycin (16) | C1-CBP-vancomycin (26) | G3-CBP-vancomycin (32) | ||||
|---|---|---|---|---|---|---|---|---|
| 300 mg/kg | 10 mg/kg | 75 mg/kg | 10 mg/kg | 50 mg/kg | 10 mg/kg | 50 mg/kg | 10 mg/kg | |
| Cmax (μg/mL) | 1665 | 62.8 | 125 | 65.0 | 58.9 | 14.1 | 152 | 35.9 |
| tmax (h) | 0.083 | 0.083 | 0.25 | 0.083 | 0.25 | 0.50 | 0.28 | 0.14 |
| AUC (μg-h/mL) | 935 | 21.8 | 575 | 135 | 430 | 81.1 | 312 | 66.0 |
| Vd (L/kg) | 0.62 | 0.34 | 1.24 | 1.28 | 1.04 | 1.25 | 0.35 | 0.41 |
| CL (L/h/kg) | 0.32 | 0.46 | 0.13 | 0.074 | 0.12 | 0.12 | 0.09 | 0.09 |
| t1/2 (h) | 1.35 | 0.52 | 6.6 | 12.0 | 6.2 | 7.0 | 4.4 | 4.3 |
Compounds administered iv @ MTD and 10 mg/kg in mice (n = 3/time point, measured at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h).
5. Conclusions and Perspective
Herein, we highlight our stepwise progress over the past two decades to overcome vancomycin resistance, including a total synthesis-based approach to first design and prepare analogues that display dual D-Ala-D-Ala/D-Lac binding affinity and directly reinstate the durable mechanism of action of vancomycin. The subsequent introduction of peripheral modifications provides additional mechanisms of action independent of D-Ala-D-Ala/D-Lac binding (Figure 14). From these studies, the maxamycins emerged that combine a binding pocket and two peripheral modifications, resulting in molecules expressing three independent synergistic mechanisms of action derived from binding and sequestration of the transpeptidase substrate, competitive inhibition of transglycosylase, and permeabilization of the cell envelope. Each mechanism of action improves the antimicrobial potency to levels well beyond that of vancomycin and suppresses the rate at which bacterial resistance emerges. This provides remarkably potent and exceptionally durable antibiotics that exceed the properties of the natural product itself. What do we mean by “durable”, the heart of what we would like to champion with this work? The mechanism of action for vancomycin is especially robust, sequestering the nearly invariant substrate of an essential enzyme-catalyzed reaction required for bacterial cell wall maturation. It is not a target subject to evolved resistance derived from single genetic mutations. Because it acts at the cell surface, vancomycin also avoids other common mechanisms of resistance derived from cytosolic bacterial metabolic deactivation, blocked entry, or efflux. Finally, the existing clinical resistance was not evolved by pathogenic bacteria, but rather is the co-opted protection mechanism found in vancomycin-producing organisms. Combined, these features suggest the binding-pocket modifications can not only reinstate activity against vancomycin-resistant organisms, but alone might likely also provide antibiotics that display clinical lifetimes approaching that of vancomycin (>60 years). Add to this two additional synergistic mechanisms of action not found in the natural glycopeptide antibiotics, both of which act independent of D-Ala-D-Ala/D-Lac binding and each of which further protects the others from resistance, it is not a stretch to suggest they may display clinical lifetimes quantitated not in decades, but rather in centuries. That is, a “durability” that substantially exceeds that of even the half century use of vancomycin.
Figure 14.
Stepwise development of vancomycin analogues.
One could make the argument that incorporation of multiple mechanisms of action into a single molecule might only express activity at the level of the most potent. However, the three mechanisms of action incorporated into the maxamycins individually display similar potencies, act on the same pathway but in different ways with each inhibiting bacterial cell wall synthesis or integrity. Combined, they ultimately display synergistic and stunning activity, acting at concentrations as much as 1000-fold lower than any individual modification.75
A challenge to moving the work forward has been the development of an efficient synthesis of the analogues, including not only maxamycin-1 (29) but others (e.g., 35), as well as the establishment of their in vivo properties to determine whether they will live up to expectations. A practical solution to the late-stage scalable and protecting group-free conversion of the aglycons to the fully glycosylated analogues is already in hand (Figure 14). What remained is work that we should be especially good at addressing - the development of a streamlined scalable total synthesis of vancomycin aglycon and its pocket-modified analogues with a considerably reduced step count, simplified implementation, more concise syntheses of the unnatural amino acid subunits, and increased overall yield.76 The quality of these recently disclosed efforts, a next-generation total synthesis of vancomycin, ensure there is now an approach capable of providing the needed supplies of materials for preclinical studies.77 Long term, it is even possible semi-synthetic or coerced biosynthetic approaches to maxamycin precursors could be developed if they live up to expectations. For the latter, it is unlikely a producing organism could survive an engineered biosynthesis of a residue 4 amidine or aminomethylene vancomycin. However, our discovery that the thioamide, [Ψ[C(=S)NH]Tpg4]vancomycin, is unable to bind D-Ala-D-Ala/D-Lac and does not display antimicrobial activity provides the foundation for such efforts given the simplicity of its late-stage conversion to the maxamycins.
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from NIH (CA041101 to D.L.B.) and a JITRI-Scripps Fellowship (Z.-C. W.). We especially wish to acknowledge and thank the Concept Acceleration Program (CAP) within The National Institute of Allergy and Infectious Diseases (NIAID), Division of Microbiology and Infectious Diseases and especially Dr. Ann Eakin for sponsoring and organizing the contract services used for some studies. We dedicate this article to Max Harmony.
Biographies
Dale L. Boger is the Richard & Alice Cramer Chair in Chemistry at The Scripps Research Institute (1990-present) where he also served as Chairman (2012–2018) of the Department of Chemistry. He received his BS in Chemistry from the University of Kansas (1975) and his Ph.D. in Chemistry from Harvard University (1980) working with Professor E. J. Corey. He was on the faculty at the University of Kansas and Purdue University before joining TSRI in 1990.
Zhi-Chen Wu received his BS in Chemistry from Tsinghua University (with Professor Mei-Xiang Wang). He is currently a PhD candidate at The Scripps Research Institute (2017–present) with Professor Boger, focusing on medicinal chemistry of glycopeptide antibiotics and synthesis of 1,2,3,5-tetrazines.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.James RC; Pierce JG; Okano A; Xie J; Boger DL Redesign of Glycopeptide Antibiotics: Back to the Future. ACS Chem. Biol 2012, 7, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okano A; Nakayama A; Wu K; Lindsey EA; Schammel AW; Feng Y; Collins KC; Boger DL Total Synthesis and Initial Examination of [Ψ[C(=S)NH]Tpg4]Vancomycin, [Ψ[C(=NH)NH]Tpg4]Vancomycin, [Ψ[CH2NH]Tpg4]Vancomycin and their (4-Chlorobiphenyl)methyl Derivatives: Synergistic Binding Pocket and Peripheral Modifications for the Glycopeptide Antibiotics. J. Am. Chem. Soc 2015, 137, 3693–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okano A; Isley NA; Boger DL Total Synthesis of Vancomycin Related Glycopeptide Antibiotics and Key Analogues. Chem. Rev 2017, 117, 11952–11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okano A; Isley NA; Boger DL Peripheral Modifications of [Ψ[CH2NH]Tpg4]Vancomycin with Added Synergistic Mechanisms of Action Provide Durable and Potent Antibiotics. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E5052–E5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019. (https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf).
- 6.Jernigan JA; Hatfield KM; Wolford H; Nelson RE; Olubajo B; Reddy SC; McCarthy N; Paul P; McDonald LC; Kallen A; Fiore A Multidrug-resistant Bacterial Infections in US Hospitalized Patients, 2012–2017. N. Engl. J. Med 2020, 382, 1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCormick MH Vancomycin, A New Antibiotic. I. Chemical and Biologic Properties. Antibiot. Annu 1956, 3, 606–611. [PubMed] [Google Scholar]
- 8.Perkins HR Vancomycin and Related Antibiotics. Pharmacol. Ther 1982, 16, 181–197. [DOI] [PubMed] [Google Scholar]
- 9.Van Bambeke F; Van Laethem Y; Courvalin P; Tulkens PM Glycopeptide Antibiotics. Drugs 2004, 64, 913–936. [DOI] [PubMed] [Google Scholar]
- 10.Kahne D; Leimkuhler C; Lu W; Walsh C Glycopeptide and Lipoglycopeptide Antibiotics. Chem. Rev 2005, 105, 425–448. [DOI] [PubMed] [Google Scholar]
- 11.Levine D Vancomycin: A History. Clin. Infect. Dis 2006, 42, S5–S12. [DOI] [PubMed] [Google Scholar]
- 12.Glycopeptide Antibiotics; Nagarajan R, Ed.; Marcel Dekker, Inc.: New York, 1994. [Google Scholar]
- 13.Cooper GL; Given DB The Development of Vancomycin. In Vancomycin, A Comprehensive Review of 30 Years of Clinical Experience; Park Row Publications: Indianapolis, IN, 1986; pp 1–5. [Google Scholar]
- 14.Barna JCJ; Williams DH The Structure and Mode of Action of Glycopeptide Antibiotics of the Vancomycin Group. Annu. Rev. Microbiol 1984, 38, 339–357. [DOI] [PubMed] [Google Scholar]
- 15.Hubbard BK; Walsh CT Vancomycin Assembly: Nature’s Way. Angew. Chem., Int. Ed 2003, 42, 730–765. [DOI] [PubMed] [Google Scholar]
- 16. The mechanism of action of vancomycin is inhibition of cell wall synthesis by binding to the D-Ala-D-Ala terminal of the growing peptidoglycan chain during cell wall synthesis. This results in inhibition of the transpeptidase, which prevents cross-linking and further elongation of the peptidoglycan matrix. Vancomycin also indirectly inhibits the action of transglycosylase, a second enzyme responsible for extending the polysaccaride, although this appears to be a less important mechanism compared to inhibition of transpeptidase. The mechanism for transglycosylase inhibition is unclear but, unlike the direct action of the CBP modification, it requires D-Ala-D-Ala binding for expression of this activity. The net result is a mechanism of similar to that of the beta-lactam antibiotics but occurs by binding to a different target (substrate D-Ala-D-Ala vs enzyme transpeptidase). See reference 10.
- 17.Parker MT; Jevons MP A Survey of Methicillin Resistance in Staphylococcus aureus. Postgrad. Med. J 1964, 40, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayden MK; Rezai K; Hayes RA; Lolans K; Quinn JP; Weinstein RA Development of Daptomycin Resistance in vivo in Methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol, 2005, 43, 5285–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsiodras S; Gold HS; Sakoulas G; Eliopoulos GM; Wennersten C; Venkataraman L; Moellering RC Jr; Ferraro MJ Linezolid Resistance in a Clinical Isolate of Staphylococcus aureus. Lancet 2001, 358, 207–208. [DOI] [PubMed] [Google Scholar]
- 20.Leclercq R; Derlot E; Duval J; Courvalin P Plasmid-mediated Resistance to Vancomycin and Teicoplanin in Enterococcus faecium. N. Engl. J. Med 1988, 319, 157–161. [DOI] [PubMed] [Google Scholar]
- 21.Weigel LM; Clewell DB; Gill SR; Clark NC; McDougal LK; Flannagan SE; Kolonay JF; Shetty J; Killgore GE; Tenover FC Genetic Analysis of a High-level Vancomycin-resistant Isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571. [DOI] [PubMed] [Google Scholar]
- 22.Courvalin P Vancomycin Resistance in Gram-positive Cocci. Clin. Infect. Dis 2006, 42, S25–S34. [DOI] [PubMed] [Google Scholar]
- 23.Walsh TR; Howe RA The Prevalence and Mechanisms of Vancomycin Resistance in Staphylococcus aureus. Annu. Rev. Microbiol 2002, 56, 657–675. [DOI] [PubMed] [Google Scholar]
- 24.Marshall CG; Lessard IAD; Park I-S; Wright GD Glycopeptide Antibiotic Resistance Genes in Glycopeptide-producing Organisms. Antimicrob. Agents Chemother 1998, 42, 2215–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Healy VL; Lessard IA; Roper DI; Knox JR; Walsh CT Vancomycin Resistance in Enterococci: Reprogramming of the D-Ala-D-Ala Ligases in Bacterial Peptidoglycan Biosynthesis. Chem. Biol 2000, 7, 109–119. [DOI] [PubMed] [Google Scholar]
- 26.Bugg TDH; Wright GD; Dutka–Malen S; Arthur M; Courvalin P; Walsh CT Molecular Basis of Vancomycin Resistance in Enterococcus faecium BM4147: Biosynthesis of a Depsipeptide Peptidoglycan Precursor by Vancomycin Resistance Proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. [DOI] [PubMed] [Google Scholar]
- 27.Walsh CT Vancomycin Resistance: Decoding the Molecular Logic. Science 1993, 261, 308–310. [DOI] [PubMed] [Google Scholar]
- 28.Guan D; Chen F; Xiong L; Tang F; Qiu Y; Zhang N; Gong L; Li J; Lan L; Huang W Extra Sugar on Vancomycin: New Analogues for Combating Multidrug-resistant Staphylococcus aureus and Vancomycin-resistant Enterococci. J. Med. Chem 2018, 61, 286–304. [DOI] [PubMed] [Google Scholar]
- 29.Yarlagadda V; Konai MM; Manjunath GB; Ghosh C; Haldar J, Tackling Vancomycin-resistant Bacteria with ‘Lipophilic-Vancomycin-Carbohydrate Conjugates’. J. Antibiot 2015, 68, 302–312. [DOI] [PubMed] [Google Scholar]
- 30.Antonoplis A; Zang X; Huttner MA; Chong KK; Lee YB; Co JY; Amieva MR; Kline KA; Wender PA; Cegelski L, A Dual-function Antibiotic-transporter Conjugate Exhibits Superior Activity in Sterilizing MRSA Biofilms and Killing Persister Cells. J. Am. Chem. Soc 2018, 140, 16140–16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yarlagadda V; Sarkar P; Samaddar S; Haldar J A Vancomycin Derivative with a Pyrophosphate-binding Group: A Strategy to Combat Vancomycin-resistant Bacteria. Angew. Chem., Int. Ed 2016, 55, 7836–7840. [DOI] [PubMed] [Google Scholar]
- 32.Yarlagadda V; Samaddar S; Paramanandham K; Shome BR; Haldar J Membrane Disruption and Enhanced Inhibition of Cell-wall Biosynthesis: A Synergistic Approach to Tackle Vancomycin-resistant Bacteria. Angew. Chem., Int. Ed 2015, 54, 13644–13649. [DOI] [PubMed] [Google Scholar]
- 33.Long DD; Aggen JB; Chinn J; Choi SK; Christensen BG; Fatheree PR; Green D; Hegde SS; Judice JK; Kaniga K; Krause KM Exploring the Positional Attachment of Glycopeptide/p-lactam Heterodimers. J. Antibiot 2008, 61, 603–614. [DOI] [PubMed] [Google Scholar]
- 34.Yarlagadda V; Akkapeddi P; Manjunath GB; Haldar J Membrane Active Vancomycin Analogues: A Strategy to Combat Bacterial Resistance. J. Med. Chem 2014, 57, 4558–4568. [DOI] [PubMed] [Google Scholar]
- 35.Leadbetter MR; Adams SM; Bazzini B; Fatheree PR; Karr DE; Krause KM; Lam BM; Linsell MS; Nodwell MB; Pace JL; Quast K Hydrophobic Vancomycin Derivatives with Improved ADME Properties. J. Antibiot 2004, 57, 326–336. [DOI] [PubMed] [Google Scholar]
- 36.Corey GR; Stryjewski ME; Weyenberg W; Yasothan U; Kirkpatrick P Telavancin. Nat. Rev. Drug Discov 2009, 8, 929–930. [DOI] [PubMed] [Google Scholar]
- 37.Anderson VR; Keating GM Dalbavancin. Drugs 2008, 68, 639–648. [DOI] [PubMed] [Google Scholar]
- 38.Markham A Oritavancin: First Global Approval. Drugs 2014, 74, 1823–1828. [DOI] [PubMed] [Google Scholar]
- 39.Boger DL; Miyazaki S; Loiseleur O; Beresis RT; Castle SL; Wu JH; Jin Q Thermal Atropisomerism of Aglucovancomycin Derivatives: Preparation of (M,M,M)- and (P,M,M)-Aglucovancomycin. J. Am. Chem. Soc 1998, 120, 8920–8926. [Google Scholar]
- 40.Boger DL; Miyazaki S; Kim SH; Wu JH; Loiseleur O; Castle SL Diastereoselective Total Synthesis of the Vancomycin Aglycon with Ordered Atropisomer Equilibrations. J. Am. Chem. Soc 1999, 121, 3226–3227. [Google Scholar]
- 41.Boger DL; Miyazaki S; Kim SH; Wu JH; Castle SL; Loiseleur O; Jin Q Total Synthesis of the Vancomycin Aglycon. J. Am. Chem. Soc 1999, 121, 10004–10011. [Google Scholar]
- 42.Boger DL; Kim SH; Miyazaki S; Strittmatter H; Weng J-H; Mori Y; Rogel O; Castle SL; McAtee JJ Total Synthesis of the Teicoplanin Aglycon. J. Am. Chem. Soc 2000, 122, 7416–7417. [DOI] [PubMed] [Google Scholar]
- 43.Boger DL; Kim SH; Mori Y; Weng J-H; Rogel O; Castle SL; McAtee JJ First and Second Generation Total Synthesis of the Teicoplanin Aglycon. J. Am. Chem. Soc 2001, 123, 1862–1871. [DOI] [PubMed] [Google Scholar]
- 44.Crowley BM; Mori Y; McComas CC; Tang D; Boger DL; Total Synthesis of the Ristocetin Aglycon. J. Am. Chem. Soc 2004, 126, 4310–4317. [DOI] [PubMed] [Google Scholar]
- 45.Garfunkle J; Kimball FS; Trzupek JD; Takazawa S; Shimamura H; Tomishima M; Boger DL Total Synthesis of Chloropeptin II (Complestatin) and Chloropeptin I. J. Am. Chem. Soc 2009, 131, 16036–16038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimamura H; Breazzano SP; Garfunkle J; Kimball FS; Trzupek JD; Boger DL Total Synthesis of Complestatin: Development of a Pd(0)-mediated Indole Annulation for Macrocyclization. J. Am. Chem. Soc 2010, 132, 7776–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wan KK; Shenvi RA Conjuring a Supernatural Product – DelMarine. Synlett 2016, 27, 1145–1164. [Google Scholar]
- 48.Wu Z-C; Boger DL The Quest for Supernatural Products: The Role of Total Synthesis in Complex Natural Products Medicinal Chemistry. Nat. Prod. Rep submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McComas CC; Crowley BM; Boger DL Partitioning the Loss in Vancomycin Binding Affinity for D-Ala-D-Lac into Lost H-bond and Repulsive Lone Pair Contributions. J. Am. Chem. Soc 2003, 125, 9314–9315. [DOI] [PubMed] [Google Scholar]
- 50.Crowley BM; Boger DL Total Synthesis and Evaluation of [Ψ[CH2NH]Tpg4]Vancomycin Aglycon: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J. Am. Chem. Soc 2006, 128, 2885–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie J; Okano A; Pierce JG; James RC; Stamm S; Crane CM; Boger DL Total Synthesis of [Ψ[C(=S)NH]Tpg4]Vancomycin Aglycon, [Ψ[C(=NH)NH]Tpg4]Vancomycin Aglycon and Related Key Compounds: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J. Am. Chem. Soc 2012, 134, 1284–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie J; Pierce JG; James RC; Okano A; Boger DL A Redesigned Vancomycin Engineered for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding Exhibits Potent Antimicrobial Activity Against Vancomycin-resistant Bacteria. J. Am. Chem. Soc 2011, 133, 13946–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okano A; James RC; Pierce JG; Xie J; Boger DL Silver(I)-promoted Conversion of Thioamides to Amidines: Divergent Synthesis of a Key Series of Vancomycin Aglycon Residue 4 Amidines that Clarify Binding Behavior to Model Ligands. J. Am. Chem. Soc 2012, 134, 8790–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayama A; Okano A; Feng Y; Collins JC; Collins KC; Walsh CT; Boger DL Enzymatic Glycosylation of Vancomycin Aglycon: Completion of a Total Synthesis of Vancomycin and N- and C-terminus Substituent Effects of the Aglycon Substrate. Org. Lett 2014, 16, 3572–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Losey HC; Peczuh MW; Chen Z; Eggert US; Dong SD; Pelczer I; Kahne D; Walsh CT Tandem Action of Glycosyltransferases in the Maturation of Vancomycin and Teicoplanin Aglycones: Novel Glycopeptides. Biochemistry 2001, 40, 4745–4755. [DOI] [PubMed] [Google Scholar]
- 56.Thayer DA; Wong C-H Vancomycin Analogues Containing Monosaccharides Exhibit Improved Antibiotic Activity: A Combined One-pot Enzymatic Glycosylation and Chemical Diversification Strategy. Chem.-Asian J 2006, 1, 445–452. [DOI] [PubMed] [Google Scholar]
- 57.Okano A; Nakayama A; Schammel AW; Boger DL Total Synthesis of [Ψ[C(=NH)NH]Tpg4]Vancomycin and its (4-Chlorobiphenyl)methyl Derivative: Impact of Peripheral Modifications on Vancomycin Analogs Redesigned for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding, J. Am. Chem. Soc 2014, 136, 13522–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagarajan R; Schabel AA; Occolowitz JL; Counter FT; Ott JL; Felty-Duckworth AM Synthesis and Antibacterial Evaluation of N-alkyl Vancomycins. J. Antibiot 1989, 42, 63–72. [DOI] [PubMed] [Google Scholar]
- 59.Nagarajan R Structure-Activity Relationships of Vancomycin-type Glycopeptide Antibiotics. J. Antibiot 1993, 46, 1181–1195. [DOI] [PubMed] [Google Scholar]
- 60.Allen NE; Hobbs JN; Nicas TI Inhibition of Peptidoglycan Biosynthesis in Vancomycin-susceptible and -resistant Bacteria by a Semisynthetic Glycopeptide Antibiotic. Antimicrob. Agents Chemother 1996, 40, 2356–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ge M; Chen Z; Russell H; Kohler J; Silver LL; Kerns R; Fukuzawa S; Thompson C; Kahne D Vancomycin Derivatives that Inhibit Peptidoglycan Biosynthesis without Binding D-Ala-D-Ala. Science 1999, 284, 507–511. [DOI] [PubMed] [Google Scholar]
- 62.Chen L; Walker D; Sun B; Hu Y; Walker S; Kahne D Vancomycin Analogues Active Against VanA-resistant Strains Inhibit Bacterial Transglycosylase without Binding Substrate. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 5658–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malabarba A; Goldstein BP Origin, Structure, and Activity in vitro and in vivo of Dalbavancin. J. Antimicrob. Chemother 2005, 55, ii15–ii20. [DOI] [PubMed] [Google Scholar]
- 64.This name has a special meaning for the senior author. The work has been and continues to be driven forward by a commitment to provide my grandchildren with at least a foundation, if not the actual drug, that could fundamentally overcome the challenge of antibiotic resistance. One, at age 10, asked me to explain the work to him shortly after we completed our work on C1-CBP-[Ψ[CH2NH]Tpg4]vancomycin (29) in 2017. At the end of our discussion which he understood, he was beaming and told me that he had a name for the compound. That name, maxamycin, perfectly embodied the compound, the objectives of the studies, the path taken to discover it, and the remarkable properties of the molecule. As the inaugural molecule in the class, we have been referring to it as maxamycin-1 since that time. It is also no accident that my grandson’s name is fittingly Max Harmony to whom we dedicate this article. Additionally, and perhaps it is fate, our colleague Maxwell Moore is spearheading the ongoing efforts to make additional maxamycin analogues accessible and available.
- 65.Wu Z–C; Isley NA; Boger DL N-Terminus Alkylation of Vancomycin: Ligand Binding Affinity, Antimicrobial Activity, and Site-specific Nature of Quaternary Trimethylammonium Salt Modification. ACS Infect. Dis 2018, 4, 1468–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Z–C; Boger DL Exploration of the Site-specific Nature and Generalizability of a Trimethylammonium Salt Modification on Vancomycin: A-ring Derivatives. Tetrahedron 2019, 75, 3160–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Z–C; Isley NA; Okano A; Weiss WJ; Boger DL C1-CBP-Vancomycin: Impact of a Vancomycin C-Terminus Trimethylammonium Cation on Pharmacological Properties and Insights into Its Newly Introduced Mechanism of Action. J. Org. Chem 2020, 85, 1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Z–C; Cameron MD; Boger DL Vancomycin C-Terminus Guanidine Modifications and Further Insights into an Added Mechanism of Action Imparted by a Peripheral Structural Modification. ACS Infect. Dis 2020, 6, 2169–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Percy MG; Grundling A Lipoteichoic Acid Synthesis and Function in Gram-positive Bacteria. Annu. Rev. Microbiol 2014, 68, 81–100. [DOI] [PubMed] [Google Scholar]
- 70.Brown S; Santa Maria JP Jr.; Walker S Wall Teichoic Acids of Gram-positive Bacteria. Annu. Rev. Microbiol 2013, 67, 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Höltje J-V; Tomasz A Lipoteichoic Acid: A Specific Inhibitor of Autolysin Activity in Pneumococcus. Proc. Natl. Acad. Sci. U. S. A 1975, 72, 1690–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bierbaum G; Sahl H-G Induction of Autolysis of Staphylococci by the Basic Peptide Antibiotics Pep 5 and Nisin and their Influence on the Activity of Autolytic Enzymes. Arch. Microbiol 1985, 141, 249–254. [DOI] [PubMed] [Google Scholar]
- 73.Bierbaum G; Sahl H-G Autolytic System of Staphylococcus simulans 22: Influence of Cationic Peptides on Activity of N-acetylmuramoyl-L-alanine Amidase. J. Bacteriol 1987, 169, 5452–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fetterly GJ; Ong CM; Bhavnani SM; Loutit JS; Porter SB; Morello LG; Ambrose PG; Nicolau DP Pharmacokinetics of Oritavancin in Plasma and Skin Blister Fluid Following Administration of a 200-milligram Dose for 3 Days or a Single 800-milligram Dose. Antimicrob. Agents Chemother 2005, 49, 148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Silver LL Multi-targeting by Monotherapeutic Antibacterials. Nat. Rev 2007, 6, 41–55. [DOI] [PubMed] [Google Scholar]
- 76.Wright PM; Seiple IB; Myers AG The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew. Chem., Int. Ed 2014, 53, 8840–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moore MJ; Qu S; Tan C; Cai Y; Mogi Y; Keith DJ; Boger DL Next-generation Total Synthesis of Vancomycin. J. Am. Chem. Soc 2020, 142, 16039–16050. [DOI] [PMC free article] [PubMed] [Google Scholar]