

Abstract
Export of lactic acid from glycolytic cancer cells to the extracellular tumor milieu has been reported to enhance tumor growth and suppress antitumor immunity. In this study, a pH‐activatable nanodrug is reported for tumor‐targeted inhibition of monocarboxylate transporter‐1 (MCT1) that reverses lactic acid‐induced tumor immunosuppression. The nanodrug is composed of an MCT1 inhibitor (AZD3965) loaded inside the ultra‐pH‐sensitive nanoparticles (AZD‐UPS NPs). AZD‐UPS NP is produced by a microfluidics method with improved drug loading efficiency and optimal nanoparticle size over sonication methods. The nanodrug remains as intact micelles at pH 7.4 but rapidly disassembles and releases payload upon exposure to acidic pH. When combined with anti‐PD‐1 therapy, AZD‐UPS NP leads to potent tumor growth inhibition and increases survival in two tumor models over oral administration of AZD3965 at dramatically reduced dose (>200‐fold). Safety evaluations demonstrate reduced drug distribution in heart and liver tissues with decrease in toxic biomarkers such as cardiac troponin by the nanodrug. Increased T‐cell infiltration and reduced exhaustive PD1+Tim3+ T cells are found in tumors. These data illustrate that tumor‐targeted inhibition of MCT1 can reverse the immune suppressive microenvironment of solid tumors for increased safety and antitumor efficacy of cancer immunotherapy.
Keywords: cancer immunotherapy, lactic acid exportation, pH‐activatable drug release, tumor‐targeted drug delivery, ultra‐pH‐sensitive micelles
A pH‐activatable nanodrug is established for tumor‐targeted inhibition of MCT1 that reverses lactic acid‐induced immunosuppression in solid tumors. The nanodrug is composed of an MCT1 inhibitor (AZD3965) loaded inside the ultra‐pH‐sensitive nanoparticles (AZD‐UPS NPs), and allows for tumor‐specific inhibition of lactic acid exportation. Combining with anti‐PD‐1, the nanodrug synergistically inhibits tumor growth by enhancing CD8 T cell infiltration with reduced exhaustion inside tumors.
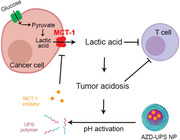
Cancer cells have upregulated glucose metabolism that are reprogrammed towards aerobic glycolysis (also known as Warburg effect), resulting in the rapid production and exportation of lactic acid into the tumor microenvironment.[ 1 ] Lactate transport across the cancer cell membrane is mediated by monocarboxylate transporter proteins, which are coupled to the co‐transportation of protons.[ 2 ] Export of lactic acid and subsequent low extracellular tumor pH has been reported to increase tumor growth and metastasis,[ 3 ] angiogenesis,[ 4 ] and immune suppression.[ 5 ] Lactic acid is reported as a potent inhibitor of function and survival of T and NK cells leading to blunting of immune surveillance.[ 6 ] Therefore, targeting monocarboxylate transporter proteins to reduce lactic acid export from the cancer cells has the potential to rescue tumor metabolism‐induced immune evasion (Scheme 1 ).
Scheme 1.
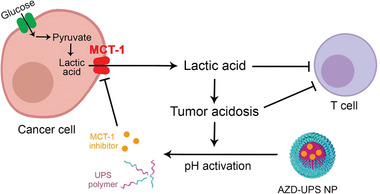
Tumor‐targeted inhibition of monocarboxylate transporter 1 (MCT1) by AZD3965‐loaded ultra‐pH‐sensitive nanoparticles (AZD‐UPS NPs) primes the tumor microenvironment for enhanced T‐cell immunity against cancer.
AZD3965 is a small molecule drug developed by AstraZeneca that specifically inhibits monocarboxylate transporter 1 (MCT1) (Figure 1a). Previous report on the mechanism of action was mainly attributed to the accumulation of intracellular lactic acid, which leads to a decrease of intracellular pH and feedback inhibition of glycolysis, thereby deterring the proliferation of tumor cells.[ 7 ] The effect of MCT1 inhibition on the tumor microenvironment and resulting influence on antitumor immunity is not clear. A challenge in the clinical use of AZD3965 arises from dose‐limiting toxicities in the heart and/or eye tissues, where MCT1 expression is high, due to nonspecific drug distribution from oral administration. Adverse side effects such as rise of cardiac troponin levels and electroretinogram changes were observed.[ 8 ] Tumor‐targeted delivery of AZD3965 exploiting tumor acidotic metabolism has the potential to increase the therapeutic window of the drug while allowing the investigation of MCT1 inhibition on antitumor immunity.
Figure 1.
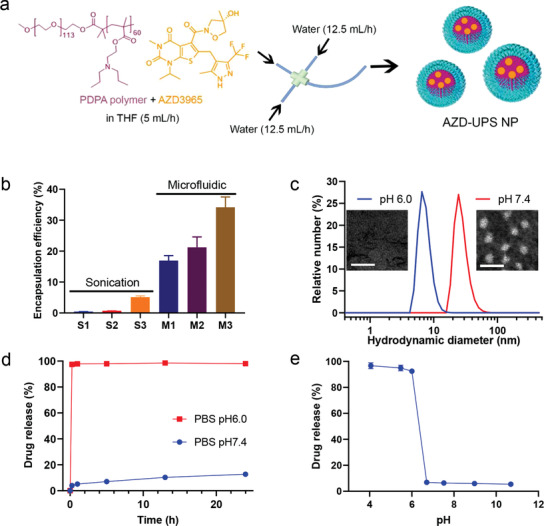
Preparation and characterization of AZD‐UPS NPs. a) Schematic of microfluidic method to produce AZD‐UPS NPs. b) Microfluidic method increases encapsulation efficiency of AZD3965 in UPS nanoparticles over sonication method with different UPS/AZD3965 ratios. c) Dynamic light scattering and transmission electron microscopy (TEM) analysis of AZD‐UPS NPs at pH 7.4 and its dissociation into unimers at pH 6.0. The apparent pKa of the UPS polymer is 6.1. Scale bars = 50 nm in the TEM images. d) pH‐dependent drug release from AZD‐UPS NPs in phosphate buffered saline (PBS) over 24 h at 37 °C. At pH 6.0, instantaneous release of AZD3965 drug was observed after micelle dissociation whereas majority of the drug was kept in the micelles at pH 7.4. e) A binary drug release profile across the micelle transition pH of the PDPA copolymer was observed from AZD‐UPS NPs after 15 min incubation in the PBS solution. All the data are presented as mean ± SD, n = 3.
Our lab has previously synthesized a library of ultra‐pH‐sensitive (UPS) micelle nanoparticles[ 9 ] for tumor imaging and therapeutic applications.[ 10 ] UPS nanoparticles remain as intact micelles at physiological pH (7.4) during blood circulation but disassembles when the environmental pH is dropped below the micelle transition pH (pHt) upon exposure to tumor acidic milieu. Recently, positron emission tomography (PET) imaging of a radionucleotide (64Cu)‐functionalized UPS nanoprobe demonstrate dramatic increase of nanoparticle accumulation through acid‐triggered “capture and integration mechanism” in multiple tumor models over non‐pH sensitive, passive targeting PEG‐b‐poly(d,l‐lactic acid) micelles. In this study, we investigate the use of UPS nanoparticles for tumor‐targeted delivery and release of AZD3965 to prime the tumor microenvironment for T‐cell therapy of cancer. We hypothesized that UPS NP would maximize the efficacy of AZD3965 by targeted delivery to tumor areas of greatest MCT1 activity and thus most susceptible to inhibition. We also hypothesized that this selective, self‐limiting delivery approach would allow decreases in the overall dose of AZD3965 leading to improved safety outcomes.
Because of its mild hydrophobicity (partition coefficient log P = 1.78[ 11 ]), loading of AZD3965 in a representative UPS micelle, PEG‐b‐(poly(dipropylaminoethyl methacrylate) (PDPA, Figure 1a) is difficult using conventional encapsulation methods such as sonication. Microfluidics‐based methods have been reported to improve drug loading efficiency[ 12 ] by precise control of liquid streams for mixing.[ 12 , 13 ] We set up a microfluidic device according to the IDEX method[ 12 , 13 ] with one central flow, two side flows and one outlet with a curved channel structure (Figure 1a). AZD3965 and PDPA polymer in different ratios were dissolved in tetrahydrofuran with 9.1% dimethyl sulfoxide, and introduced to the microfluidic device through the central channel at a flow rate of 5 mL h−1. Water was introduced through the two side channels at 12.5 mL h−1. Upon mixing in the junction, AZD3965‐loaded UPS nanoparticles (AZD‐UPS NPs) are generated in a single nanoprecipitation step. The encapsulation efficiency after purification is 34% (formulation M3 with a PDPA/AZD3965 ratio of 10:1, analyzed by HPLC), which is higher than the sonication method (5.1% in formulation S3, Figure 1b, Table S1 and Figure S1, Supporting Information). The diameter of AZD‐UPS NPs by microfluidics method is 33.3 ± 0.2 nm, with a polydispersity index (PDI) of 0.15 ± 0.01, which is smaller than the drug‐loaded micelles from the sonication method (82.8 ± 2.0 nm in diameter) (Table S1, Supporting Information). This result is consistent with the reported literature that rapid mixing in a microfluidic device reduced nanoparticle aggregation resulting in smaller and more homogeneous nanoparticles.[ 14 ]
AZD‐UPS NPs exhibit similar ultra‐pH sensitivity across the transition pH of 6.1 for the PDPA polymer (Figure S2a, Supporting Information) to the drug‐free PDPA micelles,[ 15 ] likely due to the neutral and mildly hydrophobic (log P = 1.78) characteristic of the drug. We analyzed the particle size and morphology by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Figure 1c). At pH 7.4, spherical micelles were formed with hydrodynamic diameter of 33.3 ± 0.2 nm; at pH 6.0, which is below the apparent pKa of PDPA (6.1), micelles dissociated into unimers (9.9 ± 2.6 nm in diameter). The pH‐dependent release kinetics of the AZD‐UPS micelles was quantified by HPLC after micelle incubation with phosphate buffered saline (PBS) solution at pH 7.4 and 6.0 over 24 h (Figure 1d, Figure S3a, Supporting Information). At pH 7.4, less than 10% of drugs leaked out of the PDPA micelles over 24 h. In contrast, majority (>95%) of the drug was instantaneously released from the micelles at pH 6.0 within 15 min. Further pH‐dependent drug release studies show a binary off/on drug release phenotype after 15 min incubation across the apparent pKa (6.1) of the PDPA polymer (Figure 1e, Figure S3b, Supporting Information). At pH higher than 6.1, drugs were stably encapsulated inside the PDPA micelles; when pH dropped below 6.1, micelle disassembly led to rapid release and dose dumping.
To investigate the drug activity, we quantified the changes of lactate and pH in the culture medium of TC‐1 cancer cells. Free AZD3965 (1 × 10−6 m) significantly decreased the secretion of lactate and inhibited the lowering of pH in the medium compared to the untreated control over 24 h (p < 0.001, Figure 2a,b). Intact AZD‐UPS NPs (1 × 10−6 m effective AZD3965 concentration) had no significant effect (p = 0.068) on medium pH or lactate concentration primarily due to drug encapsulation in the micelles. Pretreatment of AZD‐UPS NPs in pH 6.0 buffer to induce pH‐activated drug release followed by addition into the cell culture medium exhibited effects similar to free AZD3965. Seahorse assay further supported the micelle‐modulated drug effect in response to glucose addition (Figure S2b, Supporting Information). These data illustrate AZD‐UPS NPs effectively blocked the drug effect in the micelle state; upon pH‐triggered drug release, AZD3965 was able to inhibit the export of lactic acid from cancer cells.
Figure 2.
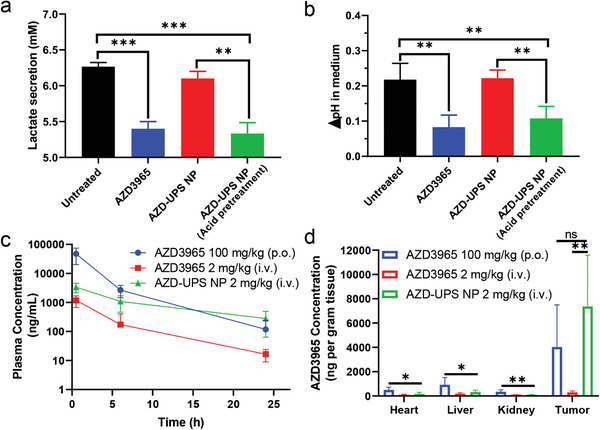
AZD‐UPS NPs inhibited export of lactic acid from cancer cells and improved drug pharmacokinetics in a mouse tumor model. AZD3965 alone or pretreatment of AZD‐UPS NP at pH 6.0 effectively decreased lactate secretion a) and pH change b) in the cell culture medium at 24 h. Intact AZD‐UPS NPs at pH 7.4 did not affect the lactate secretion or pH change compared to the control groups. c) Plasma concentration of AZD3965 following oral administration (100 mg kg − 1), intravenous administration of AZD3965 alone and AZD‐UPS NP at the same drug dose (2 mg kg−1). d) AZD3965 distribution in heart, liver, kidney and tumor tissues 24 h after administration. AZD3965 concentrations were normalized per gram of tissue and expressed as ng g−1. ***P < 0.001, **P < 0.01, *P < 0.05 (nonparametric Student's t‐test). All the data are presented as mean ± SD, a,b) n = 4; c,d) n = 5–6.
We utilized TC‐1 tumor‐bearing mice to evaluate the pharmacokinetics and biodistribution of AZD3965 drug from different formulations. AZD3965 by oral administration (100 mg kg−1, dose recommended by AstraZeneca) exhibited rapid oral absorption and high peak drug concentration of 47 ± 27 µg mL−1 at 30 min (Figure 2c), similar to the value from literature.[ 11 ] In contrast, intravenous injection of AZD‐UPS NP at much reduced dose (2 mg kg−1) resulted in significantly decreased peak concentration (3.3 ± 1.2 µg mL−1) at 30 min but comparable plasma concentrations at 24 h. Intravenous injection of free AZD3965 drug at the same dose as AZD‐UPS‐NP (i.e., 2 mg kg−1) showed rapid clearance of the drug. The area under the concentration‐time (AUC) value of AZD‐UPS NP is 24 ± 7 µg mL−1 h−1, approximately 4.5‐fold over free AZD3965 (5.4 ± 2.5 µg mL−1 h−1). Tissue distribution analysis was performed in the heart, liver, kidney, and tumor tissues 24 h after the administration (Figure 2d). AZD‐UPS NP achieved significantly higher levels of tumor accumulation of the AZD3965 drug (7.4 ± 4.2 µg g−1 of tissue) over free drug (0.29 ± 0.12 µg g−1, p = 0.0049). Remarkably, AZD‐UPS NP delivered even higher drug dose to the tumors over oral administration of a 50‐fold higher dose of AZD3965 (4.0 ± 3.5 µg g−1). In contrast, drug distribution in the heart and liver tissues was significantly reduced in AZD‐UPS NP group over oral administration of AZD3965. Safety evaluations show AZD‐UPS NP and oral administration of AZD3965 did not cause weight loss during two weeks of treatment (Figure S4a,b, Supporting Information), while oral administration of AZD3965 significantly increased the levels of cardiac troponin‐I (cTnI), alanine aminotransferase (ALT), and aspartate aminotransferase (AST), indicating that encapsulating AZD3965 into UPS NP can reduce liver and cardiac toxicities (Figure S4c,d, Supporting Information).
We investigated the antitumor efficacy of AZD‐UPS NP in combination with anti‐PD‐1 therapy to evaluate the effect of priming the tumor microenvironment for T‐cell immunotherapy (Figure 3 ). We employed two animal tumor models, TC‐1 tumors and B16F10 melanoma for the study. Three intravenous injections of AZD‐UPS NPs (each at 2 mg kg−1 AZD3965 dose) were administered on day 7, 10, and 13 after tumor inoculation. For the free drug control, oral administration at 50 mg kg−1 AZD drug was performed twice a day from day 7 to 13 (a total of 14 doses). The cumulative dose is 234‐fold greater in the oral AZD3965 group. In the combination groups, anti‐PD‐1 was i.p. injected on day 5 and 8 after tumor inoculation.
Figure 3.
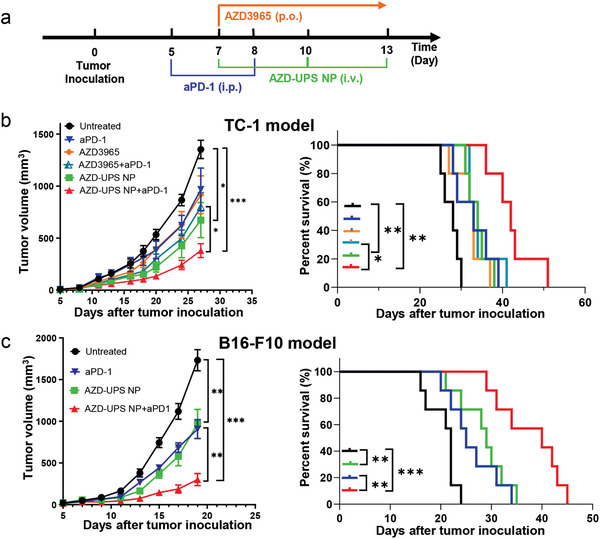
AZD‐UPS NP showed antitumor efficacy and synergy with anti‐PD‐1 therapy. a) Treatment regimen in TC‐1 and B16F10 tumor models. b) In TC‐1 model, C57BL/6 mice (n = 5 per group) were inoculated with 2 × 105 TC‐1 tumor cells and treated with different drugs. Tumor growth and Kaplan–Meier survival curves are shown. c) In the B16F10 tumor model, tumor growth inhibition and survival data in C57BL/6 mice (n = 7 per group) were analyzed. In (b,c), data are presented as means ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05.
Results show in the TC‐1 model, treatment with AZD‐UPS NP led to significant tumor growth inhibition (tumor volume = 6.7 ± 3.8 × 102 mm3) over untreated group (1.4 ± 0.2 × 103 mm3, p = 0.01) on day 27 after tumor inoculation, whereas orally administrated AZD3965 (9.2 ± 4.1 × 102 mm3, p = 0.09) showed insignificant differences over control (Figure 3b). The synergistic effect of AZD‐UPS NPs with anti‐PD‐1 treatment was also detected in the TC‐1 model (3.8 ± 1.5 × 102 mm3), which is significantly better than the combination of oral AZD3965 and anti‐PD‐1 (8.0 ± 3.1 × 102 mm3, p = 0.025). Long‐term survival was also improved (p = 0.023) between the two groups. The therapeutic synergy in combining AZD‐UPS NP with anti‐PD‐1 therapy was further validated in the B16F10 melanoma tumor model. Reduction in tumor growth (p < 0.01) and prolonged survival (p < 0.01) were observed in the combination group over any single arm control (Figure 3c). These results support the premise that inhibiting tumor acidosis can improve response to anti‐PD‐1 treatment.[ 16 ]
We investigated the changes of immune profiles after treatment with anti‐PD‐1 alone, AZD‐UPS NP alone or combination therapy in the TC1 tumors (Figure 4a). We first examined the ratio of tumor‐infiltrating T cells over cancer cells (CD45−) from different groups. Tumor tissues were dissociated into single cells and the percentage of infiltrating T cells were analyzed by flow cytometry. Compared to the untreated mice, AZD‐UPS NPs alone or anti‐PD‐1 alone did not significantly increase tumor infiltrating T cells (CD45+CD3+ cells, p > 0.05) or antigen‐specific CD8+ T cells (E7 tetramer+ CD8+ CD3+ cells, p > 0.05) compared to untreated group (Figure 4b,c, Figure S5, Supporting Information). In contrast, AZD‐UPS NPs with anti‐PD‐1 treatment results in significant increase of both tumor infiltrating T cells (p = 0.022) and antigen‐specific CD8+ T cells (p = 0.0004), which supports the correlation of antitumor efficacy with T cell infiltration inside the tumors.[ 17 ] Furthermore, we evaluated the exhaustion status of T cells in tumors.[ 6 ] Previous studies reported that PD1+Tim3− CD8+ T cells have better cytotoxic functions whereas coexpression of PD1 and TIM‐3 is associated with T‐cell exhaustion.[ 18 , 19 ] AZD‐UPS NPs with anti‐PD‐1 treatment showed distinctively increased PD1+Tim3− effector and decreased PD1+Tim3+ CD8 T cell responses, compared to single arm controls (Figure 4d,e). The number of CD4+ T cells and Tregs between different treatment groups exhibited no significant changes (Figure S6, Supporting Information). These results illustrate anti‐PD‐1 alone or AZD‐UPS NP alone were not able to prime a durable immune response to TC‐1 tumors due to the immunosuppressive effects of tumor acidosis, while the combination led to a significant improvement in CD8 T cell responses against tumors.
Figure 4.
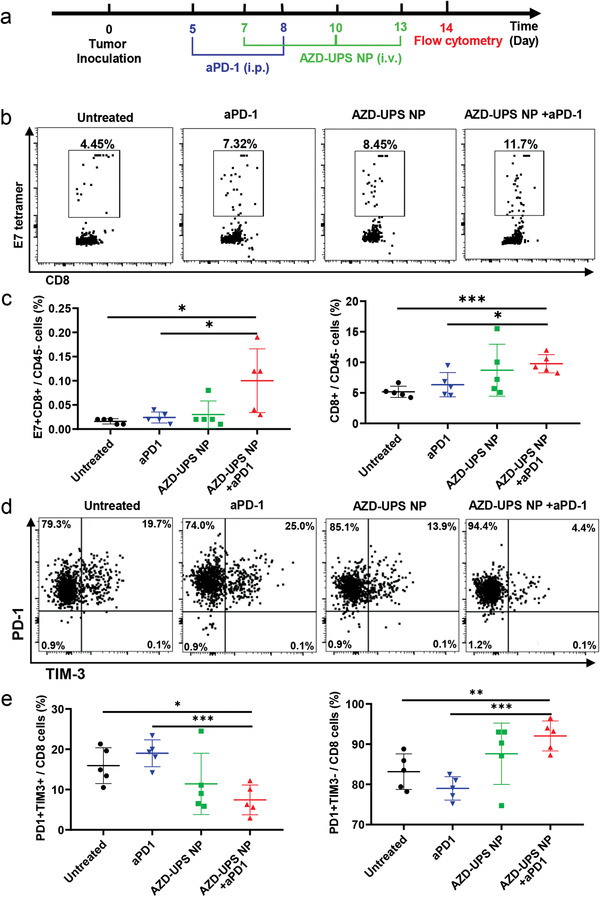
Immune profiles of the TC1 tumor after different treatment regimens. a) Scheme of the flow cytometry analysis of TC‐1 tumors. b) Representative flow dot plots of H‐2Db HPV16 E7 (RAHYNIVTF) tetramer staining of CD8+ T cells in the tumor. c) Flow cytometry data show the significant increase of E7‐specific CD8+ T cells after combined treatment of AZD‐UPS NP with anti‐PD‐1. d) Representative flow dot plots of PD1 and TIM‐3 of CD8+ T cells in the tumor after treatment. e) Flow cytometry data show the decrease of PD1+Tim3+ CD8 T cells and increase of PD1+Tim3− CD8 T cells in TC‐1 tumors after combined treatment. In (c,e), data are presented as means ± SD, n = 5. Statistical significance was calculated by Student's t‐test: ***P < 0.001, **P < 0.01, *P < 0.05.
In summary, we report a novel nanodrug composition consisting of an MCT1 inhibitor (AZD3965) encapsulated in ultra‐pH sensitive micelles for tumor‐specific priming of microenvironment to overcome tumor immune evasion. Microfluidics method enabled successful encapsulation of AZD3965 into nanoparticles. A binary drug release profile was observed across the phase transition pH of the polymer carrier with deposition of the drug dependent on tumor acidosis and thus MCT1 activity. This specific targeting of active MCT1 sites by AZD‐UPS nanodrug allows for tumor‐selective inhibition of lactic acid exportation, which primes antitumor immunity with reduced systemic toxicity at dramatically reduced drug dose. In combination with checkpoint blockade, AZD‐UPS nanodrug effectively inhibited tumor growth by enhancing antigen‐specific CD8 T cell immunity against tumors. This study establishes the preclinical proof of concept to target monocarboxylate transporters to prime tumor microenvironment to augment cancer immunotherapy and to exploit the biological activity of a protein target to improve drug delivery specificity and efficacy.
Conflict of Interest
J.G. and B.D.S. are scientific founders and advisors for OncoNano Medicine, Inc.
Supporting information
Supporting Information
Acknowledgements
T.H. and Q.F. contributed equally to this work. This work was supported by the Cancer Moonshot Initiative from the National Cancer Institute (U54CA244719). J.G. acknowledges the support from the Mendelson‐Young Endowment in Cancer Therapeutics. The authors thank T. C. Wu for kindly providing the TC‐1 tumor cells and P. Hwu for the B16F10 cancer cells. All animal procedures were performed with ethical compliance and approval by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Huang T., Feng Q., Wang Z., Li W., Sun Z., Wilhelm J., Huang G., Vo T., Sumer B. D., Gao J., Tumor‐Targeted Inhibition of Monocarboxylate Transporter 1 Improves T‐Cell Immunotherapy of Solid Tumors. Adv. Healthcare Mater. 2021, 10, 2000549. 10.1002/adhm.202000549
The copyright line for this article was changed on 18 June 2020 after original online publication.
References
- 1.a) Zu X. L., Guppy M., Biochem. Biophys. Res. Commun. 2004, 313, 459; [DOI] [PubMed] [Google Scholar]; b) Mookerjee S. A., Goncalves R. L. S., Gerencser A. A., Nicholls D. G., Brand M. D., Biochim. Biophys. Acta 2015, 1847, 171. [DOI] [PubMed] [Google Scholar]
- 2.a) Halestrap A. P., Wilson M. C., IUBMB Life 2012, 64, 109; [DOI] [PubMed] [Google Scholar]; b) Halestrap A. P., Compr. Physiol. 2013, 3, 1611. [DOI] [PubMed] [Google Scholar]
- 3.a) Estrella V., Chen T., Lloyd M., Wojtkowiak J., Cornnell H. H., Ibrahim‐Hashim A., Bailey K., Balagurunathan Y., Rothberg J. M., Sloane B. F., Johnson J., Gatenby R. A., Gillies R. J., Cancer Res. 2013, 73, 1524; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tasdogan A., Faubert B., Ramesh V., Ubellacker J. M., Shen B., Solmonson A., Murphy M. M., Gu Z., Gu W., Martin M., Kasitinon S. Y., Vandergriff T., Mathews T. P., Zhao Z., Schadendorf D., DeBerardinis R. J., Morrison S. J., Nature 2020, 577, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fukumura D., Xu L., Chen Y., Gohongi T., Seed B., Jain R. K., Cancer Res. 2001, 61, 6020. [PubMed] [Google Scholar]
- 5.a) Brand A., Singer K., Koehl G. E., Kolitzus M., Schoenhammer G., Thiel A., Matos C., Bruss C., Klobuch S., Peter K., Kastenberger M., Bogdan C., Schleicher U., Mackensen A., Ullrich E., Fichtner‐Feigl S., Kesselring R., Mack M., Ritter U., Schmid M., Blank C., Dettmer K., Oefner P. J., Hoffmann P., Walenta S., Geissler E. K., Pouyssegur J., Villunger A., Steven A., Seliger B., Schreml S., Haferkamp S., Kohl E., Karrer S., Berneburg M., Herr W., Mueller‐Klieser W., Renner K., Kreutz M., Cell Metab. 2016, 24, 657; [DOI] [PubMed] [Google Scholar]; b) Fischer K., Hoffmann P., Voelkl S., Meidenbauer N., Ammer J., Edinger M., Gottfried E., Schwarz S., Rothe G., Hoves S., Renner K., Timischl B., Mackensen A., Kunz‐Schughart L., Andreesen R., Krause S. W., Kreutz M., Blood 2007, 109, 3812. [DOI] [PubMed] [Google Scholar]
- 6. Brand A., Singer K., Koehl G. E., Kolitzus M., Schoenhammer G., Thiel A., Matos C., Bruss C., Klobuch S., Peter K., Kastenberger M., Bogdan C., Schleicher U., Mackensen A., Ullrich E., Fichtner‐Feigl S., Kesselring R., Mack M., Ritter U., Schmid M., Blank C., Dettmer K., Oefner P. J., Hoffmann P., Walenta S., Geissler E. K., Pouyssegur J., Villunger A., Steven A., Seliger B., Schreml S., Haferkamp S., Kohl E., Karrer S., Berneburg M., Herr W., Mueller‐Klieser W., Renner K., Kreutz M., Cell Metab. 2016, 24, 657. [DOI] [PubMed] [Google Scholar]
- 7.a) Doherty J. R., Yang C., Scott K. E., Cameron M. D., Fallahi M., Li W., Hall M. A., Amelio A. L., Mishra J. K., Li F., Tortosa M., Genau H. M., Rounbehler R. J., Lu Y., Dang C. V., Kumar K. G., Butler A. A., Bannister T. D., Hooper A. T., Unsal‐Kacmaz K., Roush W. R., Cleveland J. L., Cancer Res. 2014, 74, 908; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Halestrap A. P., Meredith D., Pflugers Arch. 2004, 447, 619. [DOI] [PubMed] [Google Scholar]
- 8. Halford S. E. R., Jones P., Wedge S., Hirschberg S., Katugampola S., Veal G., Payne G., Bacon C., Potter S., Griffin M., Chenard‐Poirier M., Petrides G., Holder G., Keun H. C., Banerji U., Plummer E. R., J. Clin. Oncol. 2017, 35, 2516. [Google Scholar]
- 9. Ma X., Wang Y., Zhao T., Li Y., Su L. C., Wang Z., Huang G., Sumer B. D., Gao J., J. Am. Chem. Soc. 2014, 136, 11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Wang Y., Zhou K., Huang G., Hensley C., Huang X., Ma X., Zhao T., Sumer B. D., DeBerardinis R. J., Gao J., Nat. Mater. 2014, 13, 204; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang G., Zhao T., Wang C., Nham K., Xiong Y., Gao X., Wang Y., Hao G., Ge W. P., Sun X., Sumer B. D., Gao J., Nat. Biomed. Eng. 2020, 4, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guan X., Morris M. E., Pharm. Res. 2019, 37, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun J., Xianyu Y., Li M., Liu W., Zhang L., Liu D., Liu C., Hu G., Jiang X., Nanoscale 2013, 5, 5262. [DOI] [PubMed] [Google Scholar]
- 13.a) Feng Q., Zhang L., Liu C., Li X., Hu G., Sun J., Jiang X., Biomicrofluidics 2015, 9, 052604; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fang C., Shao L., Zhao Y., Wang J., Wu H., Adv. Mater. 2012, 24, 94; [DOI] [PubMed] [Google Scholar]; c) Feng Q., Sun J., Jiang X., Nanoscale 2016, 8, 12430. [DOI] [PubMed] [Google Scholar]
- 14. Karnik R., Gu F., Basto P., Cannizzaro C., Dean L., Kyei‐Manu W., Langer R., Farokhzad O. C., Nano Lett. 2008, 8, 2906. [DOI] [PubMed] [Google Scholar]
- 15. Zhou K., Wang Y., Huang X., Luby‐Phelps K., Sumer B. D., Gao J., Angew. Chem., Int. Ed. 2011, 50, 6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pilon‐Thomas S., Kodumudi K. N., El‐Kenawi A. E., Russell S., Weber A. M., Luddy K., Damaghi M., Wojtkowiak J. W., Mule J. J., Ibrahim‐Hashim A., Gillies R. J., Cancer Res. 2016, 76, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li J., Byrne K. T., Yan F., Yamazoe T., Chen Z., Baslan T., Richman L. P., Lin J. H., Sun Y. H., Rech A. J., Balli D., Hay C. A., Sela Y., Merrell A. J., Liudahl S. M., Gordon N., Norgard R. J., Yuan S., Yu S., Chao T., Ye S., Eisinger‐Mathason T. S. K., Faryabi R. B., Tobias J. W., Lowe S. W., Coussens L. M., Wherry E. J., Vonderheide R. H., Stanger B. Z., Immunity 2018, 49, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Fourcade J., Sun Z., Benallaoua M., Guillaume P., Luescher I. F., Sander C., Kirkwood J. M., Kuchroo V., Zarour H. M., J. Exp. Med. 2010, 207, 2175; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wherry E. J., Kurachi M., Nat. Rev. Immunol. 2015, 15, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Sakuishi K., Apetoh L., Sullivan J. M., Blazar B. R., Kuchroo V. K., Anderson A. C., J. Exp. Med. 2010, 207, 2187; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Granier C., Dariane C., Combe P., Verkarre V., Urien S., Badoual C., Roussel H., Mandavit M., Ravel P., Sibony M., Biard L., Radulescu C., Vinatier E., Benhamouda N., Peyromaure M., Oudard S., Mejean A., Timsit M. O., Gey A., Tartour E., Cancer Res. 2017, 77, 1075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information