

Abstract
ApoB Lipoproteins (apoB-Lp) are produced in hepatocytes and their secretion requires the cargo receptor sortilin. We examined the secretion of apoB-Lp-containing very low-density lipoprotein (VLDL), an LDL progenitor. Sortilin also regulates the trafficking of the subtilase PCSK9, which when secreted binds the LDL receptor (LDLR) resulting in its endocytosis and destruction at the lysosome. We show that the Site-2 binding compound (cpd984) has multiple effects in hepatocytes including: 1) Enhanced Apo-Lp secretion; 2) Increased cellular PCSK9 retention; and 3) Augmented levels of LDLR at the plasma membrane. We postulate that cpd984 enhances apoB-Lp secretion in part through binding the lipid PIP3, which is present at higher levels on circulating VLDL form fed rats relative to after fasting. We attribute the elevated VLDL secretion to its increased binding affinity to sortilin Site-1 induced by cpd984 binding Site-2. This hinders PCSK9 binding and secretion, which would subsequently prevent its binding to LDLR leading to its degradation. This suggests that Site-2 is an allosteric regulator of Site-1 binding. This effect is not limited to VLDL, as cpd984 augments binding of the neuropeptide neurotensin (NT) to sortilin Site-1. Molecular dynamics simulations demonstrate that the NT C-terminus (Ct-NT) stably binds Site-1 through an electrostatic interaction. This was bolstered by the ability of Ct-NT to disrupt lower affinity interactions between sortilin and the Site-1 ligand PIP3. Together these data show that binding cargo at sortilin Site-1 is allosterically regulated through Site-2 binding, with important ramifications for cellular lipid homeostasis involving proteins such as PCSK9 and LDLR.
Keywords: Sortilin, Neurotensin, PCSK9, apo B, LDL receptor
INTRODUCTION
Human sortilin is an ancient protein transporter, which derived its origin from primitive eukaryotes. The gene for the sortilin orthologue in Saccharomyces cerevisiae is VPS101, which codes for a protein mainly responsible for trafficking vacuolar proteases to the vacuole. Yeast Vps10 is predicted to contain two separate and unique binding domains for protein sorting2 increasing its molecular weight to ~173 kDa, whereas sortilin in humans is only synthesized as a ~93 kDa protein with one luminal beta propeller binding domain. The canonical Vps10 pathway in yeast traffics the lysosomal protease carboxypeptidase Y (CPY) from the Golgi to the vacuole, after which Vps10 is taken back to the Golgi through a retrograde pathway defined by the retromer protein complex. Vps10 interaction with the retromer complex is facilitated through in interactions with the cytoplasmic tail of Vps10 and the Rab Ypt73. The mammalian orthologue sortilin traffics proteins from the Golgi where it is activated by furin mediated cleavage of its pro-domain4, and is responsible for trafficking its ligands between secretory pathways and lysosomal protein pathways5. Both Vps10 and sortilin release cargo upon entering acidic endosomal compartments, which may be followed by dimerization of the luminal beta-propeller domains6. Subsequently, sortilin like Vps10 can be recycled back to the Golgi by the Retromer pathway7.
In higher eukaryotes, sortilin is expressed in diverse cell types where it serves as a tissue specific cargo receptor. In neuronal cells, sortilin mediates the secretion of neurotensin (NT) and other neuromodulators8, while in adipose tissue, sortilin traffics the type 4 glucose transporter Glut4 to the plasma membrane9. That said, sortilin has of recent been highly studied in hepatocytes where it primarily regulates apo B containing lipoproteins (Apo-Lp) sorting through apo B binding, however roles for apo E binding and PCSK9 regulation in hepatocytes have been examined8, 10.
Through GWAS analysis, SORT1 mutations have been identified that correlate with cardiovascular disease outcomes11, 12. Sortilin has complex relationships with lipoprotein metabolism13 and in lipid accumulation in arteries10. This is illustrated by the buildup of circulating LDL, which can be elevated when LDLR is endocytosed and degraded by binding PCSK9. At the plasma membrane, sortilin is found in clathrin-coated pits similar to the low-density lipoprotein (LDL) receptor (LDLR), both of which bind ApoB-Lp14. Differences in function and ligand specificity for the two receptors is unknown. Further complicating understanding of sortilin function is its additional role in hepatic very low-density lipoprotein (VLDL) secretion, where hepatic knockouts of sortilin have been described to either increase or decrease VLDL secretion15.
Sortilin is a multiligand receptor that can interact with the various ligands found on VLDL including B10013, apo E16, 17, and phosphatidylinositol 3,4,5-trisphosphate (PIP3)18. The PIP3 content of circulating VLDL is enriched relative to high density lipoproteins (HDL) and low-density lipoprotein (LDL)18. The paradox for the ability of sortilin to either increase or decrease VLDL secretion under different conditions could relate to the relative concentration and position of ligands on the VLDL surface. Presence of PIP3 on lipoproteins relates to insulin signaling where PIP3 is considered as a principal mediator of insulin signal transduction. The presence of PIP3 on VLDL could reveal a mechanism for short-term modulation of VLDL interaction with sortilin, though we cannot at present rule out the role for other apo B-Lp ligands for sortilin binding to VLDL such as apo E19, 20. Overall, our results favor the concept that VLDL composition determines secretion and post-secretion pathways for VLDL catabolism.
An additional ligand transported by sortilin is PCSK9 (proprotein convertase subtilisin-kexin type 9), which has been shown to bind to human sortilin with high affinity21. Importantly, another study showed that NT, a natural neuronal ligand for sortilin, did not inhibit PCSK9 binding, even at saturating concentrations22, suggesting the presence of an additional binding site on sortilin. Once PCSK9 is released by sortilin extra-cellularly, it binds to LDLR triggering its internalization and degradation of the associated complex at the lysosome. This results in decreased expression of LDLR on the cell surface, which can lead to eventual increases in circulating LDL.
In this study we present data showing that binding sortilin Site-2 with compounds such as cpd984 increases PCSK9 cellular retention while increasing VLDL secretion. Conversely, the binding of sortilin Site-1 with cpd541 decreases VLDL secretion. We suggest that this pathway is a complex and dynamic system for regulating the metabolism of apo B containing lipoproteins including LDL and its precursor VLDL utilizing both binding sites on sortilin for regulation. We propose that Site-2 allosterically regulates binding to Site-1 to affect the co-regulation of apo B and PCSK9 secretion, with the balances of this mode of modulation translating to changes in LDLR expression at the plasma membrane.
METHODS AND EXPERIMENTAL DETAILS
Cell culture, materials and reagents
McArdle RH-7777 cells (McA cells) were cultured as previously described in serum containing complete Dulbecco’s Modified Eagle’s Medium (cDMEM)23, 24. Human sortilin (sortilin) (S78-N755) with C-terminal 6-His tag was from R&D Systems. Rabbit polyclonal PCSK9 (Ab125251, 1:2000) and LDLR (LS.C146979, 1:2000) antibody was from LSBio. Mouse anti-glyceralde-hyde phosphate dehydrogenase (GAPDH) from Santa Cruz Biotechnology (sc32233 (6C5), 1:200). Lipofectamine 2000, Plus™ Reagent was from (Thermo Fisher). Anti-rabbit (NA934, 1:10,000) and anti-mouse (NA931, 1:10,000) horseradish peroxidase (HRP)-linked IgG and Hyperfilm™ were purchased from GE Healthcare. Horseradish peroxidase linked donkey anti-rabbit IgG (NA9340, 1:10,000), sheep anti-mouse IgG (NXA931, 1:10,000) and ECL Prime Western Blotting Detection Reagent (RPN2232) were from GE Healthcare. Compound 98477898 (2S)-1-methylN-3-[(3-phenypropanoyl)-amino]phenylpyrrolidine-2-carboxamied (cpd984) and compound 54122218 ([2-([(1R)-1-phenylethyl]amino)carbonyl)-phenyl]amino}acetic acid were obtained from ChemBridge Corp. Stock solutions of cpd984 and cpd541 (10 mM) were prepared in DMSO, and stored in aliquots at −20°C. DPPC (Dipalmitoyl-sn-glycero-3-phosphatidylcholine) and POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine) were purchased from Avanti Polar Lipids (Alabaster, AL) as chloroform stock solutions and stored at 20°C. DiC16 PIP3 (Dipalmitoyl phosphatidylinositol 3, 4, 5, trisphosphate) and diC8-PIP3 (Dioctanoyl PIP3) were from Echelon (Salt Lake City, UT). CM7 and Ni-NTA (standard and S series) sensor chips, and regeneration buffers (glycine pH 1–3) were procured from GE Healthcare (Buckinghamshire UK). Monolith NT.115 standard treated capillaries for thermophoresis were purchased from Nanotemper (München Germany). Membrane scaffold protein 1SD1 (MSP) was prepared as described25.
Cell Culture.
Rat hepatocytes (RH) were isolated from Sprague-Dawley rat livers and were cultured on collagen-coated dishes in Waymouth’s 751/1 medium containing 0.2% (w/v) BSA as described previously26. Wild-type McA cells were maintained in culture in complete DMEM (cDMEM)23. Inhibitors were used at reported concentrations for cpd984 and cpd541. Inhibitors were validated in RH where cell toxicity was minimal as determined by LDH release.
Knockdown of Sortilin in McA cells using siRNA.
McA cells were transfected using Fugene6 according to manufacturer’s protocol (Promega Corp.) using three different pGIPZ based vectors expressing shRNAi targeting rat Sort1 mRNA (V2LMM_58553, V3LMM_450660, V3LMM_ 450662), and one scrambled, non-silencing control (GE Healthcare Dharmacon) as previously described27. Puromycin selection was performed on McA cells. Sortilin knockdown from each cell line was examined by immunoblotting.
Immunoblotting.
McA cell lysates were prepared and denatured proteins were separated by SDS-PAGE, transferred to PVDF and incubated with primary antibodies overnight at 4°C in blocking buffer with antibody binding detected with species specific secondary HRP-linked antibodies and developed using Amersham™ Prime reagent (GE Healthcare). Chemiluminescence was measured with ChemiDocXRS+ system (Bio-Rad) and quantified using Image Lab 3.0.1 software from BioRad.
Lipoprotein Preparation and PIP3 nanodisc assembly.
Plasma VLDL, LDL, and high-density lipoprotein (HDL) from fasted rats were isolated by sequential density ultracentrifugation as previously described28. Nanodiscs were prepared as described18. Nanodiscs were composed of 3.023 μmol DPPC, 0.098 μmol diC16-PIP3, and 0.78 μmol POPE, which were combined, dried, and desiccated overnight. Lipids were dissolved in 20 mM sodium deoxycholate in TBS (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.02% (w/v) sodium azide and sonicated, after which membrane scaffold protein 1D1 (MSP1D1) was added at a ratio of 70:1 lipid to protein and detergent removed with Bio-Beads® SM-2 (Bio-Rad). Nanodiscs were isolated using size exclusion chromatography. Nanodiscs concentration was quantified using a NanoDrop and extinction coefficient of 21,000 L mol−1 cm−1 for MSP1D1, and resultant mg/ml divided by two.
Computational modeling and compound screening.
Schrödinger’s Maestro program (version 9.3.5) was used as the primary graphical user interface and Maestro version 10.2 (Schrödinger, LLC) was used for ligand interaction diagramming. Virtual screening was performed on compounds contained in ChemBridge libraries (www.chembridge.com) that were prepared with Schrödinger’s LigPrep program (Schrödinger, LLC). The virtual screening method was performed using Schrödinger’s GLIDE software29 on the sortilin crystal structure PDB ID: 4PO7 27. Compounds were docked on grids generated with Glide with cpd541 docked at a box determined by C-terminal NT and cpd984 docked at a box determined by the N-terminal fragment of NT. Grids were then adapted from alignment of PDB ID:6EHO to PDB ID: 4PO7 and docking performed for all grids using Glide XP setting with results exported into GraphPad Prism.
MD Simulations of apo and holo Sortilin.
Using the aforementioned crystal structure of sortilin (PDB ID:4PO7) molecular dynamics simulations were done using NAMD 2.1230 using the CHARMM36m force field31. Prior to simulating, the system was prepared using the CHARMMGUI solution builder, with a salt concentration of 150 mM NaCl. Simulation parameters included constant pressure of 1 atm via Langevin dynamics, as well as a constant temperature of 310 K using Langevin piston Nosé−Hoover methods32, 33.
Long-range electrostatic forces were evaluated using the particle mesh Ewald (PME) with a 1 Å grid spacing34, 35. Van der Waals interactions were calculated using a 12 Å cutoff with a force-based switching scheme after 10 Å, as well as a 2 fs time step integration via the SETTLE algorithm36. Visualization and analysis were done using VMD 1.9.337. The system was equilibrated for 20 ns restraining the Cα atoms of the protein (1.0 kcal/mol/Å2) to allow for solvation. This was followed by a production run of 50 ns without restraints for 4 poses taken from ensemble docking, 3 poses with the highest affinity pose for Site-1 of sortilin run in duplicate for each, and the pose for Site-2 that represented the predicted pose as shown previously27. Compounds were docked using GLIDE at the site where the N-terminal fragment of NT is found in the crystal structure and cpd984 was chosen for biological screening based on its docking score. Schrödinger’s PRIME software was used to generate missing side chains and loops of this crystal structure predicting the NT peptide spanning the cavity of sortilin as in27. MD simulations were performed on full length NT as well as the three potential peptides derived from this structure using Schrödinger Maestro to manipulate the N-terminal density from this structure converting it from a carboxylate to an amide. Schrödinger protein preparation wizard was run on the C-terminal density as well as the two N-terminal densities and all 3 potential structures minimized. 50 ns simulations as described above were performed on all 3 NT fragments and RMSD of all 3 simulations and final poses from each simulation exported.
Ensemble Molecular Docking of cpd541 and cpd984.
To probe cpd541 and cpd984 interactions with sortilin, ensemble molecular docking was employed as described38. Using snapshots from the 50 ns production simulation to sample protein dynamics, snapshots were taken every 200 ps. Each of the resultant 250 snapshots were used to dock cpd541 and cpd984 using a 100Å by 90Å by 70Å grid box. Docking was done with an exhaustiveness of 10, yielding a total of 2500 docked poses. Resultant poses were clustered using a hybrid K-centers/K-medoids algorithm, utilizing an RMSD method39, 40. Representative poses with highest scoring affinities in clusters closest to Site-1 and Site-2 were selected for further 50 ns simulations. The resultant drug bound simulations were analyzed with VMD as well as MDAnalysis41, 42.
Microscale Thermophoresis.
Thermophoresis measurements were performed using a Monolith NT.115 labeled thermophoresis instrument43. Sortilin-His6 was labeled with Ni-NTA Atto-488 according to the manufacturer’s protocol as previously described44, 45. M.O. Control software was used for operation of MST. Target protein concentrations were 50 nM for sortilin labeled protein. LED excitation power was set to 90% and MST set to high allowing 3 sec prior to MST on to check for initial fluorescence differences, 25 sec for thermophoresis, and 3 sec for regeneration after MST off. Analysis was performed using M.O. Affinity Analysis Software as the difference between initial fluorescence measure in the first 5 sec as compared with thermophoresis at 15 sec. All measurements were performed in PBS buffer (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4, pH 7.4) without Tween and binding affinity was generated using Graphpad Sigmoidal 4PL fit from points exported from M.O. Affinity Analysis software using KD Model with target concentration fixed at 50 nM.
Surface Plasmon Resonance.
SPR measurements were performed on a Biacore T200 instrument equipped with CM5 sensor chips with ~2000 response units (RU) of sortilin covalently immobilized to the surface for VLDL and LDL binding, (6C and 6E), ~3500 RU crosslinked sortilin for small molecule binding (6A), a CM5 with ~6500 RU crosslinked sortilin for NT binding (5A) and a CM7 chip as in18 (7A and 7C). HBS-DMSO running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 1% DMSO) was used at a flow rate of 30 μl/min and injections performed with times for association of 90 sec, and dissociation of 300 sec, followed by injection of buffer to regenerate the sortilin surface. Regeneration for CM5 NHS/EDC crosslinked sortilin required a 30 sec injection of 10 mM NaOH as described18. Binding was expressed in relative RU; the difference in response between the immobilized protein flow cell and the corresponding control flow cell. Results were exported from BiaEvaluate software into GraphPad prism (GraphPad Software). Cpd984 and cpd541 saturation curves were fit using a specific binding equation with hill slope, whereas all other SPR saturation curves were fit using a 1:1 specific binding model.
Statistics.
Unless noted, results are expressed as the mean ± S.E.M., where n equals the number of independent experiments in which replicate analyses were performed in each experiment. Significant differences were assessed using one-way ANOVA with p-values ≤ 0.05 considered significant.
RESULTS
Cpd984 and Cpd541 Bind at Different Locations of Human Sortilin.
In a previous study we defined the interaction of a small molecule compound (cpd984) with a newly defined second binding site (Site-2) on the luminal beta-propeller domain of sortilin, and its effects on the secretion of NT and VLDL27. Here we define a small molecule (cpd541) that specifically targets Site-1 on the opposite side of the beta-propeller relative to Site-2 (cpd984). Figure 1A shows the structures of both cpd984 and cpd541. Using SPR we titrated cpd984 and showed that it bound to sortilin with high affinity, having a KD of 118 nM ± 38 nM using the GraphPad Prism v. 8.4.3 using specific binding with hill slope model (Fig. 1B). Next, we determined the binding affinity of sortilin for cpd541 and found the KD to be 6.9 μM ± 1.1 μM using the same model (Fig. 1C). To determine whether binding Site-2 was independent of Site-1, we measured the binding of cpd984 in the presence of a nearly saturating concentration of cpd541 (10 μM) showing that cpd984 bound to sortilin with a similar affinity in the presence as in the absence of cpd541 just looking at the saturation curves visually (Fig. 1D). This indicated site specificity of cpd984 for Site-2 of sortilin and that its binding was independent of Site-1 interactions when coupled to previous results utilizing this compound27. These results are consistent with our previous study showing that cpd984 bound sortilin in the presence of the C-terminal portion of NT18.
Figure 1. Experimental and Computational Characterization of cpd541 and cpd984.
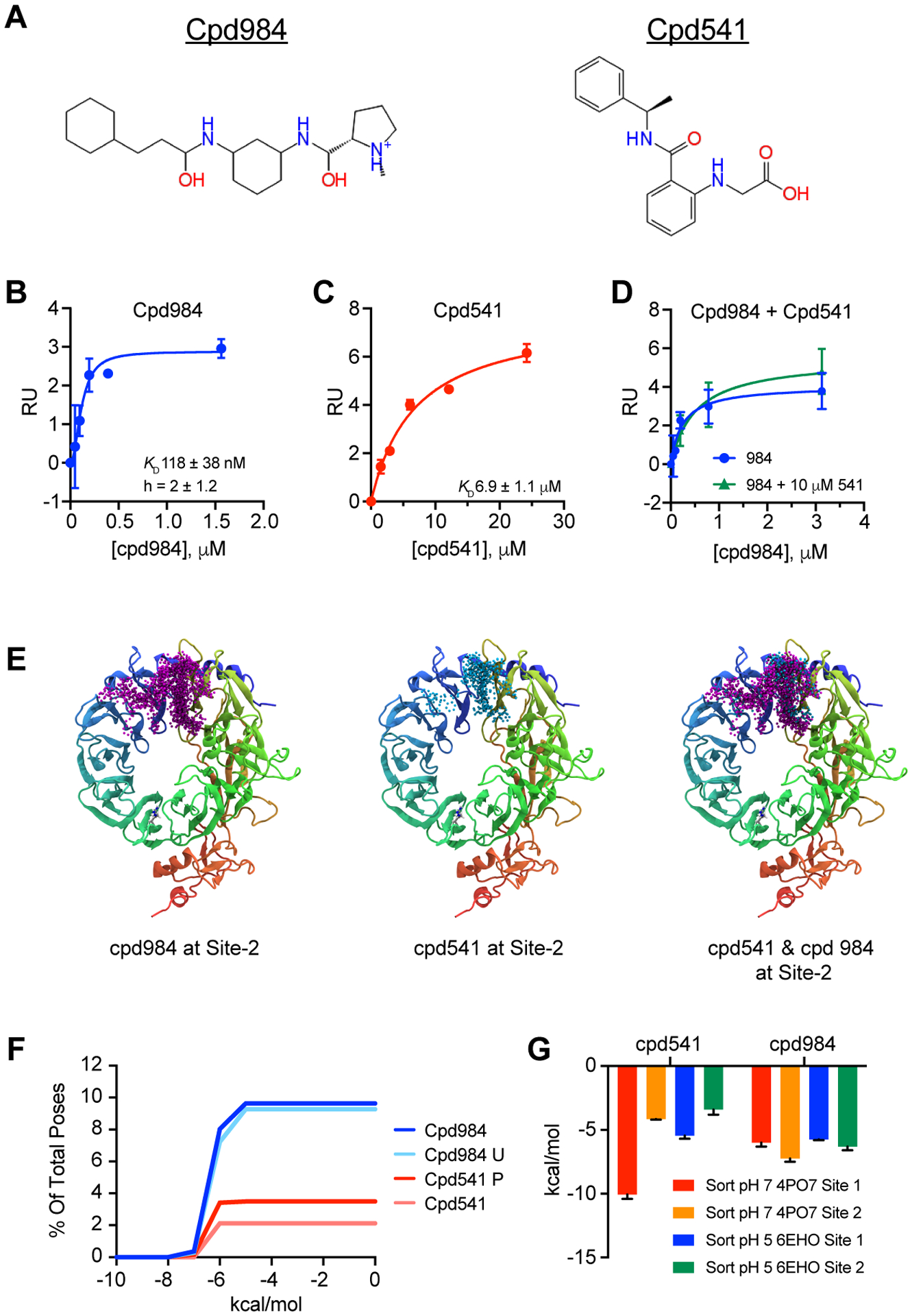
(A) Structures of cpd984 and cpd541. (B) SPR determined binding affinity of cpd984 to a CM5 chip loaded with ~3000 RU of human sortilin with saturation curve (blue) indicating KD 118 nM ± 38 nM. (C) SPR of cpd541 binding to sortilin with saturation curve (red) KD 6.9 μM ± 1.1 μM. (D) SPR of binding a cpd984 titration in the presence (green) and absence (blue) of 10 μM cpd541 with saturation curves. (E) Resultant poses from ensemble docking for cpd984 (purple) and cpd541 (cyan) at Site-2 of sortilin alone and superimposed. (F) Percent of overall poses from ensemble docking versus relative docking score for cpd984 (red) and cpd541 (blue) where the Y axis is the percent of all poses for ensemble docking that bind Site-2 of sortilin, with cpd984 having about 9 % percent of all poses in site 2 and cpd541 having about 3 percent of all poses. (G) Schrödinger Glide Xp scores of cpd541 and cpd984 docked in Site-1 and Site-2 of ~pH7 PDB ID: 4PO7 and ~pH5.5 PDB ID: 6EHO.
We employed state of the art computational techniques to further explore site specificity of these molecules to sortilin. Using ensemble molecular docking we showed the association of cpd984 and cpd541 to regions of sortilin Site-2. This showed that cpd984 associated with Site-2 with a higher rate of contacts than cpd541 (Fig. 1E). When quantitated, cpd984 was found to contact Site-2 by greater than 2-fold relative to cpd541 (Fig. 1F). Furthermore, we found that cpd984 with an unprotonated pentaamine ring had a basic pH of 8.4 (± 1.2), as determined by ab initio calculations using Jaguar46. The protonation state of cpd984 had no effect on Site-2 binding, whereas cpd541 protonation of the carboxylate resulted in the enrichment of cpd541 to Site-2 of sortilin. To test whether pH dependent conformational changes impacted Site-1 or Site-2 binding, using Schrödinger Glide XP, we computationally docked both cpd541 and cpd984 in both Site-1 and Site-2 of a crystal structure of sortilin at neutral pH (PDB ID: 4PO7) and acidic pH (PDB ID: 6EHO) (Fig. 1G). Cpd541 showed dramatically reduced binding to sortilin Site-1 at low pH as compared to cpd984, indicating that cpd541 is similar to other Site-1 ligands that bind to the NT binding site of sortilin, and lose affinity in acidic environments6. Furthermore, docking results confirm that cpd984 shows little discrimination between acidic and neutral pH structures of sortilin.
Using MD, six 50 ns simulations were run for cpd541 and cpd984 to both Sites-1 and −2 of sortilin totaling 24 simulations and over 1 microsecond of total simulation time (Fig. 2A). The three best binding poses of cpd984 to Site-1 were chosen using ensemble poses generated from clusters of Site-1, whereas the three best poses with a salt bridge from cpd541 to R292 with the best autodock docking score were chosen and all three poses run in duplicate for 50 ns each. For Site-2, we used the cpd984 pose with the closest match to our previously determined pose for cpd984 binding to sortilin Site-227. In comparison, the three best poses of cpd541 to Site-2 neatest to previously reported L539 were used27. We found that cpd984 remained within 10 Å from L539 throughout the simulation (Fig 2A). In contrast, simulations of cpd984 to Site-1 showed that it did not stay within 10 Å of R292 for Site-1. In addition to the stable association of cpd984 with Site-2, we found that cpd984 buries itself into the hydrophobic sortilin beta-propeller, between blades 1 and 10. Using F555 as a reference amino acid nearing the end of blade 10 of sortilin, we show that cpd984 on average over six 50 ns simulations comes closer to this hydrophobic cavity over time (Fig. 2C). A representative endpoint of one of these simulations is presented with a ligand interaction diagram in Figure 2B. Taken together, we hypothesized that the hydrophobic cpd984 was more likely to stay bound to Site-2 than to Site-1. Additionally, as cpd984 does not carry a negative charge at acidic or neutral pH and has greater association with Site-2 than Site-1 of sortilin, it was likely that cpd984 binding to sortilin was pH independent in the cell.
Figure 2. Molecular Dynamics Simulations of cpd984 to Sortilin Site-1 and Site-2.
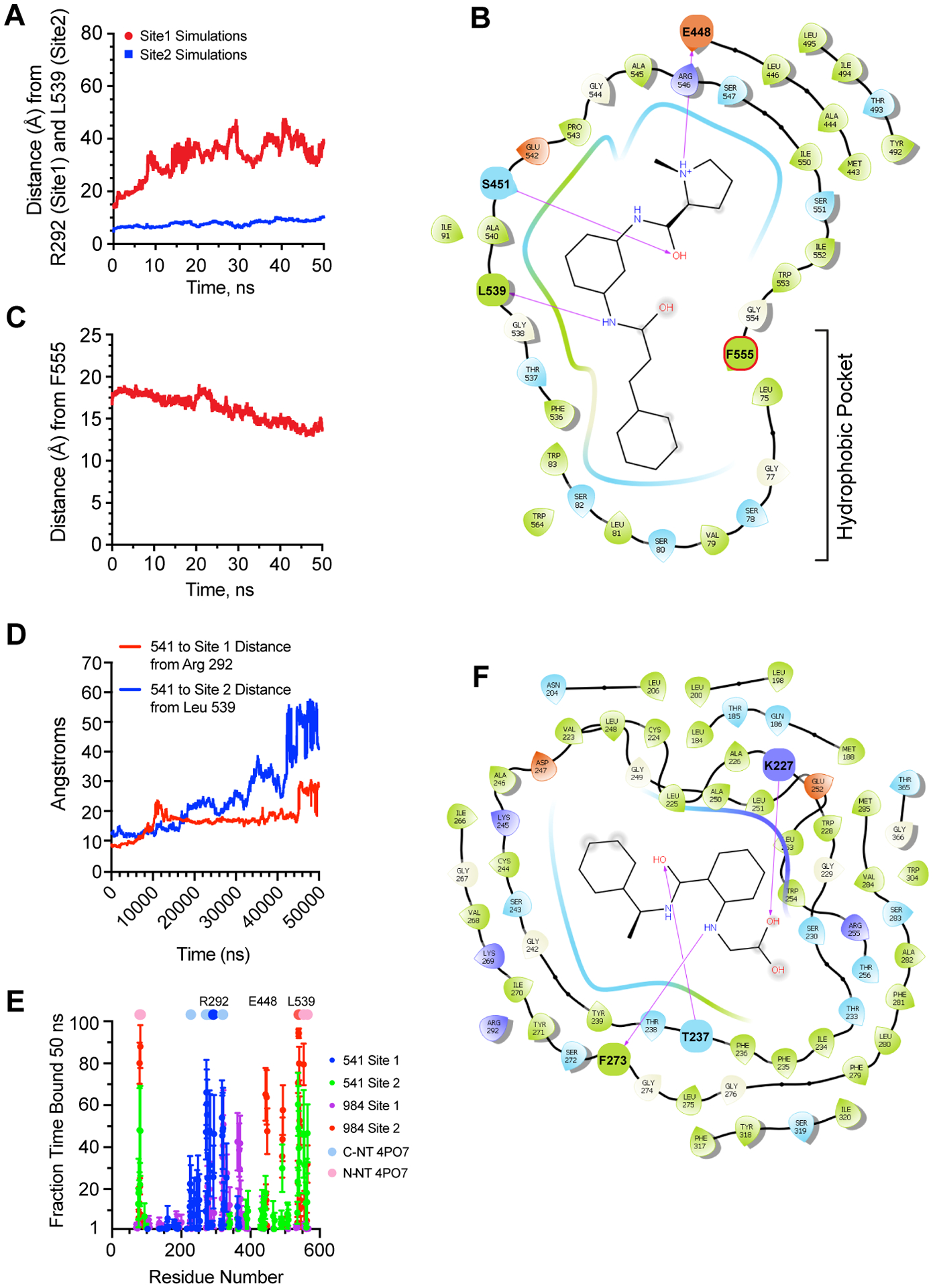
(A) Simulations of bound cpd984 poses taken from ensemble docking as in Figure 1D, 1E with Site-2 pose taken from closest ensemble docking pose to pose (n=6) and the three highest scoring poses for cpd984 to Site-1 (n=2 each) based on ensemble docking. Center of mass difference over time (50 ns) between cpd984 and representative residue of Site-1 R292 for the three duplicate simulations of cpd984 to Site-1 are shown in red, whereas center of mass differences between cpd984 and representative residue of Site-2 L539 for the six duplicate simulations of cpd984 to Site-2 are shown in blue. (B) Ligand interaction diagram taken from a representative simulation from Figure 2A of cpd984 to Site-2 of sortilin at the end of the 50 ns MD simulation. (C) Center of mass difference over time as in Figure 2a between cpd984 and a representative residue of the hydrophobic ligand binding pocket of sortilin adjacent to Site-2 F555 of six simulations of Site-2 bound ligands). (D) Simulations of bound cpd541 as in Fig. 2A, with center of mass taken from R202 and L539 as in Fig. 2A, from top 3 cpd541 ensemble docking poses in Site-2 (n=2) and cpd541 poses from Site-1 with a salt bridge between cpd541 and R292. (E) Residue analysis for all 24 50 ns simulations of cpd984 and cpd541 in Site-1 and Site-2 of sortilin with fraction time bound calculated by determining number of frames (5000) where a residue of sortilin was within 3.5 Å of either cpd984 or cpd541. C-term and N-term NT used as a legend at 100% over the one frame taken from PDB coordinates of PDB ID: 4PO7. (F) Ligand interaction diagram taken from a representative simulation from Figure 2D of cpd541 to Site-1 of sortilin at the end of the 50 ns MD simulation.
Regarding cpd541, we hypothesized that it required some electrostatic binding to form a salt bridge with R292 and stably interact with Site-1 via its carboxylate. Our data show that cpd541 did not stay as tightly associated to Site-2 of sortilin as compared to cpd984 (Fig. 2D). In contrast, cpd541 stably associated with Site-1. That said, not all cpd541 simulations remained associated with R292. A fraction of cpd541 simulations started with a salt bridge to R292, eventually stably forming a salt bridge with K227 depicted in one of the final frames of a cpd541 simulations to Site-1 of sortilin (Fig. 2F).
To visualize the interactions of cpd984 and cpd541 with different residues on sortilin, we analyzed individual contacts over time in all MD simulations (n = 24) to determine whether either cpd541 or cpd984 stayed within 3.5 Å of a given residue of sortilin. Here, each of the 50,000 frames analyzed represent 1 ps. From this it was clear that cpd984 had a greater fraction bound in Site-2 relative to cpd541 using this rubric, and that cpd541 had a greater fraction time bound to Site-1 (Fig. 2E). Furthermore, the residues of interest for these interactions corresponded well to the NT peptide binding modes of sortilin from PDB ID: 4PO7, which we have hypothesized represent the full NT peptide across the sortilin cavity connecting Site-1 and Site-2 of sortilin27.
Modulation of PCSK9 and LDLR by cpd984.
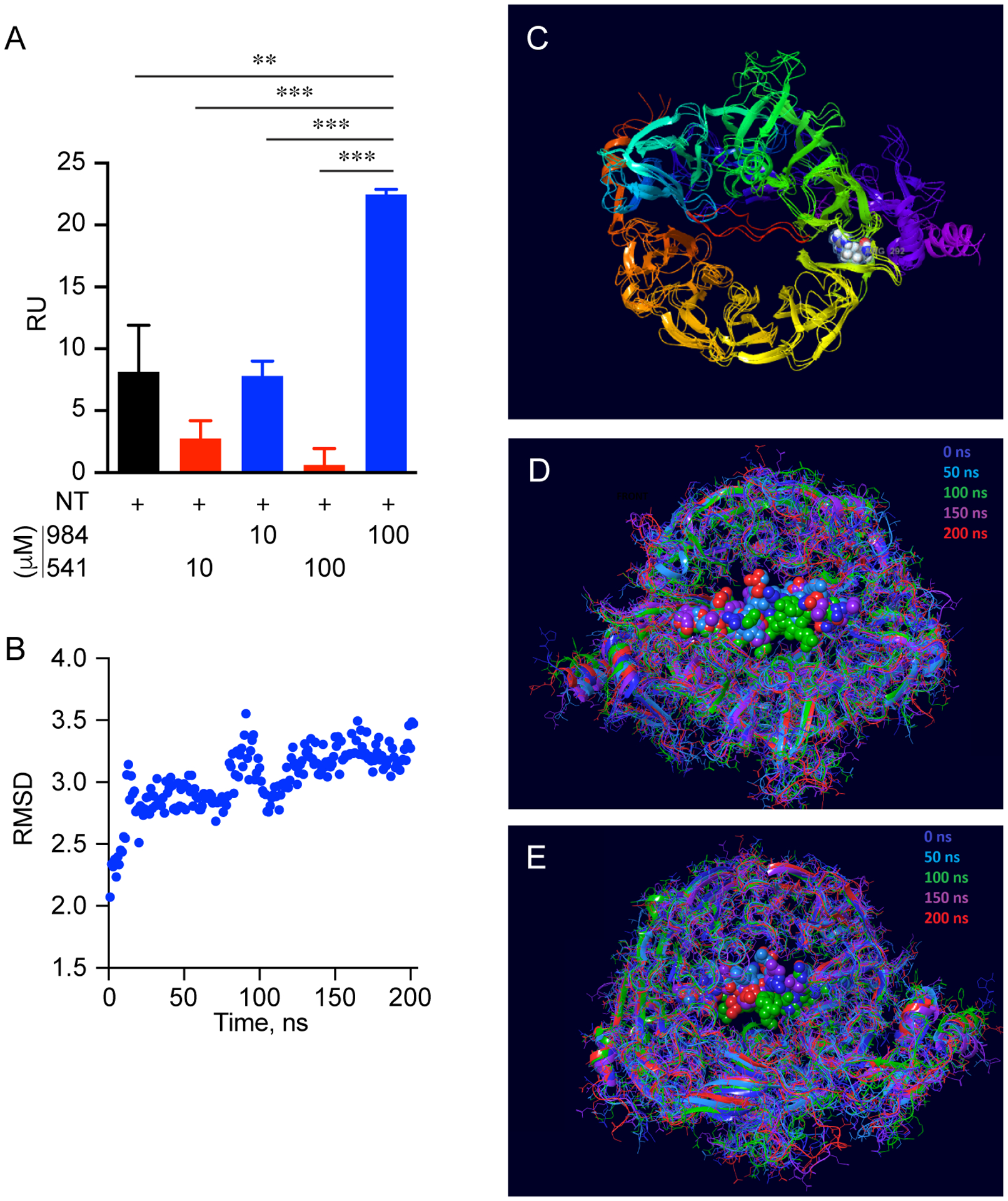
While the above computational work was essential to see how and where these compounds interact with sortilin, it was important to investigate the downstream effects on biological sortilin ligands. For this purpose, we used the McCardle hepatocyte (McA) cell line to examine sortilin-mediated trafficking of PCSK9 and LDLR when cells were treated with cpd984 and cpd541. We found that treatment with 10 μM cpd984 increased the level of intracellular PCSK9, suggesting that its secretion was reduced by cpd984-sortilin interactions (Fig. 3A). This also showed a significant increase in the total amount of LDLR by greater than 2-fold. Because LDLR is membrane anchored and is known to be degraded through the endolysosomal pathway, the effect of cpd984 can be attributed to inhibition of degradation of PCSK9 by the lysosome. Importantly, the administration of 10 μM cpd541 had little effect on the levels of PCSK9 or LDLR. These results led us to hypothesize that increased cellular PCSK9 corresponded to its decreased secretion, which would serve as an explanation for subsequent reduction of the endocytosis and degradation of LDLR-PCSK9 complexes. Based on these observations we posit that cpd984 administration results in increased sortilin-VLDL binding, reducing the availability of sortilin to bind to PCSK9.
Figure 3. Biological Effect of cpd984 and cpd541 Treatment in McCardle on Cellular PCSK9 and LDLR Levels.
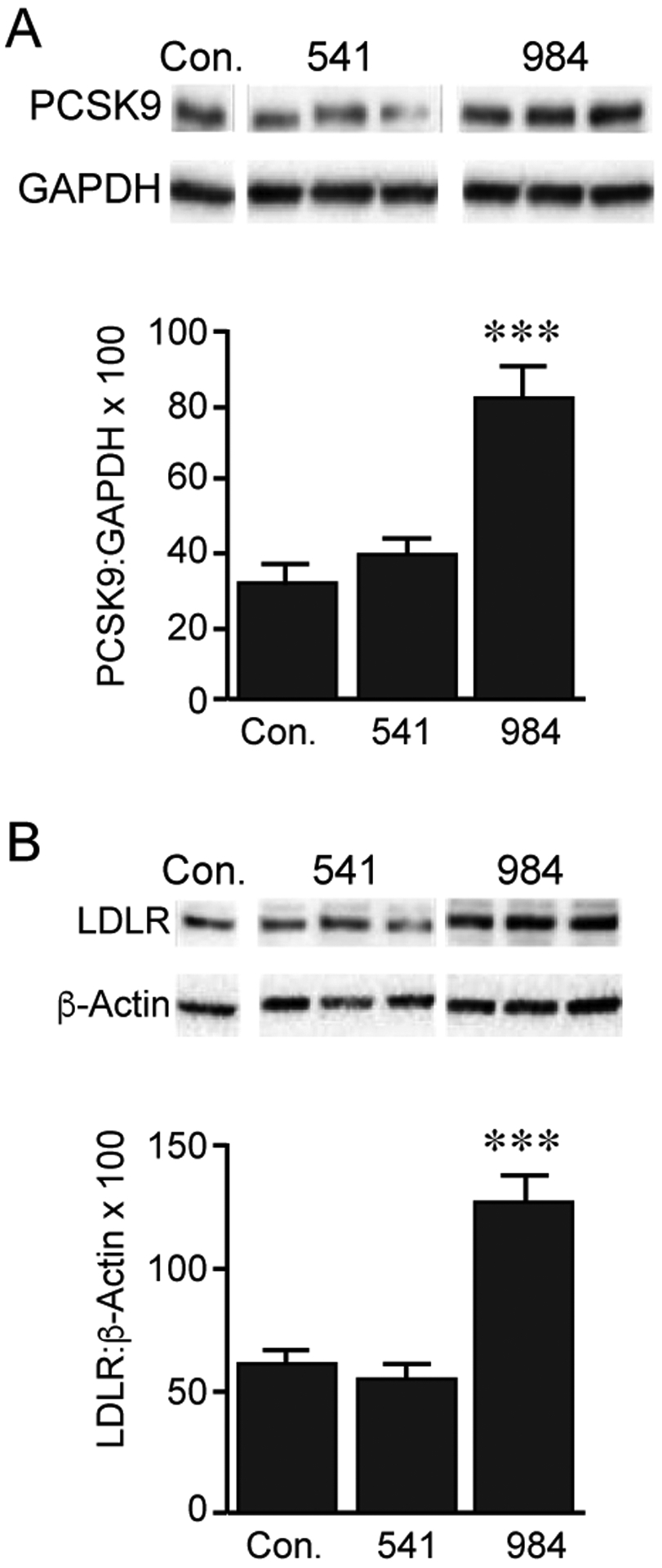
McA cells were incubated in 1% BSA/DMEM with DMSO, cpd541 (10 μM) or cpd984 (10 μM) for 18 h. Cellular proteins were extracted and analyzed by immunoblotting for LDLR (LS-C146979, 1:2000) (A), or PCSK9 (Ab125251, 1:2000) (B) using HRP-linked secondary antibody and ECL detection. Loading controls included – beta-Actin (Rockland ph600-401-886, 1:1,000) and GAPDH (sc-32233 (6C5), 1:200). Band intensities were measured using ChemiDocXRS+ system and evaluated using ImageLab 5.1 software. Error bars represent SEM (n=3). *** p<0.001 (one-way ANOVA with multiple comparisons).
Sortilin-targeting Small Molecule alters NT Binding and apo B Secretion.
The effect of cpd984 on PCSK9 binding suggested that other sortilin cargo could be affected by allosteric conformational changes to sortilin. We first examined the dependence of sortilin levels on the effect of cpd984. This was done utilizing siRNA to differentially knockdown sortilin expression in McA cells. Sortilin knockdown cell lines 1–4 expressed 95%, 70%, 40% and 10% of sortilin, respectively, relative to the scrambled siRNA control cells (Fig. 4A). Using these McA cells lines we measured apo B secretion under conditions of 10 μM cpd984 and found that cpd984 enhanced apo B secretion in proportionally with sortilin expression. We hypothesized that binding Site-2 with cpd984 allosterically enhanced VLDL binding at Site-1, which would explain increased secretion of VLDL. This also shows that sortilin is the rate limiting ligand and that overall cellular effects of site-specific sortilin modulation represent trafficking balances within the cell. In parallel, we tested whether this trend held when cells containing normal levels of sortilin were incubated with increasing concentrations of the Site-1 and Site-2 specific molecules. To do this, we varied the concentrations of cpd984 and cpd541 with cells expressing wild type levels of sortilin. These results showed that cpd984 enhanced apo B secretion in a dose dependent manner (Fig. 4B). Cpd541 on the other hand, showed a does dependent inhibition of apo B secretion. This is in keeping with the notion that binding Site-2 with cpd984 allosterically affected Site-1 interactions with apo B.
Figure 4. Cpd984 Increased LDLR levels and decreases PCSK9.
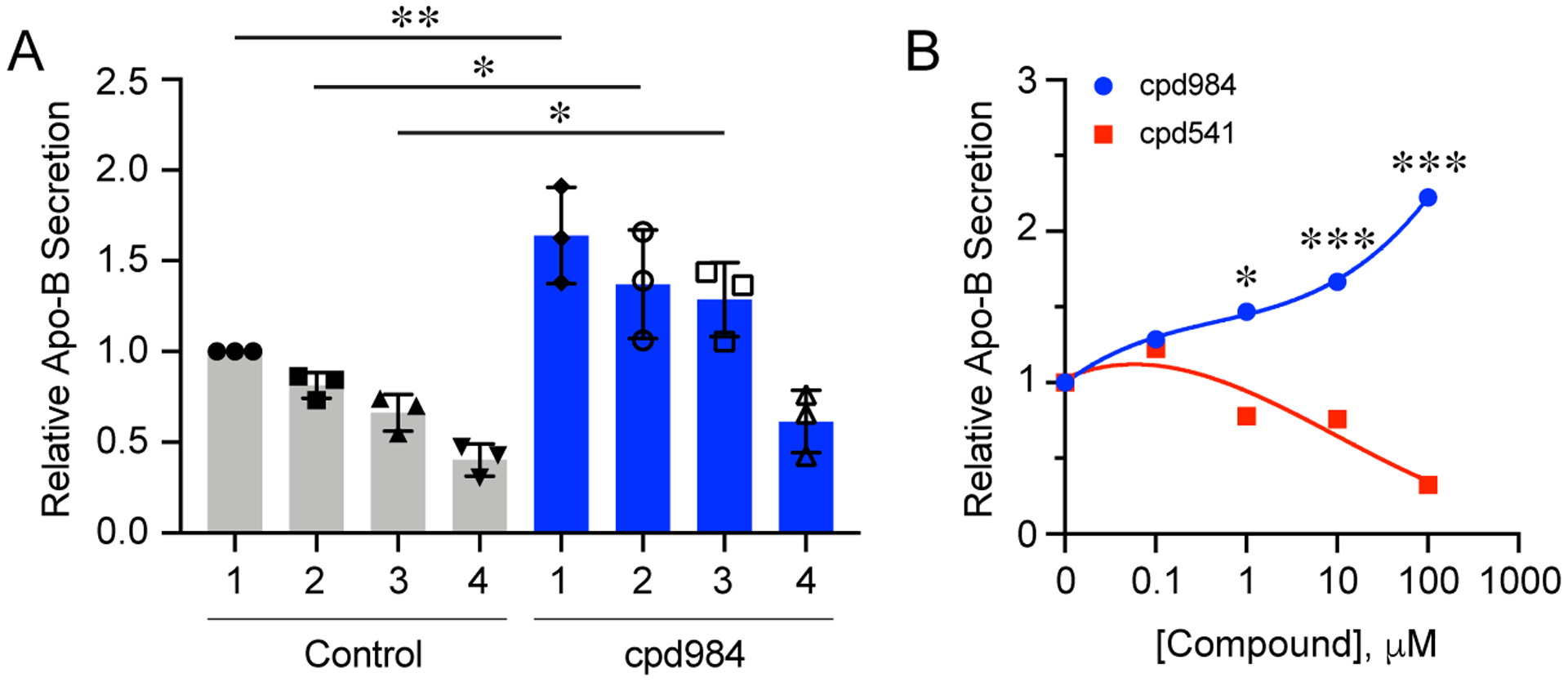
(A) McA knockdowns with varying levels of sortilin were measured for secretion of VLDL-B100 into media and assessed by immuno-slot blot. Sortilin knockdown cell lines 1–4 expressed 95%, 70%, 40% and 10% of sortilin, respectively, relative to the scrambled siRNA control cells. Cells were treated with 10 μM cpd984 or media alone (1% BSA/DMEM). Results are the average of triplicate plates for each condition. Error bars represent SEM (n=3). * p<0.05, ** p<0.01 (one-way ANOVA with multiple comparisons). (B) Insulin sensitive McA were incubated with increasing concentrations of cpd541 or cpd984 and secretion of VLDL-B100 into media was assessed by immune-slot blotting. Each curve was normalized to 0 μM treatment that was set to 1. Error bars represent SEM (n=3). * p<0.05, *** p<0.001 (two-way ANOVA with multiple comparisons).
Characterization of VLDL and LDL Sortilin Affinity.
Having concluded previously that PIP3 binds to Site-1 of sortilin17, we tested the effects of cpd541 and cpd984 on PIP3 binding to sortilin to determine whether PIP3 binding to sortilin could be allosterically modulated through Site-2 on sortilin. SPR experiments showed that soluble short chain diC8-PIP3 bound with high affinity to crosslinked sortilin with a KD of 4.2 μM ± 0.4 μM using the GraphPad Prism v. 8.4.3 specific binding with hill slope model (Fig. 5A). When we tested the same diC8-PIP3 concentration curves in the presence of 10 μM cpd541 and 10 μM cpd984, we found that cpd541 abolished PIP3 binding, whereas cpd984 enhanced diC8-PIP3 binding to sortilin by 10-fold with a KD of 474 nM ± 85 nM using the same fit in Figure 5A. These data suggest that PIP3 binding to Site-1 can be regulated by Site-2 conformational changes and demonstrates that the effect of cpd984 is not limited to the apo B ligand present on the VLDL surface. To determine if these effects held true for more native forms of PIP3 that might be available for sortilin binding, we used nanodiscs containing 2.5% diC16-PIP3. Sortilin bound PIP3 nanodiscs with an increased binding affinity over diC8-PIP3 for sortilin with a KD of 55 nM ± 13 nM indicating that a full bilayer is important for the interaction between PIP3 and sortilin binding, wherecpd984 augmented the affinity of this interaction by 10-fold for a KD of 5.4 nM ± 0.8 nM using GraphPad Prism v. 8.4.3 one-site specific binding model for both fits (Fig. 5B).
Figure 5. SPR of Sortilin Lipoprotein Binding.
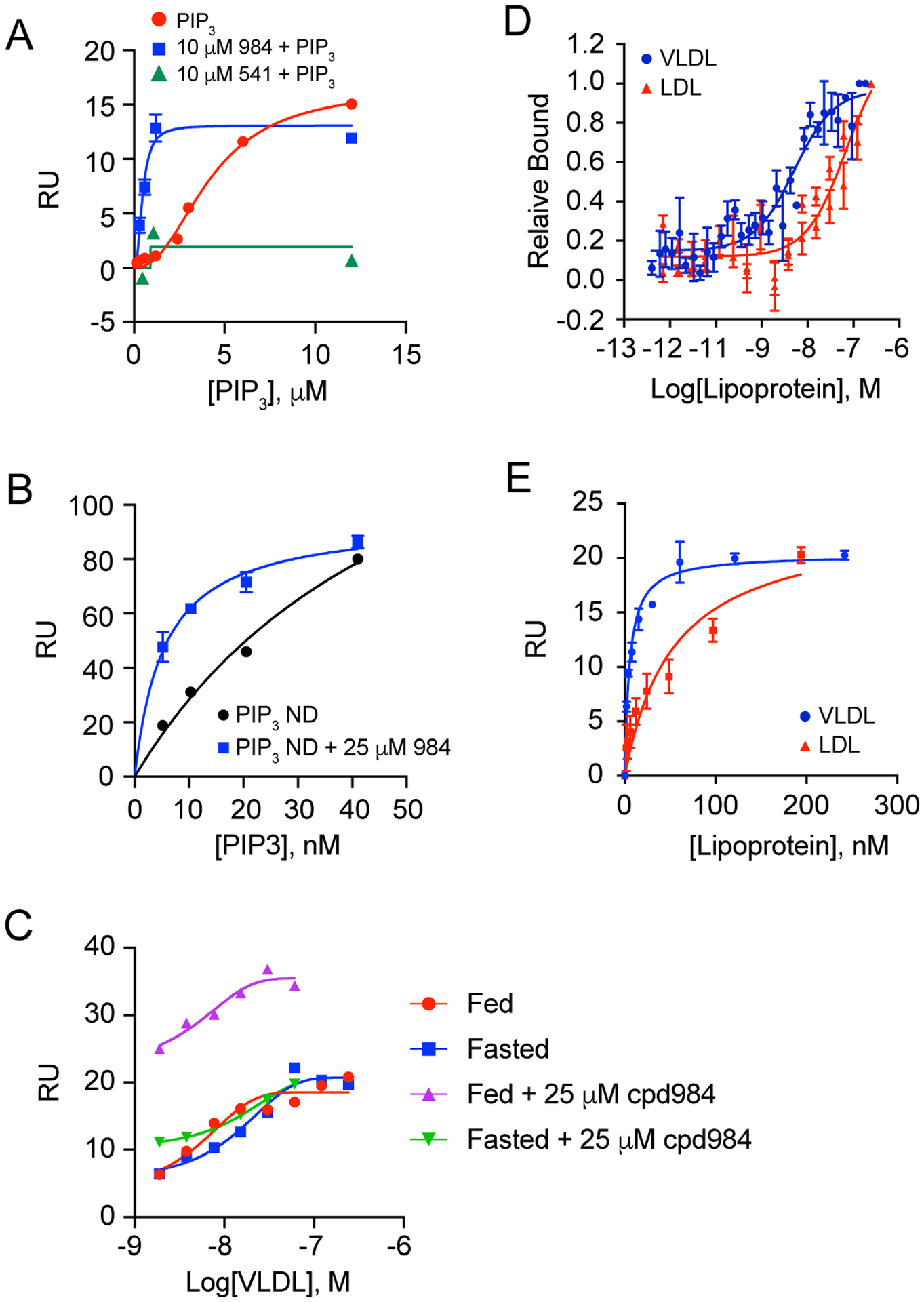
(A) SPR as in Figure 1A of diC8-PI(3,4,5)P3 binding measurements in the presence and absence (red) of 10 μM cpd984 (blue) and cpd541 (green) indicating Di-C8 PIP3 bound with a KD of 4.2 μM ± 0.4 μM, which was enhanced to a KD of 474 nM ± 85 nM in the presence of 984. (B) SPR analysis using PIP3 nanodiscs was performed using a CM7 chip containing ~27,000 RU of hsortilin indicating binding affinity of KD 54 nM, which was enhanced roughly 10-fold in the presence of 25 μM 984. (C) SPR of sortilin binding to VLDL fed fraction in the presence of cpd984 and otherwise no difference in the saturation curves for VLDL binding to sortilin harvested from rats under both a fed and fasted diet. (D) Labeled microscale thermophoresis (MST) using Atta-647 Ni-NTA labeled sortilin (12.5 nM) measured with 32 different concentrations of VLDL and LDL run in triplicate and analyzed using Nanotemper M.O. Affinity Analysis software with KD for VLDL of ~4 nM and for LDL of ~74 nM. (E) SPR performed using a CM5 chip containing ~2000 RU of sortilin of circulating rat VLDL lipoprotein fed and fasted fractions pooled for overall KD for VLDL of ~5 nM and for LDL of ~54 nM.
In vivo, levels of PIP3 are increased under conditions of insulin stimulation. We hypothesized that VLDL harvested under conditions of insulin stimulation would show increased binding to sortilin as compared to VLDL isolated under conditions of decreased insulin. To test this, we used VLDL purified from fed and fasted rats. VLDL samples were titrated (n=2 on two different chips, also used a CM7 data not shown) over sortilin attached to a CM5 chip with ~2000 RU sortilin where we found that the KD for fed VLDL using a 95% confidence interval and GraphPad Prism v. 8.4.3 one-site specific binding model to be between 2.87 and 5.27 nM and between 3.96 and 10.8 nM for fasted VLDL (Fig. 5C). In the presence of cpd984 binding affinities were shifted higher for both fed and fasted with 95% confidence intervals of 404 pM to 1.47 nM for Fed VLDL and 400 pM to 4.71 nM for fasted VLDL in the presence of 25 micromolar cpd984. The difference in measured binding affinity between fed and fasted VLDL fractions in the presence of cpd984 showed doubled the amount of bound VLDL to sortilin harvested from fed rats (Fig. 5C). This data lends support to the hypothesis that insulin signaling might affect VLDL particle composition resulting in altered trafficking patterns that depend on protein sorting chaperone sortilin. This data is consistent with the hypothesis that sortilin binding to VLDL was increased by the presence the Site-1 ligand PIP3 as cpd984 increased binding only to the fed fraction of VLDL, which we propose to be a condition for generation of this signaling molecule nearby where Apo B and VLDL is synthesized and incorporated into the ER in liver cells (Fig. 5A, B).
We previously showed that circulating VLDL contains more PIP3 than LDL18. We thus predicted that VLDL would bind sortilin with higher affinity compared to LDL. For this purpose, we purified VLDL and LDL from rat plasma and tested sortilin binding by SPR and MST. The KD for VLDL was between 4–5 nM and the KD for LDL was between 54–74 nM using fits generated from GE Biacore T200 Evaluation Software v.3.2 (Fig. 5E) and this was comparable with the average KD obtained using Nanotemper M.O. Affinity Analysis Software v.2.1.5 (Fig. 5D) which are plotted in GraphPad Prism v. 8.4.3 using a one-site specific binding model and a sigmoidal model for SPR and MST respectively. The lipoproteins were analyzed for PIP3 content and determined to be consistent with previous results18 (Fig. 5D, E). We hypothesized that this binding affinity difference was due to differences in particle composition, where VLDL acquired PIP3 during co-synthesis with apo B. These data suggest the differences between LDL and VLDL binding was independent of apo B, as both particle types contain the protein, suggesting that the primary causative factor for binding affinity difference to be the presence of another ligand, which we hypothesize to be PIP3.
Cpd984 and Neurotensin binding to Sortilin Site-1.
Thus far this study has focused on the effect of cpd984 on hepatocyte cargo binding to sortilin. To test if cpd984 had similar effects to cargo from another tissue such as neuronal, we tested the effect of this compound on the characterized binder of sortilin, neurotensin or NT. Using SPR we monitored NT binding to sortilin and found that cpd541 competed for NT binding to sortilin in a dose dependent fashion (Fig. 6A). In contrast, cpd984 enhanced NT binding to sortilin in a dose dependent fashion27. As NT binding has been shown to be mainly guided by Site-1 interactions, we hypothesize that cpd541 functions as an inhibitor by binding to Site-1 to compete off NT binding to sortilin47. The effects of cpd984 increasing NT binding to sortilin suggest that the primary interaction of NT was with Site-1 of sortilin.
Figure 6. Dynamics of Full-Length NT Binding Sortilin Luminal Domain.
(A) SPR analysis following administration of 100 nM NT alone and in the presence of either cpd984 or cpd541 at concentrations of 10 and 100 μM. RU subtractions of cpd984 and cpd541 alone were performed for injections at the corresponding concentrations in the presence of neurotensin in order to depict the effect of these compounds on the binding of neurotensin to sortilin. Canonical binding to sortilin Site-1 demonstrated by competition of cpd541 for NT binding to sortilin. Non-canonical binding of Site-2 by cpd984 enhancement NT binding on sortilin. (B) RMSD of 200 ns MD simulation of full-length NT across the central cavity of sortilin based on PDB ID: 4PO7. (C) NT backbone strands taken at 0, 100 and 200 ns. (D) Space filling model of NT bound to sortilin taken from vantage opposite transmembrane attachment site of aligned slices of sortilin molecular dynamics simulations with 0 ns in gray, 50 ns in light blue, 100 ns in light green, 150 ns in purple, and 200 ns in red. (E) Space filling model of same poses as in taken from vantage facing transmembrane attachment site of sortilin. Error bars represent SEM (n=3). ** p<0.01, ***, p<0.001 (one-way ANOVA with multiple comparisons).
We previously hypothesized that NT binds across the sortilin beta-propeller cavity connecting Site-1 with Site-2 of sortilin in line with previous observations from the structural biology groups responsible for determining the structure of sortilin at various concentrations of NT27, 48. We now show the results of an MD simulation utilizing our previously predicted NT-Sortilin bound structure. Over the course of a 200 ns simulation, full length NT stayed stably bound across the sortilin beta-propeller throughout this and two other 100 ns simulations. The RMSD over the course of the 200 ns trajectory is presented (Fig. 6B). To visualize the movement of NT we took NT poses from 0, 100 and 200 ns slices of the simulation and overlaid them to show their proximity to R292 demonstrating that NT stayed stably bound to R292 over the course of the 200 ns simulation (Fig. 6C). While the strands differed in positioning across the central cavity of the beta-propeller, they stayed stably bound to the hydrophobic pocket of Site-2 in addition to Site-1 of sortilin. Variability in NT alignment across the beta propeller central cavity was found when 5 slices of the 200 ns simulation were exported at 50 ns intervals, and the strands of NT were colored according to the time of the simulation. These simulations were aligned using Schrödinger’s Maestro and results shown for both the front and back of the propeller central cavity (Fig. 6D, E) bound to full length NT indicating that both halves of NT stayed bound across the central cavity over the course of 100 ns simulation. These results indicated that there was variability in the conformation of NT in the center of the cavity of sortilin. We propose that the additional amino acids connecting the C-terminal and N-terminal portions of NT are not required for physical binding to sortilin and therefore could have an additional role in modulating sortilin. These simulations taken together support that NT stays stably bound to Site-1 and that it serves as a bona fide ligand for Site-1 binding as previously suggested, though it may include some cooperative binding from Site-2 as C-terminal NT we showed to bind with lower affinity than full length NT and we now show stable Site-2 binding of NT over the course of a medium length MD simulation18.
Neurotensin Fragments Utilized to Define Site Specific Sortilin probes.
MD simulations showed that NT stably associates to both Site-1 and Site-2 of sortilin. To determine the binding dynamic of both ends of NT with sortilin, we generated 4 amino acid constructs of the N- and C-termini of NT (Fig. 7A). First, we examined the effects of these NT truncations on the binding of PIP3 by sortilin using nanodiscs containing 1% PIP3. This showed PIP3 binding to sortilin was inhibited by the C-terminal fragment of NT (Ct-NT) with an IC50 of 157 nM using GraphPad Prism v. 8.4.3 and a 95% confidence interval of 43 to 557 nM in line with our previously reported KD for Ct-NT to sortilin of 138 nM18. This is in contrast to essentially no inhibition at similar concentration administration of the N-terminal fragment of NT (Nt-NT) (Fig. 7B). This is in agreement with simulations showing that the C-terminus of NT interacts with Site-1, where PIP3 binds. The lack of an effect by Nt-NT was in keeping with our previous report as well as a report from Quistgaard et al., which indicated that Nt-NT did not bind to sortilin18, 47. While this was similar to other findings, it did not reflect the modeling showing that full-length NT interacted with both Site-1 and Site-2 of sortilin. We theorized that the C-terminal carboxylate of Nt-NT could prevent interactions with the central portion of the beta-propeller. To test this, we modified the C-terminal end (PDB ID: 4PO7) to replace the terminal carboxylate with an amide, which we term Nt-NT-amide. This allowed the Nt-NT-amide to bind Site-2 with a KD of ~170 nM using a one-site specific binding model in GraphPad Prism v. 8.4.3, which was close to the affinity of Ct-NT for binding Site-1, with a KD of ~120 nM using the same model (Fig. 7C).
Figure 7. NT as a Small Molecule Probe and Development of Novel NT Sortilin Probe.
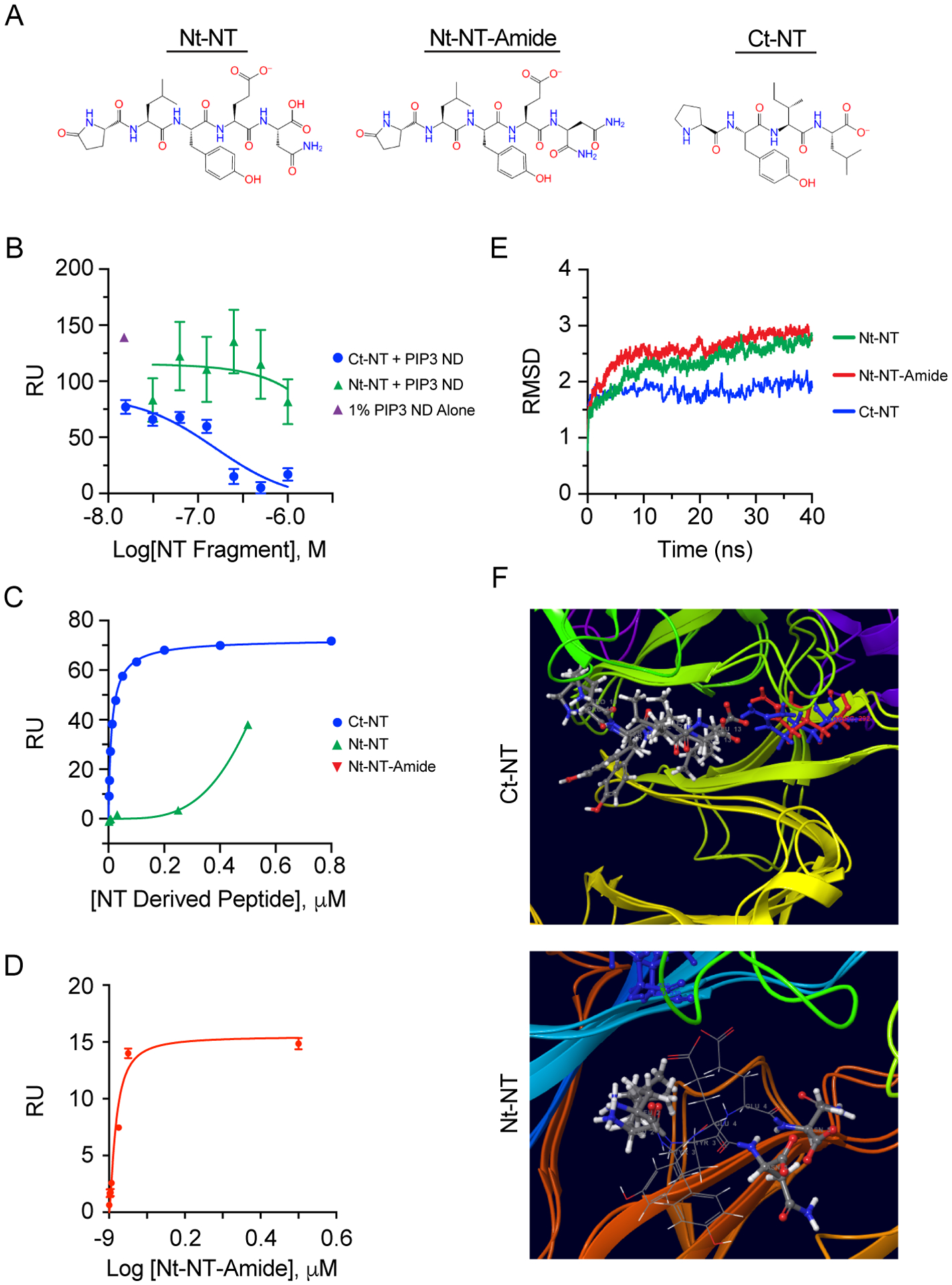
(A) Structures of NT fragments. (B) Sortilin SPR using NT fragments from PDB ID: 4PO7 showing Ct-NT inhibits sortilin binding to 79/20/1 PC/PE/PIP3 containing nanodiscs whereas the Nt-NT shows no binding. (C) SPR traces of Nt-NT and Ct-NT binding to immobilized sortilin. (D) SPR trace of Nt-NT-amide to sortilin. (E) RMSD of MD of triplicate 50 ns simulations of NT fragments. (F) Representative pre/post MD simulation with both structures aligned, and R292 for start of simulation in shown in blue and red at the end of the simulation, indicating maintenance of salt bridge with Ct-NT. (G) Representative pose of Nt-NT-amide fragment bound to the hydrophobic pocket of Site-2 with the amide bond facing away from the hydrophobic pocket of sortilin.
We extended our MD analysis of these NT fragments to determine if they stably bound to sortilin, and whether the N-terminal pose generated in Site-2 of sortilin from PDB ID: 4PO7 was biologically relevant. This showed that the Nt-NT, Nt-NT-amide and Ct-NT fragments were stably bound to sortilin over the course of 50 ns simulations (n=3 for each type of simulation). Interestingly there was more overall flux in the backbone of sortilin for the two Site-2 N-terminal NT simulations (n=3 for each) on average across these 6 simulations as compared to the Ct-NT simulations, with a slight increase for the Nt-NT-amide fragment over the Nt-NT-carboxylate (Fig 7E). Final poses from all three C-terminal (Fig. 7F) and N-terminal NT (Fig. 7G) simulations are shown indicating stable salt bridge formation of Arg292 with the C-terminal carboxylate of Ct-NT and burying of the N-terminal portion of NT opposite the end of the C-terminal carboxylate or amide. These data indicate the usefulness of NT fragments for analysis of site-specific binding of sortilin and validate our previous observations of small molecule probe specificity to sortilin.
A model for allosteric regulation sortilin binding.
This study has shown that engaging Site-2 of sortilin results in demonstrable consequences for sorting of sortilin cargo that depend on Site-1 of sortilin for secretion. We now propose a model for site specific regulation of sortilin trafficking including a model for how the administration of Site-1 and Site-2 specific sortilin binding molecules result in altered hepatic protein trafficking (Fig.8). In this model, cpd984 binds to Site-2 and induces conformational changes allowing greater binding of VLDL to sortilin through Site- 1 interactions. Increased binding of VLDL to sortilin results in decreased binding of PCSK9 to sortilin due to crowding at the sortilin binding interface. The result of decreased binding of sortilin to PCSK9 is that PCSK9 is diverted away from secretion resulting in increased cellular PCSK9 retention reflected by increased LDLR at the plasma membrane. Modulation of LDLR by inhibiting PCSK9 binding to LDLR is currently a frontline treatment for diseases such as hypercholesterolemia. We believe that the finding that LDLR can be modulated by targeting sortilin to be an important fundamental consideration in future treatments targeted towards achieving healthy circulation in patients with high cholesterol.
Figure 8. Model for allosteric regulation of sortilin binding.
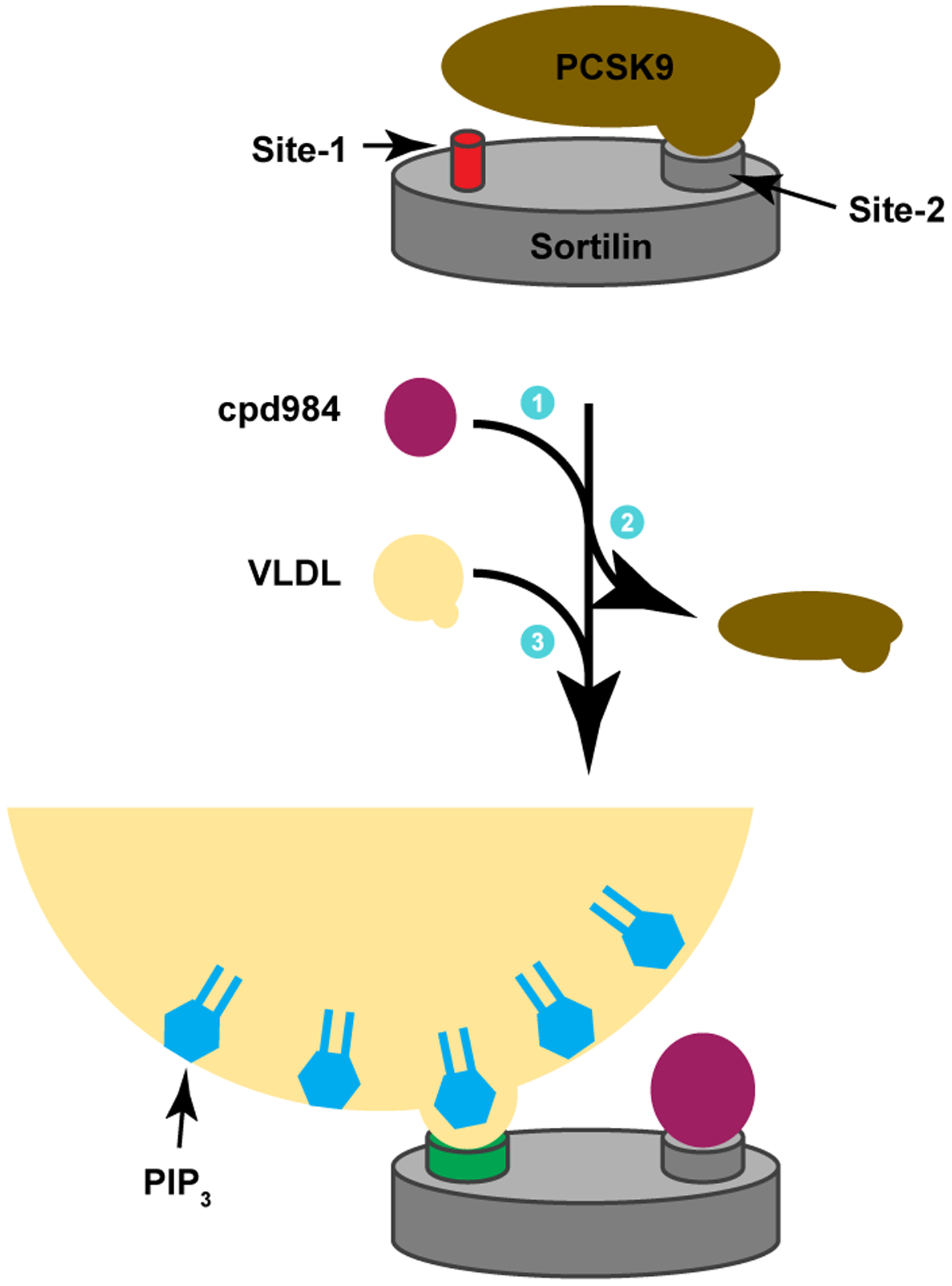
Shown is a schematic representing the allosteric effect of binding Site-2 with cpd984 and its effects on Site-1 binding by VLDL (yellow). The sortilin beta-propeller is shown with Sites-1 and −2. Site-1 is shown as low affinity (red) for VLDL binding. Once cpd984 (purple) binds Site-2, PCSK9 (brown) is released, and Site-1 undergoes a conformational change (green) to enhance VLDL binding. VLDL is shown to have PIP3 (cyan) to improve Sit-2 binding.
DISCUSSION
The endocytic pathway for ApoB-Lp uptake by LDLR in hepatocytes has been studied extensively and forms much of the basis for current therapies controlling high LDL cholesterol in humans49. That said, little is known about the role of sortilin in this process or how the relative concentrations of receptors are regulated by sortilin50, 51. With this understanding, we undertook studies to examine the role of sortilin in LDL/VLDL trafficking. Considering that PCSK9 is an additional ligand for sortilin-mediated secretion in hepatocytes50, we also explored its binding to sortilin. As part of this effort, we introduce a small molecule (cpd541) that inhibited binding of NT to sortilin at the canonical binding site (Site-1). In addition, we show that the Site-2 binding molecule cpd984 enhances NT-sortilin interactions in vitro, and that treatment of McA cells with cpd984 resulted in increased surface expression of LDLR as a reporter for reduced PCSK9 secretion. The absence of these effects with the Site-1 ligand cpd541 suggests that Site-2 of sortilin regulates PCSK9 trafficking.
Our findings indicate that VLDL secretion is oppositely affected by administration of these two compounds. Cpd984 administration led to increases in VLDL secretion in McA, whereas cpd541 inhibited VLDL secretion. These results indicate the potential for site specific modulation of sortilin chaperone activity being a primary determinant for sortilin-dependent VLDL trafficking outcomes in hepatocytes by blocking the interaction of sortilin with PCSK9 resulting in cellular accumulation of PCSK9. We hypothesize that cpd984 treatment of McA results in the cellular accumulation of PCSK9. This hypothesis is supported by increased cellular LDLR, which reflects inhibited PCSK9 secretion, as decreased PCSK9 secretion would prevent the internalization and degradation of LDLR. These results are consistent with what is known about the effect of PCSK9 on reducing the amount of LDLR available to remove circulating LDL from the blood stream. However, it is not clear what effect this will truly have on circulating LDL because of the balance between secretion and degradation rates. Overall, these results suggest that Site-2 is the binding site for PCSK9 trafficking in hepatic cells. We hypothesize that sortilin in the secretory pathway is rate limiting so when more is bound to VLDL, less is available for PCSK9 secretion.
Together, these results present a complex interconnected regulatory system for hepatic apo B lipoprotein metabolism that is rooted in the mechanisms of protein sorting by the orthologue of yeast Vps10, sortilin. Considering the complexity of this system, our observations on sortilin regulation in hepatocytes may serve to support new paradigms for sortilin trafficking in other cell types including neuronal and adipose. Specifically, these studies may help in understanding insulin secretion11, 18, 27, Glut4 transport52–55, and plaque formation in Alzheimer’s disease56–58.
In hepatocytes, sortilin plays a central role in apo B-Lp metabolism, yet its exact role remains enigmatic15. Significant questions remain such as how sortilin knockdowns both increase and decrease hepatic VLDL secretion13, and whether separate VLDL ligands interact on the VLDL surface when binding to sortilin? Considering the complexity of these questions, we propose a model where sortilin contains at least two interactive sites for ligand binding. We also suggest that an allosteric conformational change exists where binding Site-2 induces tighter binding of cargo to Site-1. This was exhibited by the effects on cpd984 on the binding of NT to Site-1 and could also explain why full length NT binds sortilin stronger than Ct-NT. Overall, we think that the use of Site-2 directed sortilin-binding molecules such as cpd984 may be useful clinically to increase LDLR expression, however care should be taken with respect to increased VLDL secretion. It is envisioned by the authors that a Site-2 molecule may be used in a cocktail with PCSK9/LDLR disruptors such as the monoclonal antibody treatment Repatha® though no experiments were conducted to this effect. Furthermore, approaches could be developed to selectively regulate involved pathways by specifically targeting either site of sortilin. Such strategies could potentially reduce apo B 100 secretion to lower the risk of atherosclerosis or increase VLDL secretion in an attempt to modulate hepatic triglyceride secretion to reduce hepatic steatosis27. To help delineate Site-1 and Site-2 of sortilin we developed a novel NT based probe to Site-2 of sortilin and showed that cpd984 behaves similarly to this compound. We hope this will help researchers further understand the complicated nature of sortilin trafficking with respect to site-specific modulation of trafficking patterns dependent on the protein sortilin
An appealing hypothesis based on these observations is that VLDL particle composition directs trafficking of PCSK9 in hepatocytes. The discovery of a second ligand binding site on sortilin that regulates PCSK9 binding and cell retention when bound is novel. The allosteric effect of binding to Site-2 in increasing ligand binding to Site-1 offers therapeutic potential in regulating VLDL secretion while enhancing VLDL remnant clearance by LDLR, which would be augmented by decreased LDLR degradation.
Acknowledgements
This paper is dedicated to Janet DeHoff Sparks. Following training at the University of Pennsylvania, Janet went on to become a Full Professor at University of Rochester Medical Center in the Department of Pathology. Janet loved to get her work done early in the morning, go to Midtown Athletic Center, and then go right back to work to perform experiments. Janet continued this routine until her death in Sarasota, Fl from an unexpected heart attack. Many of the experiments in this paper were performed by Janet herself.
Funding
This research was supported by grants from the National Institutes of General Medical Sciences (R01GM101132 to RAF, and P41GM104601, U01GM111251 and U54GM087519 to ET), the National Science Foundation - Molecular and Cellular Biology (MCB 1818310 to RAF), and the Office of Naval Research (ONR N00014-16-1-2535 to E.T.). Computational resources were provided by XSEDE (XSEDE MCA06N060) and Blue Waters (ACI1440026). SPR was aided by the help of Dr. Jermaine Jenkins at the University of Rochester Structural Biology & Biophysics Facility with support from National Center for Research Resources (1S10 RR027241), as well as National Institute of Allergy and Infectious Diseases (P30AI078498) and the University of Rochester School of Medicine and Dentistry.
Abbreviations used:
- apo B-Lp
apo B containing lipoproteins
- NT
neurotensin
- MST
microscale thermophoresis
- VLDL
very low-density lipoprotein
- B100
apolipoprotein B
- SPR
surface plasmon resonance
- CPY
carboxypeptidase Y
- McA
McCardle cells
- PIP3
phosphatidylinositol 3,4,5 trisphosphate
Footnotes
The authors declare that they do not have conflicts of interest with the contents of this article.
References
- [1].Heitman J (2015) On the Discovery of TOR As the Target of Rapamycin., PLoS Pathog 11, e1005245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fitzgerald I, and Glick BS (2014) Secretion of a foreign protein from budding yeasts is enhanced by cotranslational translocation and by suppression of vacuolar targeting., Microb Cell Fact 13, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arlt H, Reggiori F, and Ungermann C (2015) Retromer and the dynamin Vps1 cooperate in the retrieval of transmembrane proteins from vacuoles., J Cell Sci 128, 645–655. [DOI] [PubMed] [Google Scholar]
- [4].Nielsen MS, Madsen P, Christensen EI, Nykjaer A, Gliemann J, Kasper D, Pohlmann R, and Petersen CM (2001) The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein., EMBO J 20, 2180–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Staudt C, Puissant E, and Boonen M (2016) Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View., Int J Mol Sci 18, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leloup N, Lössl P, Meijer DH, Brennich M, Heck AJR, Thies-Weesie DME, and Janssen BJC (2017) Low pH-induced conformational change and dimerization of sortilin triggers endocytosed ligand release., Nat Commun 8, 1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Itoh S, Mizuno K, Aikawa M, and Aikawa E (2018) Dimerization of sortilin regulates its trafficking to extracellular vesicles., J Biol Chem 293, 4532–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nykjaer A, and Willnow TE (2012) Sortilin: a receptor to regulate neuronal viability and function., Trends Neurosci 35, 261–270. [DOI] [PubMed] [Google Scholar]
- [9].Kandor KV (2018) The role of sortilin in the “Glut4 Pathway”, Commun Integr Biol 11, e1393592. [Google Scholar]
- [10].Goettsch C, Kjolby M, and Aikawa E (2018) Sortilin and Its Multiple Roles in Cardiovascular and Metabolic Diseases., Arterioscler Thromb Vasc Biol 38, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gao A, Cayabyab FS, Chen X, Yang J, Wang L, Peng T, and Lv Y (2017) Implications of Sortilin in Lipid Metabolism and Lipid Disorder Diseases., DNA Cell Biol 36, 1050–1061. [DOI] [PubMed] [Google Scholar]
- [12].Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, and Rader DJ (2010) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus., Nature 466, 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Strong A, Patel K, and Rader DJ (2014) Sortilin and lipoprotein metabolism: making sense out of complexity., Curr Opin Lipidol 25, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Martínez-Oliván J, Arias-Moreno X, Velazquez-Campoy A, Millet O, and Sancho J (2014) LDL receptor/lipoprotein recognition: endosomal weakening of ApoB and ApoE binding to the convex face of the LR5 repeat., FEBS J 281, 1534–1546. [DOI] [PubMed] [Google Scholar]
- [15].Sparks CE, Sparks RP, and Sparks JD (2015) The enigmatic role of sortilin in lipoprotein metabolism., Curr Opin Lipidol 26, 598–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Carlo AS (2013) Sortilin, a novel APOE receptor implicated in Alzheimer disease., Prion 7, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Carlo AS, Gustafsen C, Mastrobuoni G, Nielsen MS, Burgert T, Hartl D, Rohe M, Nykjaer A, Herz J, Heeren J, Kempa S, Petersen CM, and Willnow TE (2013) The proneurotrophin receptor sortilin is a major neuronal apolipoprotein E receptor for catabolism of amyloid-β peptide in the brain., J Neurosci 33, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sparks RP, Jenkins JL, Miner GE, Wang Y, Guida WC, Sparks CE, Fratti RA, and Sparks JD (2016) Phosphatidylinositol (3,4,5)-trisphosphate binds to sortilin and competes with neurotensin: Implications for very low density lipoprotein binding., Biochem Biophys Res Commun 479, 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Haas ME, Attie AD, and Biddinger SB (2013) The regulation of ApoB metabolism by insulin., Trends Endocrinol Metab 24, 391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ijuin T, and Takenawa T (2012) Regulation of insulin signaling by the phosphatidylinositol 3,4,5-triphosphate phosphatase SKIP through the scaffolding function of Pak1., Mol Cell Biol 32, 3570–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kjolby M, Nielsen MS, and Petersen CM (2015) Sortilin, encoded by the cardiovascular risk gene SORT1, and its suggested functions in cardiovascular disease., Curr Atheroscler Rep 17, 496. [DOI] [PubMed] [Google Scholar]
- [22].Mazella J, Zsürger N, Navarro V, Chabry J, Kaghad M, Caput D, Ferrara P, Vita N, Gully D, Maffrand JP, and Vincent JP (1998) The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor., J Biol Chem 273, 26273–26276. [DOI] [PubMed] [Google Scholar]
- [23].Chamberlain JM, O’Dell C, Sparks CE, and Sparks JD (2013) Insulin suppression of apolipoprotein B in McArdle RH7777 cells involves increased sortilin 1 interaction and lysosomal targeting., Biochem Biophys Res Commun 430, 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sparks JD, Magra AL, Chamberlain JM, O’Dell C, and Sparks CE (2016) Insulin dependent apolipoprotein B degradation and phosphatidylinositide 3-kinase activation with microsomal translocation are restored in McArdle RH7777 cells following serum deprivation., Biochem Biophys Res Commun 469, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Denisov IG, Grinkova YV, Lazarides AA, and Sligar SG (2004) Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size, J Am Chem Soc 126, 3477–3487. [DOI] [PubMed] [Google Scholar]
- [26].Sparks JD, Chamberlain JM, O’Dell C, Khatun I, Hussain MM, and Sparks CE (2011) Acute suppression of apo B secretion by insulin occurs independently of MTP., Biochem Biophys Res Commun 406, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sparks RP, Guida WC, Sowden MP, Jenkins JL, Starr ML, Fratti RA, Sparks CE, and Sparks JD (2016) Sortilin facilitates VLDL-B100 secretion by insulin sensitive McArdle RH7777 cells., Biochem Biophys Res Commun 478, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sparks JD, Phung TL, Bolognino M, Cianci J, Khurana R, Peterson RG, Sowden MP, Corsetti JP, and Sparks CE (1998) Lipoprotein alterations in 10- and 20-week-old Zucker diabetic fatty rats: hyperinsulinemic versus insulinopenic hyperglycemia., Metabolism 47, 1315–1324. [DOI] [PubMed] [Google Scholar]
- [29].Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, and Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening., J Med Chem 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- [30].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, and Schulten K (2005) Scalable molecular dynamics with NAMD., J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins., Nat Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Feller SE, Zhang Y, Pastor RW, and Brooks BR (1995) Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method, J Chem Phys 103, 4613–4621. [Google Scholar]
- [33].Martyna GJ, Tobias DJ, and Klein ML (1994) Constant Pressure Molecular Dynamics Algorithms, J Chem Phys 101, 4177–4189. [Google Scholar]
- [34].Darden T, York D, and Pedersen LG (1993) Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems, J Chem Phys 98, 10089–10092. [Google Scholar]
- [35].Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995) A Smooth Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems., J Chem Phys 103, 8577–8593. [Google Scholar]
- [36].Miyamoto S, and Kollman PA (1992) SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Molecules, J Comput Chem 13, 952–962. [Google Scholar]
- [37].Humphrey W, Dalke A, and Schulten K (1996) VMD − Visual Molecular Dynamics, J Mol Graphics 14, 22–28. [DOI] [PubMed] [Google Scholar]
- [38].Trott O, and Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J Comput Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Beauchamp KA, Bowman GR, Lane TJ, Maibaum L, Haque IS, and Pande VS (2011) MSMBuilder2: Modeling Conformational Dynamics at the Picosecond to Millisecond Scale., J Chem Theory Comput 7, 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pande VS, Beauchamp K, and Bowman GR (2010) Everything you wanted to know about Markov State Models but were afraid to ask., Methods 52, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gowers RJ, Linke M, Barnoud J, Reddy TJE, Melo MN, Seyler SL, Dotson DL, Domanski J, Buchoux S, Kenney IM, and Beckstein O (2016) MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations, Proceedings of the 15th Python in Science Conference doi: 10.25080/majora-629e541a-00e, 98–105. [DOI] [Google Scholar]
- [42].Michaud-Agrawal N, Denning EJ, Woolf TB, and Beckstein O (2011) MDAnalysis: a toolkit for the analysis of molecular dynamics simulations., J Comput Chem 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sparks RP, and Fratti R (2019) Use of Microscale Thermophoresis (MST) to Measure Binding Affinities of Components of the Fusion Machinery., Methods Mol Biol 1860, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sparks RP, Arango AS, Starr ML, Aboff ZL, Hurst LR, Rivera-Kohr DA, Zhang C, Harnden KA, Jenkins JL, Guida WC, Tajkhorshid E, and Fratti RA (2019) A small-molecule competitive inhibitor of phosphatidic acid binding by the AAA+ protein NSF/Sec18 blocks the SNARE-priming stage of vacuole fusion., J Biol Chem 294, 17168–17185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Starr ML, Sparks RP, Arango AS, Hurst LR, Zhao Z, Lihan M, Jenkins JL, Tajkhorshid E, and Fratti RA (2019) Phosphatidic acid induces conformational changes in Sec18 protomers that prevent SNARE priming., J Biol Chem 294, 3100–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bochevarov AD, Harder E, Hughes TF, Greenwood JR, Braden DA, Philipp DM, Rinaldo D, Halls MD, Zhang J, and Friesner RA (2013) Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences, Int. J. Quanum Chem 113, 2110–2142. [Google Scholar]
- [47].Quistgaard EM, Grøftehauge MK, Madsen P, Pallesen LT, Christensen B, Sørensen ES, Nissen P, Petersen CM, and Thirup SS (2014) Revisiting the structure of the Vps10 domain of human sortilin and its interaction with neurotensin., Protein Sci 23, 1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Trabjerg E, Abu-Asad N, Wan Z, Kartberg F, Christensen S, and Rand KD (2019) Investigating the Conformational Response of the Sortilin Receptor upon Binding Endogenous Peptide-and Protein Ligands by HDX-MS., Structure 27, 1103–1113.e3. [DOI] [PubMed] [Google Scholar]
- [49].Nair P (2013) Brown and Goldstein: the cholesterol chronicles., Proc Natl Acad Sci U S A 110, 14829–14832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gustafsen C, Kjolby M, Nyegaard M, Mattheisen M, Lundhede J, Buttenschøn H, Mors O, Bentzon JF, Madsen P, Nykjaer A, and Glerup S (2014) The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion., Cell Metab 19, 310–318. [DOI] [PubMed] [Google Scholar]
- [51].Wolf DH, and Schäfer A (2005) CPY* and the power of yeast genetics in the elucidation of quality control and associated protein degradation of the endoplasmic reticulum., Curr Top Microbiol Immunol 300, 41–56. [DOI] [PubMed] [Google Scholar]
- [52].Kioumourtzoglou D, Pryor PR, Gould GW, and Bryant NJ (2015) Alternative routes to the cell surface underpin insulin-regulated membrane trafficking of GLUT4., J Cell Sci 128, 2423–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Morris NJ, Ross SA, Lane WS, Moestrup SK, Petersen CM, Keller SR, and Lienhard GE (1998) Sortilin is the major 110-kDa protein in GLUT4 vesicles from adipocytes., J Biol Chem 273, 3582–3587. [DOI] [PubMed] [Google Scholar]
- [54].Pan X, Zaarur N, Singh M, Morin P, and Kandror KV (2017) Sortilin and retromer mediate retrograde transport of Glut4 in 3T3-L1 adipocytes., Mol Biol Cell 28, 1667–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shi J, and Kandror KV (2005) Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes., Dev Cell 9, 99–108. [DOI] [PubMed] [Google Scholar]
- [56].Capsoni S, Carlo AS, Vignone D, Amato G, Criscuolo C, Willnow TE, and Cattaneo A (2013) SorLA deficiency dissects amyloid pathology from tau and cholinergic neurodegeneration in a mouse model of Alzheimer’s disease., J Alzheimers Dis 33, 357–371. [DOI] [PubMed] [Google Scholar]
- [57].Ruan CS, Liu J, Yang M, Saadipour K, Zeng YQ, Liao H, Wang YJ, Bobrovskaya L, and Zhou XF (2018) Sortilin inhibits amyloid pathology by regulating non-specific degradation of APP., Exp Neurol 299, 75–85. [DOI] [PubMed] [Google Scholar]
- [58].Zhao Y, Cui JG, and Lukiw WJ (2007) Reduction of sortilin-1 in Alzheimer hippocampus and in cytokine-stressed human brain cells., Neuroreport 18, 1187–1191. [DOI] [PubMed] [Google Scholar]