

Abstract
Radiation-induced acute intestinal injury is a common and serious occurrence following abdominal and pelvic irradiation. The Nod-like receptor protein 3 (NLRP3)-dependant inflammasome and inflammation activation is crucial in this process. In a pre-experimental design of radiation-induced intestinal injury, we found that rosiglitazone inhibited caspase-1 which is a key marker of inflammasome activation. The purpose of the present study was to clarify the inhibitory effect of rosiglitazone on the NLRP3 inflammasome both in vivo and in vitro. Radiation-induced intestinal injury after rosiglitazone treatment, and the expression of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), caspase-1 and NLRP3 in a radiation-induced intestinal injury model in a rat and macrophages were observed. We found that rosiglitazone ameliorated radiation-induced intestinal injury in rats by suppressing the expression of caspase-1, NLRP3, IL-1β and TNF-α. Treatment with rosiglitazone in vitro reduced the expression of NLRP3, and the NLRP3 activator monosodium urate (MSU) reversed the inhibition of IL-1β and TNF-α by rosiglitazone in macrophages. MSU reversed the protective effect of rosiglitazone on radiation-induced intestinal injury in rats by reversing the rosiglitazone-induced inhibition of IL-1β and TNF-α. Taken together, these findings indicate that the peroxisome proliferator-activated receptor gamma (PPARγ) agonist, rosiglitazone, ameliorates radiation-induced intestine inflammation in rats via inhibiting the induction of the NLRP3-dependent inflammasome in macrophages.
Keywords: Rosiglitazone, inflammasome, inflammation, radiation-induced intestinal injury, TNF-α
INTRODUCTION
Radiotherapy is an important component of multimodal cancer therapy. However, radiation-induced acute intestinal injury following abdominal and pelvic irradiation is a common and serious occurrence [1]. Since exposure to radiation induces massive cell death, severe tissue damage and inflammation in the intestine [2], it is important to prevent the occurrence of inflammation and injury of the small intestine during radiotherapy.
The intestinal environment is a complex ecosystem. Intestinal immune cells that mediate tolerance-inducing responses and participate in host defense localize to inductive and effector sites within the intestinal mucosa [3]. The intestine is enriched with a wide variety of immune effector cells, including T cells, B cells, dendritic cells, and macrophage/monocyte lineage cells [4]. Moreover, the mutual regulation of various immune cells affects inflammasome activation, and overactivation of the inflammasome can cause intestinal injury [5, 6]. Seo et al. reported that some commensals can induce interleukin-1β (IL-1β) via the nod-like receptor protein 3 (NLRP3) inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury [7]. Besides, it has also been demonstrated [8] that colchicine prevents Nonsteroidal Antiinflammatory Drug (NSAID)-induced small intestinal injury by inhibiting NLRP3 inflammasome activation.
The NLRP3 inflammasome is activated in response to the detection of pathogens or pro-inflammatory signals via specialized receptors, which are critical regulators of intestinal tissue homeostasis [5]. It regulates intestinal homeostasis by modulating the intestinal microbial ecology, inflammation and tissue repair [6]. NLRP3 is a kind of pattern recognition receptor. As the main component of the NLRP3 inflammasome, NLRP3 assembles the inflammasome, a protein complex responsible for the proteolytic maturation and secretion of the pro-inflammatory cytokine, IL-1β [9]. IL-1β promotes inflammation through the recruitment and activation of immune cells and inducing the production of pro-inflammatory cytokines [10]. Therefore, regulation of the NLRP3 inflammasome may be a key process related to inflammation-mediated injury of the intestine.
Peroxisome proliferator-activated receptor gamma (PPARγ) belongs to the nuclear receptor family of ligand-inducible transcription factors and has been found to exhibit pervasive effects on the transduction of metabolic, anti-inflammatory, anti-fibrotic and antineoplastic signals [11]. The interactions between PPARγ and the inflammasome have been widely demonstrated. Li et al. [12] suggested that the PPARγ modulator, apigenin, ameliorates chronic mild stress-induced depressive behavior by inhibiting NLRP3 inflammasome activation and IL-1β production in brains. Besides, the endogenous PPARγ ligand, 15d-PGJ2, is found to inhibit NLRP1 and NLRP3 inflammasomes in a murine anthrax infection model [13]. Similar findings were documented in endothelials cells [14]. Moreover, PPARγ is widely expressed in macrophages and has been identified as a putative target for the development of novel therapies against inflammatory bowel disease [15]. Although it has been demonstrated that PPARγ agonists suppress the inflammatory process in the intestine, the effect and mechanism of rosiglitazone on radiation-induced intestinal injury remains unknown. The aim of the present study was to clarify the inhibitory effect of rosiglitazone on the NLRP3 inflammasome in vitro and in vivo.
METHODS
In vivo experiments
Animals
Male Sprague–Dawley (SD) rats (250–350 g) were housed in a temperature-controlled, specific-pathogen-free environment with a 12 h light/dark cycle, and fed standard chow and water. The animals were placed on a restrainer and abdominal irradiation (ABI) was performed on the rats using a vertical 137Cs γ-ray irradiator (Atomic Energy of Canada, Chalk River, Ontario, Canada), from which the rest of the body was protected by a 3-cm-thick lead shield. The animals were anesthetized with (intraperitoneally (i.p.)) pentobarbital (40 mg/kg body weight) prior to irradiation. Rats were exposed to 7, 9 and 11 Gy at 2.5 Gy/min for 4 min at room temperature, and 9 Gy was selected as the optimal irradiation dose for further experiments.
Rats were randomly divided into the following four groups: (i) control group (no ABI); (ii) irradiation group (ABI only); (iii) rosiglitazone group (ABI and rosiglitazone, 3 mg/kg body weight, i.p.; 5 min, 6 h and 24 h after ABI); and (iv) rosiglitazone + monosodium urate (MSU) group (ABI + rosiglitazone + MSU, 2.5 mg per rat, oral administration, after ABI) [16].
All the procedures regarding animal experiments comply with the Principles of Laboratory Animal Care (NIH publication No. 85–23, revised 1985).
IL-1β and tumor necrosis factor-α enzyme linked immunosorbent assay (ELISA)
Blood samples from the rats in each group were collected from the femoral artery at 24 h after ABI. The blood was centrifuged at 3000 × g for 15 min. The supernatant was collected and frozen in a refrigerator at −80°C. The concentration of IL-1β and tumor necrosis factor-α (TNF-α) in the serum samples was determined by a commercial ELISA (abcam, American). The detection process was carried out in strict accordance with the ELISA kit instructions.
Hematoxylin and eosin staining
The rats from each group were anesthetized with chloral hydrate (10%; 0.33 mL/kg) and euthanized at 24 and 72 h after ABI. A 5-cm section of the small intestine near the ileocecal region was harvested from the rats. The samples were then fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (HE). The morphological changes of the intestinal mucosa and villus were observed under a light microscope.
Western blot analysis of caspase-1 and NLRP3 expression
The rats from each group were anesthetized with chloral hydrate (10%; 0.33 mL/kg) and euthanized 24 h after ABI. A 5-cm section of the small intestine located near the ileocecal area was harvested from the rats, cut open and rinsed in phosphate buffered saline (PBS). An appropriate amount of tissue was placed into a mortar followed by the addition of liquid nitrogen. The tissue was ground into powder form and a small amount of Radio Immunoprecipitation Assay (RIPA) (containing l mmol/L Phenylmethylsulfonyl fluoride) was mixed with the lysate before transferring the lysate into an EP tube. The eppendorf (EP) tube was placed into an ice bath and an ultrasonic cell disrupter was used to interrupt the ultrasound for 5 s 25–30 times. The samples were then centrifuged at 14 000 × g at 4°C for 30 min. The supernatant from the EP tube was carefully collected to avoid precipitation and stored at −80°C until future use. The protein was quantified using a bicinchoninic acid protein assay kit (Thermo Scientific) and 10 μg of protein was loaded into each lane. The samples were electrophoresed onto a 12% sodium dodecyl sulfate/polyacrylamide (SDS-PAGE) gel, and transferred to polyvinylidene fluoride membranes (Millipore, MA, USA). After blocking with 10% BSA in TBST (25 mM Tris-HCl, 0.15 M saline and 1% Tween 20) at room temperature for 2 h, the membranes were incubated with the following primary antibodies at 4°C overnight: anti-actin (1:500; Santa Cruz), anti-NLRP3 (1:700; Abcam) and anti-caspase-1 (1:600; Abcam). The membranes were then washed and incubated with horseradish peroxidase-conjugated secondary antibody (1:2000; Abcam) for 2 h at 4 °C. Throughout the experiment, the low-temperature environment was strictly maintained, and protease inhibitors were added to avoid protein degradation.
The membranes were developed using the ChemiDoc-It TM TS2 Imaging System, and the relative optical density was analyzed using Quantity one v4.4 software (Bio-Rad).
In vitro experiments
Collection and culture of peritoneal macrophages
Resident macrophages of radiation-induced intestine injury SD rats were isolated by washing the peritoneal cavity with 10 mL of ice-cold PBS (pH 7.4) supplemented with 2% fetal calf serum (FCS, Gibco, Grand Island, NY, USA). Individual cell suspensions were washed twice with PBS/2% FCS and adjusted to 1 × 107 cells/mL.
Aliquots of cells were adjusted to 1 × 106 cells/mL in RPMI 1640 medium (Gibco, Invitrogen Corporation, Carlsbad, CA, USA) supplemented with heat-inactivated 5% FCS, 2 mM L-glutamine, 50 µM 2-mercaptoethanol, 100 μg/mL penicillin and 100 U/mL streptomycin (RPMI) and plated into 24-well flat-bottomed tissue culture plates (NUNC, Roskilde, Denmark) for cytokine assays.
The cells were cultured for 2 h at 37°C in a moist atmosphere of 5% CO2 in air to allow the cells to adhere to the plates. The non-adherent cells were removed by washing the plates twice with warm RPMI. The remaining adherent cells were highly enriched for macrophages and further incubated at 37°C and 5% CO2 in RPMI (control) or RPMI supplemented with 1 μg/mL Lipopolysaccharide (LPS) (Sigma-Aldrich Chemie, Taufkirchen, Germany), 1 ng/mL Granulocyte-macrophage Colony Stimulating Factor (GM-CSF) (ImmunoTools, Friesoythe, Germany) or 1 ng/mL IL-4 (ImmunoTools, Friesoythe, Germany) [17]. Following a 24 h incubation in 24-well plates, the culture supernatants were collected and frozen at −70°C until they were assayed for TNF-α and IL-1β. The macrophages were divided into the following three groups: (i) control group, untreated; (ii) rosiglitazone group, treated with rosiglitazone [1 μmol/L in (Dimethylsulfoxide (DMSO) (Abcam, ab142179)]; and (iii) rosiglitazone + MSU group, treated with rosiglitazone (1 μmol/L in DMSO) for 3 h, cultured overnight, and stimulated with MSU (150 μg/mL) for 5 h [18]. Purity of macrophage was identified by anti-CD68, CD68 antibody using flow cytometry.
Detection of NLRP3 via western blot
The total cellular protein was extracted and 25 μg was used for SDS-PAGE. After blocking the membrane for 30 min, rinsing three times with TBST buffer and incubating with anti-NLRP3 (1:800; Abcam) for 12 h at 4°C, the membranes were washed three times in wash buffer (0.2% gelatin, 0.05% Tween 20 in TBS). The membranes were then incubated for 1 h with a horseradish peroxidase-labeled secondary antibody, washed again, and exposed to electrochemiluminescence (ECL) color development reagents. The membranes were developed using the ChemiDoc-It TM TS2 Imaging System (Bio-Rad), and relative optical density was analyzed using ImageJ2x software (National Institute of Health, Bethesda, MD, USA).
IL-1β and TNF-α ELISA
The production of IL-1β and TNF-α was determined by ELISA following rosiglitazone treatment. The supernatants from the macrophage cell cultures were harvested, and the level of IL-1β and TNF-α was determined using a commercial ELISA kit (GBD Co.: sensitivity for IL-1β < 12 pg/ml; sensitivity for TNF-α 1.04 pg/ml) in accordance with the manufacturer’s instructions.
Statistical analysis
All statistical analyses were conducted using SPSS 14.0 software. Normally distributed data are expressed as mean ± SD. Comparisons between groups were performed using a one-way analysis of variance (ANOVA) followed by a Tukey–Kramer multiple comparisons post hoc test.
RESULTS
Rosiglitazone ameliorates radiation-induced intestinal injury
Radiotherapy induced a time-dependent injurious effect on the jejunal mucosa. In the control group, the villus structure was slender and the crypt structure was clear. In the irradiation group, the villus and crypt structure was damaged, there was inflammatory cell infiltration, and spotted hemorrhage or necrosis was observed. The villus and crypt structures in the rosiglitazone group were improved compared with the radiation group; however, the villus remained low, wide and barbed with epithelial cell shedding and gland atrophy (Fig. 1).
Fig. 1.
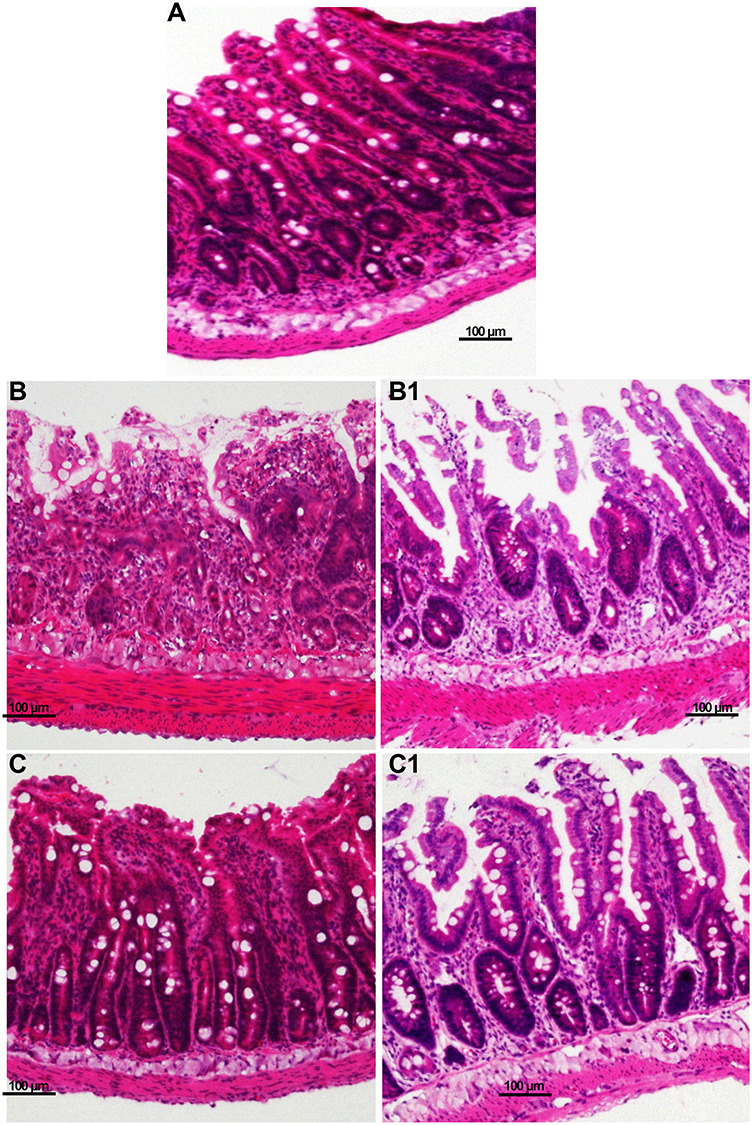
HE staining of rat small intestines. (A) control group; (B) irradiation (IR) 24 h post ABI; (B1) IR group 72 h post ABI; (C) IR + rosiglitazone group 24 h post ABI; (C1) IR + rosiglitazone group 72 hours post ABI. Each experiment was repeated three times.
Compared with the irrigation group, improved villus height, crypts and inflammatory cell infiltration was detected in the rosiglitazone-treated rats (Table 1).
Table 1.
Villus height and the number of crypts and inflammatory cells in each of the three groups. Results are given as mean ± SD; each experiment was repeated at least five times (one-way ANOVA followed by a Tukey–Kramer multiple comparisons post hoc test)
| Villus height (height) | Number of crypts | Number of inflammatory cells (fold increase compared with control) | |
|---|---|---|---|
| Control | 377.43 ± 37.81 | 89.59 ± 10.30 | 1.00 ± 0.15 |
| Irradiation | 254.32 ± 29.62* | 22.35 ± 4.35* | 10.33 ± 0.77* |
| Rosiglitazone | 308.47 ± 31.25*,** | 48.21 ± 5.62*,** | 5.75 ± 0.42*,** |
* P < 0.05 vs control; **
P < 0.05 vs IR.
Rosiglitazone treatment reduced the expression of caspase-1, NLRP3, IL-1β and TNF-α in radiation-induced intestinal injury rats
The level of IL-1β and TNF-α in the blood collected from rats was detected by ELISA. The level of IL-1β in the irradiation group (41.56 ± 7.39 pg/ml) was significantly higher compared with that of both the control (12.31 ± 2.89 pg/ml) and rosiglitazone groups (15.71 ± 3.85 pg/ml). The control group had the lowest level of IL-1β. The level of TNF-α in the irradiation group (47.61 ± 5.42 pg/ml) was higher than that of the control (24.37 ± 4.56 pg/ml) and rosiglitazone groups (32.43 ± 5.12 pg/ml). There was no significant difference between the rosiglitazone and control groups (Fig. 2A and C).
Fig. 2.
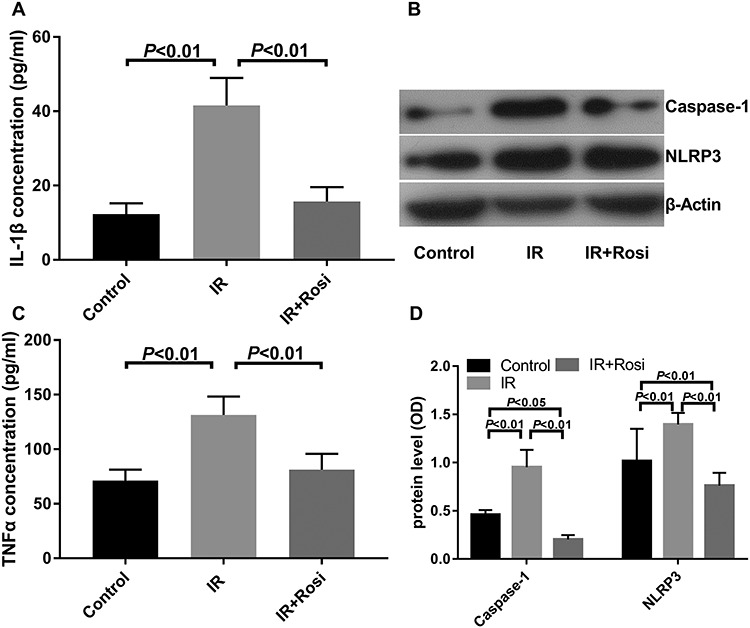
The protein expression of caspase-1 and NLRP3, and the concentration of IL-1β and TNF-α in control, irradiation (IR) and IR + rosiglitazone (Rosi) groups. (A) IL -1β expression level; (B) representative western blots showing the effects of rosiglitazone on caspase-1 and NLRP3; (C) TNF-α expression level; (D) relative caspase-1 and NLRP3 protein level. Each experiment was repeated at least three times (one-way ANOVA followed by a Tukey–Kramer multiple comparisons post test).
In the WB experiment, the expression of caspase-1 in the irradiation group (0.95 ± 0.18) was higher compared with that of the control group (0.20 ± 0.05), whereas caspase-1 expression was lower in the rosiglitazone group (0.46 ± 0.05) compared with the irradiation group. There was no significant difference in the expression of caspase-1 between the rosiglitazone and control groups. The expression of NLRP3 in the small intestine was also detected, and the results showed that the level of NLRP3 in the control group (0.76 ± 0.12) was the lowest. The expression of NLRP3 in the rosiglitazone group (1.02 ± 0.335) was lower than that of the irradiation group (1.40 ± 0.12) (Fig. 2B and D).
Taken together, these results may suggest that rosiglitazone ameliorates radiation-induced intestinal injury by suppressing caspase-1, NLRP3, IL-1β and TNF-α.
PPARγ activation reduces the expression of NLRP3 in macrophages, and the NLRP3 activator, MSU, reversed the inhibition of IL-1β and TNF-α by rosiglitazone in vitro
In vitro experiments were performed using macrophages. The purity of the obtained macrophages was confirmed using flow cytometry. Results showed that CD68-positive cells accounted for nearly 90% (Fig. 3).
Fig. 3.
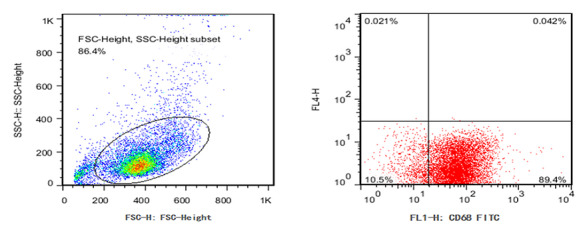
Identification of macrophages using flow cytometry.
To confirm the effect of NLRP3 activation on the ability of rosiglitazone to inhibit inflammation, we compared the expression of IL-1β and TNF-α between the control, rosiglitazone, and rosiglitazone + NLRP3 agonist (MSU) groups by ELISA. The level of IL-1β in the rosiglitazone group (151.25 ± 27.91 pg/ml) was lower than that of both the control (228.78 ± 38.36) and rosiglitazone + MSU groups (502.34 ± 64.95 pg/ml). This indicates that the activation of NLRP3 reversed the inhibitory effect of rosiglitazone on IL-1β. The expression of TNF-α in the rosiglitazone group (101.72 ± 19.46 ρg/ml) was lower than that of the control group (155.23 ± 21.91 ρg/ml). There was no significant difference in the level of TNF-α expression between the rosiglitazone and the rosiglitazone + MSU groups (119.48 ± 17.72 pg/ml) (Fig. 4A and C). This finding suggests that the activation of NLRP3 had little effect on the inhibition of TNF-α induced by rosiglitazone.
Fig. 4.
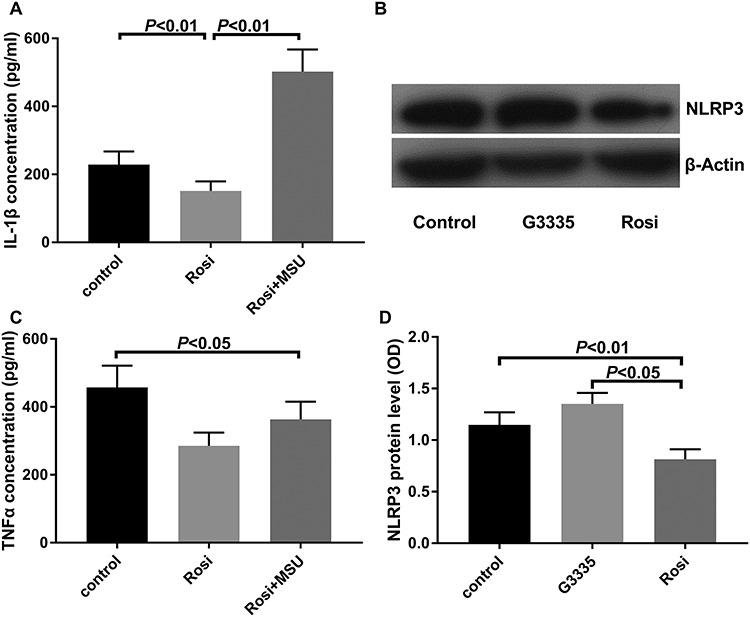
The protein expression of NLRP3 and the concentration of IL-1β and TNF-α in control, MSU and rosiglitazone groups. (A) IL -1β expression level; (B) representative western blots showing the effects of rosiglitazone on NLRP3; (C) TNF-α expression level; (D) relative NLRP3 protein level. Each experiment was repeated at least three times (one-way ANOVA followed by a Tukey–Kramer multiple comparisons post test).
The level of NLRP3 in macrophages was detected by western blot. The level of NLRP3 expression in the rosiglitazone group (0.82 ± 0.10) was lower than that of the control (1.15 ± 0.12) and the G3335 (PPAR-γ antagonist) groups (1.35 ± 0.11). This finding suggests that the expression of NLRP3 was related to the activation status of PPARγ (Fig. 4B and D).
The NLRP3 activator, MSU, reverses the protective effect of rosiglitazone on radiation-induced intestinal injury rats
In both the irradiation and rosiglitazone + MSU groups, damage to the villus and crypt structures, inflammatory cell infiltration, and spotted hemorrhage or necrosis were observed. The villus and crypt structures of the rosiglitazone group were improved compared with the other two groups (Fig. 5).
Fig. 5.
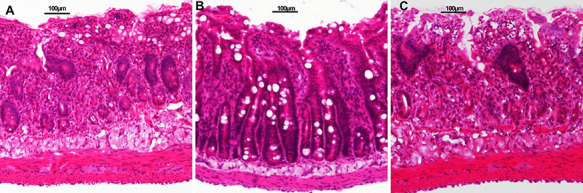
HE staining of rat small intestines 24 h post ABI. (A) Irradiation (IR) group; (B) rosiglitazone group; (C) rosiglitazone + MSU group. Each experiment was repeated three times.
Compared with the irrigation and rosiglitazone + MSU groups, the rosiglitazone-treated rats displayed improved villus height, crypts and inflammatory cell infiltration (Table 2).
Table 2.
Villus height, and the number of crypts and inflammatory cells in the irradiation, rosiglitazone and rosiglitazone + MSU groups. Each experiment was repeated at least five times (one-way ANOVA followed by a Tukey–Kramer multiple comparisons post hoc test).
| Villus height | Number of crypts | Number of Inflammatory cells (fold increase vs control) | |
|---|---|---|---|
| Irradiation | 257.44 ± 32.54 | 19.22 ± 3.21 | 1 ± 0.12 |
| Rosiglitazone | 332.58 ± 34.37* | 47.39 ± 5.87* | 0.11 ± 0.02* |
| Rosiglitazone + MSU | 271.61 ± 35.57** | 20.23 ± 2.75** | 1.12 ± 0.14** |
* P < 0.05 compared with control group;
** P < 0.05 compared with rosiglitazone group.
MSU reversed the inhibition of IL-1β and TNF-α by rosiglitazone in vivo
The IL-1β in the blood of rats was detected by ELISA. The level of IL-1β in the rosiglitazone group (16.82 ± 4.12 pg/ml) was significantly lower than that in the irradiation group (37.56 ± 6.39 pg/ml) and the rosiglitazone + MSU group (53.77 ± 6.83 pg/ml; (Fig. 6). This suggests that the NLRP3 agonist, MSU, can reverse the rosiglitazone-induced inhibition of IL-1β in rats.
Fig. 6.
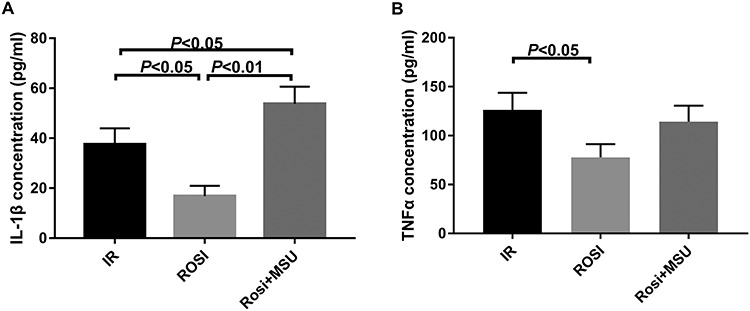
The concentration of IL-1β and TNF-α in control, rosiglitazone and rosiglitazone + MSU groups. (A) IL -1β expression level; (B) TNF-α expression level. Each experiment was repeated at least three times (one-way ANOVA followed by a Tukey–Kramer multiple comparisons post test).
The level of TNF-α in the blood was detected. The level of TNF-α in the rosiglitazone group (32.51 ± 4.93 pg/ml) was lower than that in the irradiation group (46.72 ± 5.53 pg/ml), but was not significantly different from the rosiglitazone + MSU group (37.28 ± 4.61 pg/ml). This result, combined with the effect of MSU on TNF-α in macrophages, suggests that rosiglitazone may alter the level of inflammation both through the NLRP3 inflammasome as well as via another method.
DISCUSSION
Rosiglitazone is a clinical drug for type 2 diabetes that exhibits the potential to inhibit the inflammatory response [19]. It has been reported that rosiglitazone has a positive effect in reducing intestinal inflammation in various experimental models [20]. The aim of the present study was to clarify the inhibitory effect of rosiglitazone on the NLRP3 inflammasome in a rat model of radiation-induced intestinal injury and in vitro. Our findings indicate that rosiglitazone could attenuate inflammation in radiation-induced intestinal injury by inhibiting the NFRP3 inflammasome and TNF-α expression in macrophages.
In our in vivo experiments, we found that radiotherapy caused harmful effects on the small intestines of rats. Moreover, rosiglitazone treatment could significantly ameliorate the structural damage to the mucosal villi and crypts, as well as inflammatory cell infiltration after radiation. Although the role of PPARγ activation in inhibiting inflammasomes has been described in various animal experimental models [21], the effect of PPARγ activation on the inflammasome in the intestine remains unclear. Thus, we further examined the expression of caspase-1 and NLRP3 in the small intestine. We found that rosiglitazone decreased the expression of caspase-1 and NLRP3, and reduced the levels of serum IL-1β and TNF-α associated with radiation-induced intestinal injury. It has been well-established that the NLRP3 inflammasome is a multi-protein complex composed of NLRP3, adaptor protein (ASC) and pro-caspase1. Activated NLRP3 recruits ASC which binds to pro-caspase1 and thus assembly of the NLRP3 inflammasome is completed. [22] The inflammasome mediates the activation of caspase-1, which is a self-cleaved protease, and activates pro-inflammatory cytokines (e.g. Pro-IL1β and Pro-IL18) [23]. Therefore, caspase-1 is a key signal required for the activation of NLRP3, and NLRP3 is a constituent of the inflammasome. The results of the present study suggest that rosiglitazone ameliorates radiation-induced intestinal injury by suppressing caspase-1, NLRP3 and TNF-α.
Macrophages are important components of protective immunity [24] and a major constituent of the intestinal stroma, which contains the largest pool of macrophages in the body. In the present study, the activation of PPARγ reduced the expression of NLRP3 in macrophages, which was reversed when PPARγ was blocked by G3335. Moreover, the level of IL-1β in macrophages increased when NLRP3 and PPARγ were activated simultaneously. As previously mentioned, the maturation and secretion of IL-1β are controlled by the inflammasome. In addition, IL-1β has potent proinflammatory activities which direct the host response to infection and injury [25]. Thus, our in vitro results may indicate that the rosiglitazone-mediated inhibition of the inflammatory response in macrophages was through NLRP3.
We also wondered whether treatment with the NLRP3 agonist, MSU, could reverse the rosiglitazone-mediated inhibition of inflammation. In the present study, to probe the interaction between rosiglitazone and the NLRP3 inflammasome in vivo, the NLRP3 agonist, MSU, was applied. MSU can activate the NLRP3 inflammasome and drives IL-1β production [26]. Compared with the rosiglitazone group, MSU treatment improved the level of inflammation and injury observed in the small intestine tissue. In addition, the increase in serum IL-1β was consistent with changes in the small intestine tissue in vivo. This finding in conjunction with the above analysis may suggest that the PPARγ agonist, rosiglitazone, could ameliorate the level of inflammation and injury in rats with radiation-induced intestinal injury by inhibiting the NLRP3-dependent inflammasome.
TNF-α is a cell signaling protein involved in systemic inflammation and one of the cytokines that comprise the acute phase reaction [27]. TNFα is produced chiefly by activated macrophages [28]. Moreover, the inflammatory changes in the intestine have been shown to be partially mediated by TNF-α [29], and TNF-α has been shown to significantly contribute to the complications associated with radiotherapy [30]. Mangoni et al. [31] found that rosiglitazone exerts a protective effect on normal tissues, and reduces the bowel inflammation in a radiation-induced bowel toxicity model by inhibiting proinflammatory cytokines, including TNF-α. Although TNF-α was detected in the present study, the activation of NLRP3, both in vivo and in vitro, appeared to have little effect on the reduction of TNF-α, which is inhibited by rosiglitazone. This finding may suggest that rosiglitazone-mediated inhibition of inflammation is mediated both via the NLRP3 inflammasome pathway, as well as other pathways (e.g. TNFα/NF-κB signaling). Furthermore, several studies have found that PPARγ activation can inhibit Nuclear factor-kappaB (NF-κB) [32]. However, this hypothesis requires further exploration in the context of radiation-induced intestinal injury.
The PPARγ agonist, rosiglitazone, ameliorates radiation-induced intestinal inflammation in rats, which may be mediated via inhibition of NLRP3-dependent inflammasome activation in macrophages, as well as the suppression of TNF-α.
Contributor Information
Liqiong Hu, Department of Intensive Care Unit of Guangzhou Red Cross hospital, Medical College, Jinan University, Guangzhou 51000, China.
Hao Chen, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, China.
Xingliang Zhang, Shenzhen Children’s Hospital, Shenzhen, 518000, China.
Zhencheng Feng, Guangzhou institute of traumatic surgery, Guangzhou Red Cross hospital, Medical College, Jinan University, Guangzhou 510000, China.
Haifeng Zhang, Department of Cardiology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou 51000, China.
Qingqi Meng, Guangzhou institute of traumatic surgery, Guangzhou Red Cross hospital, Medical College, Jinan University, Guangzhou 510000, China.
FUNDING
This work was supported by the Guangdong Medical Foundation (grant numbers A2020103) and the Guangzhou Medical Foundation (grant number 20201A011017), National Natural Science Foundation of China (Grant No. 81300279, 81741067), and High-level Hospital Construction Project (Grant No. DFJH201803 and KJ012019099).
CONFLICT OF INTEREST
None declared.
References
- 1. Peach MS, Showalter TN, Ohri N. Systematic review of the relationship between acute and late gastrointestinal toxicity after radiotherapy for prostate cancer. Prostate Cancer 2015;2015:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Delgado ME, Grabinger T, Brunner T. Cell death at the intestinal epithelial front line. FEBS J 2016;283:2701–19. [DOI] [PubMed] [Google Scholar]
- 3. Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell 2010;140:859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol Rev 2014;260:86–101. [DOI] [PubMed] [Google Scholar]
- 5. Hirota SA, Ng J, Lueng A et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis 2010;17:1359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strowig T, Henao-Mejia J, Elinav E et al. Inflammasomes in health and disease. Nature 2012;481:278. [DOI] [PubMed] [Google Scholar]
- 7. Seo S-U, Kamada N, Muñoz-Planillo R et al. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 2015;42:744–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Otani K, Watanabe T, Shimada S et al. Colchicine prevents NSAID-induced small intestinal injury by inhibiting activation of the NLRP3 inflammasome. Sci Rep 2016;6:32587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014;157:1013–22. [DOI] [PubMed] [Google Scholar]
- 10. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009;27:519–50. [DOI] [PubMed] [Google Scholar]
- 11. Lehrke M, Lazar MA. The many faces of PPARγ. Cell 2005;123:993–9. [DOI] [PubMed] [Google Scholar]
- 12. Li R, Wang X, Qin T et al. Apigenin ameliorates chronic mild stress-induced depressive behavior by inhibiting interleukin-1β production and NLRP3 inflammasome activation in the rat brain. Behav Brain Res 2016;296:318–25. [DOI] [PubMed] [Google Scholar]
- 13. Maier NK, Leppla SH, Moayeri M. The cyclopentenone prostaglandin 15d-PGJ2 inhibits the NLRP1 and NLRP3 inflammasomes. The Journal of Immunology 2015;194:2776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baselet B, Driesen RB, Coninx E et al. Rosiglitazone protects endothelial cells from irradiation-induced mitochondrial dysfunction. Front Pharmacol 2020;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hontecillas R, Horne WT, Climent M et al. Immunoregulatory mechanisms of macrophage PPAR-γ in mice with experimental inflammatory bowel disease. Mucosal Immunol 2011;4:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu D-Z, Chu P-Y, Chen S-J et al. Mast cell stabilizer ketotifen inhibits gouty inflammation in rats. Am J Ther 2016;23:e1009–e15. [DOI] [PubMed] [Google Scholar]
- 17. Dimitrijevic M, Stanojevic S, Blagojevic V et al. Aging affects the responsiveness of rat peritoneal macrophages to GM-CSF and IL-4. Biogerontology 2016;17:359–71. [DOI] [PubMed] [Google Scholar]
- 18. Wu Y-H, Kuo W-C, Wu Y et al. Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ 2014;21:451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meng Q-q, Lei W, Chen H et al. Combined rosiglitazone and Forskolin have neuroprotective effects in SD rats after spinal cord injury. PPAR Research 2018;2018:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yi J-H, Park S-W, Brooks N et al. PPARγ agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res 2008;1244:164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cuzzocrea S, Pisano B, Dugo L et al. Rosiglitazone and 15-deoxy-Δ12, 14-prostaglandin J2, ligands of the peroxisome proliferator-activated receptor-γ (PPAR-γ), reduce ischaemia/reperfusion injury of the gut. Br J Pharmacol 2003;140:366–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu A, Wu H. Structural mechanisms of inflammasome assembly. FEBS J 2015;282:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Broz P, Dixit VM. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016;16:407. [DOI] [PubMed] [Google Scholar]
- 24. Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev 2014;260:102–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–32. [DOI] [PubMed] [Google Scholar]
- 26. Hari A, Zhang Y, Tu et al. Activation of NLRP3 inflammasome by crystalline structures via cell surface contact. Sci Rep 2014;4:7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan FK-M, Siegel RM, Lenardo MJ. Signaling by the TNF receptor superfamily and T cell homeostasis. Immunity 2000;13:419–22. [DOI] [PubMed] [Google Scholar]
- 28. Bode JG, Nimmesgern A, Schmitz J et al. LPS and TNFα induce SOCS3 mRNA and inhibit IL-6-induced activation of STAT3 in macrophages. FEBS Lett 1999;463:365–70. [DOI] [PubMed] [Google Scholar]
- 29. Shen Z-Y, Zhang J, Song H-L et al. Bone-marrow mesenchymal stem cells reduce rat intestinal ischemia-reperfusion injury, ZO-1 downregulation and tight junction disruption via a TNF-α-regulated mechanism. World J Gastroenterol: WJG 2013;19:3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Linard C, Grémy O, Benderitter M. Reduction of peroxisome proliferation-activated receptor γ expression by γ-irradiation as a mechanism contributing to inflammatory response in rat colon: Modulation by the 5-aminosalicylic acid agonist. Journal of Pharmacology and Experimental Therapeutics 2008;324:911–20. [DOI] [PubMed] [Google Scholar]
- 31. Mangoni M, Sottili M, Gerini C et al. A PPAR-gamma agonist protects from radiation-induced intestinal toxicity. United European Gastroenterology Journal 2017;5:218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H-B, Zhang Y, Chen C et al. Pioglitazone inhibits advanced glycation end product-induced matrix metalloproteinases and apoptosis by suppressing the activation of MAPK and NF-κB. Apoptosis 2016;21:1082–93. [DOI] [PubMed] [Google Scholar]