

Abstract
The liver is home to five known human hepatitis viruses (hepatitis A virus–hepatitis E virus). Despite being phylogenetically unrelated, these viruses replicate and spread in the liver without causing apparent cytopathic effects, and all have evolved strategies to counteract antibody-mediated inhibition of virus spread. In this review, we discuss the current understanding regarding the spread mechanisms for these viruses with an attempt to extract common principles and identify key questions for future studies.
Keywords: : antibody neutralization, cell-to-cell transmission, hepatitis A virus, hepatitis B virus, hepatitis C virus, hepatitis D virus, hepatitis E virus, quasi-envelopment, virus entry, virus spread
The liver performs many essential roles in the body including detoxification, regulation of glycogen storage and production of bile. The hepatocytes are the main cell type in the liver, comprising approximately 80% of the liver cells. They are of epithelial origin, but differ from columnar epithelial cells by nature of their multipolar organization. Their basolateral membranes face the sinusoid blood and their apical membranes form the bile canaliculi through which the bile is secreted [1]. Hepatocytes are the main and probably only site for replication for the five known hepatitis viruses, each belonging to a different family (Figure 1). While hepatitis A virus (HAV) and hepatitis E virus (HEV) are usually transmitted via a fecal–oral route and generally cause self-limited infections, hepatitis B virus (HBV), hepatitis C virus (HCV) and hepatitis D virus (HDV) are transmitted by body fluids and usually cause persistent infections that can lead to liver cirrhosis and hepatocellular carcinoma [2,3]. Together they represent a major public health burden, affecting hundreds of millions of people globally and causing significant morbidity and mortality [3]. Remarkably, all of these viruses can replicate in and spread between hepatocytes without causing apparent cytopathic effects. This review seeks to provide a current understanding on the spread mechanisms for these viruses, with a goal to identify common principles and gaps in our knowledge. A brief summary of the spread mechanisms for these viruses is provided in Figure 2. For the detailed life cycle of individual hepatitis viruses, the readers are referred to other recent reviews [4–8].
Figure 1. . The liver is a common target for five phylogenetically distinct human hepatitis viruses (hepatitis A virus–hepatitis E virus).

Virus particles are depicted based on their basic structural components (envelope/quasi-envelope, envelope glycoproteins, capsid/nucleocapsid, and genome). HAV and HEV are transmitted via a fecal–oral route, and both exist in two virion types: naked virions that are shed into feces and quasi-enveloped virions that circulate in the blood. HBV, HCV and HDV are blood-borne pathogens and transmitted via contaminated body fluids. Note that HDV is a satellite virus of HBV and uses HBV's envelope for its extracellular release.
HAV: Hepatitis A virus; HBV: Hepatitis B virus; HCV: Hepatitis C virus; HDV: Hepatitis Dvirus; HEV: Hepatitis E virus.
Figure 2. . Spread mechanisms for hepatitis viruses.
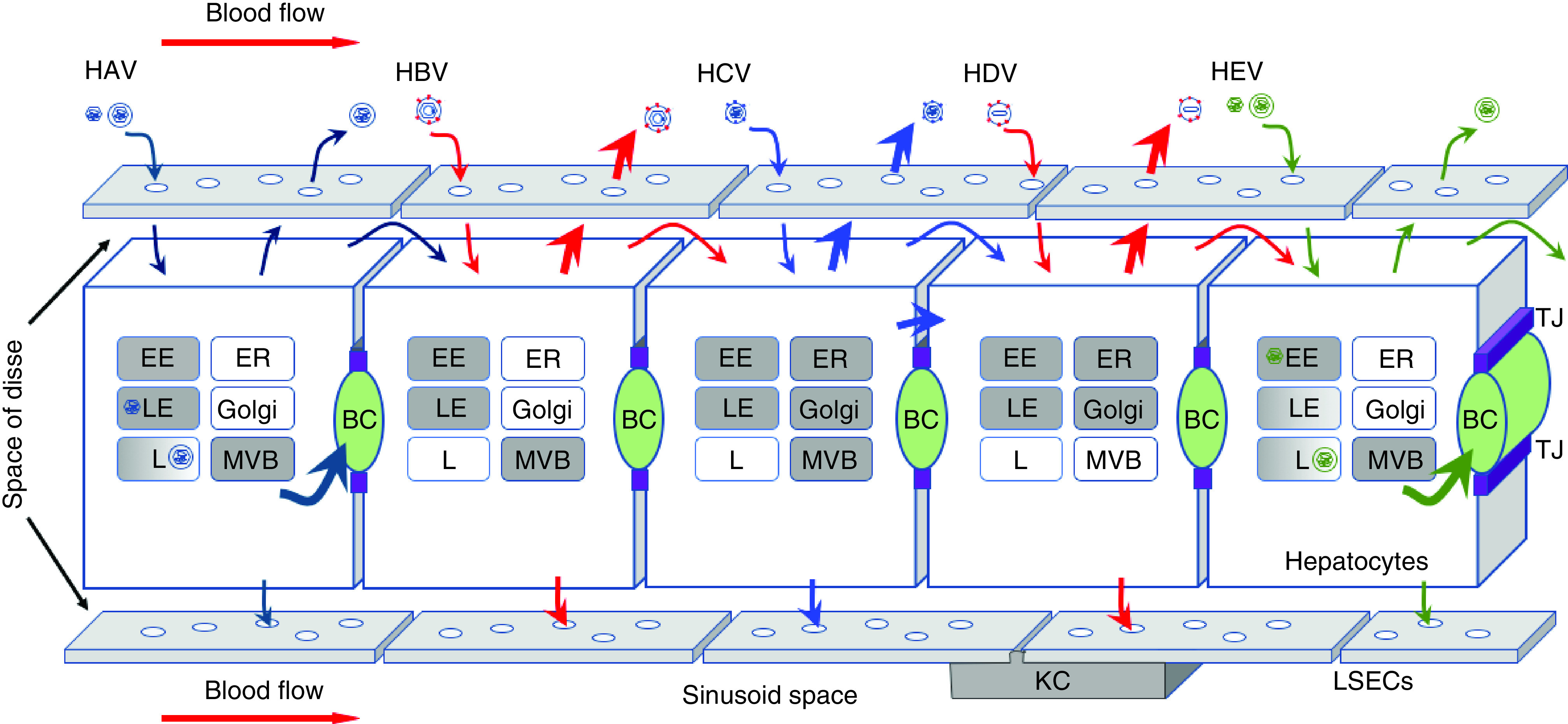
Hepatocytes and surrounding LSECs are depicted. The tight junctions (purple) separate bile canaliculi (green) formed by apical membranes of adjacent hepatocytes from the space of Disse and sinusoid blood. Circulating virus particles enter the space of Disse through fenestrated LSECs (white empty circles) to gain access to hepatocytes. All five human hepatitis viruses infect hepatocytes via the basolateral membrane and propagate in a noncytolytic fashion. Progeny virions are released either at the basolateral surface or apical surface as indicated by arrows. HCV also spreads via direct cell-to-cell transmission. Cellular organelles involved in entry and egress of particular viruses are shaded in gray. Note that HAV and HEV have two types of virions that upon internalization travel to distinct compartments for uncoating.
BC: Bile canaliculi; EE: Early endosomes; ER: Endoplasmic reticulum; HAV: Hepatitis A virus; HBV: Hepatitis B virus; HCV: Hepatitis C virus; HDV: Hepatitis Dvirus; HEV: Hepatitis E virus; KC: Kupffer cell; L: Lysosomes; LE: Late endosomes; LSEC: Liver sinusoid endothelial cell; MVB: Multivesicular bodies; TJ: Tight junction.
Spread mechanism for HAV & HEV
Both HAV and HEV are small positive-strand RNA viruses that are transmitted enterically and have been long considered as nonenveloped. However, recent studies show that both circulate in the blood as membrane-associated, ‘quasi-enveloped’ particles [9–11]. The biogenesis of quasi-enveloped HAV and HEV particles is similar to that of exosomes, small extracellular vesicles that mediate intercellular communication [12]. In doing so, HAV and HEV usurp the cellular endosomal sorting complex required for transport (ESCRT) machinery to bud into the multivesicular bodies (MVBs). Fusion of the MVBs with the plasma membrane results in the release of single membrane-encased viral particles to the extracellular space. Cargos destined for exosomal release typically contain one or more late domains, motifs recognized by specific ESCRT proteins [13]. For HAV, two tandem YPX3L late domains present in the viral capsid protein VP2 specifically interact with cellular ALIX (ALG2-interacting protein X) to facilitate the budding of newly assembled capsids into the MVBs [9]. A recent study shows that pX, a C-terminal extension of the VP1 protein, can also interact with ALIX and perhaps plays a redundant role in HAV release [14]. For HEV, the budding is mediated by a specific interaction between the viral protein ORF3 and an ESCRT-I protein TSG101 (tumor susceptibility gene-101) [15–17]. ORF3 is a membrane-associated protein and possesses an ion channel activity, properties that are also important for HEV release [18,19].
Since quasi-enveloped particles are the only form detected in the serum from patients infected with HAV or HEV [9,10], they most likely mediate virus spread within an infected host. However, neither HAV nor HEV has an envelope protein, and biochemical evidence suggests that no viral antigens are exposed on the surface of quasi-enveloped HAV or HEV particles [9,20]. As such, these particles cannot not use the traditional mechanisms of virus entry used by other viruses. Studies have shown that quasi-enveloped HAV and HEV particles enter cells via clathrin-mediated endocytosis [21–23], although the molecules that mediate cell attachment and the signal(s) that triggers endocytosis remain unclear. Cell attachment of quasi-enveloped HEV particles is tenfold less efficient than the naked HEV particles [21], and feces-derived HEV is more infectious than serum-derived HEV in humanized mice [24,25]. Following internalization, the quasi-enveloped particles travel to the endolysosome where the viral membrane becomes destroyed by lysosomal enzymes, exposing the capsid to its yet-to-be-identified receptor [22]. Since the receptors for HAV and HEV are unknown, the uncoating process for these viruses is poorly understood.
The discovery of quasi-enveloped HAV and HEV has fundamentally changed the view of the life cycle of these viruses and how the body reacts to them. Since no viral antigens are exposed on their surface, these particles probably cannot distinguish their hepatocyte target cells from other cells. If this is the case, then how is their hepatotropism maintained? Also puzzling is the role of antibodies in these infections. While it is well known that preexisting antibodies can protect against hepatitis A and hepatitis E, whether antibodies also play a role in the primary infection is not entirely clear. Indeed, virus-specific antibodies coexist with infectious particles in the blood of HAV- or HEV-infected patients [10,20,26]. Intriguingly, adding neutralizing anti-HAV antibodies to the culture medium after HAV inoculation can reduce HAV replication up to 90% [9], and passive transfer of anti-HAV can protect against hepatitis A as long as the antibodies were administered within two weeks of exposure [27]. These observations led to the hypothesis that anti-capsid antibodies that are internalized by cells may bind to the capsid and prevent uncoating once the viral membrane becomes disrupted in the endolysosome [11].
Spread mechanism for HBV & HDV
HBV is an enveloped DNA virus in the Hepadnaviridae family [28]. Although a DNA virus, HBV uses RNA as a replication intermediate. Replication occurs inside the capsid in the cytoplasm, and the virus acquires its envelope by budding into the MVBs [29]. The mature HBV particles, also known as ‘Dane’ particles, contain three envelope proteins: L, M and S. These proteins differ only in their N-terminal sequences, with L being the longest. HBV-infected cells also release abundant subviral particles (SVPs) that contain viral envelope proteins but not genomes. The SVPs are thought to play a role in immune evasion [30].
To initiate an infection, HBV first attaches to the cells via heparan sulfate proteoglycans (HSPGs) [31], followed by a specific interaction with its receptor sodium taurocholate cotransporting polypeptide (NTCP) [32]. A myristoylated peptide located at the N terminus of the HBV L protein is critical for this interaction. Binding to NTCP triggers virus particle internalization in a process that is also dependent on the EGFR [33]. HBV enters cells through clathrin-mediated endocytosis [34]. Fusion of viral membrane with the endosomal membrane releases the nucleocapsid into the cytoplasm, followed by its intracytoplasmic transport along the microtubules to the nucleus. Within the nucleus, the HBV genome is converted to covalently closed circular DNA (cccDNA) to establish a persistent infection.
HDV, classified as a Deltaviridae, is a satellite virus that uses HBV's envelope to exit from cells. HDV has a small, 1.7 kb circular RNA genome that encodes two proteins, a large hepatitis delta antigen (L-HDAg) and a small delta antigen (S-HDAg) [35]. These two proteins are produced by an RNA editing process and differ only in their C-terminal sequences. While S-HDAg is involved in HDV replication, L-HDAg is required for virus particle assembly. L-HDAg binds to the HDV genome, forming a rod-shaped nucleoprotein complex. Through an interaction between the C terminus of L-HDAg and HBV envelope proteins, HDV acquires its envelope in the endoplasmic reticulum (ER) and exits from cells via the secretory pathway. It is not clear why HBV and HDV acquire their envelope at different locations.
Although significant progress has been made toward understanding HBV/HDV egress and entry [36–38], none of the current cell culture systems support efficient HBV/HDV spread. The infectivity of HBV and HDV in cell culture is extremely low, even in the presence of 4% polyethene glycol (a chemical routinely used to enhance HBV cell attachment) and with overexpressed NTCP. Better cell culture and animal models are clearly needed. Intriguingly, mice engineered to express human NTCP are susceptible to HDV infection, but not to HBV [39]. This result suggests that additional host factor(s) are required for HBV entry. The intracellular life cycle of HBV is more complicated than HDV, and unique host factors may be required to facilitate HBV nuclear entry following endosomal escape.
A recent study shows that envelope proteins of unrelated viruses (e.g., the vesicular stomatitis virus and HCV) can functionally substitute for the HBV envelope proteins in HDV release [40]. This finding suggests there is a certain degree of flexibility in HDV morphogenesis and raises an interesting question about the origin and evolution of HDV.
Spread mechanism for HCV
HCV is a positive-strand RNA virus and a member of the Flaviviridae family. Since its discovery in 1989, substantial work has been done to understand HCV molecular biology and pathogenesis due to its ability to cause chronic infection and liver cancer [41]. In 2005, three independent groups reported the establishment of a robust cell culture system [42–44], making the study of the entire HCV life cycle possible. In many ways, HCV is unique, including its virion morphogenesis and cell entry [45,46]. HCV virions exist as lipoviroparticles (LVPs) due to their association with low-density and very-low-density lipoproteins (LDL and VLDL) [47]. LVPs exhibit low buoyant density and have limited exposure of the viral envelope proteins E1 and E2. This feature is thought to help shield the virus from neutralizing antibodies. HCV particles contain high levels of apolipoprotein E (apoE) and B (apoB), and apoE is important for efficient HCV egress and cell entry [48]. The assembly of HCV particles is initiated at the ER and cytoplasmic lipid droplets (cLDs) interface [49]. The newly synthesized HCV genome binds to the core proteins transferred from cLDs to form a nucleocapsid that then buds into the ER. Maturation and release of HCV virions is coupled to the VLDL pathway, and particles are produced as LVPs, which exit cells via the classic secretory pathway [50].
HCV entry is among the most complex and best studied mechanisms of viral entry [46,51]. A cascade of receptor interactions is necessary for HCV entry into the cell [52,53]. Attachment factors including the LDL receptor and syndecan-1 concentrate HCV at the cell surface [46]. Entry factors are necessary receptors for viral entry and include scavenger receptor class B type 1 (SR-B1), cluster of differentiation 81 (CD81), claudin-1 (CLDN1) and occludin (OCLN) [54]. HCV envelope 2 (E2) protein first binds SR-B1, CD81 and the epidermal growth factor receptor (EGFR) at the basolateral membrane. The complex next migrates to tight junctions in an actin dependent, where the virus particle interacts with CLDN1 and then OCLN. EGFR is subsequently activated and the virus particle is engulfed via clathrin-mediated endocytosis [53]. HCV entry is dependent on endosomal low pH, and fusion of the viral envelope to the endosomal membrane results in viral genome release into the cytoplasm.
Because HCV enters cells via the tight junctions, newly released virions can immediately bind to and infect adjacent cells. This direct cell-to-cell transmission provides the most efficient way for its spread. In addition, the confined space also limits the access of antibodies to the transferring virions [52]. Some enveloped viruses can also spread via direct cell-to-cell transmission without envelope proteins, but this hasn't been found to be significant in HCV, although low efficiency, exosome-mediated transfer of replication-competent HCV replicase complexes has been described [55,56].
Conclusion & future perspective
Viral spread, like all biological processes, is governed by several overarching principles. What appears to be common for the five hepatitis viruses discussed above is their noncytolytic spread and strategies to counteract antibody-mediated blockade of spread. The nonlytic spread is particularly remarkable for HAV and HEV, which have been considered as nonenveloped viruses for decades. HAV is the only picornavirus that can replicate in the liver. To adapt to grow in liver cells, HAV has evolved unique features to avoid cell lysis. This includes using highly deoptimized codons to slow the speed of protein translation [57], and hijacking cellular ESCRT components for nonlytic release [9]. HEV likely originated from ancient recombination events between an alpha-like virus and a picorna-like virus at the nonstructural/structural junction [58], and has evolved a unique protein ORF3 to facilitate its release via the MVB pathway. The ESCRT machinery is also involved in HBV release and exosomal transfer of HCV RNA between cells, pointing to a central role of the MVB pathway in hepatitis virus spread.
Also notable is that all five human hepatitis viruses have evolved strategies to evade host antibody responses to maximize their spread. While HAV and HEV hide themselves in host membranes from circulating antibodies, HCV is secreted as lipoviroparticles with minimal exposure of its envelope glycoproteins. HCV can also spread via direct cell-to-cell transmission to avoid neutralizing antibodies. HBV and HDV adopt a different strategy by producing high quantities of subviral particles that act as a ‘sink’ for neutralizing antibodies. Interestingly, HEV produces a secreted form of the ORF2 protein that appears to play a similar role [59].
So, where do we go from here? Here are a number of areas/questions that we think will be important for future studies.
What determines the hepatotropism of HAV & HEV?
Tissue and cell tropisms are often determined at the receptor level. As discussed above, quasi-enveloped particles probably cannot distinguish hepatocytes from other cell types during cell attachment. Thus, identifying the receptors for HAV and HEV will be an important step towards understanding their tropism.
Are there extrahepatic replication sites for HAV & HEV?
Although both HAV and HEV are transmitted enterically, there is no compelling evidence that either of them replicates in the gut. And, because the mechanism by which they penetrate the gut epithelium is poorly understood, we do not know what form these viruses take when leaving the gut and entering the bloodstream. The recently developed human intestinal organoids or ‘enteroids’ support infection and replication of many enteric viruses [60,61], providing an exciting tractable system for answering these important questions. HEV infections have been associated with various forms of extrahepatic manifestations, including neurological, hematological and renal conditions [62,63]. Whether direct infection/replication contributes to these diseases is also an area of interest [64].
What is missing in HBV entry & spread?
Mice expressing human NTCP support HDV infection and spread, but not HBV [39]. Since direct delivery of the HBV genome into the mouse liver supports replication, there likely exist unknown host factors critical for HBV entry at a step after membrane fusion (e.g., getting the genome out of the nucleocapsid, or converting the partially double-stranded DNA genome to cccDNA). Also puzzling is the poor HBV spread in cell culture. Is it simply due to inefficient quantities of infectious particles being released, or is there something missing in the released particles? A better understanding of the HBV entry and release process is necessary for developing tractable animal models for HBV which are desperately needed in the field.
How important is cell-to-cell transmission in HCV infection?
Among the five known hepatitis viruses, only HCV has been shown to spread via direct cell-to-cell transmission. Direct cell-to-cell transmission is the most efficient way for local spread, and has the advantages of evading the humoral response and achieving a higher multiplicity. The extent of this type of spread is in part affected by local interferon (IFN) responses, as is the case for HCV [65]. On the other hand, high quantities of free HCV virions are produced and enter the blood (approximately 1012 particles per day) [66]. Free virions have the ability to infect cells at distal sites in the liver that are less influenced by the local IFN response at the original site. Thus, both free virion-mediated spread and direct cell-to-cell transmission may be important for the establishment and persistence of HCV infection. It is worth noting that most studies on HCV used Huh7 cells, which are not polarized as primary hepatocytes are. Thus, the nature of cell-to-cell transmission in polarized hepatocytes and its relative contribution to HCV spread in vivo require further investigation.
Does cell death contribute to hepatitis virus spread?
While there is no compelling evidence that this is the case, this possibility has not been formally ruled out either. Hepatocytes are terminally differentiated cells with a slow turnover rate in a healthy liver. However, since their growth is asynchronous, with a fraction of cells undergo cell death as a part of the normal physiological process. In addition, virus-infected cells may have an altered physiological status causing accelerated cell death. Given their potential to release a much higher quantity of intracellular progeny virions, lysis of even a minor fraction of infected cells could have a significant impact on local and distal virus spread. Since most early studies on hepatitis viruses were conducted in cancer cell lines, the importance of this issue remains to be addressed. The recent development of new models that more closely mimic in vivo conditions such as liver organoids and animal models provide new tools to revisit this issue.
Finally, what are the roles of LSECs & Kupffer cells in hepatitis virus spread?
LSECs and Kupffer cells are hepatic scavenger cells that play a role in clearing apoptotic cells and bloodborne pathogens. The anatomic location of these cells presents a physical barrier to virus spread via the blood (Figure 2). Phosphatidylserines present on the surface of quasi-enveloped HAV and HEV particles may make Kupffer cells a preferential target for binding. In addition, liver nonparenchymal cells including LSECs and Kupffer cells have been shown to restrict HBV spread through production of IFNs and transferring antiviral effector proteins directly into infected cells [67]. It is foreseeable that advances in the development of liver organoids and coculture systems using isolated primary cells or stem-derived cells will provide ample opportunities for studying these important questions.
Executive summary.
Liver structure/common models of liver
The largest metabolic organ, the liver mostly consists of hepatocytes, a type of epithelial cell specific to it.
Hepatocytes differ from other epithelial cells by nature of their multipolar organization.
Common models for studying the liver include primary hepatocytes, hepatic and nonhepatic immortalized cell cultures, and liver-humanized mice.
Hepatitis A virus
Hepatitis A virus (HAV) is a positive-sense RNA virus with exceptional stability in the environment.
As a quasi-enveloped virus, HAV appears to have originated from a nonenveloped virus that ‘hijacked’ a lipid envelope from host cell membranes.
eHAV is released nonlytically at both basolateral and apical membranes as small extracellular vesicles (EVs) using the MVB pathway mediated by the ESCRT machinery.
Hepatitis B virus
Hepatitis B virus (HBV) is a circular, partially double-stranded para-retrovirus.
Sodium taurocholate cotransporting polypeptide is a necessary cell surface receptor for the entry of HBV.
Infectious HBV particles exit cells using the MVB pathways while HBV sub-viral particles are released via the classic secretory pathway.
Hepatitis C virus
Hepatitis C virus (HCV) is a positive-sense RNA virus, consisting of a single open reading frame (ORF) and its polyprotein is processed by cellular and viral proteases into ten mature viral proteins.
HCV entry is among the most complex and best studied mechanisms of viral entry.
Maturation and release of HCV virions is coupled to the very-low-density lipoprotein pathway, and particles are produced as lipoviroparticles, which exit cells via the secretory pathway.
HCV spreads by both free virions and direct cell-to-cell transmission.
Hepatitis D virus
Hepatitis D virus (HDV) is a quasi-double stranded viroid, commonly understood to be dependent on HBV for spread.
HDV replicates via a rolling circle mechanism, and viral persistence is due to re-infection or single-stranded RNA replication.
There is recent evidence (in limited systems) that other viruses may be involved in producing infectious HDV.
Hepatitis E virus
Hepatitis E virus (HEV) is a positive-sense RNA virus with three ORFs.
Like HAV, HEV has been observed in both enveloped and nonenveloped forms.
HEV appears to have originated as an enveloped virus that adapted to be able to exist and remain infectious without one.
Acknowledgements
The authors thank M Peeples and J Zhu for critical reading the manuscript.
Footnotes
Financial & competing interests disclosure
This work is supported in part by grants from National Institutes of Health/National Institute of Allergy and Infectious Diseases (R01AI139511, R21AI137912) and the Gilead Science Research Scholars Program in Liver Disease (Z Feng). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Treyer A, Musch A. Hepatocyte polarity. Compr. Physiol. 3(1), 243–287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox AL, El-Sayed MH, Kao JH. et al. Progress towards elimination goals for viral hepatitis. Nat. Rev. Gastroenterol. Hepatol. 17(9), 533–542 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lanini S, Ustianowski A, Pisapia R, Zumla A, Ippolito G. Viral hepatitis: etiology, epidemiology, transmission, diagnostics, treatment, and prevention. Infect. Dis. Clin. North Am. 33(4), 1045–1062 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Mcknight KL, Lemon SM. Hepatitis A virus genome organization and replication strategy. Cold Spring Harb. Perspect. Med. 8(12), a033480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenney SP, Meng XJ. Cold Spring Harb. Perspect. Med. 9(1), a031724 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lucifora J, Delphin M. Current knowledge on hepatitis delta virus replication. Antiviral Res. 104812 (2020). (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 7.Dubuisson J, Cosset FL. Virology and cell biology of the hepatitis C virus life cycle: an update. J. Hepatol. 61(Suppl. 1), S3–S13 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology 479–480, 672–686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng Z, Hensley L, Mcknight KL. et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496(7445), 367–371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes the discovery and characterization of ‘quasi-enveloped’ hepatitis A virus.
- 10.Takahashi M, Yamada K, Hoshino Y. et al. Monoclonal antibodies raised against the ORF3 protein of hepatitis E virus (HEV) can capture HEV particles in culture supernatant and serum but not those in feces. Arch. Virol. 153(9), 1703–1713 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Feng Z, Lemon SM. Peek-a-boo: membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 22(2), 59–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200(4), 373–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hurley JH. The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 45(6), 463–487 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang W, Ma P, Deng L. et al. Hepatitis A virus structural protein pX interacts with ALIX and promotes the secretion of virions and foreign proteins through exosome-like vesicles. J. Extracell. Vesicles 9(1), 1716513 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagashima S, Takahashi M, Jirintai S. et al. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J. Gen. Virol. 92(Pt 2), 269–278 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Nagashima S, Takahashi M, Jirintai S. et al. Tumour susceptibility gene 101 and the vacuolar protein sorting pathway are required for the release of hepatitis E virions. J. Gen. Virol. 92(Pt 12), 2838–2848 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Yamada K, Takahashi M, Hoshino Y. et al. ORF3 protein of hepatitis E virus is essential for virion release from infected cells. J. Gen. Virol. 90(Pt 8), 1880–1891 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Gouttenoire J, Pollan A, Abrami L. et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLoS Pathog. 14(12), e1007471 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Q, Heller B, Capuccino JM. et al. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl Acad. Sci. USA 114(5), 1147–1152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi M, Tanaka T, Takahashi H. et al. Hepatitis E virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: characterization of HEV virions in blood circulation. J. Clin. Microbiol. 48(4), 1112–1125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin X, Ambardekar C, Lu Y, Feng Z. Distinct entry mechanisms for nonenveloped and quasi-enveloped hepatitis E viruses. J. Virol. 90(8), 4232–4242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes the quasi-enveloped hepatitis E virus (HEV) particles use a novel mechanism for cell entry.
- 22.Yin X, Feng Z. Hepatitis E virus entry. Viruses 11(10), 883 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera-Serrano EE, Gonzalez-Lopez O, Das A, Lemon SM. Cellular entry and uncoating of naked and quasi-enveloped human hepatoviruses. Elife 8, e43983 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Provides a detailed analysis of the entry process for both naked and quasi-enveloped hepatitis A virus (HAV) particles.
- 24.Sayed IM, Verhoye L, Cocquerel L. et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 66(5), 920–929 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Allweiss L, Gass S, Giersch K. et al. Human liver chimeric mice as a new model of chronic hepatitis E virus infection and preclinical drug evaluation. J. Hepatol. 64(5), 1033–1040 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Asher LV, Binn LN, Mensing TL, Marchwicki RH, Vassell RA, Young GD. Pathogenesis of hepatitis A in orally inoculated owl monkeys (Aotus trivirgatus). J. Med. Virol. 47(3), 260–268 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Victor JC, Monto AS, Surdina TY. et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N. Eng. J. Med. 357(17), 1685–1694 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Hu J, Protzer U, Siddiqui A. Revisiting hepatitis B virus: challenges of curative therapies. J. Virol. 93(20), (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prange R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 201(4), 449–461 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Hu J, Liu K. Complete and incomplete hepatitis B virus particles: formation, function, and application. Viruses 9(3), (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 46(6), 1759–1768 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Yan H, Zhong G, Xu G. et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1, e00049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes the identification of sodium taurocholate cotransporting polypeptide as a long-sought cellular receptor for hepatitis B and D virus.
- 33.Iwamoto M, Saso W, Sugiyama R. et al. Epidermal growth factor receptor is a host–entry cofactor triggering hepatitis B virus internalization. Proc. Natl Acad. Sci. USA 116(17), 8487–8492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrscher C, Pastor F, Burlaud-Gaillard J. et al. Hepatitis B virus entry into HepG2-NTCP cells requires clathrin-mediated endocytosis. Cell Microbiol. 22(8), e13205 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Sureau C, Negro F. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol. 64(Suppl. 1), S102–S116 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Tu T, Urban S. Virus entry and its inhibition to prevent and treat hepatitis B and hepatitis D virus infections. Curr. Opin. Virol. 30, 68–79 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Li W, Urban S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J. Hepatol. 64(Suppl. 1), S32–S40 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blondot ML, Bruss V, Kann M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 64(Suppl. 1), S49–S59 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Winer BY, Shirvani-Dastgerdi E, Bram Y. et al. Preclinical assessment of antiviral combination therapy in a genetically humanized mouse model for hepatitis delta virus infection. Sci. Transl. Med. 10(447), (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez-Vargas J, Amirache F, Boson B. et al. Enveloped viruses distinct from HBV induce dissemination of hepatitis D virus in vivo. Nat. Commun. 10(1), 2098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19(7), 837–849 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wakita T, Pietschmann T, Kato T. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11(7), 791–796 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindenbach BD, Evans MJ, Syder AJ. et al. Complete replication of hepatitis C virus in cell culture. Science 309(5734), 623–626 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Zhong J, Gastaminza P, Cheng G. et al. Robust hepatitis C virus infection in vitro. Proc. Natl Acad. Sci. USA 102(26), 9294–9299 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 8(1), e1002466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerold G, Moeller R, Pietschmann T. Hepatitis C virus entry: protein interactions and fusion determinants governing productive hepatocyte invasion. Cold Spring Harb. Perspect. Med. 10(2), a036830 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Catanese MT, Uryu K, Kopp M. et al. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl Acad. Sci. USA 110(23), 9505–9510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81(24), 13783–13793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrates the role of virion-associated apoliportein E in both entry and release of HCV.
- 49.Jones DM, Mclauchlan J. Hepatitis C virus: assembly and release of virus particles. The J. Biol. Chem. 285(30), 22733–22739 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukuhara T, Ono C, Puig-Basagoiti F, Matsuura Y. Roles of lipoproteins and apolipoproteins in particle formation of hepatitis C virus. Trends Microbiol. 23(10), 618–629 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Belouzard S, Danneels A, Feneant L, Seron K, Rouille Y, Dubuisson J. Entry and release of hepatitis C virus in polarized human hepatocytes. J. Virol. 91(18), e00478–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graw F, Perelson AS. Modeling Viral Spread. Annu. Rev. Virol. 3(1), 555–572 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baktash Y, Madhav A, Coller KE, Randall G. Single particle imaging of polarized hepatoma organoids upon hepatitis C virus infection reveals an ordered and sequential entry process. Cell Host Microb. 23(3), 382–394 e385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• An elegant study using a 3D culture system and a high-resolution imaging technique to dissect the entry pathway of HCV.
- 54.Miao Z, Xie Z, Miao J, Ran J, Feng Y, Xia X. Regulated entry of hepatitis C virus into hepatocytes. Viruses 9(5), 100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Longatti A, Boyd B, Chisari FV. Virion-independent transfer of replication-competent hepatitis C virus RNA between permissive cells. J. Virol. 89(5), 2956–2961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 10(10), e1004424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinto RM, Aragones L, Costafreda MI, Ribes E, Bosch A. Codon usage and replicative strategies of hepatitis A virus. Virus Res. 127(2), 158–163 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Kelly AG, Netzler NE, White PA. Ancient recombination events and the origins of hepatitis E virus. BMC Evol. Biol. 16(1), 210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin X, Ying D, Lhomme S. et al. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl Acad. Sci. USA 115(18), 4773–4778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ding S, Song Y, Brulois KF. et al. Retinoic acid and lymphotoxin signaling promote differentiation of human intestinal M cells. Gastroenterology 159(1), 214–226. e1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ettayebi K, Crawford SE, Murakami K. et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353(6306), 1387–1393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Webb GW, Dalton HR. Hepatitis E: an underestimated emerging threat. Ther. Adv. Infect. Dis. 6, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kamar N, Pischke S. Acute and persistent hepatitis E virus genotype 3 and 4 Infection: clinical features, pathogenesis, and treatment. Cold Spring Harb. Perspect. Med. 9(7), a031872 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng Z. Causation by HEV of extrahepatic manifestations remains unproven. Liver Int. 36(4), 477–479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wieland S, Makowska Z, Campana B. et al. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 59(6), 2121–2130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neumann AU, Lam NP, Dahari H. et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282(5386), 103–107 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Li J, Liu K, Liu Y. et al. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 14(8), 793–803 (2013). [DOI] [PubMed] [Google Scholar]