

Abstract
Recent studies have proven that long noncoding RNAs (lncRNAs) exhibit regulatory functions of both DNA damage response (DDR) and endoplasmic reticulum (ER) stress. Herein, ER stress‐induced lncRNA transcriptomic changes are reported in human oral squamous cell carcinoma (OSCC) cells and a novel lncRNA HITTERS (H ERPUD1 intronic transcript of ER stress) is identified as the most significantly upregulated lncRNA. It is shown that HITTERS is a nucleus‐located lncRNA including two transcript variants. HITTERS lacks an independent promoter but shares the same promoter with HERPUD1. HITTERS is transcriptionally regulated by Activating Transcription Factor (ATF) 6, ATF4, X‐Box Binding Protein 1 (XBP1), and DNA methylation. In human OSCC tissues, HITTERS is significantly correlated with OSCC clinicopathological features and prognosis. Gain‐ and loss‐of‐function studies reveal that HITTERS promotes OSCC proliferation and invasion via influencing the expression of growth factor receptors and the downstream pathways. Once ER stress is triggered, HITTERS significantly attenuates ER stress‐induced apoptosis both in vivo and in vitro. Mechanically, HITTERS functions as RNA scaffold to promote MRE11‐RAD50‐NBS1 complex formation in the repair of ER stress‐induced DNA damage. To sum up, this study presents a novel lncRNA, namely HITTERS, which links ER stress and DDR together in OSCC.
Keywords: apoptosis, DNA damage response, endoplasmic reticulum stress, long noncoding RNA, oral squamous cell carcinoma
HERPUD1 intronic transcript of endoplasmic reticulum (ER) stress (HITTERS) is a long noncoding RNA that is regulated by ER stress and protects oral cancer cells from ER stress‐induced apoptosis via promoting DNA repair.
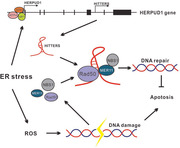
1. Introduction
The status of solid cancers, including oral squamous cell carcinoma (OSCC), inextricably links to stressful microenvironments such as hypoxia and lack of glucose or other nutrients.[ 1 ] In response to endoplasmic reticulum (ER) stress, cells will activate unfolded protein response (UPR). Once the misfolded proteins are accumulated in ER lumen, the chaperone binding‐immunoglobulin protein (BIP) departs from the UPR sensors and activates three major UPR pathways that are IRE1α‐XBP1 pathway, Activating Transcription Factor (ATF) 6α pathway, and PRKR‐like ER kinase‐EIF2α‐ATF4 pathway. These UPR pathways govern various cellular events depending on the severity and duration of the stress. These cellular events include manipulation of gene transcription and translation, mRNA decay, inhibition of global protein synthesis, promotion of unfolded proteins degradation, and finally recovery of homeostasis or initiation of apoptosis.[ 2 ]
Evidence has indicated that DNA damage response (DDR) is crucial in OSCC carcinogenesis and treatment. Specifically, genes involved in DDR, such as TP53, Ataxia Telangiectasia Mutated (ATM), and Ataxia Telangiectasia And Rad3‐Related Protein (ATR), are frequently mutated in both OSCC tissues and patient‐derived OSCC tumoroids.[ 3 ] Adjuvant chemoradiotherapy which triggers DDR is a routine treatment for OSCC patients.[ 4 ] It is well established that ER stress is closely related to cancer cell proliferation, apoptosis, angiogenesis, and metastasis.[ 1 ] However, little is known about the role of ER stress in DDR.
Long noncoding RNAs (lncRNAs) are a class of RNAs whose sequences are longer than 200 nucleotides and lack protein‐coding potential.[ 5 ] LncRNAs regulate numerous cell activities in cancer via chromatin modification, transcriptional regulation, and post‐transcriptional regulation.[ 6 ] Recent studies have proved that lncRNAs participated positively in DDR by sensing DNA damage, transducing damage signals, repairing damaged DNA, activating cell cycle checkpoints, and inducing apoptosis.[ 7 ] Besides, researchers have discovered several lncRNAs that could regulate ER stress and UPR.[ 8 , 9 , 10 ] However, little is known about how ER stress and UPR affect lncRNA transcription.
Given these issues, here we for the first time report the ER stress‐induced lncRNA transcriptomic changes in human OSCC cells and identifies thousands of differentially expressed lncRNAs. It is intriguing that only ≈10% of lncRNAs are selectively upregulated under ER stress, and among these upregulated genes the HITTERS (H ERPUD1 intronic transcript of ER stress), a novel lncRNA which is located in the sixth intron of HERPUD1, is the most altered lncRNA.
We next show that HITTERS is a nucleus‐located lncRNA including two transcript variants (TVs) which lack coding ability and are extremely sensitive to ER stress. As an intronic lncRNA, HITTERS lacks independent promoter and shares the same promoter with HERPUD1. Similar to HERPUD1, HITTERS is transcriptionally regulated by ATF6, XBP1s, ATF4, and DNA methylation. In human OSCC tissues, HITTERS shows strong coexpression with HERPUD1 and is significantly correlated with OSCC clinicopathological features and prognosis.
Finally, functional experiments show that HITTERS acts as an oncogene to promote OSCC proliferation and invasion via influencing the expression of growth factors receptors and the downstream pathways. Under basal condition, HITTERS has no impact on apoptosis; however, once ER stress is triggered, HITTERS significantly attenuates ER stress‐induced apoptosis. Mechanically, we find that the ER stress‐induced antiapoptosis function of HITTERS dose not dependent on UPR initiation or HERPUD1 expression. Instead, it functions as RNA scaffolds to promote MRE11‐RAD50‐NBS1 (MRN) complex formation to repair ER stress‐induced DNA damage.
2. Results
2.1. Identification of ER Stress‐Related lncRNAs
So far, there is no lncRNA transcriptome analysis on the impact of ER stress in human cells. To explore this question, we treated human OSCC cell lines SCC25 and CAL27 with different concentration of tunicamycin (TM)[ 11 ] and found that TM could significantly induce ER stress as determined by Western Figure 1A). We then applied quantitative polymerase chain reaction (qPCR) to test the time‐course of TM induced ER stress markers and found that at the early phase (≤6 h), TM could significantly induce the expression of ER stress markers (Figure 1B).
Figure 1.
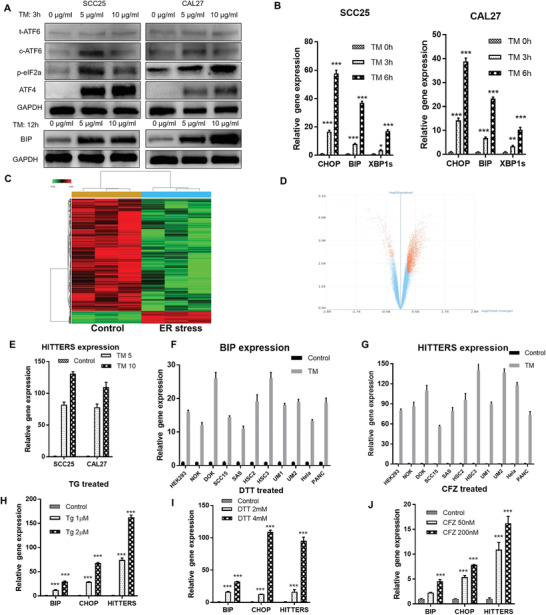
Identification of ER stress‐related lncRNAs. A) Western blot and B) qPCR confirmed that different concentrations of TM (5, 10 µg mL−1) or short time TM exposure could significantly induce ER stress of both SCC25 and CAL27 cells. C) Heatmap and D) volcano plot of HTA 2.0 lncRNA microarray for gene expression profiles of nontreated and TM‐treated SCC25 cells. E) qPCR confirmed the results of microarray that HITTERS was upregulated by treating cells with TM (5, 10 µg mL−1) for 6 h. F–J) qPCR showed F,G) treating different cells with TM (10 µg mL−1) for 6 h or H–J) treating SCC25 with different types of ER stress inducer for 6 h could upregulate both ER stress marker and HITTERS. The Student t‐test was used for analyzing the difference in (F) and (G). One‐way Analysis of Variance (ANOVA) test and Dunnett t‐test was used for (B), (E), and (H)–(J). For (E)–(G), all p < 0.001. Note: *, P < 0.05; **, P < 0.01, ***, P < 0.001.
Based on the above results, we used human transcriptome array 2.0 (HTA 2.0) microarray and analyzed the mRNA and lncRNA transcriptome change of TM treated SCC25 cells (5 µg mL−1, 6 h). 1070 (695 up, 375 down) differentially expressed mRNAs were found after TM treatment (p < 0.05, fold change ≥1.2). Both growth factor binding (GO) analysis and pathway analysis of differentially expressed mRNAs confirmed ER stress was significantly induced (Figure S1A,B, Supporting Information)
LncRNA expression profile indicated 2622 differentially expressed lncRNAs between TM treated and untreated cells, which consisted of 263 increased lncRNAs and 2359 decreased lncRNAs (p < 0.05, fold change ≥1.2; Figure 1C,D). These results indicated that at the early stage of ER stress, cells tended to repress the global lncRNA transcription, only a few lncRNAs were selectively upregulated. The transcript ENST00000570210.1 was the most significantly upregulated lncRNA under TM induced ER stress, which was further confirmed by qPCR (Figure 1E). ENST00000570210.1 is located on human chromosome 16: 56941028–56941726 forward strand, corresponding to the sixth intron of HERPUD1.
It is well known that lncRNA could regulate gene expression in cis and HERPUD1 was ranked as the second most significant upregulated gene in the mRNA expression profile (Figure S1C, Supporting Information). Moreover, lncRNA‐mRNA coexpression network analysis showed that ENST00000570210.1 was highly correlated with multiple ER stress‐related genes (Figure S1D,E, Supporting Information). Therefore, we focused on ENST00000570210.1 and named it HITTERS.
We further treated another 11 human cell lines with TM. We also treated SCC25 and CAL27 with another three types of ER stress inducer, including thapsigargin (disturbing calcium flow of ER), dithiothreitol (DTT, disturbing disulfide bonds of ER), and carfilzomib (CFZ, disturbing proteasome activity). The results revealed that once ER stress was induced, HITTERS was significantly upregulated (Figure 1F–J), indicating that the upregulation of HITTERS under ER stress was a general phenomenon.
2.2. Basic Characteristics of HITTERS
Since that HITTERS is a new lncRNA that has not been fully characterized, we first analyzed its sequence using rapid amplification of cDNA ends (RACE) assay. The 5’‐RACE showed a clear single band, the 3’‐RACE showed 3 bands, one of which was an unspecific band (Figure 2A). The RACE results revealed that HITTERS has 2 TVs. Compared to the 699 nt no‐variants lncRNA annotated in University of California Santa Cruz (UCSC) genome browser (Figure 2B), the HITTERS short TV (HITTERS‐TV1) is 793 nt, the HITTERS long TV (HITTERS‐TV2) is 1199 nt. Sequencing results mapped to the sixth intron of HERPUD1, namely the forward strand of chromosome 16: 56940933–56941725 (HITTERS‐TV1) and 56940933–56942131 (HITTERS‐TV2) (Table S4, Supporting Information). Two TVs shared the same 793 nt region and TV2 has an extra 406 nt 3’ arm (Figure 2C).
Figure 2.
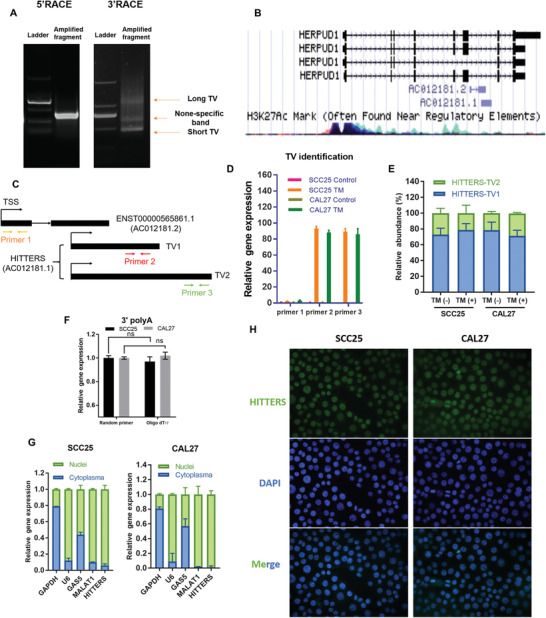
Basic characteristics of HITTERS. A) Agarose gel electrophoresis of PCR products generated by 5’ and 3’ RACE technologies. B) The schematic representation of HERPUD1 and HITTERS (annotated as AC012181.1) in UCSC Genome Browser tracks. C) The schematic representation of qPCR primers design strategy for distinguishing two HITTERS TVs and ENST00000565861.1. D) qPCR showed two HITTERS TVs changed significantly in response to TM (10 µg mL−1, 6 h), whereas ENST00000565861.1 did not change. Student t‐test was used. Only the changes of two HITTERS TVs were significant after TM treatment (p < 0.001). E) The relative abundance of two HITTERS TVs treated with or without TM (10 µg mL−1, 6 h). F) qPCR results on cDNA reverse‐transcripted by random primer or Oligo dT17 indicating that HITTERS had polyA tail. Student t‐test was used. “ns” stands for no significance. G) Nucleus‐cytoplasm fractionation qPCR and H) RNA FISH confirmed that HITTERS mainly located in cell nucleus.
Another lncRNA ENST00000565861.1 had an overlap region with HITTERS (Figure 2B). To differentiate these two genes, we designed specific primers (Figure 2C). qPCR results revealed that the expression of ENST00000565861.1 remained unchanged after inducing ER stress (Figure 2D), which confirmed that HITTERS is an independent lncRNA.
We found two TVs of HITTERS were both highly responsive to ER stress (Figure 2C,D). The ratio of their expression levels remains no change, no matter the cells were under ER stress or not, and HITTERS‐TV1 was the predominant TV, which accounts for 75% of whole HITTERS TVs (Figure 2E). We also used random primer and oligo[ 12 ] 17 primer to reverse transcribe the RNA and found the two primers showed equal efficiency (Figure 2F), indicating that HITTERS has a polyA tail.
Next, we cloned the sequence of HITTERS into pcDNA3.1(+)‐3xHA vector to explore its coding potential. None of the HITTERS recombinant plasmids showed a fusion protein (Figure S1F,G, Supporting Information), indicating that HITTERS lacks coding capability, no matter ER stress was triggered or not. Bioinformatic tools also predicted that similar to HOX Transcript Antisense RNA (HOTAIR), HITTERS had no protein coding ability (Figure S1H,I, Supporting Information). The subcellular location of lncRNA is very important for its function. By RNA fluorescent in situ hybridization (FISH) and nucleus‐cytoplasm fractionation qPCR, we found that HITTERS is predominantly located in the nucleus (Figure 2G,H). Overall, our results showed that HITTERS is a nuclear polyadenylated lncRNA with two TVs and HITTERS‐ TV1 is the major TV.
2.3. HITTERS Shares the Same Promoter with HERPUD1
Since we have identified the chromosome location of HITTERS, we next explored how HITTERS was transcribed. We cloned the potential promoter region (−2465 to +192 of transcription start sites, TSS, ≈2.6 kb DNA fragment) of HITTERS into pGL4.20 vector and found none of these DNA fragments showed promoter activity, no matter whether the ER stress was induced or not (Figure 3A). Another reporter vector pEZX‐FR01 showed identical results (Figure S1J,K, Supporting Information), which confirmed that HITTERS lacks independent promoter.
Figure 3.
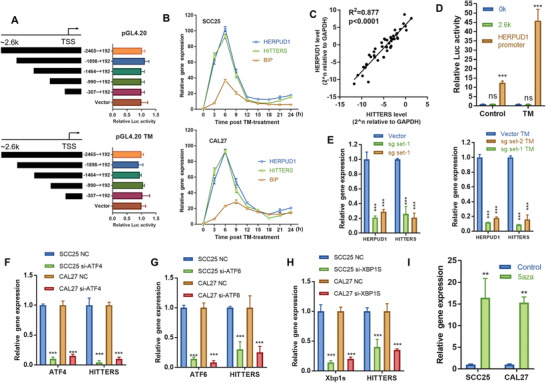
HITTERS shares the same promoter with HERPUD1. A) Dual‐luciferase reporter assay showed the potential promoter DNA fragments of HITTERS lacked transcription activity no matter threated with TM (10 µg mL−1, 6 h) or not. One‐way ANOVA and Dunnett t‐test were used, “Vector” was the control, all differences were none significant. B) qPCR results showed that time‐course change of HITTERS was identical to HERPUD1, but significant differed from BIP. Cells were treated with TM (10 µg mL−1) and measured every 3 h. C) qPCR results showed HITTERS and HERPUD1 had a strong coexpression pattern in 48 OSCC samples. Liner‐regression test was used. D) Dual‐luciferase reporter assay showed the promoter of HERPUD1 had strong transcription activity and responded obviously to TM (10 µg mL−1, 6 h). Student t‐test was used. E) qPCR results indicated that depleting HERPUD1 promoter in HEK293 by CRISPR/Cas9 system significantly suppressed the expression level of both HERPUD1 and HITTERS, no matter treated with or without TM (10 µg mL−1, 6 h). One‐way ANOVA and Dunnett t‐test were used, “Vector” was the control. qPCR results indicated knockdown of F) ATF4, G) ATF6, and H) XBP1s significantly suppressed HITTERS expression. Cells were transfected with siRNA for 48 h and then treated with TM (10 µg mL−1) for 6 h. Student t‐test was used. I) qPCR results showed treating cells with 5aza (10 × 10−6 m, 24 h) for inhibiting DNMTs significantly promoted HITTERS expression. Student t‐test was used. Note: ns, no significance; **, P < 0.01, ***, P < 0.001.
As HITTERS is an intron transcript of HERPUD1, we hypothesized that it shares the same promoter with HERPUD1. To test this hypothesis, we collected cells treated with TM every 3 h. qPCR results showed that the time‐course of changes in HITTERS and HERPUD1 RNA levels were almost identical, whereas significantly differed from another ER stress marker BIP (Figure 3B). We also found that the expression of HITTERS and HERPUD1 had a strong correlation in OSCC tissue samples (Figure 3C).
We then cloned the 338 bp promoter region (−303 to +35 of TSS) of HERPUD1 as previously described[ 13 ] and found this region had strong promoter activity and was highly responsive to ER stress (Figure 3D). We then applied the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technique and found after deleting this promoter region, the RNA levels of HERPUD1 and HITTERS were both dramatically decreased (Figure 3E). Previous study has confirmed that this promoter region had ER stress response element (ERSE)‐I, ERSE‐II, and CCAAT/enhancer binding protein‐ATF composite cis‐acting element, to which ATF6, XBP1, and ATF4 bind and upregulate the transcription of HERPUD1.[ 14 ] Also, DNA methylation was reported to suppress HERPUD1 transcription.[ 13 ] We found that similar to the transcription regulation of HERPUD1, silencing those transcript factors could decrease HITTERS RNA level (Figure 3F–H), whereas treating cells with DNA methyltransferase inhibitor 5aza could upregulate HITTERS RNA level (Figure 3I). Taken together, our results confirmed that HITTERS shares the same promoter with HERPUD1.
2.4. HITTERS Promotes OSCC Progression and Correlates with OSCC Clinicopathological Features
To explore the tumorigenic role of HITTERS in OSCC cells, we overexpressed HITTERS‐TV1 as it is the predominant TV. Since HITTERS mainly located in nucleus, we tested the knockdown efficiency of antisense oligonucleotide and siRNA and found siRNA is more effective. We selected one siRNA and constructed the corresponding shRNA lentivirus (Figure S2A–C, Supporting Information).
We found that knockdown of HITTERS significantly decreased cell proliferation, DNA replication, and colony formation in vitro (Figure 4A–C). In contrast, overexpression of HITTERS dramatically increased cell proliferation, DNA replication, and colony formation (Figure S2D–F, Supporting Information). The stably transfected OSCC cells were subcutaneously inoculated into nude mice to investigate the function of HITTERS in vivo. Results showed tumors grown from HITTERS overexpression cells were significantly larger (Figure S2G,H, Supporting Information), whereas tumors from HITTERS stable knockdown cells were significantly smaller (Figure 4D,E), comparing to their own control cells, respectively.
Figure 4.
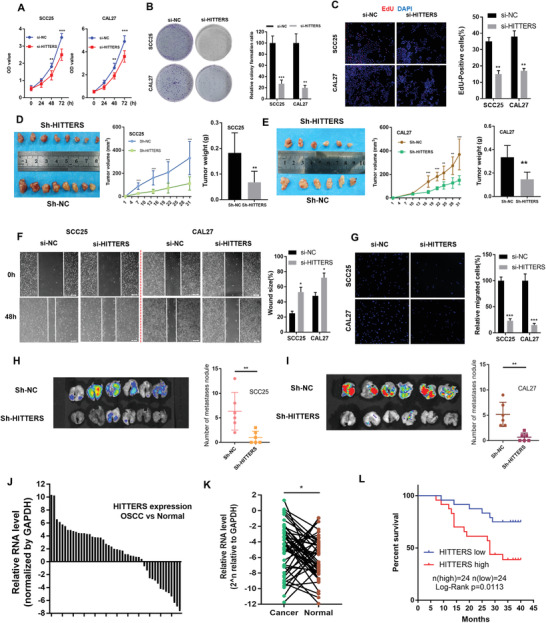
HITTERS promotes OSCC progression and correlates with OSCC clinicopathological features. A) CCK8, B) colony formation test, and C) EdU incorporation test confirmed that knockdown of HITTERS suppressed OSCC proliferation in vitro. D,E) Stably knockdown of HITTERS significantly suppressed tumor volume and tumor weight in D) SCC25 and E) CAL27 subcutaneous xenograft model. F) Wound healing test and G) transwell assay confirmed knockdown of HITTERS suppressed OSCC migration and invasion ability in vitro. Stably knockdown of HITTERS significantly suppressed H) SCC25 and I) CAL27 pulmonary metastasis nodule formation ability in vivo. The green fluorescent protein (GFP) fluorescense imaging of lungs was also presented. J) qPCR analyzed the relative fold of HITTERS in 48 OSCC tissues normalized by their paired adjacent normal tissues. K) qPCR results indicated the expression of HITTERS in OSCC was higher than their paired adjacent normal tissues. Paired t‐test was used. L) Kaplan–Meier curves showed high HITTERS expression had poor over‐all survival. Log‐rank test was used. For (A)–(I) Student t‐test was used. For (A), (D), and (E), Student t‐test was used for each time point. Note: *, P < 0.05; **, P < 0.01, ***, P < 0.001.
The effects of HITTERS on the invasion ability of OSCC cells were evaluated using wound‐healing assay and transwell assay. The results showed that overexpression of HITTERS significantly promoted cell invasion (Figure 4F,G), while HITTERS knockdown inhibited invasion (Figure S2I,J, Supporting Information). We further examined the effects of HITTERS in lung colonization by injecting OSCC cells into the tail vein of mice. HITTERS overexpression significantly increased the development of lung metastases (Figure S2K,L, Supporting Information). Mice injected with HITTERS knockdown cells exhibited a marked reduction in lung metastases (Figure 4H,I).
Next, we analyzed the correlation between HITTER expression and OSCC patient's clinicopathological features. HITTERS was significantly upregulated in OSCC tissues compared to the corresponding adjacent normal tissues (Figure 4J,K). Moreover, high expression of HITTERS in OSCC is correlated with advanced T stage, positive lymph node metastasis, high‐risk clinical stage, and poor overall survival (Table S5, Supporting Information, and Figure 4L). These results suggested that HITTERS plays an important role in OSCC progression.
2.5. Receptor Tyrosine Kinases and Transforming Growth Factor (TGF)‐β Receptors Are Downstream Targets of HITTERS
To further uncover the mechanism of how HITTERS promoted OSCC progression, we performed RNA sequencing and gene set enrichment analysis (GSEA). HITTERS knockdown significantly influenced the GO, no matter ER stress was triggered or not (Figure S3A,B, Supporting Information), which indicating it is an intrinsic function of HITTERS. We used qPCR and confirmed that some important growth factor receptors, especially the receptor tyrosine kinases (RTKs) and TGF‐β receptors, were downregulated after knocking down HITTERS (Figure S3C,D, Supporting Information). Previous study has demonstrated that RTKs could promote cell proliferation via phosphatidylinositol 3‐kinase/protein kinase B (PI3K/AKT) and mitogen activated protein kinase (MAPK) pathway,[ 15 ] and TGF‐β signaling pathway could promote cancer invasion via inducing Smad3 phosphorylation and subsequently epithelial‐mesenchymal transition (EMT).[ 16 ] In accordance with these studies, we found that HITTERS knockdown significantly decreased phosphorylation of AKT, ERK1/2, and Smad3 (Figure S3E, Supporting Information). Moreover, many downstream genes involved in cell proliferation and EMT were also regulated by HITTERS (Figure S3F,G, Supporting Information). Taken together, our results suggested that HITTERS promotes OSCC progression potentially via regulating RTKs and TGF‐β pathway.
2.6. HITTERS Attenuates ER Stress Induced Apoptosis
Severe or prolonged ER stress finally ends up with apoptosis. Since HITTERS is dramatically increased under ER stress, we explored its role in ER stress‐induced apoptosis. We found that TM treatment significantly decreased cell viability in a time‐ and dose‐dependent manner in OSCC cells (Figure 5A). The expression of apoptotic markers such as cleaved poly(ADP‐Ribose) polymerase (c‐PARP), BCL2 associated X, and cytochrome C were significantly increased after exposure to TM (Figure 5B). In order to test the effects of TM in vivo, mice were intraperitoneally injected with TM. We found that TM could significantly inhibited OSCC growth in vivo (Figure 5C,D). These results suggest that ER stress could induce OSCC apoptosis.
Figure 5.
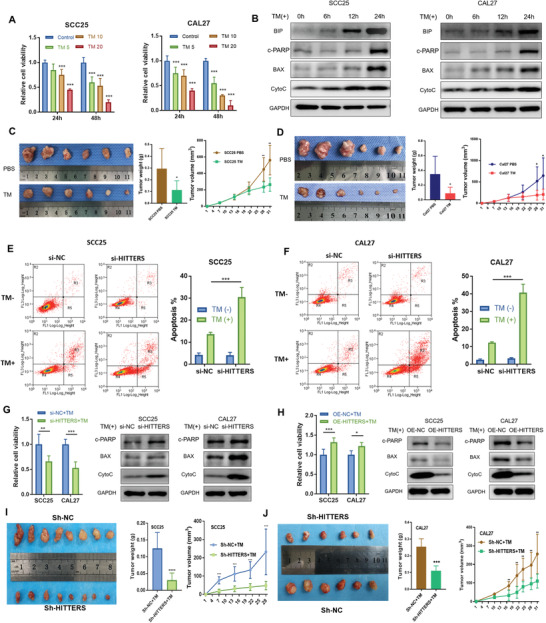
HITTERS attenuates ER stress induced apoptosis. A) Cell viability (measured by CCK8) of SCC25 and CAL27 were significantly suppressed by TM (5, 10, and 20 µg mL−1 for 24 and 48 h) in a dose‐ and time‐dependent manner. One‐way ANOVA and Dunnett t‐test were used for each time point. B) Western blot showed the ER stress marker BIP and apoptosis marker of SCC25 and CAL27 cells were significantly upregulated by TM (10 µg mL−1) in a time‐dependent manner. c‐PARP, cleaved PARP. C,D) Intraperitoneally injection of TM twice a week significantly suppressed tumor volume and tumor weight in C) SCC25 and D) CAL27 subcutaneous xenograft model. E,F) Under non‐ER stress condition, HITTERS would not affect apoptosis; however, depletion of HITTERS in TM (10 µg mL−1, 24 h) treated E) SCC25 and F) CAL27 cells significantly promoted apoptosis. Apoptosis was measured by Annexin‐V/PI double staining and flow cytometry. Proportion of R3 + R5 is considered apoptosis. Cells were transfected with siRNA for 48 h and then treated with TM (10 µg mL−1, 24 h). Two‐way ANOVA and Sidak's multiple comparisons test were used. G) Knocking‐down HITTERS significantly suppressed cell viability and promoted apoptosis marker expression; H) whereas overexpressing HITTERS obtained the opposite effect. Cells were transfected with siRNA for 48 h and then treated with TM (10 µg mL−1, 24 h). Cell viability differences were measured by CCK8 using Student t‐test. Stably knockdown of HITTERS causing I) SCC25 and J) CAL27 more sensitive to ER stress in vivo, reflected by a significantly reduction in tumor volume and tumor weight in subcutaneous xenograft model. All BALB/c nude mice were intraperitoneally injected with TM, twice a week, after tumor bearing. Student t‐test was used. Note: *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001.
We then tested whether HITTERS regulates ER stress induced apoptosis. Via Annexin V‐fluorescein isothiocyanate/propidium iodide (FITC/PI) staining, we found that HITTERS knockdown did not influence apoptosis under basal condition. Once ER stress was triggered, depletion of HITTERS significantly promoted apoptosis and decreased cell viability. In contrast, overexpression of HITTERS increased cell viability under ER stress (Figure 5E–H). We also found that HITTERS knockdown promoted the expression of apoptotic markers, whereas overexpression of HITTERS inhibited their expression (Figure 5G,H). In agreement with the in vitro studies, tumors grown from HITTERS overexpression cells were more resistant to ER stress induced apoptosis (Figure S4A,B, Supporting Information), whereas tumors from HITTERS knockdown cells were sensitive to ER stress induced apoptosis (Figure 5I,J). Taken together, these results showed that HITTERS could attenuate ER stress induced apoptosis.
2.7. HITTERS Regulates ER Stress Related DNA Damage Response
Next, we investigated how HITTERS regulates ER stress induced apoptosis. We found that HITTERS knockdown did not influence the mRNA and protein levels of HERPUD1 (Figure 6A,B). Moreover, HITTERS knockdown had little influence on the expression of important UPR molecules (Figure 6C,D). These results indicated that HITTERS may not regulate ER stress induced apoptosis through regulating HERPUD1 in cis or UPR initiation in trans. We then performed RNA sequencing and GSEA analysis. GSEA showed that HITTERS had a significant impact on OSCC apoptosis under ER stress (Figure 6E). More importantly, we found that under ER stress, depletion of HITTERS significantly affected DDR, especially the DNA repair related pathways, including homologous recombination (HR) pathway and ATR‐breast cancer susceptibility genes (BRCA) pathway (Figure 6E). Unrepaired DNA damage, especially double strand breaks, is one of the most cytotoxic DNA lesions which leads to cell death through apoptosis.[ 17 ] Since we have found HITTERS significantly inhibited apoptosis, we next focused on exploring the functional role of HITTERS in ER stress‐related DDR.
Figure 6.
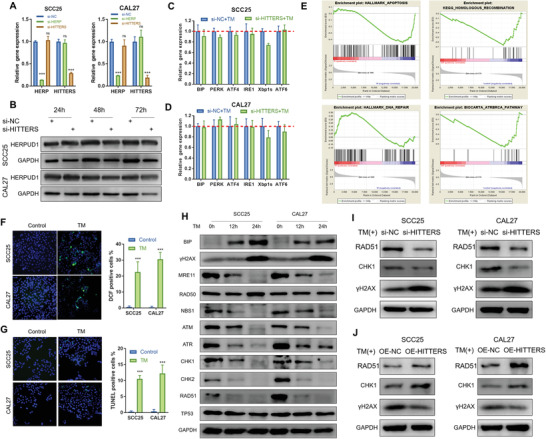
HITTERS regulates ER stress related DDR. A) qPCR and B) Western blot confirmed that knocking‐down HITTERS did not significantly change the mRNA and protein expression of HERPUD1. For qPCR, cells were transfected with siRNA for 48 h. Student t‐test was used for qPCR. C,D) Knocking‐down HITTERS did not significantly change the mRNA level of important UPR regulator in both C) SCC25 and D) CAL27 cells. Cells were transfected with siRNA for 48 h and then treated with TM (10 µg mL−1) for 6 h. E) GSEA results for RNA‐sequencing profiles. SCC25 cells were transfected with HITTERS siRNA or negative control siRNA for 48 h and then treated with TM (10 µg mL−1) for 12 h. F,G) 2',7'‐dichlorofluorescein staining and TUNEL assay indicated TM (10 µg mL−1, 24 h) treatment promoted F) ROS production and G) DNA breaks. Student t‐test was used. H) Induction of ER stress by TM (10 µg mL−1) significantly promoted the level of DNA damage marker γ‐H2AX and suppressed DNA repair proteins in a time‐dependent manner. I) Knocking‐down HITTERS significantly suppressed DNA repair protein and promoted DNA damage marker expression; J) whereas overexpressing HITTERS obtained the opposite effect. Cells were transfected with siRNA or plasmid for 48 h and then treated with TM (10 µg mL−1) for 24 h. Note: ns, no significance; ***, P < 0.001.
Previous study has shown that ER stress could induce reactive oxygen species (ROS) production and cause oxidative DNA damage.[ 18 ] We found that ER stress could significantly trigger ROS production (Figure 6F). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay directly proved that ER stress could induce DNA strand breaks (Figure 6G). Consequently, we found ER stress induced a significant upregulation of DNA damage marker γ‐H2AX (Figure 6H). HR and nonhomologous end joining (NHEJ) are two major pathways for repairing double strand breaks.[ 19 ] We then examined the expression of HR and NHEJ pathways under ER stress. We found that most HR related proteins, including MRE11, NBS1, ATM, ATR, CHK1, CHK2, and RAD51, were dramatically downregulated in a time‐dependent manner, except for RAD50 and TP53, whose expression was unaltered (Figure 6H). In contrast, we found proteins related to NHEJ remained unchanged under ER stress (Figure S4C, Supporting Information).
Next, we investigated how HITTERS affected ER stress related DDR. We found that changing the expression level of HITTERS did not influence ROS production (Figure S4D, Supporting Information). However, depletion of HITTERS significantly increased the ratio of TUNEL positive cells (Figure S4E, Supporting Information) and the level of γ‐H2AX (Figure 6I), whereas overexpression of HITTERS suppressed TUNEL positive ratio (Figure S4E, Supporting Information) and γ‐H2AX expression (Figure 6J). Moreover, HITTERS was positively associated with the expression of RAD51 and CHK1 that are involved in HR (Figure 6I,J). However, HITTERS had no influence on NHEJ pathway (Figure S4C, Supporting Information). Taken together, our findings suggested that ER stress could simultaneously induce oxidative DNA damage and suppress HR via downregulation of the crucial proteins. HITTERS acted as a protective factor in ER stress induced DNA damage, but the protective effect was not through regulating ROS production.
2.8. HITTERS Binds to and Promotes the Formation of MRE11/RAD50/NBS1 Complex under ER Stress
To identify the endogenous proteins that interact with HITTERS under ER stress, a modified comprehensive identification of RNA‐binding proteins by mass spectrometry (ChIRP‐MS) method was used (Figure 7A). The antisense probe pool retrieved about 30–50% of total HITTERS (Figure 7B). Liquid chromatograph‐MS/MS result indicated that 47 proteins, including RAD50 and MRE11, interacted with HITTERS (File S1, Supporting Information). Western blotting confirmed that under ER stress, HITTERS interacted with both RAD50 and MRE11 (Figure 7B). Interestingly, NBS1, a member of MRN complex, was not enriched by HITTERS (Figure 7B). We next performed RNA immunoprecipitation (RIP) assay and found both RAD50 and MRE11 bind to HITTERS (Figure 7C). These results confirmed that HITTERS endogenously interacted with RAD50 and MRE11.
Figure 7.
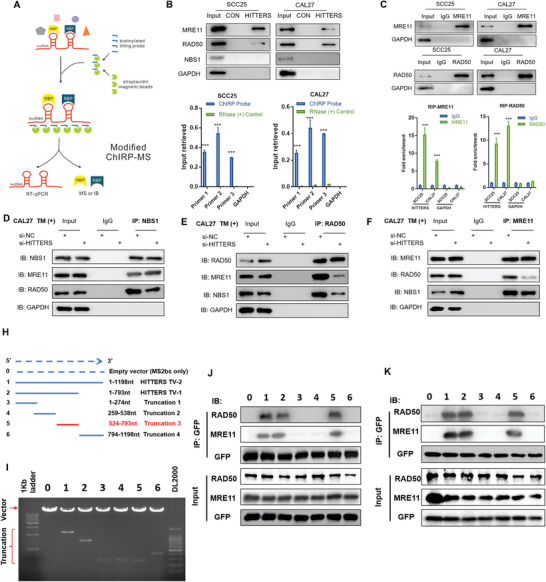
HITTERS binds to and regulates the formation of MRN complex under ER stress. A) The schematic representation of modified ChIRP‐MS. B) qPCR showed the modified ChIRP method retrieved about 30–50% of HITTERS. Western blot confirmed that HITTERS binds to both MRE11 and RAD50, but not NBS1 nor glyceraldehyde‐3‐phosphate dehydrogenase. Cells were treated with TM (10 µg mL−1, 6 h) before harvesting for ChIRP‐MS. For qPCR, Student t‐test was used for each primer. ***, P < 0.001. C) RIP assay of MRE11 and RAD50. Western blot confirmed MRE11 and RAD50 were successfully precipitated. qPCR indicated HITTERS were significantly enriched by MRE11 and RAD50. Cells were treated with TM (10 µg mL−1, 6 h) before harvesting for RIP. For qPCR, Student t‐test was used. ***, P < 0.001. D,E) Co‐IP results showed the interaction between MRE11 and RAD50 in CAL27 cells were reduced after HITTERS knockdown. The interaction between MRE11 and NBS1 remained no change after HITTERS knockdown. Cells were transfected with siRNA for 48 h and then treated with TM (10 µg mL−1) for 6 h. H) Diagrams of full‐length HITTERS and the truncations in MS2bs‐MS2bp RNA pull‐down assay. I) The reconstructed plasmids containing 12XMS2 tag and full‐length HITTERS and truncations with the correct sizes are indicated. J,K) Immunoblot analysis for RAD50 and MRE11 in the protein samples pulled down by different HITTERS truncations. J) SCC25 and K) CAL27 cells were transfected with two plasmids for 48 h and treated with TM (10 µg mL−1) for 6 h.
MRN has a central function in DNA damage repair by sensing the damaged DNA, processing broken DNA ends and activating DNA damage repair pathways.[ 17 , 20 , 21 ] We found depletion of HITTERS significantly decreased the protein level of MRE11 and NBS1 but had no effect on RAD50 expression (Figure S5A, Supporting Information). In contrast, the application of Mirin, a small molecule inhibitor of MRE11, or RAD50‐specific siRNA, could not change HITTERS expression (Figure S5B, Supporting Information). Therefore, HITTERS may act as an upstream regulator of the MRN complex. We then explored how HITTERS influence MRE11 and NBS1 protein level. We found that HITTERS had no impact on the transcription of MRE11 and NBS1, as the RNA level of MRE11 and NBS1 did not significantly change with HITTERS gain or loss of function (Figure S5C, Supporting Information). ER‐stress could suppress global protein translation via eif2α phosphorylation. However, we found that knockdown of HITTERS had no impact on eif2α phosphorylation and directly mimicking eif2α phosphorylation via Salubrinal treatment did not influence the protein level of NBS1 and MRE11 (Figure S5D, Supporting Information). Finally, we hypothesized that HITTERS might influence the protein degradation of NBS1 and MRE11. Our results proved that under ER‐stress, NBS1 and MRE11 were degraded via ubiquitin‐proteasome system rather than autophagy system (Figure S5E,F, Supporting Information). Depletion of HITTERS significantly promoted the ubiquitination of MRE11 and NBS1, and inhibiting proteasome could rescue the degradation (Figure S5I,J, Supporting Information).
We also hypothesized that HITTERS could influence the interaction between RAD50 and MRE11. We treated cells with TM for 6 h as MRE11 and NBS1 were not significantly degraded at this time‐point (Figure S4C, Supporting Information). Co‐immunoprecipitation (co‐IP) analysis (Figure 7D–F and Figure S6A–C, Supporting Information) showed that RAD50, MRE11, and NBS1 were tightly interacted with each other under ER stress. However, after knocking‐down of HITTERS, the interaction between RAD50 and MRE11 was significantly weakened. However, depletion of HITTERS did not influence the interaction between MRE11 and NBS1. These results indicated that HITTERS promoted MRN complex formation via facilitating the interaction between MRE11 and RAD50.
The binding domain of MRE11 and RAD50 in this structure is well documented.[ 21 ] Therefore, we speculate that HITTERS could interact with the domain. However, the binding sites of HITTERS with MRN need to be validated. To explore this, we used MS2bs‐MS2bp RNA pull down assay. We established a serial HITTERS RNA truncation and found that 524–793nt of HITTERS mediated its association with RAD50 and MRE11.
Since the function of MRN complex in ER stress‐related DDR is not well understood, we used siRNA or Mirin to inhibit MRN complex function. Inhibited of MRN complex resulted in elevated levels of DNA damage in OSCC, which was comparable to the levels found in HITTERS knockdown cells (Figure S4E, Supporting Information). Correspondingly, the expression of DNA damage marker γ‐H2AX and proapoptotic proteins were upregulated by HITTERS siRNA, RAD50 siRNA or Mirin, whereas cell viability and the DNA damage repair proteins RAD51 and CHK1 were downregulated (Figure S6D,E, Supporting Information). Overexpression of HITTERS enhanced cell viability by inhibiting DNA damage and promoting DNA repair. However, the protective effects were markedly abolished by dysfunction of MRN complex induced by RAD50 siRNA and Mirin (Figure S6F,H, Supporting Information). Taken together, our results showed that HITTERS functions as an RNA scaffold to promote MRE11‐RAD50 interaction and increase the protein levels of MRE11 and NBS1, also, the antiapoptosis function of HITTERS relies on MRN complex.
3. Discussion
ER stress‐triggered UPR is initially activated as a prosurvival mechanism by maintaining ER homeostasis and promoting protein folding and degradation in response to environmental challenges. However, severe or prolonged ER stress ultimately leads to cell death. Therefore, it is important to find out the gene that determines the final effects of ER stress. In the present study, we uncovered a novel lncRNA HITTERS by comprehensive characterization of the ER stress related lncRNA transcriptome. Only ≈10% of lncRNAs were selectively upregulated under ER stress and HITTERS was the most significantly upregulated lncRNA. We showed that HITTERS promoted proliferation and invasion of OSCC both in vitro and in vivo. HITTERS expression was upregulated in OSCC tissues and the upregulation of HITTERS was correlated with poor survival of patients with OSCC. More importantly, our findings demonstrated that HITTERS significantly inhibited ER stress induced apoptosis. Mechanistically, HITTERS could bind to and regulate the formation of MRN complex, and increase the expression of proteins involved in DNA damage repair, therefore alleviating ER stress‐related DNA damage. These findings underscore the potential of HITTERS as a novel target for cancer therapy.
Recent advances in RNA sequencing technology allow researchers to discover thousands of lncRNAs in eukaryotic genomes. However, the function and molecular mechanism of the majority of lncRNAs in human disease, especially in cancer, remain elusive. Many lncRNAs are reported to regulate proliferation, survival, angiogenesis, and invasion through modulating PI3K/Akt and ERK1/2 MAPK pathways.[ 22 ] Our data showed that under non‐ER stress condition, HITTERS promoted the phosphorylation of Akt and ERK1/2, and the expression of downstream targets proliferating cell nuclear antigen, Cyclin D1, and P27 was also regulated. Moreover, we found that HITTERS activated TGF‐β/Smad3 pathway, promoted transcription factors Snail and Slug, leading to increased mesenchymal markers such as Vimentin and N‐cadherin and decreased epithelial marker E‐cadherin. TGF‐β is a well‐known inducer of EMT during cancer progression, and many lncRNAs could regulate its expression. For example, lncRNA ANRIL promotes the invasion and metastasis in OSCC, prostate cancer, and thyroid cancer through regulating TGF‐β/Smad3 pathway.[ 23 ] LncRNA ELIT‐1 behaves as a Smad3 cofactor to facilitate TGF‐β/Smad3 pathway and induce EMT.[ 24 ] Therefore, the effects of HITTERS on proliferation and invasion are partly attributed to the activation of PI3K/Akt, ERK1/2 MAPK, and TGF‐β/Smad3 pathway.
As the functions of lncRNAs in physiological and pathological processes have become increasingly recognized, the association of lncRNAs with ER stress attracts more attention. To date, a few lncRNAs such as HypERlnc,[ 10 ] TUG1,[ 25 ] and MEG3 [ 9 ] are reported to be associated with ER stress. In this study, we demonstrated that ER stress significantly increased HITTERS expression. Elevated level of HITTERS, in return, inhibited ER stress induced apoptosis. Therefore, cancer cells may utilize HITTERS to overcome ER stress induced apoptosis in adverse environment. We next analyzed the mechanism of HITTERS regulated ER stress. In striking contrast to HypERlnc, TUG1, and MEG3, neither BIP expression, nor the three main UPR pathway, were affected by HITTERS. These results suggest that HITTERS modulates ER stress induced apoptosis through a novel mechanism. ER stress was reported to inhibit DNA repair by proteasomal degradation of RAD51.[ 26 ] Indeed, we found that ER stress promotes ROS production to cause DNA damage, and at the same time suppresses DNA repair, finally leading to apoptosis. Most importantly, we demonstrated that although HITTERS could not inhibit ROS production, it could significantly enhance DNA damage repair pathways under ER stress, thus protects OSCC from ER stress induced apoptosis.
Recent studies have proved that lncRNAs participated positively in DDR through varieties of mechanism.[ 7 ] LncRNAs can act as RNA scaffolds to regulate DDR. For example, lncRNA GUARDIN sustains BRCA1 stability by facilitating the BRCA1‐BARD1 complex. In this study, our work for the first time found that inhibiting MRN complex accelerated ER stress induced DNA damage and apoptosis. We demonstrated that HITTERS facilitated the formation of MRN complex under ER stress. If HITTERS was knocked down, the binding capacity between MRE11 and RAD50 was disturbed. Inhibiting MRN complex function abolished the protective effect of HITTERS, indicating that the function of HITTERS relies on MRN complex. It has been acknowledged that MRN can assemble as a heteromultimer, and the DNA binding and processing structure of the heteromultimer is formed by the MRE11 dimer and two RAD50.[ 21 ] The binding domain of MRE11 and RAD50 in this structure is well documented. Therefore, we speculate that HITTERS could interact with the domain. Our results found 24‐793 nt of HITTERS mediated its association with RAD50 and MRE11. Our results also showed that depletion of HITTERS significantly decreased the protein level of MRE11 and NBS1, via promoting proteasomal degradation of MRE11 and NBS1, even though HITTERS did not interact with NBS1. Thus, we hypothesized that under ER stress, HITTERS might influence the proteasome activity. However, the mechanism needs further exploration.
Our results found that ER‐stress could significantly increase HITTERS expression, in contrast, the DNA damage repair proteins decreased dramatically. Further analysis found if the HITTERS was depleted, the DNA damage repair proteins would decrease more significantly. These data indicated that the increment of HITTERS by TM treatment could be a protective feedback. It is intriguingly that HITTERS shares the same promotor with HERPUD1. However, we found that HITTERS knockdown does not influence the mRNA and protein levels of HERPUD1. This raises doubt on why they share the same promoter? Whether HERPUD1 could act like HITTERS, by regulating PI3K/Akt, ERK1/2 MAPK, and TGF‐β/Smad3 pathway, or participating ER stress induced DDR, are still unclear. It is reasonable that the function of these two genes may cooperate as their transcription regulation are identical. Another possible explanation is that HITTERS is independently processed from the pre‐mRNA of HERPUD1. In support of this hypothesis, our ChIRP‐MS results showed HITTERS could pull‐down DDX39B which is closely related to pre‐mRNA splicing. However, further studies will be necessary to verify this hypothesis.
To sum up, this study presents a novel lncRNA, namely HITTERS, which links ER stress and DDR together in OSCC. Mechanisms presented here could provide viable therapeutic choices in remedying stressful microenvironment‐associated tumor progression.
4. Experimental Section
Cell Culture, Transfection, and Chemical Treatment
Human OSCC cell lines SCC25 and CAL27 were purchased from American Type Culture Collection. Other cell lines including HEK293 (human embryonic kidney cell line), Hela (human cervical cancer cell line), PANC‐1 (human pancreatic cancer cell line), NOK (human normal oral keratinocytes), DOK (human dysplastic oral keratinocytes), and other human OSCC cell lines (SAS, UM‐1, UM‐2, SCC15, HSC‐2, and HSC‐3) were all obtained from State Key Laboratory of Oral Diseases (Sichuan University, China). SCC25, SAS, UM‐1, UM‐2, and SCC15 were cultured in Dulbecco's Modified Eagle Medium / Nutrient Mixture F‐121:1, other cell lines were cultured in Dulbecco's Modified Eagle Medium. All medium was supplemented with 10% fetal bovine serum (Gibco). All cell lines were cultured at 37 °C with 5% CO2. Cell lines were authenticated by short tandem repeat profiling. All cell lines were free for mycoplasma contamination. For transient transfection of siRNA (GenePharma) or plasmid, the EndoFectin (Genecopoeia) was applied according to the manufacture's instruction. For stable transfection, the lentivirus (Genecopoeia) containing shRNA or lncRNA sequence plasmid was applied according to the manufacture's instruction. For inducing ER stress, TM (5–20 µg mL−1, Sigma), thapsigargin (Tg, 1 × 10−6–2 × 10−6 m, MedChemExpress), DTT (2 × 10−3–4 × 10−3 m, Takara), and CFZ (50 × 10−9–200 × 10−9 m, Selleck) were added. For inhibiting DNA methyltransferase, 5aza (10 × 10−6 m, Sigma) were added for 24 h. For inhibiting MRN complex, Mirin (100 × 10−6 m, Selleck) were added simultaneously with other treatment. For inhibiting proteasome, MG‐132 (10 × 10−6 m, Beyotime) were added for 12 h. For inhibiting autophagy/lysosome, hydroxychloroquine sulfate (100 × 10−6 m, Selleck) was added for 48 h. For mimicking eif2a phosphorylation, Salubrinal (50 × 10−6 m, Selleck) was added for 24 h.
ChIRP‐MS
ChIRP‐MS was carried out following Chu's protocol[ 27 ] with several modification. For each reaction, 5 × 107 cells were harvested. Micrococcal nuclease (New England Biolabs (NEB)) was used for DNA fragmentation followed by ultrasonication. The 3′‐biotin‐triethylene glycol modified ChIRP probes (Sangon) were added to streptavidin‐coated magnetic beads (Biomeg), washed, and then hybridized with cell lysates. An excessive dose of probes was used (tenfold higher concentrations). For protein extraction, the trichloroacetic acid precipitation step was skipped.
Dual‐Luciferase Reporter Assay
The targeted inserting sequence was amplified by PCR using Q5 High‐Fidelity DNA Polymerase (NEB). Quickcut (Takara) restriction endonucleases and DNA Ligation Kit (Takara) was used for vector reconstruction. The full‐length (2.5 kb around TSS) and truncated sequence of potential promoter regions were amplified by PCR. PCR products were then inserted into luciferase reporter plasmids pGL4.20 (Luc2, Promega) or pEZX‐FR01 (containing both Fluc and Rluc, Genecopoeia). When using pGL4.20, the pGL4.75 plasmid (Rluc) was cotransfected as an internal control. The dual‐luciferase reporter assay was applied in 96‐well palates at 5 × 104 cells per well. HEK293T cells were transfected with 500 ng of plasmid. Luciferase activity was determined using Luc‐Pair Duo‐Luciferase HS Assay kit (Genecopoeia).
Generation of CRISPR/Cas9 Construct
The sgRNA targeting the promoter region of HERPUD1 was designed by the online design tool Crispor. After phosphorylating and annealing of each pair of oligos using T4 polynucleotide kinase (Thermo), the oligo was inserted into BsmBI (Thermo)‐digested LentiCRISPRV2 vector (Addgene). The forward and reverse oligo pair was inserted into vectors containing puromycin and neomycin resistance, respectively. The reconstructed vector was transfected into HEK293 and selected for 2 weeks to obtain HERPUD1 promoter knockout cell lines.
High‐Throughput Analysis
Total RNA was extracted from SCC25 cells using Trizol reagent (Invitrogen). The RNA samples were subjected to mRNA and lncRNA microarray using GeneChip HTA 2.0 (Affymetrix) at Genminix Informatics Ltd., Co. HTA 2.0 covers more than 245 000 coding transcripts and 40 000 noncoding transcripts in human transcriptome. Gene Ontology (GO) analysis, pathway analysis, and lncRNA‐mRNA coexpression network analysis of the differentially expressed mRNAs and lncRNAs were performed as previously described.[ 28 ] Next generation sequencing of mRNA transcriptome and GSEA were carried out as previously described (OE biotech, China) in SCC25 cells.[ 29 ]
Animal Study
All animal studies were approved by the Animal Ethical and Welfare Committee of West China Hospital of Stomatology (WCCSIRB‐D‐2016‐075). Female BALB/C nude mice were maintained under specific pathogen free conditions in the Sichuan University Animal Center according to the institution's guidelines. For subcutaneous xenograft model, 5 × 106 OSCC cells in 200 µL phosphate buffered saline (PBS) were sub‐axillary injected. The tumor volume was documented in indicated time. For in vivo TM treatment, TM were diluted by PBS to 20 µg mL−1. Mice were intraperitoneally injected with 100 µL of PBS or TM, twice a week. At the 4th week postinjection, animals were sacrificed, and the tumors were harvested. For pulmonary metastasis model, 1 × 106 OSCC cells in 100 µL PBS were injected into the tail vein. At the 5th week postinjection, the animals were sacrificed and the lungs were harvested.
Patients’ Samples
The Institutional Ethical Committee of West China Hospital of Stomatology approved the present study (WCHSIRB‐OT‐2016‐047). All patients have signed written informed consent. All procedures were carried out in accordance with the Declaration of Helsinki. Human oral cancer tissue specimens were collected from the West China Hospital of Stomatology, Sichuan University (China). A total of 48 primary OSCC samples and paired adjacent (>1.5 cm from the tumor margin) normal tissues were obtained. After resection, the samples were snap frozen by liquid nitrogen and stored in −80 °C for up to 1 week before qPCR analysis.
Additional Methods
Cell counting kit‐8 (CCK‐8), EdU incorporation, colony formation assay, apoptosis, wound healing assay, cell invasion, TUNEL, ROS detection, nucleus‐cytoplasm fractionation, RNA FISH, RACE, RNA extraction and qPCR, Western blotting, co‐IP, RIP, and MS2bs‐MS2bp RNA pull down are detailed in the Supporting Information. All the primers, probes, oligos, and antibodies are listed in Table S1–S3 in the Supporting Information.
Statistical Analysis
GraphPad Prisma 7 software was used for statistical analysis and generating statistical figures. Data were presented from one representative experiment out of three independent experiments. For each representative experiment, at least three repetitions were measured. The statistical methods for each result were noted in figure legend. Data were presented as mean ± standard deviation. p < 0.05 was considered statistically significant.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supporting Information
Acknowledgements
C.W., W.C., and F.Y. contributed equally to this work. This work was supported by National Natural Science Foundation of China (81972538, 81672669, 81972546, and 81602373) and Graduate Student's Research and Innovation Fund of Sichuan University (2018YJSY106). The authors thank Shanghai OE Biotech Inc. (Shanghai, China) for high throughput sequencing service and bioinformatic support.
Wu C., Chen W., Yu F., Yuan Y., Chen Y., Hurst D. R., Li Y., Li L., Liu Z., Long Noncoding RNA HITTERS Protects Oral Squamous Cell Carcinoma Cells from Endoplasmic Reticulum Stress‐Induced Apoptosis via Promoting MRE11‐RAD50‐NBS1 Complex Formation. Adv. Sci. 2020, 7, 2002747 10.1002/advs.202002747
Contributor Information
Longjiang Li, Email: muzili63@163.com.
Zhe Liu, Email: liuzhe_scu@163.com.
References
- 1.a) Clarke H. J., Chambers J. E., Liniker E., Marciniak S. J., Cancer Cell 2014, 25, 563; [DOI] [PubMed] [Google Scholar]; b) Wang M., Kaufman R. J., Nat. Rev. Cancer 2014, 14, 581. [DOI] [PubMed] [Google Scholar]
- 2.a) Hetz C., Chevet E., Oakes S. A., Nat. Cell Biol. 2015, 17, 829; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walter P., Ron D., Science 2011, 334, 1081. [DOI] [PubMed] [Google Scholar]
- 3.a) Driehuis E., Kolders S., Spelier S., Lõhmussaar K., Willems S. M., Devriese L. A., de Bree R., de Ruiter E. J., Korving J., Begthel H., van Es J. H., Geurts V., He G.‐W., van Jaarsveld R. H., Oka R., Muraro M. J., Vivié J., Zandvliet M. M. J. M., Hendrickx A. P. A., Iakobachvili N., Sridevi P., Kranenburg O., van Boxtel R., Kops G. J. P. L., Tuveson D. A., Peters P. J., van Oudenaarden A., Clevers H., Cancer Discovery 2019, 9, 852. [DOI] [PubMed] [Google Scholar]
- 4.a) Cramer J. D., Burtness B., Le Q. T., Ferris R. L., Nat. Rev. Clin. Oncol. 2019, 16, 669; [DOI] [PubMed] [Google Scholar]; b) Pilié P. G., Tang C., Mills G. B., Yap T. A., Nat. Rev. Clin. Oncol. 2019, 16, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marchese F. P., Raimondi I., Huarte M., Genome Biol. 2017, 18, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kopp F., Mendell J. T., Cell 2018, 172, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Prensner J. R., Chen W., Iyer M. K., Cao Q., Ma T., Han S., Sahu A., Malik R., Wilder‐Romans K., Navone N., Logothetis C. J., Araujo J. C., Pisters L. L., Tewari A. K., Canman C. E., Knudsen K. E., Kitabayashi N., Rubin M. A., Demichelis F., Lawrence T. S., Chinnaiyan A. M., Feng F. Y., Cancer Res. 2014, 74, 1651; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang H., Hua Y., Jiang Z., Yue J., Shi M., Zhen X., Zhang X., Yang L., Zhou R., Wu S., Clin. Cancer Res. 2019, 25, 1989; [DOI] [PubMed] [Google Scholar]; c) Wan G., Hu X., Liu Y., Han C., Sood A. K., Calin G. A., Zhang X., Lu X., EMBO J. 2013, 32, 2833; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shahabi S., Kumaran V., Castillo J., Cong Z., Nandagopal G., Mullen D. J., Alvarado A., Correa M. R., Saizan A., Goel R., Bhat A., Lynch S. K., Zhou B., Borok Z., Marconett C. N., Cancer Res. 2019, 79, 3050; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hu W. L., Jin L., Xu A., Wang Y. F., Thorne R. F., Zhang X. D., Wu M., Nat. Cell Biol. 2018, 20, 492; [DOI] [PubMed] [Google Scholar]; f) Shihabudeen Haider Ali M. S., Cheng X., Moran M., Haemmig S., Naldrett M. J., Alvarez S., Feinberg M. W., Sun X., Nucleic Acids Res. 2019, 47, 1505; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Sharma V., Khurana S., Kubben N., Abdelmohsen K., Oberdoerffer P., Gorospe M., Misteli T., EMBO Rep. 2015, 16, 1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) McMahon M., Samali A., Chevet E., Am. J. Physiol. Cell Physiol. 2017, 313, C243; [DOI] [PubMed] [Google Scholar]; b) Chen Y., Chen X., Gao J., Xu C., Xu P., Li Y., Zhu Y., Yu C., FASEB J. 2019, 33, 11411; [DOI] [PubMed] [Google Scholar]; c) Bai X., Geng J., Li X., Wan J., Liu J., Zhou Z., Liu X., Antioxid. Redox Signaling 2018, 29, 355; [DOI] [PubMed] [Google Scholar]; d) Liu H., Sun H. L., Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5912. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y., Wu J., Jing H., Huang G., Sun Z., Xu S., J. Cell. Biochem. 2019, 120, 6789. [DOI] [PubMed] [Google Scholar]
- 10. Bischoff F. C., Werner A., John D., Boeckel J. N., Melissari M. T., Grote P., Glaser S. F., Demolli S., Uchida S., Michalik K. M., Meder B., Katus H. A., Haas J., Chen W., Pullamsetti S. S., Seeger W., Zeiher A. M., Dimmeler S., Zehendner C. M., Circ. Res. 2017, 121, 368. [DOI] [PubMed] [Google Scholar]
- 11. Wang F. M., Qiu K., Hu T., Wan C. X., Zhou X. D., Gutmann J. L., Int. Endod. J. 2006, 39, 477. [DOI] [PubMed] [Google Scholar]
- 12. Kowall B., Holtfreter B., Volzke H., Schipf S., Mundt T., Rathmann W., Kocher T., J. Clin. Periodontol. 2015, 42, 422. [DOI] [PubMed] [Google Scholar]
- 13. Lenz B., Bleich S., Beutler S., Schlierf B., Schwager K., Reulbach U., Kornhuber J., Bonsch D., Exp. Cell Res. 2006, 312, 4049. [DOI] [PubMed] [Google Scholar]
- 14. Ma Y., Hendershot L. M., J. Biol. Chem. 2004, 279, 13792. [DOI] [PubMed] [Google Scholar]
- 15.a) Baselga J., Science 2006, 312, 1175; [DOI] [PubMed] [Google Scholar]; b) Cocco E., Scaltriti M., Drilon A., Nat. Rev. Clin. Oncol. 2018, 15, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hao Y., Baker D., Ten Dijke P., Int. J. Mol. Sci. 2019, 20, 2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Syed A., Tainer J. A., Annu. Rev. Biochem. 2018, 87, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dvash E., Har‐Tal M., Barak S., Meir O., Rubinstein M., Nat. Commun. 2015, 6, 10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown J. S., O'Carrigan B., Jackson S. P., Yap T. A., Cancer Discovery 2017, 7, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Stracker T. H., Petrini J. H., Nat. Rev. Mol. Cell Biol. 2011, 12, 90; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rupnik A., Grenon M., Lowndes N., Curr. Biol. 2008, 18, R455. [DOI] [PubMed] [Google Scholar]
- 21. Williams G. J., Lees‐Miller S. P., Tainer J. A., DNA Repair 2010, 9, 1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Tasharrofi B., Ghafouri‐Fard S., Klin. Onkol. 2018, 31, 95; [DOI] [PubMed] [Google Scholar]; b) Peng W. X., Koirala P., Mo Y. Y., Oncogene 2017, 36, 5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Liu L., Ning S. B., Fu S., Mao Y., Xiao M., Guo B., Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6194; [DOI] [PubMed] [Google Scholar]; b) Zhao B., Lu Y.‐L., Yang Y., Hu L.‐B., Bai Y., Li R.‐Q., Zhang G.‐Y., Li J., Bi C.‐W., Yang L.‐B., Hu C., Lei Y.‐H., Wang Q.‐L., Liu Z.‐M., Cancer Biomarkers 2018, 21, 613; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhao J.‐J., Hao S., Wang L.‐L., Hu C.‐Y., Zhang S., Guo L.‐J., Zhang G., Gao B., Jiang Y., Tian W.‐G., Luo D.‐L., Oncotarget 2016, 7, 57903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakai S., Ohhata T., Kitagawa K., Uchida C., Aoshima T., Niida H., Suzuki T., Inoue Y., Miyazawa K., Kitagawa M., Cancer Res. 2019, 79, 2821. [DOI] [PubMed] [Google Scholar]
- 25. Su S., Liu J., He K., Zhang M., Feng C., Peng F., Li B., Xia X., FEBS J. 2016, 283, 1261. [DOI] [PubMed] [Google Scholar]
- 26. Yamamori T., Meike S., Nagane M., Yasui H., Inanami O., FEBS Lett. 2013, 587, 3348. [DOI] [PubMed] [Google Scholar]
- 27. Chu C., Zhang Q. C., da Rocha S. T., Flynn R. A., Bharadwaj M., Calabrese J. M., Magnuson T., Heard E., Chang H. Y., Cell 2015, 161, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu H., Wang Q., Yao Y., Fang J., Sun F., Ni Y., Shen Y., Wang H., Shao S., BMC Med. Genomics 2015, 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ou C., Sun Z., He X., Li X., Fan S., Zheng X., Peng Q., Li G., Li X., Ma J., Adv. Sci. (Weinheim, Ger.) 2020, 7, 1901380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information