

Abstract
Background: Sickle cell disease affects a significant portion of US patients with African descent. It continues to be one of the leading causes of frequent hospitalizations and high in-hospital morality risk. Until the approval of disease-modifying therapies in last two years, medical therapy has relied mostly on management of pain episodes and the use of hydroxyurea. We discuss the nationwide analysis of trends in in-hospital mortality in patients with Sickle Cell Disease from 2000 to 2014. Methods: Trends of in-hospital mortality in sickle cell patients were analyzed from a database provided by the Agency of Healthcare Research and Quality. From the data hospitalization rates and in-hospital mortality in categories by region in the US, hospital size, health insurance status, comorbidities and gender were examined. Patterns of in-hospital mortality were analyzed by logistic regression. Results: Ratio for hospitalization and mortality among the four regions described Northeast, Midwest, South, West with respective values of 0.63%, 0.65%, 0.76% and 0.89% with P = 0.008 and OR = 1.07. Odds ratio for sickle cell patients that died during hospitalization and health insurance status was OR = 0.08. Comorbidities considered in sickle cell patients; diabetes mellitus (DM), hypertension (HTN), hyperlipidemia (HLD), chronic kidney disease (CKD), smoking status. The odds ratio for comorbidities show A-fib with a value of OR = 4.47, followed by hypertension OR = 1.92, diabetes mellitus OR = 1.44 and chronic kidney disease OR = 1.29, smoking status OR = 0.60. Mortality-hospitalization ratio by gender was: males 0.77% and females 0.69% with OR = 0.87. Conclusions: In-hospital mortality by US regions, as well as health insurance status are important measurable elements that show the impact of the disease from a public health perspective. Further and more specific data of regions by states, comorbidities by states and sex, as well as health insurance status by states will provide further insight in local mortality trends.
Keywords: Delay time, polymerization, social determinants of health, sickle cell, literacy
Introduction
Sickle cell disease (SCD) encompasses a group of genetic diseases that result from a single point mutation in the position 6 of the gene that encodes hemoglobin subunit ß [1]. It includes sickle cell anemia (SCA), sickle cell hemoglobin C (HbSC) and sickle ß-thalassemia. The molecular structure of hemoglobin consists of combined globin subunits which in association with the heme cofactor confers its oxygen carrying capacity. Adult hemoglobin is made up of two α subunits and two ß subunits [1]. The substitution of valine for glutamic acid within the molecular structure of the ß-globin subunit, is the mutation which drives in the production of hemoglobin S (HbS) [2]. It is an autosomal recessive disease, where the two copies of the ß subunits need to be mutated for evident clinical manifestation. Surface charge in hemoglobin S is characterized by a variation which under conditions of deoxygenation tends to polymerize and create fibers as a consequence of intracellular crystallization. The gradually increasing fibers through crystallization contributes to the rigidity of the red cell and progressively higher viscosity of blood. Of note, there is a latency period of time or “delay time” where hemoglobin is deoxygenated, however it does not become polymerized during the initial fraction of time [3]. If transit through microvasculature is beyond the delay time, hemoglobin will be at higher risk of aggregating and will favor “sickling” [3]. As hemoglobin S polymerizes, it weakens the erythrocyte membrane, promoting a dehydrated state with a higher tendency to hemolyze. Repeated episodes of deoxygenation over time causes an irreversible stiffness of the red cell - a “sickle” shape. Acquired sickle shape and stiffness of the red cells results in higher blood viscosity, red cell membrane fragmentation and as a consequence shortened red blood cell lifespan. This is a continuous process as the higher viscosity results in a slower transit through the microvasculature causing higher oxygen extraction [3]. The lower oxygen affinity of HgS and its deoxygenation worsens the sickling of the red cells. Progressively slower transit of red cells through the capillaries increases the delay time, worsens the deoxygenation and sickling as the process continues [3].
Over time sickle cell disease leads to a chronic inflammatory state through episodic vaso-occlusion causing ischemia, pain and important organ system complications. The process of intravascular hemolysis and release of cell-free hemoglobin creates reactive oxygen species which cause nitric oxide (NO) consumption and oxidative damage [4]. As a third pathophysiologic mechanism, the reduced bioavailability of NO is thought to trigger the expression of adhesion molecules and at the same time production of the vasoconstrictor endothelin-1 [1]. Endothelial cells in activated state are associated with increased vascular tone and production of inflammatory mediators responsible for vascular damage [1]. There is an overall increase in activation of the endothelium, coagulation proteins, leukocytes and platelets with increased adhesion to endothelium mediated to some extent by P-selectin [1,5]. It is thus a predisposing factor to endothelial dysfunction and proliferative changes in the intima and smooth muscle of the vasculature, which results in eventual systemic, pulmonary hypertension and organ injury [4].
Microvascular phenomena also manifest with multiple organ damage. Beginning early in life complications that stem from microvascular changes predispose patients to vaso-occlusive pain episodes. This same mechanism can also manifest as cerebrovascular events, acute chest syndrome (due to vaso-occlusion of pulmonary vasculature). Acute chest syndrome is in fact the second most common cause of hospital admissions and most common cause of sickle cell mortality [6]. The cascade of consequences as a result of generalized vasculopathy translates into cardiopulmonary complications, in which heart failure with or without pulmonary hypertension is the primary cause of mortality [7].
Gradually there is more organ involvement in these patients, including progressive decrease in renal function [8]. Renal medulla is the most prominent site of injury as capillaries here are relatively hypoxic, acidic and hypertonic all of which favor sickling [9]. This results in greater adhesion of erythrocytes to the vascular wall which derives in greater inflammation and as a consequence an increase in vascular tone manifested as vasoconstriction [10-12]. Endothelial defective function derives in systemic hypertension. This in addition to the above stated is also an independent contributing factor for renal disease. Increased susceptibility to infections contributes to increased mortality in sickle cell disease. Microvascular changes progressively result in organ injury, which include splenic infarcts and eventual auto-splenectomy. Impaired splenic function pose a risk of severe systemic infections, while alteration in complement activation contributes to infection susceptibility in these patients [13].
Over the last two decades hydroxyurea had been the only approved medication for sickle cell, approaching the pathophysiologic mechanism by inducing fetal hemoglobin (HbF) production which reduces HbS polymerization. Newly approved disease-modifying medications over the last two years, approach other pathophysiologic pathways of the disease by either reducing oxidative stress, inhibiting endothelial activation or by allosteric modification of HbS [5]. However as these therapies have been recently approved, their impact on the sickle cell population will be subject of study over the next decade but is not reflected in this analysis.
The mortality of sickle cell patients has been influenced by several factors including the introduction of novel therapies, better follow-up care in sickle cell clinics, better outreach programs in specialized sickle cell centers, immigration of sickle cell patients etc. This study analyzes the trend in hospitalization and mortality of sickle cell patients by geographic region and hospital size. It also analyzes the medical comorbidities which are associated with death during hospitalization of sickle cell patients.
Methods
Data source
We analyzed the nationwide trend of in-hospital mortality in sickle cell patients from a database provided by Agency of Healthcare Research and Quality. This was a retrospective cross-sectional analysis of in-hospital data from the years 2000-2014 obtained from the National Inpatient Sample (NIS) database. NIS is the largest all-payer inpatient care database maintained by the Agency for Healthcare Research and Quality as part of the Healthcare Cost and Utilization Project [14]. The NIS consists of data from approximately a 20% sampling of inpatient admissions to acute care hospitals in the United States. It is stratified by geographic region, urban/rural location, teaching status, and hospital bed size to minimize sampling bias. Each admission is weighted to make NIS representative of nationwide hospital systems. Patient’s demographics, primary and secondary diagnoses, Clinical Modification (ICD-9-CM) diagnosis and procedure codes, resource utilization and clinical outcomes are reported in the database. The data within the NIS is publicly available and does not contain any identifying information, making this retrospective study exempt from review by the Institutional Review Board. Inclusion criteria for patients were age more than equal to 18 years and with a diagnosis of sickle cell disease. Exclusion criteria included patients less than 18 years of age and with any diagnosis other than sickle cell disease.
Statistical analysis
Chi square test using excel software by Microsoft has been used to analyze them.
Logistic regression analysis was used to interpret patterns of mortality during hospitalization in this population. We compared it to data provided by the statistical brief from the Healthcare Cost and Utilization Project, which followed the characteristics of in-patients with diagnosis of sickle cell from 2000 to 2014.
Results
Geographical factors
Data on sickle cell patients hospitalized from year 2000 to 2014 was divided by categories of region, hospital size defined as small, medium or large hospital, medical health insurance status, sex, comorbidities and analyzed for their total in-hospital mortality for every category. Total deaths and hospitalizations ratio by regions described as Northeast (329/52,254 = 0.63%), Midwest (/332/50,872 = 0.65%), South (1,013/133,438 = 0.76%), West (207/23,370 = 0.89%) with P = 0.008 and OR = 1.07. Ratio of death and health insurance status for Medicaid (558/116,109 = 0.48%), Medicare (782/77,748 = 1.00), private (394/45,837 = 0.85%) and self-pay (76/12,012 = 0.63%) in-hospital deaths with OR = 0.84. Ratio according to death during hospitalization and hospital size; small (159/26,070 = 0.61%), medium (463/65,077 = 0.71%), large (1247/167,572 = 0.74%). These trends are represented in Figures 1 and 2.
Figure 1.
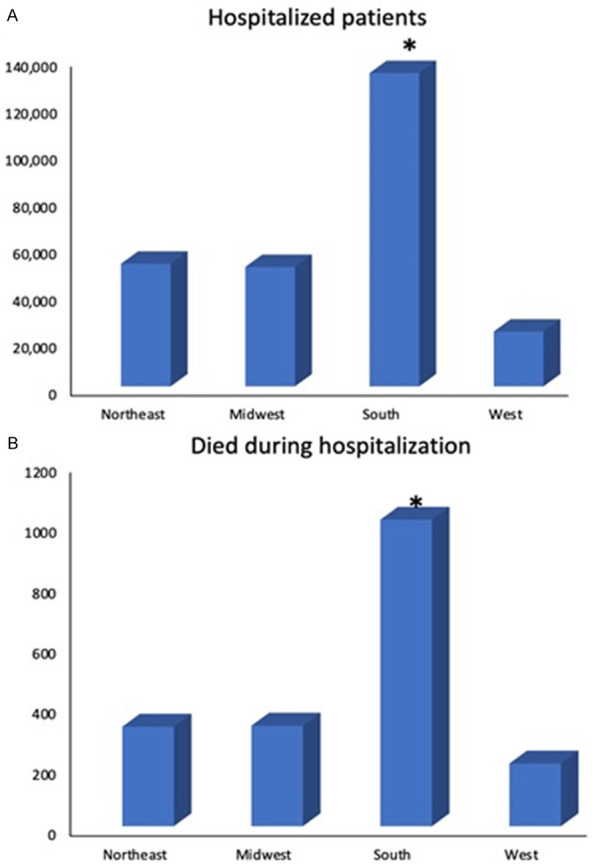
A: Hospitalization of sickle cell patients by region from 2000-2014. All data pertaining to region and mortality were extracted from NIS database. * denotes a p value <0.05. B: Mortality during hospitalization for sickle cell patients by region from 2000-2014. All data pertaining to region and mortality were extracted from NIS database. * denotes a p value <0.05.
Figure 2.
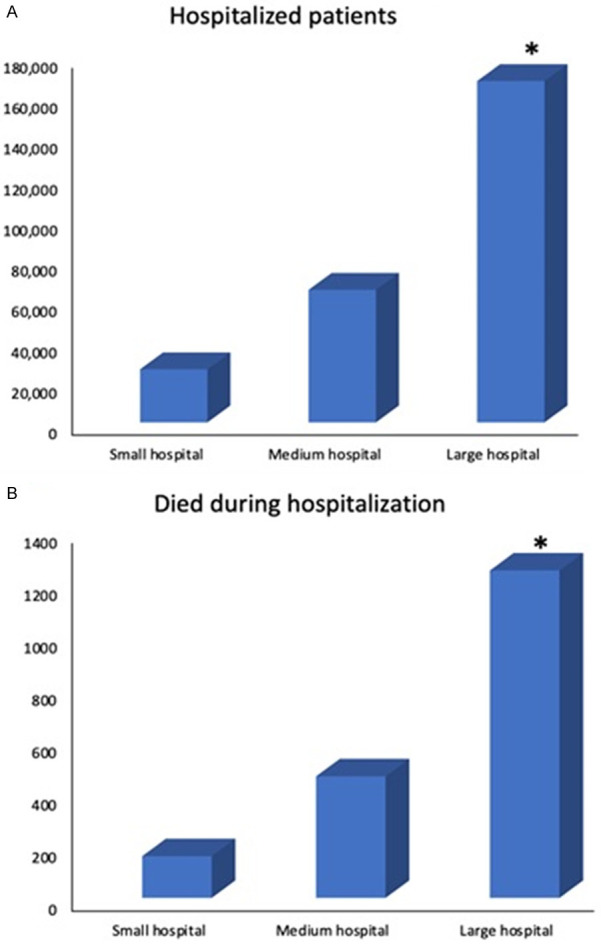
A: Hospitalization of sickle cell patients by hospital size from 2000-2014. All data pertaining to region and mortality were extracted from NIS database. * denotes a p value <0.05. B: Mortality during hospitalization for sickle cell patients by hospital size from 2000-2014. All data pertaining to region and mortality were extracted from NIS database. * denotes a p value <0.05.
Mortality and co-morbidity trends
Deaths and comorbidities ratio considered with their respective odds ratio for mortality during hospitalization; A-fib (155/3685 = 4.2%, OR = 4.47), diabetes mellitus (171/11,789 = 1.45%, OR = 1.44), hypertension (592/43,310 = 1.36%, OR = 1.92), hyperlipidemia (61/4,209 = 1.44%, OR = 1.07), chronic kidney disease (99/6,594 = 1.50%, OR = 1.29), smoking status (157/34,592 = 0.45%, OR = 0.60). In ratio of mortality and hospitalization by sex category males (859/111,475 = 0.77%) and females (1,022/148,059 = 0.69%) with OR = 0.87. This is shown in Table 1.
Table 1.
Prevalence and Odds ratios of sickle cell patients who died during hospitalization according to comorbidities and health insurance status
| Comorbidity | Prevalence | Odds Ratio |
|---|---|---|
| A-fib | 4.2% | 4.4788 |
| Hyperlipidemia | 1.44% | 1.0748 |
| Smoking | 0.45% | 0.6064 |
| Diabetes Mellitus | 1.45% | 1.4423 |
| Hypertension | 1.36% | 1.9245 |
| Chronic Kidney Disease | 1.5% | 1.2949 |
| Health Insurance | 0.8482 | |
| Hospital Region | 1.106 | |
| Gender | 0.8770 | |
Discussion
Increased mortality in sickle cell disease is a consequence of the interaction of different pathophysiologic mechanisms inducing immunologic compromise and vascular phenomena which results in complications due to multiple organ injury. Non-medical barriers add complexity to the management, monitoring and mortality as social determinants of health pose significant challenge requiring a comprehensive approach. In general terms sickle cell patients have a significantly fragmented care and different factors may aggravate that fragmentation. Non-medical barriers can have a significant impact on the medical management and evolution of the disease. Although painful episodes are the most characteristic clinical presentation as a result of vaso-occlusive ischemic episodes, sickle cell disease has become a chronic disease with associated progressive organ damage and declining quality of life.
Sickle cell disease (SCD) is significantly more prevalent in patients of African descent in the United Sates where approximately 100,000 have the diagnosis [15]. Arabic, Indian, certain Mediterranean countries, South, Central America and the Caribbean can also be affected [2]. Advancements over the last years have resulted in improved treatment of complications and decreased mortality in pediatric patients nearly to 95% who now reach 18 years of age [15]. Management of complications and life expectancy in adults has improved moderately although is still challenging. Currently life expectancy for adult individuals with sickle cell disease is 54 years [16]. Geographic and social distribution of the disease adds complexity to the follow up and management as it directly impacts quality of life and mortality.
It is worth considering the level of literacy of sickle cell disease treatment guidelines among healthcare professionals. This may also be hindering adequate monitoring and management of the disease, which would contribute to better prevention of complications. There has been an increased effort by the Department of Health and Human Services to not only identify cures aside from the already established bone marrow transplant (not widely available and is currently for a selected group of sickle cell patients) [15]. There had also been an increased effort to educate healthcare professionals on identifying and treating sickle cell patients in pain crisis, as well as on how to transition adequately from pediatric into adult care [15].
The statistical brief presented by the Healthcare Cost and Utilization Project (HCUP) and referenced in this analysis, described characteristics of inpatient hospital stays involving sickle cell patients from 2000 to 2016. The majority of hospital admissions involving patients with sickle cell disease, occurred in lowest income communities and in metropolitan areas. Correspondingly the readmission rates after sickle cell disease admissions were lower as community-level income increased [15]. This study reported, sickle cell disease hospital admissions were 4.1% more likely to result in discharge against medical advice, as opposed to 1.2% in non-sickle cell disease patients from 2000 to 2016 [15]. Of note 33.5% of sickle cell disease hospital admissions demonstrated a 30 day readmission as opposed to 12.5% of hospital admission for patients without the disease [15]. This must also be considered as a contributing factor for in-hospital mortality. In 2016 three-fourths of hospital admissions of sickle cell patients had a public expected payer (Medicare 25.7% and Medicaid 49.6%) [15].
Findings in this statistical brief correlate with our results from Nationwide Analysis of Trends in In-hospital Mortality in patients with Sickle Cell Disease from 2000 to 2014. Mortality during hospital stay was analyzed in categories of geographical region in the US, health insurance status comorbidities, sex and hospital size. Statistically significant difference was reported in the rate of mortality between small, medium or large hospitals. However the hospitalization and mortality ratio was different when comparing Northeast, Midwest, South and West geographical regions (0.63%, 0.65%, 0.76% and 0.89% respectively) with P = 0.008 confirming that the statistically significant difference in ratio among regions was not determined by chance. This suggests that there may be a factor affecting the mortality outcome by regions. It is worth investigating in a new study if social determinants of health are confounding these results. The overall odds ratio for the category of region OR = 1.07 however suggests that although hospital region and death are correlated, it was not a strong determinant for death during hospitalization. The data on the geographical regions does not report distance or access to healthcare, as in whether the referred populations were located in a metro area, rural adjacent to metro area, or rural-remote area. This component may shed light into whether higher in-hospital mortality in certain geographical regions is secondary to complications developed over time, and a possible role social determinants of health in disease progression. Inadequate follow up and literacy on sickle cell disease by both patients and medical providers may be contributing to these results. It is worth mentioning that there is no statistical description of socioeconomic status by region. This may as well be considered in view of higher hospitalization and mortality in South and West regions, possibly correlated with lower quality of life, remote access to health services and consequently higher comorbidity burden. A description of mortality by each state encompassed in per region and season of the year, may also be contributory to assess possible environmental triggers and particular regional needs. Of note extremes in temperatures is a recognized pain trigger and have been associated with acute complications, however studies have not provided evidence to establish correlation between acute pain episodes during such temperature conditions and hospital admissions [17]. Pain triggers must be considered in the context of their interaction with other contributing factors altering cycles of dehydrations and sickling which may underlie these results.
This study provided data of health insurance status from public expected payers (Medicaid, Medicare), private health insurance and self-pay showing statistically significant difference. Odds ratio for sickle cell patients that died during hospitalization and health insurance status was OR = 0.84. This value OR<1 suggests having a health insurance may be a protective factor from dying during hospitalization. In-hospital mortality of self-pay patients with 0.63% may underrepresent this group as it is reasonable to consider that many uninsured patients might not seek medical service or may die before admission. Cumulative organ damage over time in sickle cell patients, results a consequence of the interaction between the different pathophysiologic pathways. All of the comorbidities analyzed in this study are common in the general population as a consequence of dietary habits and lifestyle. Vascular injury and dysfunction underlie the pathophysiologic changes in all of these chronic diseases. Dietary habits and lifestyle can be confounding factors in the evolution and overall health deterioration of sickle cell patients. However, the pathophysiology of the microvascular damage, multi-organ disfunction and chronic inflammation in sickle cell is an unavoidable reality of these patients. The odds ratio for comorbidities show A-fib with a value of OR = 4.47, followed by hypertension OR = 1.92, diabetes mellitus OR = 1.44 and chronic kidney disease OR = 1.29. All of these values >1 suggest a strong correlation of having these comorbidities and dying during hospitalization. Higher OR values for A-fib and hypertension suggest having these two comorbidities contribute most strongly to higher risk of in-hospital mortality. It may reflect the cardiopulmonary component as leading cause of death in these patients [7]. It is reasonable to investigate if the patients with more comorbidities correlated with rural or remote areas to medical services. A comprehensive approach to management is required to bridge the gaps in the various components of the disease and patient care. Common comorbidities in the general population, in the context of these patients can be confounding the natural course of the disease. Patient management and monitoring must meet a perspective of sickle cell disease. Chronic liver disease, chronic periodic transfusion patterns and iron overload, were not considered among analyzed comorbidities and mortality. Many sickle cell patients receive chronic transfusions and may develop iron overload and should be included as important risk factors in future studies.
This study reported lower in-hospital mortality in females compared to males. It is worth investigating comorbidities by sex. Although menstrual cycles can be a trigger for pain it may also provide prospective protection to organ damage secondary to iron overload. Also, the physiologically and relatively lower hematocrit in females may confer a minimal protective factor in vaso-occlusion and organ damage if we assume there is an associated comparatively lower viscosity. Lower in-hospital mortality may be reflecting these factors.
Hydroxyurea, despite potential toxicities is very well tolerated and does reduce disease associated complications [18]. It may have contributed to the improvements in life expectancy and quality of life in sickle cell patients over the last 2 decades, however its’ use and compliance was not reported in this study. Advanced organ injury and complications can be confounded by limited access to medical services, lack of adherence to medication and appointments and even lack of knowledge from the medical services. The perception from the medical providers on the use of hydroxyurea and the side effect profile can be adding a barrier of perceived complexity to the already challenging care of these patients. Awareness that the management is a combined effort relying in the primary-care physician as much as in the hematologist is key in the overall evolution and management. An adequate level of literacy on sickle cell disease by primary-care physicians is crucial in reducing barriers that could potentially prevent patients from considering hydroxyurea and also improving these population’s quality of life [15]. This aspect is worth investigating as it may also provide insight on the patterns of in-hospital mortality by region, in addition to the proposed description of location in terms of proximity to medical services.
In-hospital mortality trends discussed in this study are representative of the complex interaction of pathophysiologic, medical and non-medical components of sickle cell in a time frame where hydroxyurea had been the only approved medical treatment. Newer disease-modifying medications are not represented in this study will eventually be discussed in future studies over the next decade. The different pathophysiologic pathways of the disease are being targeted by new approved medications. L-glutamine reduces vaso-occlusive episodes by protecting the red blood cell against oxidative damage. A different anti-sickling agent (voxelotor) increases oxygen affinity of HbS by allosteric modification [5]. Crinalizumab is a third recently approved medication that target abnormal cellular adhesion and vascular dysfunction [5]. The reduction in mortality and morbidity in sickle cell disease patients due to introduction of these newer pharmacological agents will be apparent in the next few years and will warrant further follow-up studies.
Conclusions
Socioeconomic background as well as previously proposed detailed geographic distribution of people with the disease by state, in addition to distribution by region may reflect patterns of social and health disparities in the care of sickle cell disease patients. This can help address the disparities between urban and rural population as well as health insurance status and disease knowledge among patients and medical providers. Recently approved disease modifying medications may also bridge the gaps in sickle cell treatments to a certain extent. Improved access to medical service as well as alternatives in treatment can have direct impact in disease evolution, development of complications and in-hospital mortality risk. A multidisciplinary approach is desired for improving quality of life, prevention and management of complications as addressing modifiable risk factors can improve in-hospital mortality.
Disclosure of conflict of interest
None.
References
- 1.Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, Vichinsky EP. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi: 10.1038/nrdp.2018.10. [DOI] [PubMed] [Google Scholar]
- 2.Banks M, Shikle J. Hyperhemolysis syndrome in patients with sickle cell disease. Arch Pathol Lab Med. 2018;142:1425–1427. doi: 10.5858/arpa.2017-0251-RS. [DOI] [PubMed] [Google Scholar]
- 3.Ferrone FA. The delay time in sickle cell disease after 40 years: a paradigm assessed. Am J Hematol. 2015;90:438–445. doi: 10.1002/ajh.23958. [DOI] [PubMed] [Google Scholar]
- 4.Maitra P, Caughey M, Robinson L, Desai PC, Jones S, Nouraie M, Gladwin MT, Hinderliter A, Cai J, Ataga KI. Risk factors for mortality in adult patients with sickle cell disease: a meta-analysis of studies in North America and Europe. Haematologica. 2017;102:626–636. doi: 10.3324/haematol.2016.153791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carden MA, Little J. Emerging disease-modifying therapies for sickle cell disease. Haematologica. 2019;104:1710–1719. doi: 10.3324/haematol.2018.207357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farooq S, Abu Omar M, Salzman GA. Acute chest syndrome in sickle cell disease. Hosp Pract (1995) 2018;46:144–151. doi: 10.1080/21548331.2018.1464363. [DOI] [PubMed] [Google Scholar]
- 7.Wood KC, Gladwin MT, Straub AC. Sickle cell disease: at the crossroads of pulmonary hypertension and diastolic heart failure. Heart. 2020;106:562–568. doi: 10.1136/heartjnl-2019-314810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle SM, Jacobs B, Sayani FA, Hoffman B. Management of the dialysis patient with sickle cell disease. Semin Dial. 2016;29:62–70. doi: 10.1111/sdi.12403. [DOI] [PubMed] [Google Scholar]
- 9.Naik RP, Derebail VK. The spectrum of sickle hemoglobin-related nephropathy: from sickle cell disease to sickle trait. Expert Rev Hematol. 2017;10:1087–1094. doi: 10.1080/17474086.2017.1395279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vichinsky E. Chronic organ failure in adult sickle cell disease. Hematology Am Soc Hematol Educ Program. 2017;2017:435–439. doi: 10.1182/asheducation-2017.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haymann JP, Stankovic K, Levy P, Avellino V, Tharaux PL, Letavernier E, Grateau G, Baud L, Girot R, Lionnet F. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol. 2010;5:756–761. doi: 10.2215/CJN.08511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cook JA, Hislop J, Altman DG, Fayers P, Briggs AH, Ramsay CR, Norrie JD, Harvey IM, Buckley B, Fergusson D, Ford I, Vale LD DELTA group. Specifying the target difference in the primary outcome for a randomised controlled trial: guidance for researchers. Trials. 2015;16:12. doi: 10.1186/s13063-014-0526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: a review. Int J Infect Dis. 2010;14:e2–e12. doi: 10.1016/j.ijid.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Arabi YM, Mandourah Y, Al-Hameed F, Sindi AA, Almekhlafi GA, Hussein MA, Jose J, Pinto R, Al-Omari A, Kharaba A, Almotairi A, Al Khatib K, Alraddadi B, Shalhoub S, Abdulmomen A, Qushmaq I, Mady A, Solaiman O, Al-Aithan AM, Al-Raddadi R, Ragab A, Balkhy HH, Al Harthy A, Deeb AM, Al Mutairi H, Al-Dawood A, Merson L, Hayden FG, Fowler RA Saudi Critical Care Trial Group. Corticosteroid therapy for critically Ill patients with middle east respiratory syndrome. Am J Respir Crit Care Med. 2018;197:757–767. doi: 10.1164/rccm.201706-1172OC. [DOI] [PubMed] [Google Scholar]
- 15.Fingar KR, Owens PL, Reid LD, Mistry KB, Barrett ML. Characteristics of inpatient hospital stays involving sickle cell disease, 2000-2016: statistical brief #251. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville (MD): 2019. [Google Scholar]
- 16.Lubeck D, Agodoa I, Bhakta N, Danese M, Pappu K, Howard R, Gleeson M, Halperin M, Lanzkron S. Estimated life expectancy and income of patients with sickle cell disease compared with those without sickle cell disease. JAMA Netw Open. 2019;2:e1915374. doi: 10.1001/jamanetworkopen.2019.15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tewari S, Brousse V, Piel FB, Menzel S, Rees DC. Environmental determinants of severity in sickle cell disease. Haematologica. 2015;100:1108–1116. doi: 10.3324/haematol.2014.120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, Xing X, Xiang N, Wu Y, Li C, Chen Q, Li D, Liu T, Zhao J, Liu M, Tu W, Chen C, Jin L, Yang R, Wang Q, Zhou S, Wang R, Liu H, Luo Y, Liu Y, Shao G, Li H, Tao Z, Yang Y, Deng Z, Liu B, Ma Z, Zhang Y, Shi G, Lam TTY, Wu JT, Gao GF, Cowling BJ, Yang B, Leung GM, Feng Z. Early transmission dynamics in wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]