

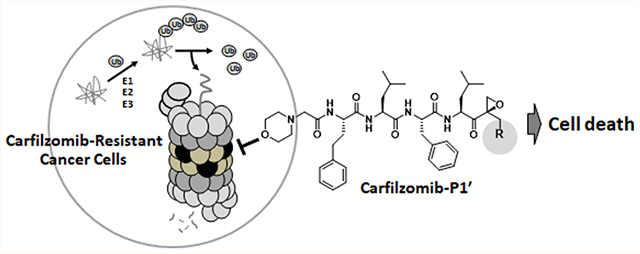
Abstract
Over the past 15 years, proteasome inhibitors (PIs), namely bortezomib, carfilzomib (Cfz) and ixazomib, have significantly improved the overall survival and quality-of-life for multiple myeloma (MM) patients. However, a significant portion of MM patients do not respond to PI therapies. Drug resistance is present either de novo or acquired after prolonged therapy through mechanisms that remain poorly defined. The lack of a clear understanding of clinical PI resistance has hampered the development of next-generation PI drugs to treat MM patients who no longer respond to currently available therapies. Here, we designed and synthesized novel epoxyketone-based PIs by structural modifications at the P1′ site. We show that a Cfz analog, 9, harboring a hydroxyl substituent at its P1′ position was highly cytotoxic against cancer cell lines displaying de novo or acquired resistance to Cfz. These results suggest that peptide epoxyketones incorporating P1′-targeting moieties may have the potential to bypass resistance mechanisms associated with Cfz and to provide additional clinical options for patients resistant to Cfz.
Graphical Abstract
INTRODUCTION
Since the FDA approval of bortezomib (Btz) in 2003, proteasome inhibitors (PIs) (Figure 1A) have become critical components of many MM treatment regimens. Compared to the pre-PI era, the use of PIs, either as single agents or in combination with an immunomodulatory agent, has significantly improved the 5-year overall survival rate for MM patients.1 Despite these remarkable successes, MM remains nearly universally incurable due to de novo or acquired drug resistance. Since 2015, the FDA has approved three non-PI drugs, daratumumab (a monoclonal antibody (mAb) targeting CD38), elotuzumab (mAb targeting SLAMF7), and panobinostat (HDAC inhibitor). Although these non-PI drugs offer additional options for MM patients relapsed on current PI-based therapies, their response rates vary widely. Results from recent clinical trials show that responses to these non-PI drugs are rather transient with a median duration of ~7–20 months before relapsing.2–4 Thus, there is an urgent unmet medical need for new agents that can yield durable responses for MM patients who have exhausted current treatment options.
Figure 1.
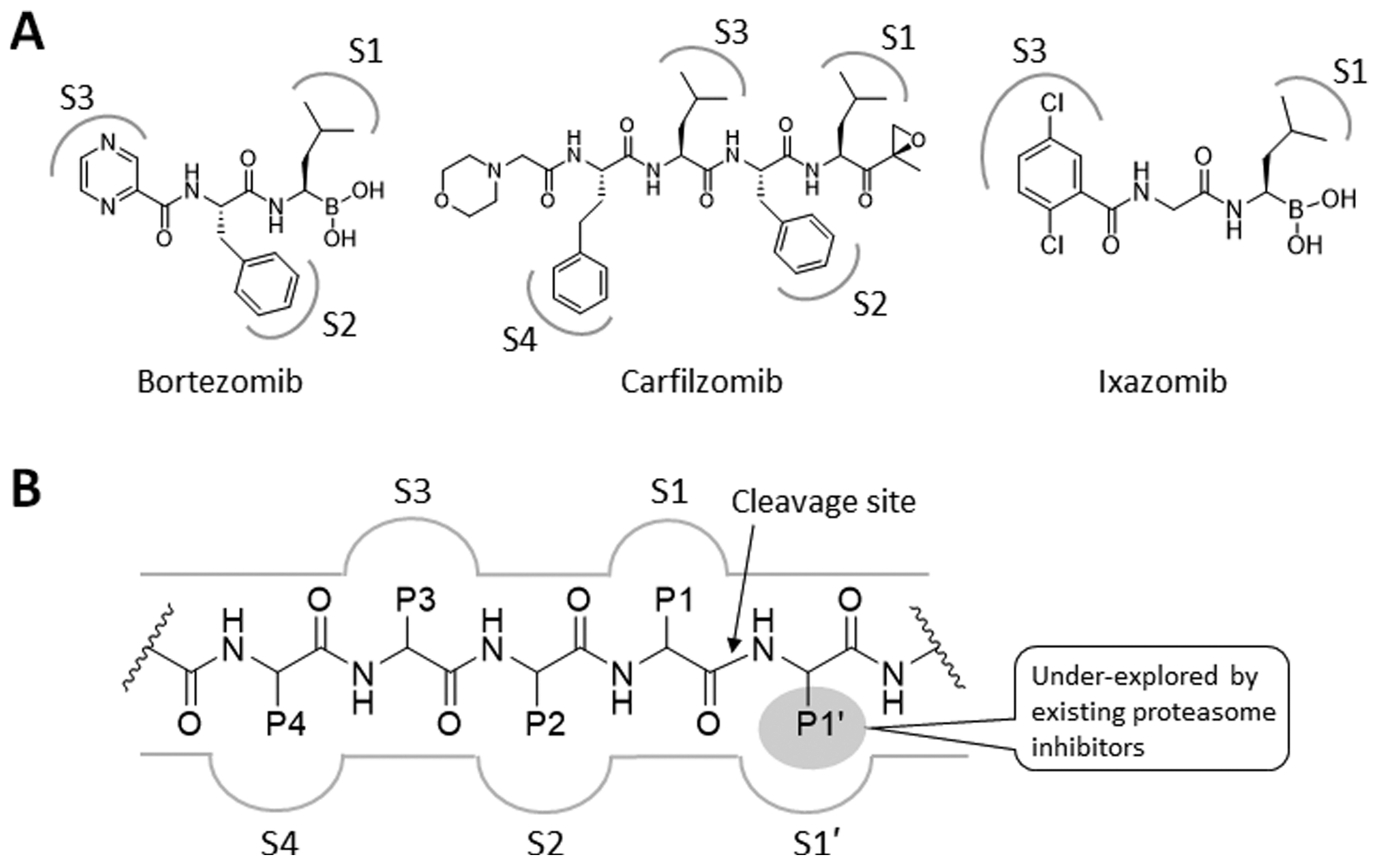
(A) Structures of proteasome inhibitors in clinical use. The gray-colored markings denote the functional groups that are proposed to form favorable interactions with the specificity pockets (S1–4) of a proteasome catalytic subunit. (B) Schematic representation of a prototypical proteasome substrate or substrate-like inhibitor bound to a proteasome catalytic subunit showing the unprimed residues (P1, P2, P3, and P4) located N-terminal to the cleavage site of the proteasome catalytic subunit (shown as an arrow) and the primed P1′ residue located C-terminal to the cleavage site.
The ubiquitin-proteasome system (UPS) is responsible for maintaining protein homeostasis via controlled degradation of numerous intracellular proteins. The UPS furthermore plays a critical role in regulating cellular signaling pathways and several key processes described as the original hallmarks of cancer by Hanahan and Weinberg (Figure 2).5 The fundamental importance of the UPS to cancer cells is further reflected by the recent inclusion of the proteotoxic stress as an additional hallmark of cancer.6
Figure 2.
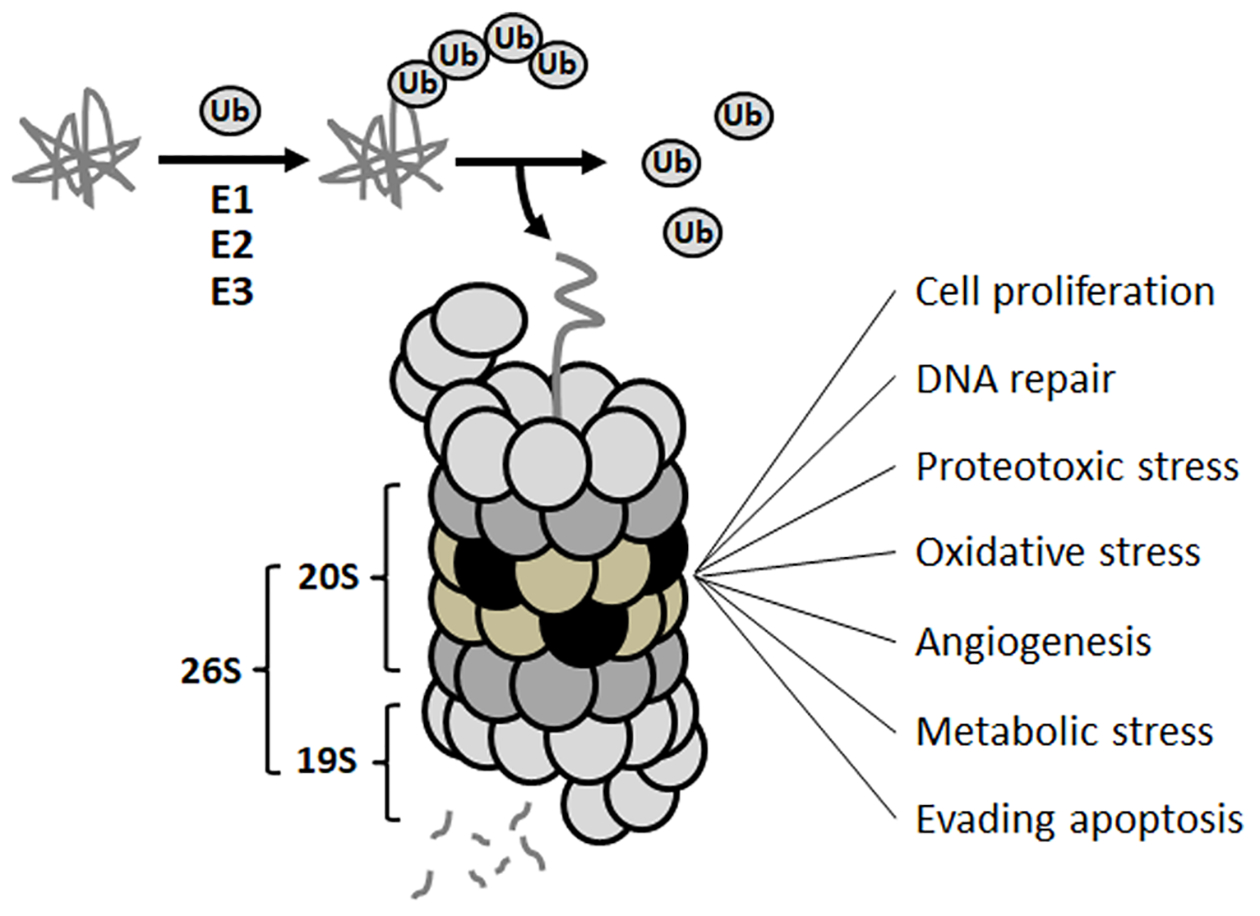
Ubiquitin-proteasome system (UPS) and its association with various hallmarks of cancer.
The UPS presumably remains essential for cancer cells regardless of resistance to existing PI drugs. The UPS components upstream of the proteasome (e.g., deubiquitinases, ubiquitin E3 ligases) are being explored as potential anticancer therapeutic targets,7–11 but clinical success is yet to be achieved. Alternatively, it remains to be seen whether the proteasome itself can be subsequently retargeted to achieve further therapeutic gains for MM patients relapsed on existing PI drugs. Although there are several new PIs in commercial development pipelines (e.g., delanzomib, oprozomib, marizomib), early clinical data indicate that those PIs may not be beneficial for patients with acquired resistance to existing PIs due to issues such as low response rates in high-risk populations and prevalent cross-resistance with existing PIs.12–17
Until now, nearly all PIs have been developed through optimization of amino acid side chains (P1, P2, P3), which interact with the conventional substrate binding pockets (S1, S2, S3, etc.) of proteasome catalytic subunits (Figure 1B) located N-terminal to the cleavage site. This binding configuration is further stabilized by the formation of an antiparallel β-sheet configuration via hydrogen bonding interactions between the inhibitor’s peptide backbone and the conserved backbone residues such as Thr21, Gly47, and Ala49 of proteasome catalytic subunits.18–20 When combined with a C-terminal warhead, which targets the catalytic Thr1 residue, this strategy typically yields potent inhibitors including the three FDA-approved PIs: Btz, Ixz (peptide boronic acids), and Cfz (a peptide epoxyketone). However, this strategy might have unintentionally contributed to an increased incidence of cross-resistance among them. A potential strategy to circumvent PI cross-resistance is to identify and exploit a structural niche not utilized by existing PIs. In that regard, previously unexplored are P1′ binding sites, which lie on the C-terminal side of the proteasome catalytic subunit’s cleavage site (Figure 1B).
Here, we have developed peptide epoxyketones bearing a P1′-targeting moiety. The anticancer efficacy of these compounds was superior to their non-P1′-targeting parent compounds when evaluated against models of intrinsic and acquired Cfz resistance. The identified lead compound, 9, an analog of Cfz containing a hydroxyl group at the P1′ position, displayed potent proteasome inhibitory activity and cytotoxicity in both Cfz-sensitive and Cfz-resistant cancer cell lines.
RESULTS
Chemistry.
The final compounds (5, 7–12) were prepared as shown in Scheme 1. TBDPS (t-butyldiphenylsilyl) and methyl-protected alkenes 1a and 1b were synthesized in three steps following a known method starting from Boc-serine methyl ester.21 Boc-serine methyl ester was treated with dimethyl methylphosphonate in the presence of n-butyllithium (n-BuLi) to yield a phosphoryl intermediate, which upon combination of Wittig-Horner and Baylis-Hillman-type one-pot reactions furnished the hydroxymethyl-substituted enone in a good yield. Protection of a hydroxyl group with TBDPS or a methyl group was carried out in TBDPS-Cl/DIEA or methyl iodide/silver oxide, respectively. Hydrogen peroxide was used to prepare epoxide 2a and 2b from the respective enones. Intermediates 3 and 4 were prepared using a conventional amide coupling reaction. Deprotection of TBDPS from the intermediate 4 in the presence of TBAF (t-butylammonium fluoride) yielded the final compound 5 as shown in Scheme 1A. For Cfz P1′ derivatives, the left-hand peptide backbone of Cfz 6 was separately prepared in three steps fromBoc-serine using a series of amide coupling reactions in good yields. Another amide coupling reaction between benzyl deprotected 6 and Boc deprotected 2a or 2b gave the final compounds 7 and 8. Deprotection of TBDPS from 7 yielded the final compound 9. Compounds 10, 11, and 12 were prepared from compound 9 using the respective alkylating agents in the presence of organic base DIEA as shown in Scheme 1B. UK101 and UK102 were used from in-house compound library.
Scheme 1. Synthesis Scheme of 5 (UK101-OH) and 9 (Cfz-OH)a.
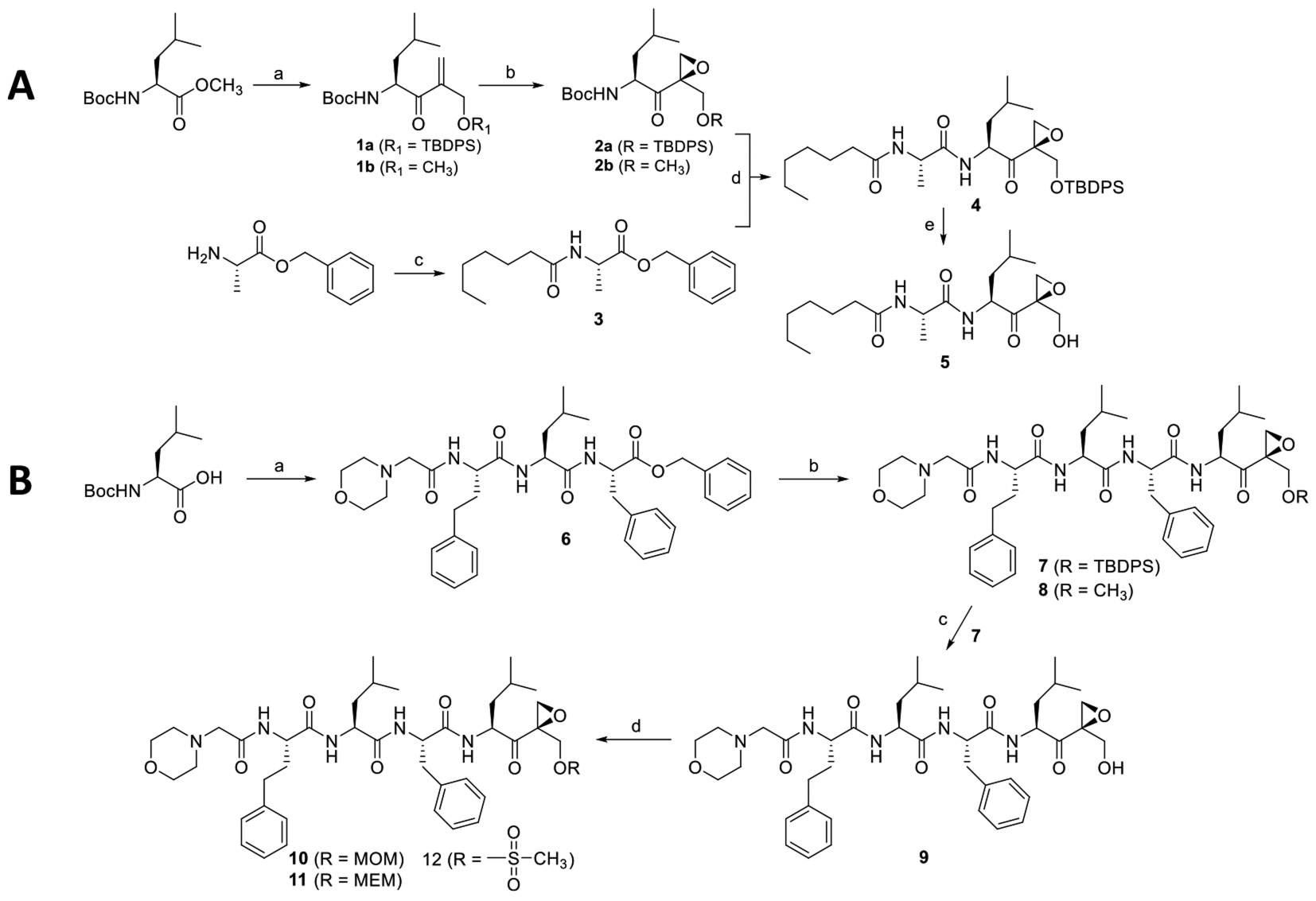
aReagents and conditions. (A) (a) (i) CH3PO(OCH3)2, n-BuLi, THF, −78 °C, 2 h, Boc-serine methylester, THF, −78 °C, 3 h, 55%; (ii) Aqueous solution of formaldehyde, K2CO3, H2O, rt, 4 h, 59%; (iii) TBDMS-Cl, imidazole, DCM, rt, 30 min, 71% or iodomethane, Ag2O, MeCN, rt, 24 h, 51%; (b) benzonitrile, H2O2, DIEA, MeOH, 0 °C, 3 h, 35–41%; (c) heptanoic acid, HBTU, HOBt, DIEA, DCM, rt, 18 h, 82%; (d) (i) H2, Pd/C, methanol, 1 h, (ii) Boc-deprotected 2a, HBTU, HOBt, DIEA, DCM, rt, 18 h, 91%; (e) TBAF, DCM, 2 h, 70%. (B) (a) (i) Phenylalanine benzyl ester hydrochloride, HBTU, HOBt, DIEA, DCM, rt, 18 h, 81%; (ii) TFA, DCM, rt, 1 h then evaporation and drying, Boc-homoPhe-OH, HBTU, HOBt, DIEA, DCM, rt, 18 h, 79%; (iii) TFA, DCM, rt, 1 h then evaporate and dried, morpholin-4-yl-acetic acid hydrochloride, HBTU, HOBt, DIEA, DCM, rt, 18 h, 65%; (b) (i) H2/Pd, C, methanol, 1 h; (ii) 2a or 2b, HBTU, HOBt, DIEA, DCM, rt, 18 h, 45% and 48%, respectively; (c) TBAF, DCM, 2 h, 64%; (d) MOM-Cl or MEM-Cl or methanesulfonyl chloride, DIEA, DCM, overnight, 60–64%.
Initial Screening for PIs That Can Overcome de Novo Resistance to Cfz.
We recently noted that H727 cells derived from human lung adenocarcinoma are intrinsically resistant to Cfz (IC50 of 611 nM, Supporting Information S1) compared to a panel of cancer cell lines (IC50’s in the low nM range). Our mechanistic investigation via chemical and genetic modulation of proteasome catalytic subunits suggests that proteasome catalytic subunit composition contributes to the de novo resistance of H727 cells to Cfz.21 Therefore, we envisioned that H727 cells can serve as an initial screening system in identifying PIs that can overcome de novo resistance to Cfz (currently there is no MM model with de novo resistance to Cfz). We hypothesized that alternative PIs with distinct structural features may interact differentially with proteasomes in H727 cells to induce proteasome inhibition and consequent cell death. As a control, we used another lung cancer cell, H23, which is highly sensitive to Cfz (IC50 of 18 nM). We compared the cytotoxic effects of PIs with differing pharmacophores including P1′-targeted peptide epoxyketones (UK101 and UK102 previously developed in our laboratory)22 in H727 and H23 cells (Figure 3).
Figure 3.
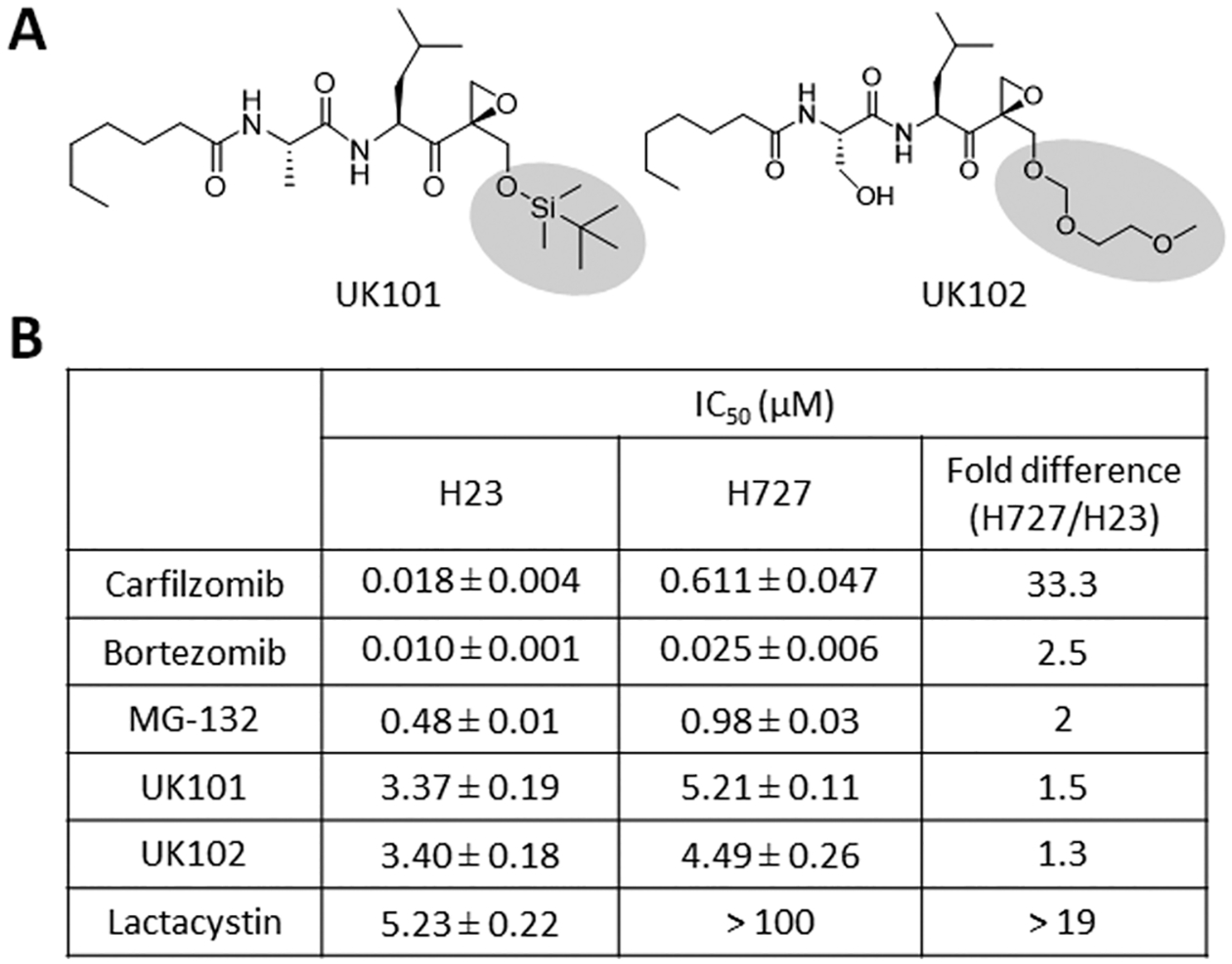
(A) Structures of UK101 and UK102. (B) Comparison of the cytotoxic potency (72-h IC50 values) of H727 and H23 cells to carfilzomib, bortezomib, MG-132, UK101, UK102, and lactacystin. Data are reported as the mean ± SD.
Btz (a peptide boronic acid) and MG-132 (a peptide aldehyde) displayed a comparable cytotoxicity in H727 and H23 cells. UK101 and UK102 were also not cross-resistant to Cfz, displaying similar IC50 values in H727 cells and H23 cells. On the other hand, lactacystin (a lactone) was not as effective in H727 cells as in H23 cells. These results suggest that proteasome inhibition using alternative compounds can still be a viable therapeutic option even in the presence of resistance to Cfz.
Anticancer Efficacy of PIs with P1′ Moieties (UK101 and UK102) Is Not Attenuated by Acquired Cfz Resistance in Cancer Cells.
As a next step to examine the effectiveness of P1′-targeting epoxyketones in cancer cells, we examined whether these compounds can overcome acquired Cfz resistance, a major clinical challenge facing Cfz-based therapies.23 We utilized a Cfz-resistant subline of RPMI8226 (derived from human MM cells), which had been adapted to escalating concentrations of Cfz over several months.
As shown in Figure 4A, the Cfz-resistant RPMI8226 cells displayed a marked (greater than 35-fold) increase in IC50 values and elevated expression of the efflux transporter P-glycoprotein (P-gp), similar to the previous reports.24,25 The use of the P-gp inhibitor reversin 121 almost completely restored Cfz sensitivity (Supporting Information S2), suggesting the predominant contribution of P-gp in the current model of acquired Cfz resistance. While both Cfz and epoxomicin (also reported as a P-gp substrate26) displayed a marked increase in IC50 values, UK101 and UK102 showed minimal changes in IC50 values in Cfz-resistant RPMI 8226 cells compared to the parental control (Figure 4B).
Figure 4.
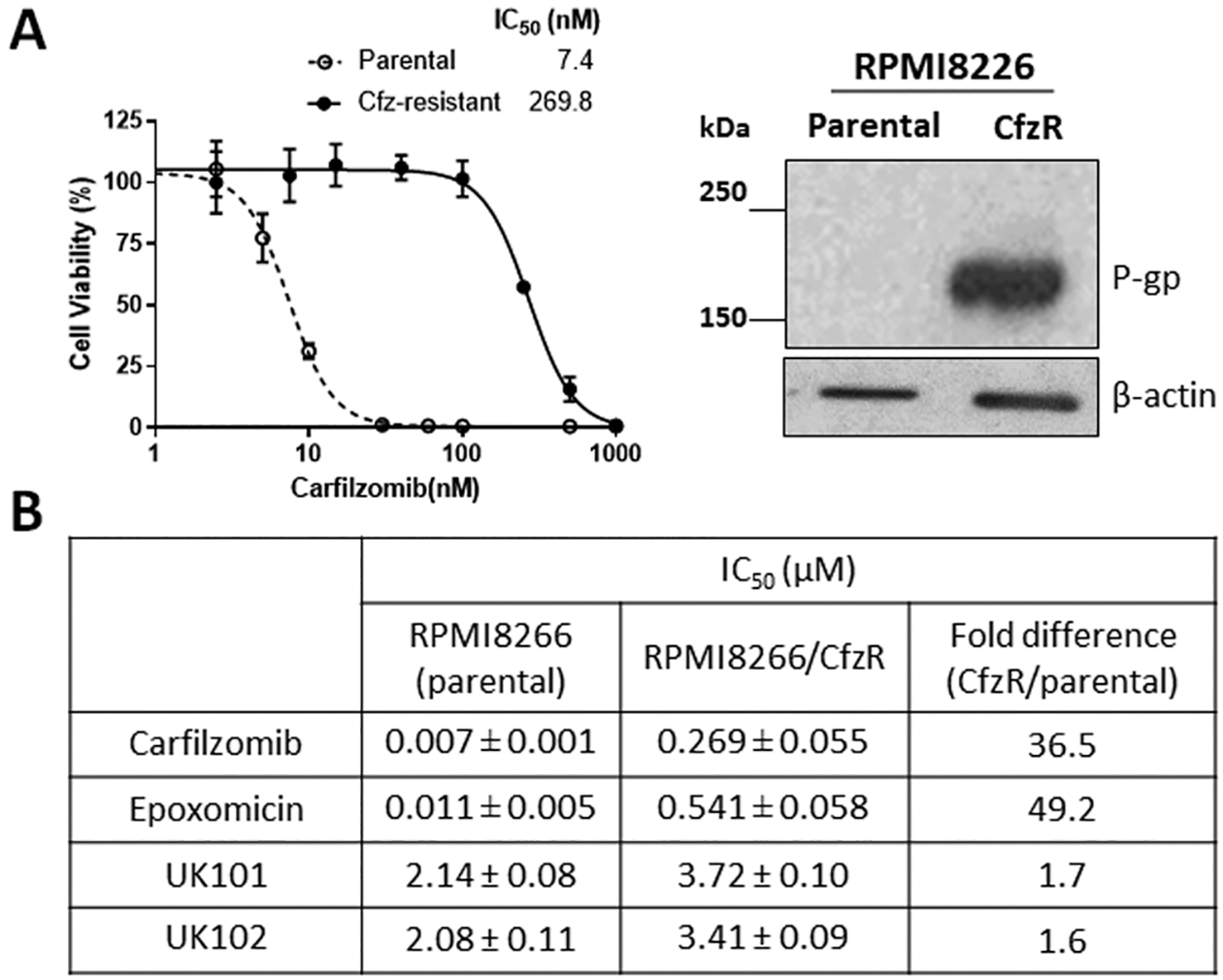
(A) Effects of carfilzomib (Cfz) on the viability of an RPMI8266 cell line with acquired Cfz resistance (RPMI8266/CfzR) in comparison to the parental cell line. Cell viability was measured by MTS assay after 72 h drug treatment (left). Immunoblotting analysis showing a marked increase of P-gp expression in RPMI8226 Cfz-resistant cells in comparison to parental cells (right). (B) Comparison of the sensitivity (72-h IC50 values) of RPMI8226 parental and Cfz-resistant cells to Cfz, epoxomicin, UK101, and UK102. Data are reported as the mean ± SD. For epoxomicin and carfilzomib, the SD values were obtained from three independent experiments. For UK101 and UK102, the SD values were from nonlinear regression analysis using three replicates.
Anticancer Efficacy of UK101 and UK102 Is Not Impaired by Acquired Btz Resistance in Primary MM Samples.
We next investigated whether UK101 and UK102 can be effective in primary MM samples from patients who do not respond to Btz (We were not able to obtain MM samples from patients who are resistant to Cfz, but it was previously reported that patients with MM refractory to or relapsed on Btz are often cross-resistant to Cfz.27). Using primary MM samples from 14 different donors (6 from PI-naïve patients and 8 from patients relapsed on Btz therapy), their sensitivity to Cfz was assessed. The results indicated varying degrees of sensitivity to Cfz, but the MM samples from the patients who relapsed on Btz tended to be less responsive to Cfz than those from patients from the PI-naïve group (Figure 5A). Because of the limited quantities of primary MM samples from five patients (#5–8 and #14), we utilized only nine primary MM samples (4 Btz-resistant and 5 PI-naïve) to examine the effect of UK101 or UK102. When nine primary MM samples treated with UK101 or UK102 for 48 h, the results indicated that cytotoxic effects of UK101 or UK102 were similar. Although the current sample size was small, the lack of apparent cross-resistance between Cfz/Btz and UK101/UK102 was encouraging.
Figure 5.
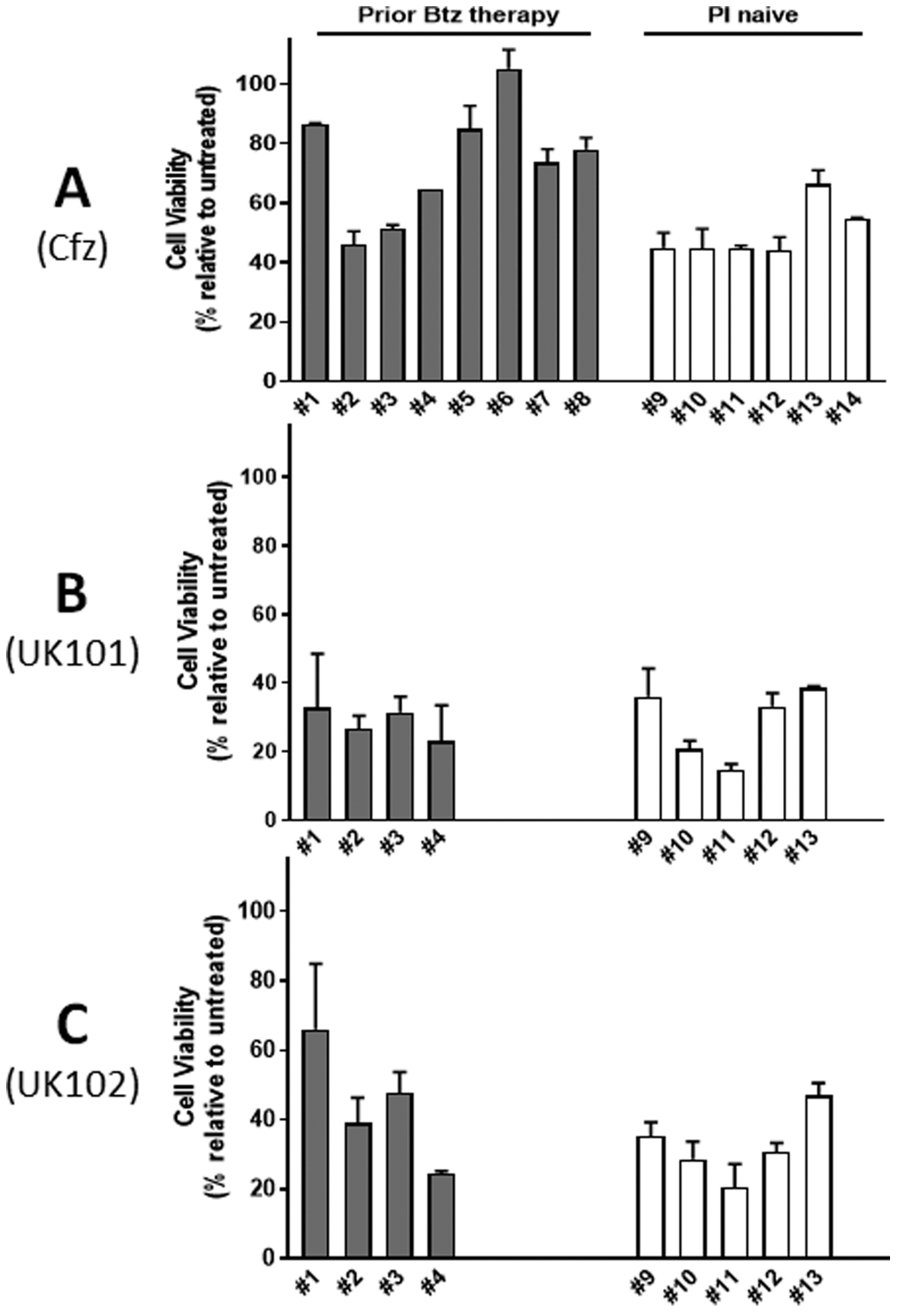
Effects of carfilzomib (Cfz), UK101, and UK102 on cell viability of primary MM samples from 14 different donors, six from Btz/Cfz-naïve patients and eight from patients relapsed on Btz therapy. Primary MM cells were treated with Cfz (A, 50 nM), UK101 (B, 10 μM), or UK102 (C, 10 μM) for 48 h. Cell viability was measured using an ATP-based luminescent assay.
Introduction of Hydrophilic Residues at P1′ Position of Peptide Epoxyketones Improves Proteasome Inhibitory Potency.
To improve the potency of UK101 and UK102 and obtain structure-activity relationship (SAR) information, we first utilized docking models of UK101 bound to β1i and β5, known targets of UK101. UK101 was initially identified as a selective inhibitor of the β1i subunit at nanomolar concentration ranges.28 Later, UK101 was also found to inhibit the β5 subunit (chymotrypsin-like (CT-L) activity) at micromolar concentration ranges.29 Docking structures shown in Figure 6 were generated by Autodock Vina using the previously reported X-ray structures from mouse 20S proteasomes complexed with ONX 091430 and were used as a guide to our optimization effort at the P1′ position. As shown in Figure 6A, the P1–P3 residues of UK101 were predicted to occupy the S1–S3 pockets located deep inside of the active sites of the β1i and β5 subunits.
Figure 6.
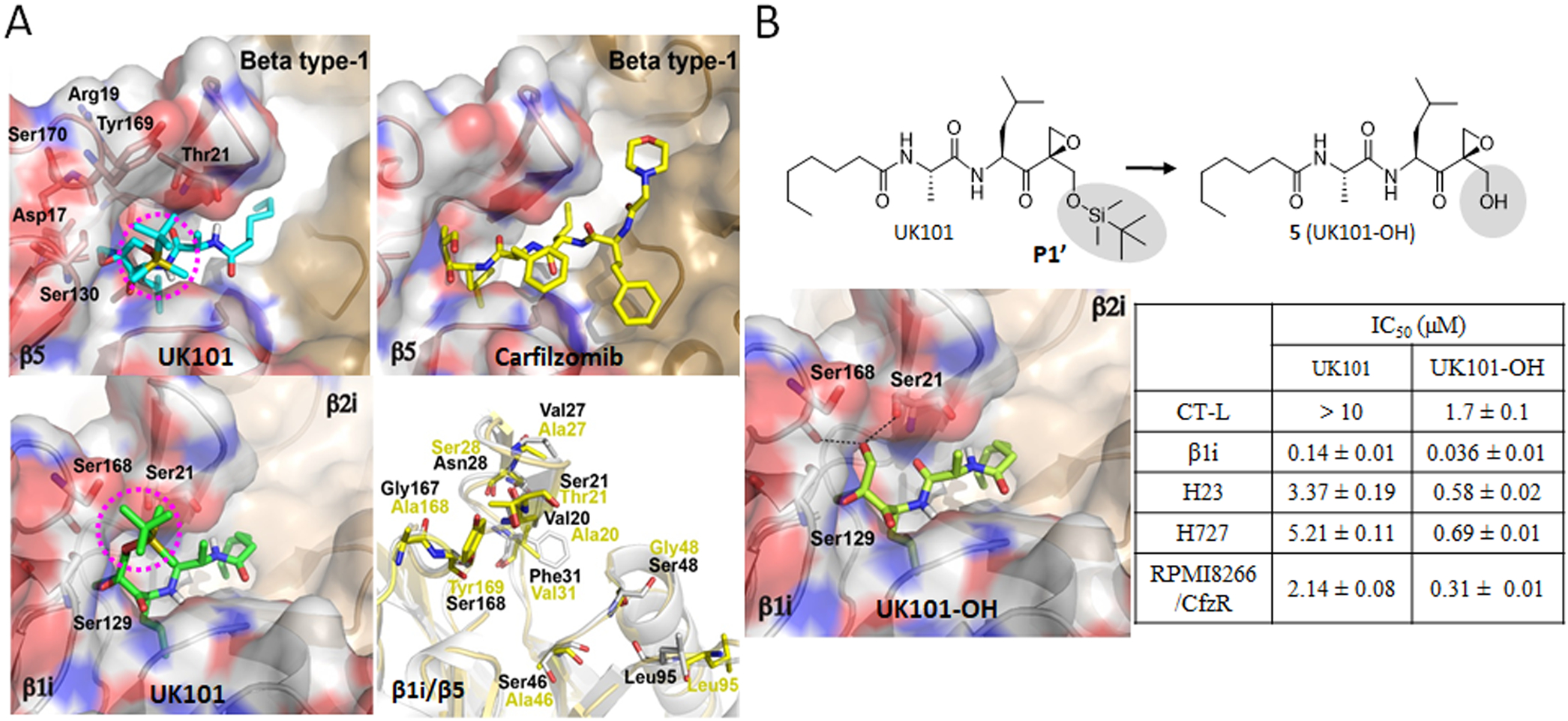
(A) Predicted docking models of UK101 and carfilzomib (Cfz) bound to β5 or β1i. The location of UK101’s TBDMS group positioned within putative P1′ pockets is highlighted using a purple-colored circle. β5 (PDB ID: 3UNB) and β1i (PDB ID: 3UNF) from mammalian 20S proteasomes were used as templates. In cartoon presentation, β1i (gray, PDB ID: 3UNF) was superposed to β5 (yellow, PDB ID: 3UNB) and only different amino acid residues are shown in stick model. (B) Comparison of UK101 and 5(UK101-OH) in terms of their potency (IC50 values) against proteasome chymotrypsin-like activity (in RPMI8226 cell lysate), β1i/LMP2 catalytic activity (in 20S purified human immunoproteasome), and against H23, H727, and Cfz-resistant RPMI8226 cells as measured by MTS cell viability assay. Data are reported as the mean ± SD. Docking model of UK101-OH bound to β1i (PDB ID: 3UNF). The P1′-OH of UK101-OH (5) is perfectly positioned to form hydrogen bonds with Ser168 and Ser21.
However, unlike Cfz bound to β5, docking models indicated that UK101 occupies an additional binding pocket (S1′) of the β5 as well as β1i subunits via its P1′ group (highlighted in purple circles, Figure 6A), which is not occupied by Cfz or Btz.31 Although the P1′ group (t-butyldimethylsilyl, TBDMS) of UK101 points toward the surface of β1i, it is predicted to contribute to a favorable hydrogen bonding interaction with the hydroxyl side chain of Ser168 within the S1′ pocket (bottom-left, Figure 6A). In the case of UK101 bound to β5, the amino acid residue changes within the S1′ pocket (especially, β1i-Ser168 → β5-Tyr169) are predicted to abolish this additional hydrogen bonding interaction with the TBDMS (top-left, Figure 6A).
The resulting change in amino acid residues of the S1′ pocket is also predicted to force the large hydrophobic TBDMS group to be surrounded by the polar amino acids (Asp17, Thr21, Ser130) within the S1′ pocket, a plausible contributing factor toward UK101’s low potency against the β5 subunit.31 On the basis of this prediction, we hypothesized that the introduction of small polar groups at the P1′ position could provide energetically favorable hydrogen bonding interactions with the polar amino acid residues within the S1′ pockets of β1i and β5, potentially improving potency against both subunits. To test this, we replaced the bulky hydrophobic P1′ substituent (TBDMS) of UK101 with a hydroxyl group (Scheme 1A) and assessed its impact. This simple P1′ substitution increased in vitro 20S proteasome inhibitory potency by ~4–10-fold (Figure 6B). This experimental result is consistent with the docking models of UK101-OH (5) bound to β1i (PDB ID: 3UNF), in which the P1′ hydroxyl group of UK101-OH (5) is perfectly positioned to form hydrogen bonds with the oxygen of Ser168 carbonyl backbone and the hydroxyl side chain of Ser21. Consequently, the P1′ hydroxyl group also considerably improved the cytotoxic activity of compound 5 in cancer cell lines, including our models of both de novo and acquired Cfz resistance. On the basis of this result, we suspect that the P1′ residue of peptide epoxyketones may play an important role in overcoming the resistance of cancer cells to Cfz.
Development of 9 (Cfz-OH) with an Improved Potency in Models of de Novo and Acquired Cfz Resistance.
In our efforts to further optimize 5, we considered that the P1–P4 groups of Cfz have already been thoroughly optimized for the S1–S4 pockets of β5 and β5i (as well as β1i, to a lesser extent, based on the largely β1i/β5 superimposed model, Figure 6A). We thus decided to attach several different P1′ moieties to Cfz (Figure 7). Our hypothesis was that Cfz analogs bearing a P1′-targeting group could overcome cross-resistance to Cfz. We also expected that a Cfz analog containing a polar P1′ moiety may have an improved inhibitory potency against both β5 and β1i compared to Cfz due to additional P1′:S1′ interactions (Figure 1B). We prepared Cfz analogs having a series of P1′ moieties varying from bulky hydrophobic to small hydrophilic residues (Scheme 1B). We subsequently measured their activity against cell lysates to measure CT-L (β5/β5i) inhibition and against 20S purified immunoproteasomes to measure β1i inhibition. As predicted by in silico docking analysis, 9 and 12 having small polar moieties were most potent in vitro with IC50 values similar to that of Cfz. When these two compounds were tested using Cfz-resistant H727 cells, 12 was less potent than Cfz against H727. On the other hand, 9 (Cfz-OH, a Cfz analog with a hydroxyl group at the P1′ position) demonstrated an improved potency by ~10-fold relative to Cfz. When tested against RPMI8226 with acquired Cfz resistance, 9 demonstrated an almost three-fold improvement in potency against Cfz-resistant RPMI8226 cells as compared to Cfz (Figure 7).
Figure 7.
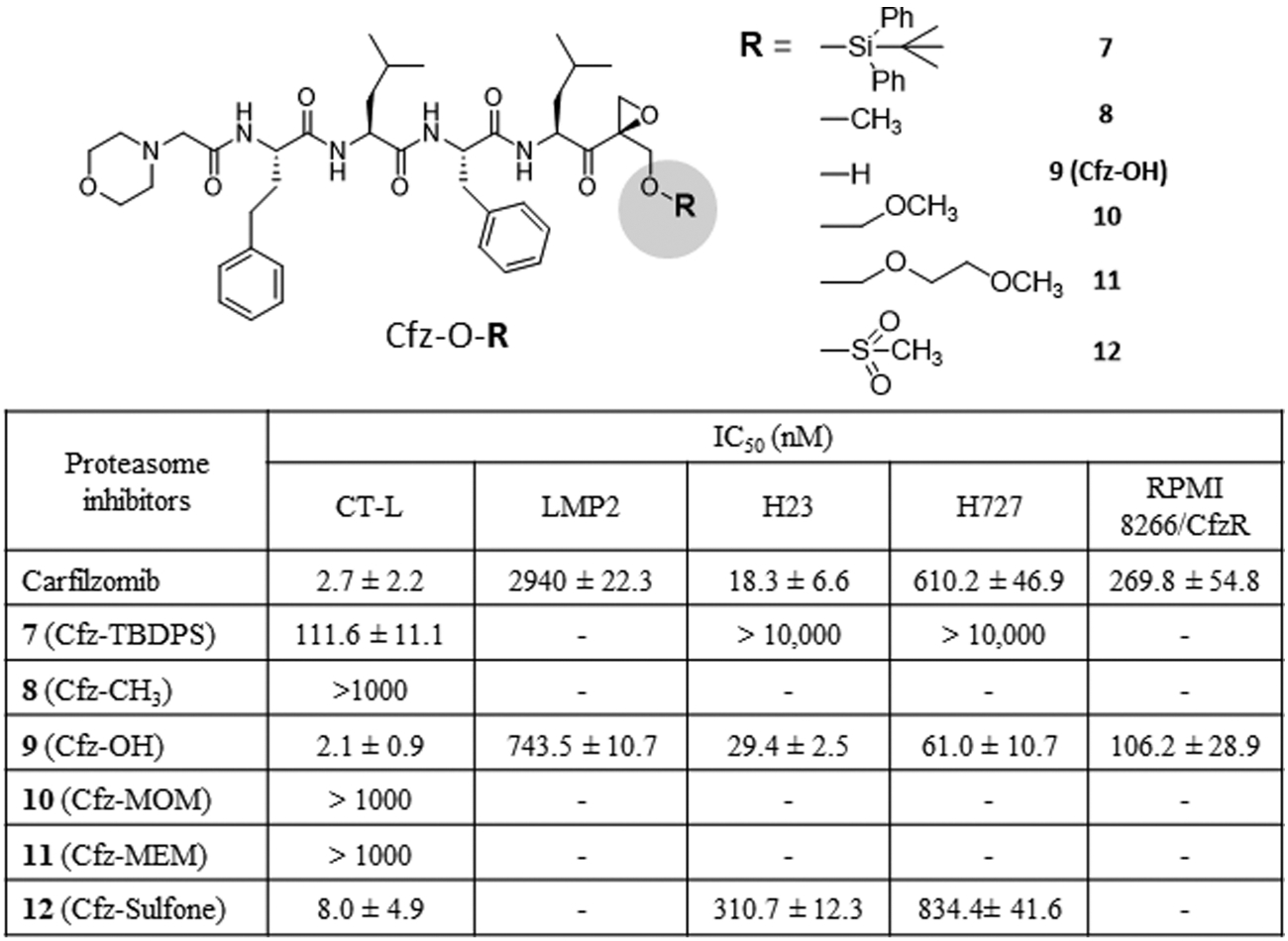
Potency (IC50 values) for compounds with various substitutions at the P1′site against proteasome chymotrypsin-like activity (RPMI8226 cell lysate), β1i/LMP2 activity (purified human 20S immunoproteasome), and cell viability of H23, H727, and Cfz-resistant RPMI8226 cells. Data reported as the mean ± SD (carfilzomib, n = 3 independent experiments) or from a single experiment (3 replicates, 7, 9, and 12).
Compound 9 (Cfz-OH) Has Improved Metabolic Stability Compared to Its Close Structural Analog, Cfz.
Cfz is rapidly metabolized in vivo with plasma half-lives of less than 1 h,32 likely contributing to its lack of efficacy against solid cancers especially those with poorly developed vasculature.33 The metabolic inactivation of Cfz is reported to be mediated mainly by microsomal epoxide hydrolase (mEH) and peptidases.34 In the case of mEH, the active site harbors two conserved tyrosine residues which may contribute to substrate specificity and orientation of substrates within the active site.35,36 The prototypical substrates of mEH include planar hydrophobic compounds such as various epoxides of polycyclic aromatic hydrocarbons and steroids.37 It is thus expected that the epoxide ring of Cfz occupies the active site of mEH with a position suitable for hydrolysis to yield Cfz-diol (Figure 8A). We hypothesized that the addition of a hydroxyl group adjacent to the epoxide ring of Cfz (compound 9) may hinder the hydrolysis of the epoxide ring by inhibiting access to the active site of mEH. When the metabolic stability of 9 was examined in rat liver homogenates, 9 was indeed more stable than Cfz (Figure 8B).
Figure 8.
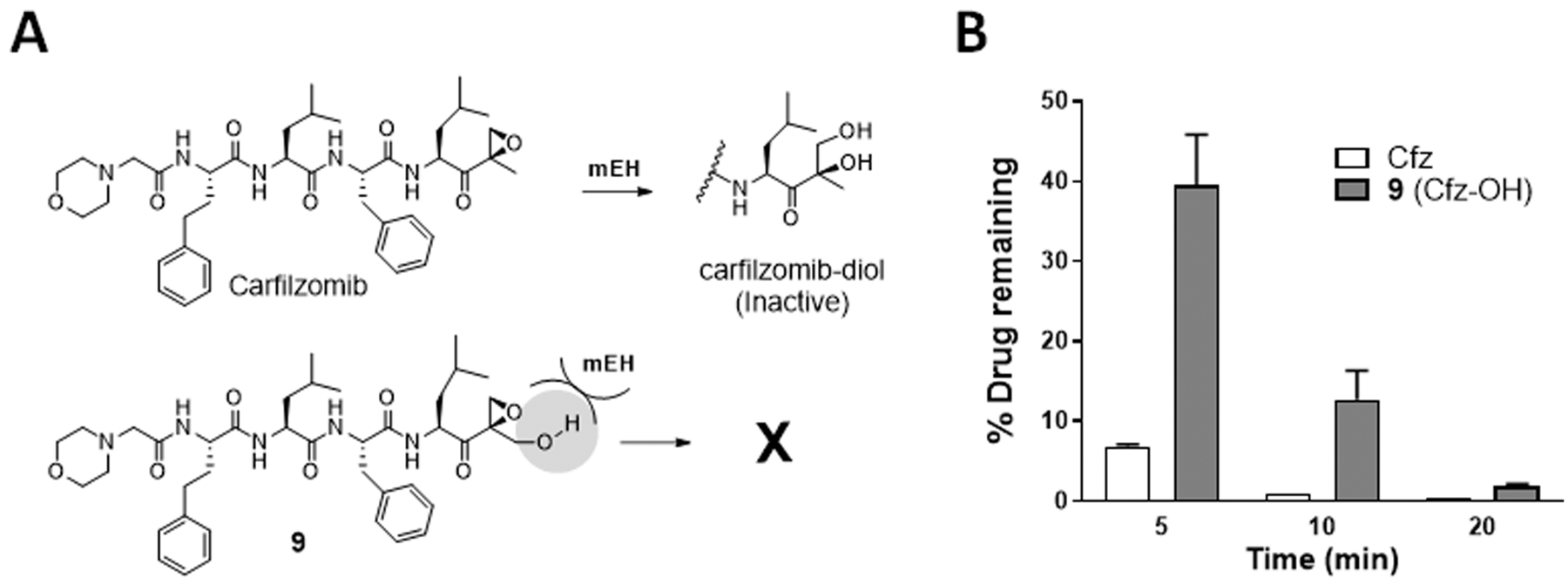
(A) Schematic depicting the rapid metabolism of Cfz by microsomal epoxide hydrolase (mEH) to the inactive diol. (B) Quantification of the remaining levels of Cfz or 9 following the incubation with rat liver homogenate containing active mEH and peptidase activities for 5, 10, and 20 min, respectively. Data presented as mean ± SD.
DISCUSSION
In expanding the clinical utility of existing PI drugs, cancer resistance, either de novo or acquired, remains a major obstacle. Currently, MM patients who do not respond to PI therapy or relapse after prolonged PI have very dismal outcomes with median survival of less than one year. To date, several mechanisms of PI resistance have been reported, including mutations/amplification of proteasome catalytic subunits, dysregulations of unfolded protein response and autophagy, overexpression of MARCKS proteins or efflux transporters, increased formation of aggresomes, and others.38–41 However, clinically relevant mechanisms of PI resistance remain to be uncovered, making it difficult to achieve an eventual goal of developing therapeutic options for patients refractory to current PI drugs.
Several reports indicate that the proteasome remains indispensable to the survival of cancer cells despite their resistance to PI drugs. The proteasome plays fundamental roles in various cellular functions and to date there appears no pathway/machinery that can fully compensate for the loss of proteasome function. Several reports showed that Btz-resistant MM cells remain susceptible to the disruption of proteasome pathways.22,42–44 For instance, an inhibitor targeting the noncatalytic α7 proteasome subunit was effective against bortezomib-resistant THP-1 cells harboring a mutated PSMB5 gene encoding the catalytic β5 subunit.45 These in vitro results require further validation in clinical settings, but they support that proteasome inhibition may offer therapeutic gains even in patients with MM relapsed/refractory to currently used PIs in clinic.
In this study, we used two established cancer cell lines with de novo or acquired resistance to Cfz and identified a novel PI compound with promising efficacy against both cell lines. Among the tested compounds, we found that epoxyketones with previously underexplored P1′ substituents can overcome both de novo and to a lesser degree, acquired Cfz resistance in cell line models. Compared to the boronic acid-based PIs, peptide epoxyketones feature a much improved selectivity in their interactions with the proteasome by forming an 1,4-oxazepane adduct with the N-terminal catalytic threonine residue of the proteasome.46 Recent extensive reviews of the literatures that describe the efficacy and toxicity profiles of carfilzomib-based regimens in clinic further supports the safety and tolerability of the peptide epoxyketone family.47,48 Peptide epoxyketones with an improved metabolic stability and an ability to bypass resistance mechanisms may offer a more promising route for further drug development efforts. Future studies will address the in vivo efficacy and metabolic stability of epoxyketones with previously underexplored P1′ substituents that can overcome both de novo and acquired Cfz resistance.
CONCLUSION
By exploring the chemical space at the P1′ position of peptide epoxyketones, we identified novel PIs effective against cell line models of de novo and acquired resistance to Cfz as well as primary MM cells. Our lead compound closely mimics the FDA-approved Cfz except for the addition of a hydroxyl group at the P1′ position. Our lead peptide epoxyketone with an S1′-occupying component displayed an improved metabolic stability over Cfz. This could provide potentially important advantages in expanding the utility of PIs to the many solid cancers in need of improved therapeutics. Further investigations regarding the contribution of P1′ substituents to metabolic stability and activity against PI-resistant patient samples are ongoing.
EXPERIMENTAL SECTION
General.
All chemical reagents starting materials and solvents were purchased from commercial vendors and used without further purification. TLC was performed on precoated plates and spots were detected by using UV or staining solution of phosphomolybdic acid hydrate (PMA) or iodine. Silica gel column chromatography was performed on 60 Å silica gel, 230–400 mesh (Merck). Proton and carbon NMR spectra were recorded on a Varian spectrometer at 400 MHz. Chemical shifts were reported in ppm units with tetramethylsilane as a reference standard. Multiplicities of NMR signals were reported using different abbreviations like singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). Chemical shifts were reported in parts per million (ppm) and coupling constant (J) is reported in Hz. Mass spectra were recorded on an Agilent Technologies 6120 Quadrupole LC/MS. The mobile phases used were A, H2O containing 0.1% formic acid and B, acetonitrile (CH3CN) containing 0.1% formic acid. The linear gradient of B/A from 5:95 to 90:10 over 18 min and then 100:0 over 7 min was used in an Agilent Eclipse XBD-C18 column (5-μm particle size), 4.6 mm diameter ×150 mm with a flow rate of 0.4 mL/min. The purity of all final compound used in bioassay was determined in an Agilent Technologies 1200 series equipped with a UV detector set at 254 nm using same column, gradient and flow rate to be ≥95%. Melting points were determined on Thermo Scientific 1001D apparatus and were uncorrected.
Synthesis. General Procedure for Deprotection of Benzyl Group.
Benzyl ester was dissolved in methanol and 5 mol % Pd/C was added. The solution was stirred under hydrogen gas environment for 30 min. Deprotected carboxylic acid was filtered through Celite and solvent was concentrated. Deprotected carboxylic acid was used in the next step without further purification.
General Procedure for Deprotection of Boc Group.
To the solution of Boc-protected amine in dichloromethane (DCM), excess of TFA was added and the reaction solution was stirred at room temperature for 1 h. Solvent was evaporated under reduced pressure and the crude TFA salt of amine was used in next step without further purification.
(S)-tert-Butyl 2-((tert-butyldiphenylsilyloxy)methyl)-6-methyl-3-oxohept-1-en-4-ylcarbamate (1a).
A solution of n-BuLi (25.46 mL, 40.76 mmol) in hexane was added dropwise to the stirred solution of dimethyl methylphosphonate (4.43 mL, 40.76 mmol) in THF at −78 °C. The reaction mixture was stirred at the same temperature for 2 h. A solution of N-(tert-butoxycarbonyl)-L-leucine methyl ester (2.5 g, 10.19 mmol) in THF was then added to the reaction mixture, and the reaction solution was further stirred for 3 h at same temperature. Reaction temperature was gradually increased up to room temperature, and the reaction was quenched with the addition of water. The product was extracted with ethyl acetate three times, and the collected organic layer was dried over sodium sulfate. It was then filtered, concentrated under reduced pressure, and purified through flash column chromatography using 50% ethyl acetate in hexane to yield pure product ((S)-tert-butyl 1-(dimethoxyphosphor-yl)-5-methyl-2-oxohexan-3-ylcarbamate) as a colorless sticky oil (1.88 g, 55% yield). 1H NMR (400 MHz, CDCl3) δ 5.18 (d, J = 8.6 Hz, 1H), 4.40–4.26 (m, 1H), 3.79 (d, J = 4.5 Hz, 3H), 3.76 (d, J = 4.4 Hz, 3H), 3.32 (dd, J = 14.2, 22.6 Hz, 1H), 3.08 (dd, J = 14.2, 22.0 Hz, 1H), 1.76–1.61 (m, 2H), 1.43 (s, 10H), 0.99–0.86 (m, 6H).
To the stirred solution of (S)-tert-butyl 1-(dimethoxyphosphoryl)-5-methyl-2-oxohexan-3-ylcarbamate (1.88 g, 5.57 mmol) and form-aldehyde (1.35 mL, 16.71 mmol) in 30 mL of water, aq. solution of potassium carbonate (0.92 g, 6.68 mmol) was added dropwise at room temperature. Reaction mixture was further stirred for 4 h at the same temperature. The crude product was extracted with ethyl acetate three times, and the collected organic layer was dried over sodium sulfate. It was then filtered, concentrated under reduced pressure, and purified by flash column chromatography using 30% ethyl acetate in hexane to yield pure product ((S)-tert-butyl 2-(hydroxymethyl)-6-methyl-3-oxohept-1-en-4-ylcarbamate) as colorless oil (0.89 g, 59% yield). 1H NMR (400 MHz, CDCl3) δ 6.22 (s, 1H), 6.17–6.06 (m, 1H), 5.03 (d, J = 12.6 Hz, 2H), 4.34 (t, J = 10.2 Hz, 2H), 1.73 (s, 1H), 1.42 (d, J = 3.4 Hz, 12H), 0.99 (d, J = 6.3 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H).
To the stirred solution of (S)-tert-butyl 2-(hydroxymethyl)-6-methyl-3-oxohept-1-en-4-ylcarbamate (0.9 g, 3.30 mmol) and imidazole (0.67 g, 9.92 mmol) in DCM at room temperature, TBDPS-Cl (2.53 mL, 9.92 mmol) was added dropwise, and the reaction solution was stirred at same temperature for 30 min. Solvent was removed under reduced pressure, and residue was purified by flash column chromatography using 5% ethyl acetate in hexane to yield pure product (1a) as colorless thick oil (1.2 g, 71% yield). 1H NMR (400 MHz, CDCl3) δ 7.66–7.58 (m, 4H), 7.44–7.31 (m, 6H), 6.33 (s, 1H), 6.24 (s, 1H), 5.10–4.93 (m, 2H), 4.50–4.27 (m, 2H), 1.68 (t, J = 7.6 Hz, 1H), 1.24 (ddd, J = 4.5, 10.0, 14.0 Hz, 2H), 1.05 (d, J = 2.2 Hz, 9H), 0.97 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H).
(S)-tert-Butyl 2-(methoxymethyl)-6-methyl-3-oxohept-1-en-4-yl-carbamate (1b).
To the solution of (S)-tert-butyl 2-(hydroxymethyl)-6-methyl-3-oxohept-1-en-4-ylcarbamate (100 mg, 0.36 mmol) and iodomethane (0.23 μL, 3.68 mmol) in acetonitrile, silver oxide (427 mg, 1.84 mmol) was added, and the reaction mixture was stirred at room temperature for 24 h. The solvent was removed under reduced pressure and residue was purified by flash column chromatography using 10% ethyl acetate in hexane. Pure product was collected as colorless sticky solid (54 mg, 51% yield). 1H NMR (400 MHz, CDCl3) δ 6.23 (s, 1H), 6.09 (s, 1H), 5.09 (d, J = 9.0 Hz, 1H), 5.00 (td, J = 3.5, 9.5 Hz, 1H), 4.09 (t, J = 1.5 Hz, 2H), 3.36 (s, 3H), 1.80–1.62 (m, 1H), 1.39 (m, 11H), 0.97 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H).
tert-Butyl (S)-1-((S)-2-((tert-butyldiphenylsilyloxy)methyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-ylcarbamate (2a) (General Procedure for Epoxidation).
The mixture of 1a (1.2 g, 2.35 mmol), benzonitrile (2.4 mL, 23.5 mmol) and hydrogen peroxide (3.99 mL, 58.75 mmol) in methanol was stirred at 0 °C, and DIEA (4.10 mL, 23.5 mmol) was added dropwise. The reaction mass was stirred at the same temperature for 3 h. Methanol was removed under reduced pressure, and the reaction mixture was extracted with ethyl acetate three times. The collected organic layer was dried under sodium sulfate, filtered, and concentrated. Crude product was purified by flash column chromatography using 10% ethyl acetate in hexane. Pure required product was collected as colorless sticky solid (500 mg, 41% yield). 1H NMR (400 MHz, CDCl3) δ 7.68 (m, 4H), 7.46–7.36 (m, 6H), 5.08 (dd, J = 7.2, 23.4 Hz, 2H), 4.47 (d,J = 10.5 Hz, 1H), 3.49 (d, J = 11.5 Hz, 1H), 3.17 (d, J = 4.9 Hz, 1H), 2.98 (d, J = 4.9 Hz, 1H), 1.23–1.11 (m, 2H), 1.00 (d, J = 1.9 Hz, 10H), 0.93 (d, J = 5.6 Hz, 6H).
tert-Butyl (S)-1-((S)-2-(methoxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-ylcarbamate (2b).
Compound 2b was prepared using same general procedure for epoxidation as 2a from (S)-tert-butyl 2-(methoxymethyl)-6-methyl-3-oxohept-1-en-4-ylcarbamate (1b) to yield colorless sticky solid (20 mg, 35% yield). 1H NMR (400 MHz, CDCl3) δ 4.89 (dd, J = 7.2, 23.4 Hz, 2H), 4.53 (d, J = 10.5 Hz, 1H), 3.51 (d, J = 11.5 Hz, 1H), 3.15 (d, J = 4.9 Hz, 1H), 2.98 (d, J = 4.9 Hz, 1H), 1.85–1.63 (m, 1H), 1.39–1.28 (m, 11H), 0.99 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H).
(S)-Benzyl 2-heptanamidopropanoate (3) (General Procedure for Amide Coupling).
To the stirred solution of alanine benzyl ester hydrochloride (1 g, 2.84 mmol) and heptanoic acid (0.44 mL, 3.13 mmol) in DCM, HBTU (2.15 g, 5.68 mmol), HOBt (0.77 g, 5.68 mmol), and DIEA (2.48 mL, 14.2 mmol) were added at room temperature, and the resulting reaction mixture was stirred at the same temperature overnight. Solvent was removed under reduced pressure, and residue was purified by flash column chromatography using 30% ethyl acetate in hexane. Pure product was collected as white sticky solid (0.68 g, 82%). 1H NMR (400 MHz, CDCl3) δ 7.63–7.31 (m, 5H), 5.95 (s, 1H), 5.16 (t, J = 4.6 Hz, 2H), 4.72–4.56 (m, 1H), 2.18 (td, J = 2.8, 8.0 Hz, 2H), 1.59 (d, J = 9.0 Hz, 2H), 1.46–1.12 (m, 9H), 0.86 (t, J = 8.6 Hz, 3H).
N-((S)-1-((S)-1-((S)-2-((tert-Butyldiphenylsilyloxy)methyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-ylamino)-1-oxopropan-2-yl)-heptanamide (4).
Benzyl ester was deprotected from compound 3 using general procedure for benzyl deprotection reaction and Boc group was deprotected from intermediate 2a using general procedure for Boc deprotection reaction. Thus, obtained free carboxylic acid and amines were coupled using general procedure for amide coupling reaction to yield intermediate 4 as colorless sticky solid (91%). 1H NMR (400 MHz, CDCl3) δ 7.74–7.59 (m, 4H), 7.38 (dt, J = 6.7, 8.9 Hz, 6H), 6.71 (d, J = 7.5 Hz, 1H), 6.09 (d, J = 7.5 Hz, 1H), 4.65–4.52 (m, 2H), 4.48 (d, J = 11.4 Hz, 1H), 3.47 (d, J = 11.5 Hz, 1H), 3.20 (d, J = 5.1 Hz, 1H), 2.97 (d, J = 5.1 Hz, 1H), 2.25–2.07 (m, 2H), 1.82–1.50 (m, 4H), 1.42–1.16 (m, 10H), 0.99 (s, 9H), 0.95 (d, J = 6.3 Hz, 3H), 0.91 (d, J = 6.3 Hz, 3H), 0.89–0.81 (m, 3H).
N-((S)-1-((S)-1-((R)-2-(Hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-ylamino)-1-oxopropan-2-yl)heptanamide (5) (General Procedure for TBDPS Deprotection).
To the solution of compound 4 (71 mg, 0.12 mmmol) in DCM, TBAF (0.25 mL, 0.25 mmol) was added at room temperature, and the reaction mass was stirred at the same temperature for 30 min. Solvent was removed under reduced pressure, and the resulting residue was purified by flash column chromatography using 5% methanol in DCM to yield pure product as colorless sticky solid (30 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ 6.97 (d, J = 7.4 Hz, 1H), 6.16 (d, J = 7.6 Hz, 1H), 4.65–4.44 (m, 2H), 4.19 (dd, J = 4.3, 12.7 Hz, 1H), 3.70 (dd, J = 4.7, 12.7 Hz, 1H), 3.31 (d, J = 5.0 Hz, 1H), 3.06 (d, J = 5.0 Hz, 1H), 2.54 (d, J = 6.6 Hz, 1H), 2.15 (dt, J = 4.3, 8.8 Hz, 2H), 1.96 (d, J = 4.6 Hz, 1H), 1.72–1.49 (m, 4H), 1.41–1.15 (m, 9H), 0.91 (t, J = 7.6 Hz, 5H), 0.87–0.80 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 207.72, 173.14, 172.51, 62.02, 61.42, 51.01, 49.31, 48.28, 39.07, 36.56, 31.46, 28.86, 25.55, 25.13, 23.30, 22.43, 21.09, 17.97, 13.98; LCMS (ES+) m/z calcd for C19H35N2O5 [M + H]+ 371.2, found: 371.2.
(S)-Benzyl 2-((S)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido) pentan-emido)-3-phenylpropanoate (6).
Intermediate 6 was prepared in three steps starting from Boc-Leu-OH. Dipeptide (S)-benzyl 2-((S)-2-(tert-butoxycarbonylamino)-4-methyl-pentanamido)-3-phenylpropanoate was prepared using the general procedure for amide coupling reaction from Boc-Leu-OH and phenylalanine benzyl ester hydrochloride as white solid (81%). 1H NMR (400 MHz, CDCl3) δ 7.34 (m, 3H), 7.30–7.22 (m, 2H), 7.19 (m, 3H), 7.00 (t, J = 4.3 Hz, 2H), 6.45 (d, J = 7.8 Hz, 1H), 5.19–5.05 (m, 2H), 4.91–4.83 (m, 1H), 4.77 (s, 1H), 3.10 (t, J = 6.7 Hz, 2H), 1.67–1.51 (m, 3H), 1.41 (s, 9H), 0.87 (dt, J = 4.0, 2.1 Hz, 6H).
The Boc deprotected above prepared dipeptide was further coupled with Boc-L-homophenylalanine using the general procedure for amide coupling reaction to prepare tripeptide (S)-benzyl 2-((S)-2-((S)-2-(tert-butoxycarbonylamino)-4-phenylbutanamido)-4-methylpentana-mido)-3-phenylpropanoate as white solid (79%). White solid (79%). 1H NMR (400 MHz, CDCl3) δ 7.33 (dd, J = 4.8, 1.8 Hz, 2H), 7.29–7.22 (m, 5H), 7.21–7.12 (m, 5H), 7.01–6.94 (m, 2H), 6.43 (d, J = 7.5 Hz, 1H), 6.37 (d, J = 8.0 Hz, 1H), 5.20–5.00 (m, 2H), 4.97–4.77 (m, 2H), 4.36 (td, J = 8.7, 4.9 Hz, 1H), 3.99 (d, J = 7.4 Hz, 1H), 3.07 (q, J = 7.9, 7.1 Hz, 2H), 2.62 (t, J = 7.9 Hz, 2H), 2.06 (dt, J = 14.2, 7.0 Hz, 1H), 1.85 (dd, J = 14.1, 7.6 Hz, 1H), 1.57 (s, 3H), 1.42 (s, 9H), 0.84 (t, J = 5.9 Hz, 6H).
Compound 6 was prepared from Boc deprotected tripeptide (S)-benzyl 2-((S)-2-((S)-2-(tert-butoxycarbonylamino)-4-phenylbutana-mido)-4-methylpentanamido)-3-phenylpropanoate and morpholin-4-yl-acetic acid using the general procedure for amide coupling reaction. White solid (65%). 1H NMR (400 MHz, CDCl3) δ 7.95 (s, 2H), 7.56 (d, J = 8.4 Hz, 1H), 7.34–7.27 (m, 2H), 7.26–7.18 (m, 3H), 7.17–7.08 (m, 5H), 6.97 (dd, J = 7.2, 2.3 Hz, 2H), 6.84 (d, J = 7.8 Hz, 1H), 6.66 (d, J = 7.7 Hz, 1H), 5.14–4.98 (m, 2H), 4.83 (dt, J = 7.6, 6.0 Hz, 1H), 4.44 (td, J = 8.1, 5.8 Hz, 1H), 4.35 (ddd, J = 9.0, 7.8, 5.3 Hz, 1H), 3.74–3.63 (m, 4H), 3.12–2.97 (m, 2H), 2.94 (d, J = 5.9 Hz, 2H), 2.57 (dq, J = 8.8, 3.1 Hz, 2H), 2.46 (q, J = 4.5 Hz, 4H), 2.17–2.03 (m, 1H), 1.97–1.84 (m, 1H), 1.62–1.39 (m, 3H), 0.81 (dd, J = 9.5, 6.1 Hz, 6H).
(S)-N-((S)-1-((S)-1-((S)-2-((tert-Butyldiphenylsilyloxy)methyl)-oxiran-2-yl)-4-methyl-1-oxopentan-2-ylamino)-1-oxo-3-phenyl-propan-2-yl)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)pentanamide (7).
Compound 7 was prepared from benzyl deprotected 6 and Boc deprotected 2a using the general procedure for amide coupling reaction. White solid (45%); melting point, 87–91 °C; 1H NMR (400 MHz, CD3OD) δ 7.69–7.66 (m, 4H), 7.47–7.34 (m, 6H), 7.29–7.09 (m, 9H), 7.09–6.98 (m, 1H), 4.69–4.50 (m, 3H), 4.41–4.33 (m, 2H), 3.69 (m, 4H), 3.35 (d, J = 11.4 Hz, 1H), 3.18–3.04 (m, 2H), 3.04–2.97 (m, 2H), 2.96–2.83 (m, 2H), 2.62–2.42 (m, 6H), 2.01–1.86 (m, 2H), 1.72–1.72 (m, 2H), 1.57–1.44 (m, 3H), 1.34 (t, J = 10.5 Hz, 1H), 0.99 (d, J = 0.7 Hz, 9H), 0.94–0.78 (m, 12H). 13C NMR (101 MHz, CD3OD) δ 135.31, 135.24, 129.58, 129.57, 128.95, 128.08, 128.05, 127.91, 127.43, 127.41, 126.23, 125.66, 66.45, 63.45, 61.02, 53.88, 53.31, 52.55, 51.07, 48.20, 47.99, 47.84, 47.77, 47.56, 47.35, 47.13, 46.92, 40.47, 38.21, 37.40, 33.99, 31.66, 25.74, 24.88, 24.39, 22.49, 21.97, 20.56, 20.12; LCMS (ES+) m/z calcd for C56H76N5O8Si [M + H]+ 974.5, found: 974.5. Purity: ≥ 98% and retention time is 14.21 min by HPLC analysis.
(S)-N-((S)-1-((S)-1-((S)-2-(Methoxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-ylamino)-1-oxo-3-phenylpropan-2-yl)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)-pentanamide (8).
Compound 8 was prepared from benzyl deprotected 6 and Boc deprotected 2b using the general procedure for amide coupling reaction. White solid (48%); melting point, 129–133 °C; 1H NMR (400 MHz, CD3OD) δ 7.28–7.22 (m, 2H), 7.22–7.08 (m, 7H), 7.08–7.00 (m, 1H), 4.66–4.57 (m, 1H), 4.51 (dd, J = 2.8, 10.9 Hz, 1H), 4.37 (ddd, J = 5.8, 9.0, 22.4 Hz, 2H), 4.19 (ddd, J = 0.7, 11.3, 16.9 Hz, 1H), 3.70 (td, J = 2.1, 4.8, 5.3 Hz, 4H), 3.29 (ddd, J = 1.3, 2.2, 4.1 Hz, 3H), 3.26–3.11 (m, 2H), 3.09 (dt, J = 1.3, 5.3 Hz, 1H), 3.04–2.99 (m, 2H), 2.99–2.93 (m, 1H), 2.93–2.77 (m, 2H), 2.64–2.44 (m, 6H), 2.08–1.82 (m, 2H), 1.73–1.41 (m, 5H), 1.29 (ddd, J = 4.5, 10.8, 14.3 Hz, 1H), 0.95–0.70 (m, 12H). 13C NMR (101 MHz, CD3OD) δ 205.96, 172.59, 172.24, 171.83, 170.93, 141.03, 136.75, 128.93, 128.08, 128.05, 127.91, 126.24, 125.67, 71.00, 66.45, 61.28, 61.02, 58.46, 53.84, 53.32, 52.54, 51.76, 50.74, 48.20, 47.99, 47.78, 47.56, 47.35, 47.14, 46.93, 40.46, 37.89, 37.35, 34.02, 31.65, 24.76, 24.37, 22.37, 21.96, 20.56, 20.05.; LCMS (ES+) m/z calcd for C41H60N5O8 [M + H]+ 750.4, found: 750.4. Purity: ≥ 96% and retention time is 10.14 min by HPLC analysis.
(S)-N-((S)-1-((S)-1-((R)-2-(Hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-ylamino)-1-oxo-3-phenylpropan-2-yl)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)-pentanamide (9).
Compound 9 was prepared from compound 7 using the general procedure for TBDPS deprotection reaction. White solid (64%); melting point, 118–122 °C; 1H NMR (400 MHz, CD3OD) δ 7.30–7.21 (m, 2H), 7.21–7.09 (m, 7H), 7.09–7.01 (m, 1H), 4.61 (dd, J = 5.4, 8.7 Hz, 1H), 4.52 (dd, J = 2.9, 11.0 Hz, 1H), 4.36 (ddd, J = 5.6, 8.8, 19.5 Hz, 2H), 4.26 (d, J = 12.6 Hz, 1H), 3.79–3.66 (m, 4H), 3.45 (d, J = 12.6 Hz, 1H), 3.14–3.05 (m, 2H), 3.00 (dd, J = 5.0, 10.1 Hz, 3H), 2.86 (dd, J = 8.7, 14.0 Hz, 1H), 2.63–2.42 (m, 6H), 2.06–1.83 (m, 2H), 1.72–1.42 (m, 5H), 1.29 (ddd, J = 3.9, 10.9, 14.5 Hz, 1H), 0.97–0.77 (m, 12H). 13C NMR (101 MHz, CD3OD) δ 206.57, 172.59, 172.26, 171.80, 170.95, 141.03, 136.74, 128.92, 128.07, 128.05, 127.90, 126.23, 125.66, 66.45, 62.40, 61.02, 60.28, 53.85, 53.31, 52.55, 51.76, 50.74, 48.19, 48.05, 47.98, 47.84, 47.76, 47.55, 47.34, 47.12, 46.91, 40.43, 37.82, 37.35, 33.98, 31.64, 24.76, 24.37, 22.36, 21.94, 20.53, 20.02; LCMS (ES+) m/z calcd for C H + 40 58N5O8 [M + H] 736.4, found: 736.4. Purity: ≥ 95% and retention time is 13.18 min by HPLC analysis.
(S)-N-((S)-1-((S)-1-((S)-2-((Methoxymethoxy)methyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-ylamino)-1-oxo-3-phenylpropan-2-yl)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)-pentanamide (10).
To the solution of compound 9 (25 mg, 0.033 mmol) in DCM, MOM-Cl (13 μL, 0.165 mmol) and DIEA (29 μL, 0.165 mmol) were added, and the reaction mixture was stirred at room temperature for 24 h. Solvent was removed under reduced pressure and residue was purified by flash column chromatography using 5% methanol in DCM to yield 16 mg (60%) white solid as pure compound. Melting point, 107–110 °C; 1H NMR (400 MHz, CD3OD) δ 7.29–7.20 (m, 2H), 7.20–7.09 (m, 7H), 7.09–6.99 (m, 1H), 4.64–4.58 (m, 1H), 4.58–4.55 (m, 2H), 4.51 (dd, J = 2.8, 10.9 Hz, 1H), 4.42–4.31 (m, 3H), 3.70 (td, J = 2.1, 4.4 Hz, 4H), 3.32 (d, J = 11.4 Hz, 1H), 3.29 (p, J = 1.6 Hz, 3H), 3.11 (dd, J = 5.2, 14.3 Hz, 2H), 3.00 (dd, J = 5.3, 7.7 Hz, 3H), 2.91–2.76 (m, 2H), 2.65–2.42 (m, 6H), 2.10–1.80 (m, 2H), 1.61–1.39 (m, 4H), 1.29 (ddd, J = 4.5, 10.9, 13.8 Hz, 2H), 0.97–0.75 (m, 12H). 13C NMR (101 MHz, CD3OD) δ 209.84, 176.54, 176.20, 175.78, 174.88, 144.97, 140.69, 132.87, 132.02, 131.99, 131.85, 130.18, 129.60, 100.10, 70.38, 69.96, 65.09, 64.95, 58.11, 57.77, 57.25, 56.48, 55.69, 54.64, 52.14, 51.97, 51.92, 51.78, 51.71, 51.50, 51.28, 51.07, 50.86, 44.38, 41.79, 41.29, 37.95, 35.59, 28.68, 28.31, 26.32, 25.90, 24.48, 23.98; LCMS (ES+) m/z calcd for C42H62N5O9 [M + H]+ 780.4, found: 780.4. Purity: ≥ 97% and retention time is 14.60 min by HPLC analysis.
(S)-N-((S)-1-((S)-1-((S)-2-(((2-Methoxyethoxy)methoxy)methyl)-oxiran-2-yl)-4-methyl-1-oxopentan-2-ylamino)-1-oxo-3-phenyl-propan-2-yl)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)pentanamide (11).
To the solution of compound 9 (25 mg, 0.033 mmol) in DCM, MEM-Cl (19 μL, 0.165 mmol) and DIEA (29 μL, 0.165 mmol) were added, and the reaction mass was stirred at room temperature for 24 h. Solvent was removed under reduced pressure and residue was purified by flash column chromatography using 5% methanol in DCM to yield 18 mg (64%) white solid as pure compound. Melting point, 85–88 °C; 1H NMR (400 MHz, CD3OD) δ 7.28–7.20 (m, 2H), 7.20–7.10 (m, 7H), 7.08–7.00 (m, 1H), 4.68–4.59 (m, 3H), 4.51 (dd, J = 2.8, 10.9 Hz, 1H), 4.42–4.30 (m, 3H), 3.70 (td, J = 2.1, 4.7 Hz, 4H), 3.66–3.57 (m, 2H), 3.57–3.49 (m, 2H), 3.37 (dd, J = 0.7, 11.4 Hz, 1H), 3.33 (dd, J = 0.8, 3.7 Hz, 1H), 3.29 (ddt, J = 1.3, 2.3, 3.3 Hz, 2H), 3.17–3.05 (m, 2H), 3.05–2.95 (m, 3H), 2.86 (dd, J = 8.7, 14.0 Hz, 1H), 2.64–2.43 (m, 6H), 2.06–1.94 (m, 1H), 1.94–1.83 (m, 1H), 1.72–1.63 (m, 1H), 1.60–1.42 (m, 4H), 1.34–1.23 (m, 2H), 0.95–0.77 (m, 12H). 13C NMR (101 CD3OD) δ 205.91, 172.59, 172.24, 171.82, 170.93, 141.04, 136.74, 128.93, 128.08, 128.06, 127.91, 126.24, 125.67, 95.20, 71.44, 66.52, 66.45, 66.07, 61.13, 61.03, 57.62, 53.83, 53.32, 52.54, 51.76, 50.69, 40.46, 37.91, 37.36, 34.02, 31.66, 24.75, 24.38, 22.38, 21.97, 20.56, 20.08; LCMS (ES+) m/z calcd for C44H66N5O10 [M + H]+ 824.4, found: 824.4. Purity: ≥ 95% and retention time is 12.66 min by HPLC analysis.
((S)-2-((4S,7S,10S,13S)-10-Benzyl-7-isobutyl-15-methyl-1-morpholino-2, 5, 8, 11-tetraoxo-4-phenethyl-3, 6, 9, 12-tetraazahexadecanecarbonyl)oxiran-2-yl)methylmethane-sulfonate (12).
A mixture of compound 9 (25 mg, 0.033 mmol), methanesulfonyl chloride (13 μL, 0.165 mmol), and DIEA (29 μL, 0.165 mmol) in DCM was stirred at room temperature for 24 h. Solvent was removed under reduced pressure and residue was purified by flash column chromatography using 5% methanol in DCM to yield 20 mg (64%) pure compound as white solid. Melting point, 87–90 °C; 1H NMR (400 MHz, CD3OD) δ 7.25 (dd, J = 6.6, 8.5 Hz, 2H), 7.21–7.11 (m, 7H), 7.11–7.01 (m, 1H), 5.04 (d, J = 11.4 Hz, 1H), 4.61 (dd, J = 5.4, 8.8 Hz, 1H), 4.47 (dd, J = 3.0, 10.8 Hz, 1H), 4.36 (ddd, J = 5.5, 8.8, 21.9 Hz, 2H), 4.03 (d, J = 11.6 Hz, 1H), 3.70 (td, J = 2.1, 4.5 Hz, 4H), 3.15–3.05 (m, 5H), 3.02 (d, J = 4.9 Hz, 2H), 2.86 (dd, J = 8.8, 14.0 Hz, 1H), 2.65–2.45 (m, 6H), 2.06–1.95 (m, 1H), 1.95–1.84 (m, 1H), 1.68 (dd, J = 3.6, 6.7 Hz, 1H), 1.62–1.40 (m, 4H), 1.39–1.22 (m, 1H), 0.96–0.81 (m, 12H). 13C NMR (101 MHz, CD3OD) δ 208.65, 176.56, 176.24, 175.85, 174.88, 144.97, 140.64, 132.86, 132.02, 132.01, 131.87, 130.21, 129.62, 72.69, 70.38, 64.94, 63.58, 57.75, 57.25, 56.52, 55.72, 54.25, 52.77, 52.14, 51.93, 51.71, 51.50, 51.29, 51.07, 50.86, 44.34, 41.77, 41.25, 39.76, 37.94, 35.60, 28.66, 28.31, 26.24, 25.90, 24.47, 23.97; LCMS (ES+) m/z calcd for C41H60N5O10S [M + H]+ 814.4, found: 814.4. Purity: ≥ 96% and retention time is 14.24 min by HPLC analysis.
Biological Assay.
Enzyme Kinetics Assays.
Purified human 20S immunoproteasome (Boston Biochem) or RPMI 8226 cell lysates were diluted in 20S proteasome assay buffer (20 mM Tris-HCl, 0.5 mM EDTA, 0.035% SDS, pH 8.0) and incubated with various concentrations of each inhibitor for 1 h in a 96-well plate. The fluorogenic substrates Suc-Leu-Leu-Val-Tyr-AMC (Bachem) or Ac-Pro-Ala-Leu-AMC (Boston Biochem) were used at the final substrate concentration of 100 μM to measure the remaining levels of chymotrypsin-like activity or LMP2-specific catalytic activity, respectively. Fluorescence signals from the release of free AMC (7-amino-4-methylcoumarin) were monitored every minute for 1 h via a SpectraMax M5 microplate reader (Molecular Devices) using excitation and emission wavelengths of 360 and 460 nm, respectively. The initial hydrolysis rates (slopes) for individual wells were calculated via linear regression and normalized to the values from vehicle-treated control wells. Nonlinear regression analysis was performed using GraphPad Prism 7 to calculate an IC50 value for each compound in inhibiting proteasomal CT-L or LMP2 activity.
Cell Culture.
Human cancer cell lines H23, H727, and RPMI8226 were obtained from the ATCC (American Type Culture Collection) and maintained in the ATCC recommended media, RPMI1640 supplemented with 10% fetal bovine serum (Gibco, and Atlanta Biologicals). RPMI8226 cells with acquired resistance to Cfz were established by adapting them in the presence of stepwise increasing concentrations of Cfz up to 80 nM over a period of approximately 6 months. Cfz-resistant cells were maintained in 80 nM Cfz and then grown in the absence of Cfz for approximately 1 week prior to the experiments.
Isolation of Primary MM Cells.
Bone marrow (BM) aspirates were obtained from patients after approval by the UC Cancer Institute Institutional Review Board followed by positive selection with miltenyi CD138 microbeads developed for the isolation of plasma cells. Immunophenotyping by flow cytometry was performed to confirm the purity and quantity of selected CD138+ plasma cells.
Cell Viability Assay. Established Cancer Cell Lines.
H23 cells or H727 cells were plated at 5000 or 10 000 cells per well, respectively. RPMI8226 cells and Cfz-resistant RPMI8226 sublines growing in suspension were plated at 10 000 cells per well. Twenty-four hours after plating, media containing the test compounds were added to each well to deliver the intended final concentration. After 72 h, cell viability was determined using the assay protocol recommended by the manufacturer (CellTiter 96 Aqueous One Solution Cell Proliferation assay, Promega). The resulting signals were measured using a SpectraMax M5 microplate spectrophotometer (Molecular Devices). Nonlinear regression analysis was performed using GraphPad Prism 7 to calculate an IC50 value for each compound to incur cell death.
Primary MM Cells.
Purified primary MM cells were plated on 96-well plates at a density of 20 000 cells per well in IMDM media (Gibco) supplemented with 10% FBS. After cells were treated with test compounds for 48 h, the percentage of viable cells was determined using the CellTiter Glo Luminescent Cell Viability Assay (Promega) and a Veritas microplate luminometer (Promega).
Immunoblotting.
Cell lysates were prepared using ice-cold RIPA lysis buffer (50 mM Tris-Cl, 150 mM NaCl, 1% NP-40, 1% Triton X-100) supplemented with 1% protease inhibitor cocktail (Sigma-Aldrich). After centrifugation at 14 000g at 4 °C for 20 min, the resulting supernatant was collected and subject to the total protein assay using Protein Assay Dye Reagent Concentrate (Bio-Rad). Proteins were resolved by 7.5% SDS-PAGE and transferred onto PVDF membranes (Bio-Rad) via semidry transfer. After blocking in 5% nonfat dry milk, membranes were incubated with primary antibodies (anti-P-gp (Abcam) or anti-β-actin (Enzo)) at 4 °C overnight. Membranes were washed and incubated with appropriate peroxidase-conjugated secondary antibodies for 1 h at room temperature. Proteins were visualized on Kodak BioMax XAR Films (Sigma-Aldrich) using ECL.
In Vitro Metabolic Stability of Cfz-OH.
To assess whether Cfz-OH has indeed an improved metabolic stability over Cfz, we compared the rate by which Cfz-OH or Cfz disappears in the presence of rat liver homogenates, as previously reported.49 Briefly, the liver was obtained from male Sprague-Dawley rats (8 week-old, Nara Biotech Co. Ltd., Seoul, Korea) using the protocol approved by the Seoul National University Institutional Animal Care and Use Committee (approval No. SNU-160512-5-1). The harvested liver was washed with phosphate-buffered saline (PBS, pH 7.4) and homogenized using five-fold excess volume of PBS per gram of tissue. After preincubation at 37 °C, an aliquot of liver homogenates was spiked with the stock solution of Cfz or Cfz-OH to achieve the final concentration of 1 μM (total volume of 400 μL, n = 3). At the predesignated time (0, 5, 10, and 20 min), an aliquot (40 μL) was collected and mixed with four-fold excess volume of ice-cold acetonitrile containing chlorpropamide (an internal standard, IS, 0.5 μM). After vortexing and centrifugation, the drug levels in the resulting supernatant were analyzed via HPLC interfaced with mass spectrometry (LC-MS/MS).
The analytical conditions for Cfz were reported previously.50 Cfz-OH was quantified using the slightly modified analytical conditions via LC-MS/MS (1260 infinity HPLC system interfaced with 6430 Triple Quad LC-MS system, Agilent Technologies, Palo Alto, CA) in a positive ion mode. The chromatic separation was performed using a Poroshell 120 EC-C18 column (4.6 × 50 mm2, 2.7 μm, Agilent Technologies, Palo Alto, CA) and an isocratic mobile phase composed of acetonitrile and water (75:25, v/v) at a flow rate of 0.3 mL/min. The retention time of Cfz-OH was 1.7 min, and the gas temperature was set at 300 °C. The source-dependent parameters, the fragment voltage, collision energy, and cell accelerator voltage were set as follows: 170, 75, and 1 V for Cfz-OH and 90 V, 30 and 7 V for chlorpropamide (IS). Quantification was performed in the selected reaction monitoring (SRM) mode using the following transitions: m/z 736.2 > 99.9 for Cfz-OH and m/z 276.9 > 110.8 for chlorpropamide (IS). The calibration samples were prepared in the range of 1 to 200 nM and the signals showed linearity with the r2 value greater than 0.98. The data were processed using the MassHunter Workstation Software Quantitative Analysis (vB.05.00; Agilent Technologies).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank the National Institutes of Health (R01 CA188354 to K.B.K. and R01 GM111084 to S.L.), the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (No. 1520250 to W.L.), and Creative-Pioneering Researchers Program through Seoul National University (to W.L.) for financially supporting this work.
ABBREVIATIONS USED
- PI
proteasome inhibitor
- Btz
bortezomib
- Cfz
carfilzomib
- Ixz
ixazomib
- UPS
ubiquitin-proteasome system
- CT-L
chymotrypsin-like
- MM
multiple myeloma
- P-gp
P-glycoprotein transporter
- mAb
monoclonal antibody
- IFN-γ
interferon-gamma
- TBDMS
t-butyldimethylsilyl
- TBDPS
t-butyldiphenylsilyl
- TFA
trifluoroacetic acid
- mEH
microsomal epoxide hydrolase
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.8b01943.
Cell viability data of H727 and H23 cells for carfilzomib; effect of reversin-121 on carfilzomib sensitivity in RPMI 8226 with acquired carfilzomib resistance; 1H NMR, 13C NMR spectra; LC-MS profiles and molecular formula strings of 5 and 7–12 (PDF)
Molecular docking models in PDB format (PDF) Compound data (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Bergsagel PL Where we were, where we are, where we are going: progress in multiple myeloma. Am. Soc. Clin Oncol Educ Book 2014, 34, 199–203. [DOI] [PubMed] [Google Scholar]
- (2).Lonial S; Durie B; Palumbo A; San-Miguel J Monoclonal antibodies in the treatment of multiple myeloma: current status and future perspectives. Leukemia 2016, 30, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sanchez L; Wang Y; Siegel DS; Wang ML Daratumumab: a first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol 2016, 9, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).San-Miguel JF; Hungria VT; Yoon SS; Beksac M; Dimopoulos MA; Elghandour A; Jedrzejczak WW; Gunther A; Nakorn TN; Siritanaratkul N; Schlossman RL; Hou J; Moreau P; Lonial S; Lee JH; Einsele H; Sopala M; Bengoudifa BR; Binlich F; Richardson PG Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): a randomised, placebo-controlled, phase 3 trial. Lancet Haematol 2016, 3, e506–e515. [DOI] [PubMed] [Google Scholar]
- (5).Hanahan D; Weinberg RA Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- (6).Luo J; Solimini NL; Elledge SJ Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Anderson DJ; Le Moigne R; Djakovic S; Kumar B; Rice J; Wong S; Wang J; Yao B; Valle E; Kiss von Soly S; Madriaga A; Soriano F; Menon MK; Wu ZY; Kampmann M; Chen Y; Weissman JS; Aftab BT; Yakes FM; Shawver L; Zhou HJ; Wustrow D; Rolfe M Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 2015, 28, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tian Z; D’Arcy P; Wang X; Ray A; Tai YT; Hu Y; Carrasco RD; Richardson P; Linder S; Chauhan D; Anderson KC A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Selvaraju K; Mazurkiewicz M; Wang X; Gullbo J; Linder S; D’Arcy P Inhibition of proteasome deubiquitinase activity: a strategy to overcome resistance to conventional proteasome inhibitors? Drug Resist. Updates 2015, 21–22, 20–29. [DOI] [PubMed] [Google Scholar]
- (10).Chauhan D; Tian Z; Nicholson B; Kumar KG; Zhou B; Carrasco R; McDermott JL; Leach CA; Fulcinniti M; Kodrasov MP; Weinstock J; Kingsbury WD; Hideshima T; Shah PK; Minvielle S; Altun M; Kessler BM; Orlowski R; Richardson P; Munshi N; Anderson KC A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gu D; Wang S; Kuiatse I; Wang H; He J; Dai Y; Jones RJ; Bjorklund CC; Yang J; Grant S; Orlowski RZ Inhibition of the MDM2 E3 ligase induces apoptosis and autophagy in wild-type and mutant p53 models of multiple myeloma, and acts synergistically with ABT-737. PLoS One 2014, 9, No. e103015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Moreau P; Masszi T; Grzasko N; Bahlis NJ; Hansson M; Pour L; Sandhu I; Ganly P; Baker BW; Jackson SR; Stoppa AM; Simpson DR; Gimsing P; Palumbo A; Garderet L; Cavo M; Kumar S; Touzeau C; Buadi FK; Laubach JP; Berg DT; Lin J; Di Bacco A; Hui AM; van de Velde H; Richardson PG; Group T-MS Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med 2016, 374, 1621–1634. [DOI] [PubMed] [Google Scholar]
- (13).Gallerani E; Zucchetti M; Brunelli D; Marangon E; Noberasco C; Hess D; Delmonte A; Martinelli G; Bohm S; Driessen C; De Braud F; Marsoni S; Cereda R; Sala F; D’Incalci M; Sessa C A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur. J. Cancer 2013, 49, 290–296. [DOI] [PubMed] [Google Scholar]
- (14).Infante JR; Mendelson DS; Burris HA 3rd; Bendell JC; Tolcher AW; Gordon MS; Gillenwater HH; Arastu-Kapur S; Wong HL; Papadopoulos KP A first-in-human dose-escalation study of the oral proteasome inhibitor oprozomib in patients with advanced solid tumors. Invest. New Drugs 2016, 34, 216–224. [DOI] [PubMed] [Google Scholar]
- (15).Richardson PG; Zimmerman TM; Hofmeister CC; Talpaz M; Chanan-Khan AA; Kaufman JL; Laubach JP; Chauhan D; Jakubowiak AJ; Reich S; Trikha M; Anderson KC Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052–101 Part 1. Blood 2016, 127, 2693–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rajan AM; Kumar S New investigational drugs with single-agent activity in multiple myeloma. Blood Cancer J. 2016, 6, No. e451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vogl DT; Martin TG; Vij R; Hari P; Mikhael JR; Siegel D; Wu KL; Delforge M; Gasparetto C Phase I/II study of the novel proteasome inhibitor delanzomib (CEP-18770) for relapsed and refractory multiple myeloma. Leuk. Lymphoma 2017, 58, 1–11. [DOI] [PubMed] [Google Scholar]
- (18).Harshbarger W; Miller C; Diedrich C; Sacchettini J Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 2015, 23, 418–424. [DOI] [PubMed] [Google Scholar]
- (19).Adams GM; Crotchett B; Slaughter CA; DeMartino GN; Gogol EP Formation of proteasome-PA700 complexes directly correlates with activation of peptidase activity. Biochemistry 1998, 37, 12927–12932. [DOI] [PubMed] [Google Scholar]
- (20).Micale N; Scarbaci K; Troiano V; Ettari R; Grasso S; Zappala M Peptide-based proteasome inhibitors in anticancer drug design. Med. Res. Rev 2014, 34, 1001–1069. [DOI] [PubMed] [Google Scholar]
- (21).Lee MJ; Miller Z; Park JE; Bhattarai D; Lee W; Kim KB H727 cells are inherently resistant to the proteasome inhibitor carfilzomib, yet require proteasome activity for cell survival and growth. Sci. Rep 2019, 9, 4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ho A; Cyrus K; Kim KB Towards immunoproteasome-specific inhibitors: an improved synthesis of dihydroeponemycin. Eur. J. Org. Chem 2005, 2005, 4829–4834. [Google Scholar]
- (23).Chen R; Chen B; Zhang X; Gao C Efficacy of carfilzomib in the treatment of relapsed and (or) refractory multiple myeloma: a meta-analysis of data from clinical trials. Discov Med. 2016, 22, 189–199. [PubMed] [Google Scholar]
- (24).Hawley TS; Riz I; Yang W; Wakabayashi Y; Depalma L; Chang YT; Peng W; Zhu J; Hawley RG Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluripotent stem cell fluorescent dye CDy1. Am. J. Hematol 2013, 88, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ao L; Wu Y; Kim D; Jang ER; Kim K; Lee DM; Kim KB; Lee W Development of peptide-based reversing agents for p-glycoprotein-mediated resistance to carfilzomib. Mol. Pharmaceutics 2012, 9, 2197–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gutman D; Morales AA; Boise LH Acquisition of a multidrug-resistant phenotype with a proteasome inhibitor in multiple myeloma. Leukemia 2009, 23, 2181–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Siegel DS; Martin T; Wang M; Vij R; Jakubowiak AJ; Lonial S; Trudel S; Kukreti V; Bahlis N; Alsina M; Chanan-Khan A; Buadi F; Reu FJ; Somlo G; Zonder J; Song K; Stewart AK; Stadtmauer E; Kunkel L; Wear S; Wong AF; Orlowski RZ; Jagannath S A phase 2 study of single-agent carfilzomib (PX-171–003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ho YK; Bargagna-Mohan P; Mohan R; Kim KB LMP2-specific inhibitors: Novel chemical genetic tools for proteasome biology. Chem. Biol 2007, 14, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).de Bruin G; Huber EM; Xin BT; van Rooden EJ; Al-Ayed K; Kim KB; Kisselev AF; Driessen C; van der Stelt M; van der Marel GA; Groll M; Overkleeft HS Structure-based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J. Med. Chem 2014, 57, 6197–6209. [DOI] [PubMed] [Google Scholar]
- (30).Huber EM; Basler M; Schwab R; Heinemeyer W; Kirk CJ; Groettrup M; Groll M Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [DOI] [PubMed] [Google Scholar]
- (31).Lei B; Abdul Hameed MD; Hamza A; Wehenkel M; Muzyka JL; Yao XJ; Kim KB; Zhan CG Molecular basis of the selectivity of the immunoproteasome catalytic subunit LMP2-specific inhibitor revealed by molecular modeling and dynamics simulations. J. Phys. Chem. B 2010, 114, 12333–12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang Z; Yang J; Kirk C; Fang Y; Alsina M; Badros A; Papadopoulos K; Wong A; Woo T; Bomba D; Li J; Infante JR Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab. Dispos 2013, 41, 230–237. [DOI] [PubMed] [Google Scholar]
- (33).Papadopoulos KP; Burris HA 3rd; Gordon M; Lee P; Sausville EA; Rosen PJ; Patnaik A; Cutler RE Jr.; Wang Z; Lee S; Jones SF; Infante JR A phase I/II study of carfilzomib 2–10-min infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol 2013, 72, 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang Z; Fang Y; Teague J; Wong H; Morisseau C; Hammock BD; Rock DA; Wang Z In vitro metabolism of oprozomib, an oral proteasome inhibitor: role of epoxide hydrolases and cytochrome P450s. Drug Metab. Dispos 2017, 45, 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Saenz-Mendez P; Katz A; Perez-Kempner ML; Ventura ON; Vazquez M Structural insights into human microsomal epoxide hydrolase by combined homology modeling, molecular dynamics simulations, and molecular docking calculations. Proteins: Struct., Funct., Genet 2017, 85, 720–730. [DOI] [PubMed] [Google Scholar]
- (36).Shen HC; Hammock BD Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J. Med. Chem 2012, 55, 1789–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hosagrahara VP; Rettie AE; Hassett C; Omiecinski CJ Functional analysis of human microsomal epoxide hydrolase genetic variants. Chem.-Biol. Interact 2004, 150, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Verbrugge SE; Al M; Assaraf YG; Niewerth D; van Meerloo J; Cloos J; van der Veer M; Scheffer GL; Peters GJ; Chan ET; Anderl JL; Kirk CJ; Zweegman S; Dijkmans BA; Lems WF; Scheper RJ; de Gruijl TD; Jansen G Overcoming bortezomib resistance in human B cells by anti-CD20/rituximab-mediated complement-dependent cytotoxicity and epoxyketone-based irreversible proteasome inhibitors. Exp. Hematol. Oncol 2013, 2, 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Verbrugge SE; Assaraf YG; Dijkmans BA; Scheffer GL; Al M; den Uyl D; Oerlemans R; Chan ET; Kirk CJ; Peters GJ; van der Heijden JW; de Gruijl TD; Scheper RJ; Jansen G Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)-proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J. Pharmacol. Exp. Ther 2012, 341, 174–182. [DOI] [PubMed] [Google Scholar]
- (40).Leung-Hagesteijn C; Erdmann N; Cheung G; Keats JJ; Stewart AK; Reece DE; Chung KC; Tiedemann RE Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Niewerth D; Jansen G; Assaraf YG; Zweegman S; Kaspers GJ; Cloos J Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist. Updates 2015, 18, 18–35. [DOI] [PubMed] [Google Scholar]
- (42).Kuhn DJ; Hunsucker SA; Chen Q; Voorhees PM; Orlowski M; Orlowski RZ Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009, 113, 4667–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kraus M; Ruckrich T; Reich M; Gogel J; Beck A; Kammer W; Berkers CR; Burg D; Overkleeft H; Ovaa H; Driessen C Activity patterns of proteasome subunits reflect bortezomib sensitivity of hematologic malignancies and are variable in primary human leukemia cells. Leukemia 2007, 21, 84–92. [DOI] [PubMed] [Google Scholar]
- (44).Kraus M; Bader J; Geurink PP; Weyburne ES; Mirabella AC; Silzle T; Shabaneh TB; van der Linden WA; de Bruin G; Haile SR; van Rooden E; Appenzeller C; Li N; Kisselev AF; Overkleeft H; Driessen C The novel beta2-selective proteasome inhibitor LU-102 synergizes with bortezomib and carfilzomib to overcome proteasome inhibitor resistance of myeloma cells. Haematologica 2015, 100, 1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Li X; Wood TE; Sprangers R; Jansen G; Franke NE; Mao X; Wang X; Zhang Y; Verbrugge SE; Adomat H; Li ZH; Trudel S; Chen C; Religa TL; Jamal N; Messner H; Cloos J; Rose DR; Navon A; Guns E; Batey RA; Kay LE; Schimmer AD Effect of noncompetitive proteasome inhibition on bortezomib resistance. J. Natl. Cancer Inst 2010, 102, 1069–1082. [DOI] [PubMed] [Google Scholar]
- (46).Schrader J; Henneberg F; Mata RA; Tittmann K; Schneider TR; Stark H; Bourenkov G; Chari A The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [DOI] [PubMed] [Google Scholar]
- (47).Mushtaq A; Kapoor V; Latif A; Iftikhar A; Zahid U; McBride A; Abraham I; Riaz IB; Anwer F Efficacy and toxicity profile of carfilzomib based regimens for treatment of multiple myeloma: a systematic review. Crit Rev. Oncol Hematol 2018, 125, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Federspiel JD; Codreanu SG; Goyal S; Albertolle ME; Lowe E; Teague J; Wong H; Guengerich FP; Liebler DC Specificity of protein covalent modification by the electrophilic proteasome inhibitor carfilzomib in human cells. Mol. Cell. Proteomics 2016, 15, 3233–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ao L; Reichel D; Hu D; Jeong H; Kim KB; Bae Y; Lee W Polymer micelle formulations of proteasome inhibitor carfilzomib for improved metabolic stability and anticancer efficacy in human multiple myeloma and lung cancer cell lines. J. Pharmacol. Exp. Ther 2015, 355, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Park JE; Chun SE; Reichel D; Min JS; Lee SC; Han S; Ryoo G; Oh Y; Park SH; Ryu HM; Kim KB; Lee HY; Bae SK; Bae Y; Lee W Polymer micelle formulation for the proteasome inhibitor drug carfilzomib: anticancer efficacy and pharmacokinetic studies in mice. PLoS One 2017, 12, No. e0173247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.