

Abstract
Since 2019, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) causing coronavirus disease 2019 (COVID‐19) has infected 10 millions of people across the globe, and massive mutations in virus genome have occurred during the rapid spread of this novel coronavirus. Variance in protein sequence might lead to a change in protein structure and interaction, then further affect the viral physiological characteristics, which could bring tremendous influence on the pandemic. In this study, we investigated 20 nonsynonymous mutations in the SARS‐CoV‐2 genome in which incidence rates were all ≥ 1% as of September 1st, 2020, and then modeled and analyzed the mutant protein structures. The results showed that four types of mutations caused dramatic changes in protein structures (RMSD ≥ 5.0 Å), which were Q57H and G251V in open‐reading frames 3a (ORF3a), S194L, and R203K/G204R in nucleocapsid (N). Next, we found that these mutations also affected the binding affinity of intraviral protein interactions. In addition, the hot spots within these docking mutant complexes were altered, among which the mutation Q57H was involved in both Orf3a–S and Orf3a–Orf8 protein interactions. Besides, these mutations were widely distributed all over the world, and their occurrences fluctuated as time went on. Notably, the incidences of R203K/G204R in N and Q57H in Orf3a were both over 50% in some countries. Overall, our findings suggest that SARS‐CoV‐2 mutations could change viral protein structure, binding affinity, and hot spots of the interface, thereby might have impacts on SARS‐CoV‐2 transmission, diagnosis, and treatment of COVID‐19.
Keywords: mutation, protein structure, protein–protein interaction, SARS‐CoV‐2
1. INTRODUCTION
The ongoing outbreak of coronavirus disease 2019 (COVID‐19), caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), has been characterized as a pandemic by the World Health Organization (WHO). SARS‐CoV‐2 is a positive‐sense single‐stranded RNA virus and belongs to betacoronavirus, 1 whose genome has ~30,000 nucleotides, containing 12 open‐reading frames (ORFs), encoding 4 structural and 22 nonstructural proteins. The structural proteins include spike protein (S), envelope protein (E), membrane protein (M), and nucleocapsid protein (N), while the nonstructural proteins (nsp) contain nsp1–16 encoded by ORF1ab and six accessory proteins, which are Orf3a, 6, 7a, 7b, 8, and 9b. 2 It is known a virus that undergo evolution and natural selection, and most of them evolve rapidly. As reported, the evolution rate of a typical RNA virus is about 10−4 substitutes per year per site, 3 and mutations could occur during each replication cycle. The high mutation rates of RNA viruses, coupled with short generation times and large population sizes, allow viruses to evolve rapidly and adapt to the host environment. The rapidity of viral mutation also causes problems in the development of successful vaccines and antiviral drugs. With the continued spread of SARS‐CoV‐2 around the world, thousands of mutations have been identified, some of which have relatively high incidences, but their potential impacts on virus characteristics still remain unknown.
Among all the mutations, nonsynonymous mutations cause amino acid substitutions, then could alter virus protein structures, which might affect viral reproduction and give rise to false‐negative diagnoses and drug resistance. It is reported that, after depletion of nine amino acids in SARS‐CoV‐2 Orf6, its protein structure was dramatically changed and its transmembrane localization was also shifted, which would lead to interferon (IFN) resistance during antiviral therapy. 4 Additionally, along with the occurrence of S139A and F140A mutations, the structure of SARS‐CoV 3CLpro altered remarkably, followed by the decrease of its enzyme activity. 5 Furthermore, modified protein structure begotten by mutations can also affect protein–protein binding, and intraviral protein–protein interactions are often indispensable in assembly and release of coronavirus. In SARS‐CoV, the interactions between structural proteins are essential for its maturation, 6 while the binding between nonstructural proteins guarantees the completion of virus replication. 7 Besides, mutations I529T and D510G in MERS‐CoV S reduced the binding affinity of the receptor‐binding domain (RBD)‐CD26, respectively, which impaired the virulence of the virus. 8 However, it is still unclear whether some SARS‐CoV‐2 mutations can lead to the changes in protein structures, protein–protein interactions, protein function, and even virus infection, which could strike on COVID‐19 epidemic control.
In this study, we selected the nonsynonymous mutations of SARS‐CoV‐2 with ≥1% incidence, predicted and compared their protein structures, and found that four types of mutations had significant impacts on protein structures, which resulted in remarkable changes in binding affinities and hot spots between virus proteins. Besides, statistical analyses on mutation rates exhibited dynamic changes in the process of time, and also demonstrated that these four SARS‐CoV‐2 variants were widely distributed and had relatively high incidences in certain countries. These findings on SARS‐CoV‐2 mutations would be very helpful for better understandings of this virus and dealing with the complicated situations in COVID‐19 prevention, diagnosis, and treatment.
2. METHODS
2.1. Online resources
All information about SARS‐CoV‐2 genomes were obtained from the China National Center for Bioinformation 2019 Novel Coronavirus Resource (CNCB 2019nCoVR; https://bigd.big.ac.cn/ncov).
2.2. Protein structure prediction
Protein structure of control SARS‐CoV‐2 S was from C‐I‐TASSER (https://zhanglab.ccmb.med.umich.edu/C-I-TASSER/2019-nCoV/), 9 while other SARS‐CoV‐2 protein structure models were predicted by I‐TASSER. 10 For each protein, five models were generated and the model with the highest C‐score was selected as the best one and used for the following analysis.
2.3. Protein structure alignment
The mutant protein structures were aligned to corresponding control ones by using the TM‐align web‐server (https://zhanglab.ccmb.med.umich.edu/TM-align/). 11 Random structural similarity was determined by TM‐score between 0.0 and 0.3 and root‐mean‐square deviation (RMSD) ≥ 5.0 Å. 11 , 12
2.4. Molecular docking and hot spots prediction
Protein–protein docking was performed with the HADDOCK web‐server (http://haddock.chem.uu.nl/). 13 The structure was chosen according to the HADDOCK score, and a complex binding affinity was calculated by PRODIGY. 14 , 15 Hot spots within protein–protein interfaces were predicted using Knowledge‐based FADE and Contacts Server (KFC, https://mitchell-web.ornl.gov/KFC_Server/index.php). 16 Data visualization was accomplished by PyMOL.
3. RESULTS
3.1. SARS‐CoV‐2 mutations led to altered protein structures
As the COVID‐19 pandemic is spreading around the world, more than 10,000 SARS‐CoV‐2 mutations have been evolved. To investigate these SARS‐CoV‐2 nucleotide polymorphisms, first, one of the earliest reported SARS‐CoV‐2 genome sequences (GenBank accession number: MN908947.3) was chosen as a control, then 20 nonsynonymous mutations with frequency ≥1% were selected from CNCB 2019nCoVR database (see Section 2), as of September 1st, 2020, which were located in 10 different SARS‐CoV‐2 protein‐coding regions (Table S1). Specifically, there was one amino acid substitution in nsp5, nsp6, or nsp7. Meanwhile, two single substitutions were observed in nsp2, nsp12, nsp13, S, Orf3a, or Orf8, among which mutation D614G in S had the highest incidence (43.27%), and five single ones were found in N. In particular, there were also some mutation combinations, such as P504L/Y541C in nsp13 and R203K/G204R in N (Table S1). Briefly, there were 17 types of mutants composed of 20 mutations with ≥1% incidence, which included one nsp5, nsp6, nsp7, or nsp13 mutant, two nsp2, nsp12, S, Orf3, or Orf8 mutants and three N mutants.
To examine whether these mutations had effects on protein structures, three‐dimensional (3D) structures of each mutant and its reference protein were predicted with I‐TASSER (Table S1), 10 and the structural similarity between mutant and control protein were measured by RMSD. 11 After structural alignment, we found that four mutants exhibited a significant difference in protein structural morphology from their control ones (RMSD ≥ 5.0 Å), which were Q57H Orf3a, G251V Orf3a, S194L N, and R203K/G204R N (Figure 1 and Table S1).
Figure 1.
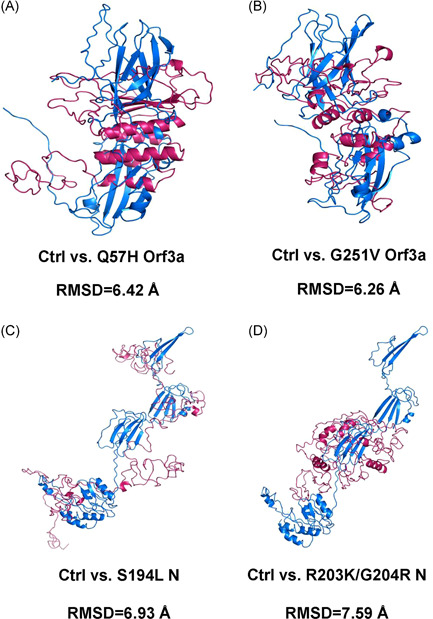
Comparisons of protein structures between SARS‐CoV‐2 control and mutant proteins. The aligned structures of control ones and Q57H Orf3a (A), G251V Orf3a (B), S194L N (C), and R203K/G204R N (D) are shown in blue (control, Ctrl) and warm pink (mutant) with the value of RMSD below. N, nucleocapsid protein; RMSD, root‐mean‐square deviation; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
3.2. SARS‐CoV‐2 mutations resulted in the changes in protein–protein interactions
The SARS‐CoV‐2 proteins have been shown to display characteristic SARS‐CoV features. 17 , 18 So here, we referenced SARS‐CoV protein combined patterns and explored the effect of these SARS‐CoV‐2 mutations related to mutant structure alterations on intraviral protein interaction, which was known as a rate‐limited procedure for virus reproduction. 19 , 20 , 21 , 22 Among 16 mutant docking pairs, five mutant complexes had significantly higher binding affinity than control ones (Figure 2). Differently, another four mutant pairs showed weakened binding affinity in comparison with the control, and there was no obvious change in the remaining seven groups in which absolute values of ΔΔG (=G control − ΔG mutant) were <1 kcal/mol (Figure 2). Strikingly, the binding affinity of Q57H Orf3a–S complex showed the greatest increase (ΔΔG = 4.2 kcal/mol), while the most dramatic decrease was observed in the binding affinity of G251V Orf3a–M complex (ΔΔG = −2.3 kcal/mol; Figure 2A). Moreover, in the process of Orf3a interacting with M or S, Q57H mutant pairs showed enhanced binding affinity, while G251V mutant pairs had attenuated ones as just mentioned (Figure 2A), suggesting that diverse amino acid replacements in the same protein have given different consequences on binding affinity.
Figure 2.
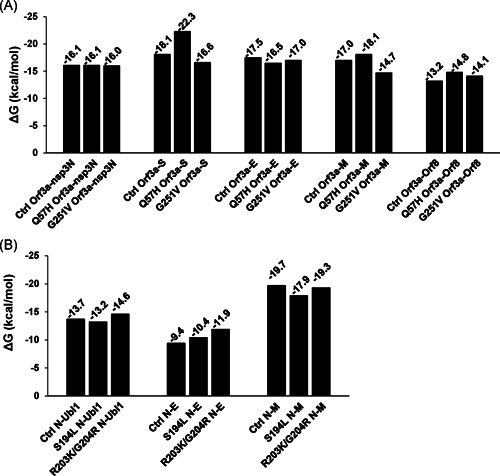
Protein‐binding affinity in SARS‐CoV‐2 mutant complexes. Bars show Gibbs‐free energy (ΔG) for representing the binding affinity in Orf3a (A) and N (B) relevant complexes. Nsp3N: 1‐595aa of nsp3, Ubl1: 1‐112aa of nsp3. E, envelope protein; M, membrane protein; N, nucleocapsid protein; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; S, spike protein
3.3. SARS‐CoV‐2 mutations caused varied hot spots within protein complexes
Hot spots are functional sites within protein‐interacting interfaces, which are conservative and often taken as attractive drug targets via preventing protein–protein interactions. To further study the influence of SARS‐CoV‐2 mutations on protein‐binding hot spots, we predicted the hot spots in nine mutant complexes with altered bind affinity using the KFC2 server (Figure 3, S1, S2, S3, and S4). Surprisingly, the results showed that the binding hot spots on mutant protein–protein interfaces were notably different from control ones, and few identical ones were shared by both the mutant and control complexes. In particular, Q57 residue in control Orf3a was not involved in the protein‐binding interfaces, but amino acid substitution Q57H became a hot spot in both Orf3a–S and Orf3a–Orf8 complexes (Figure 3). These results indicated that SARS‐CoV‐2 mutations might destroy drug‐targeting sites and lead to therapy failure by shifting the protein‐binding interface.
Figure 3.
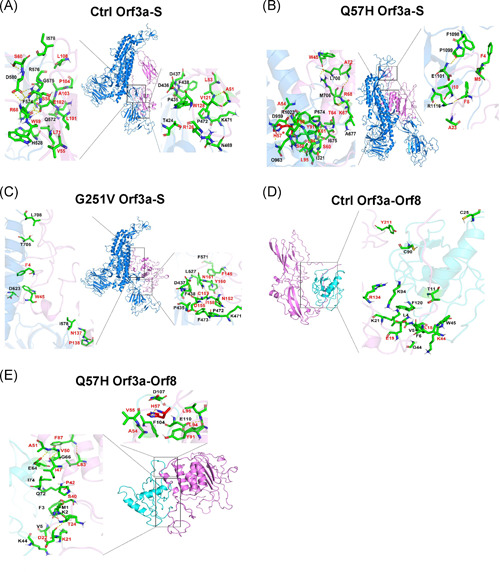
Hot spots within interactions between SARS‐CoV‐2 Orf3a and S or Orf8. The hot spots in Ctrl Orf3a–S (A), Q57H Orf3a–S (B), G251V Orf3a–S (C), Ctrl Orf3a–Orf8 (D), or Q57H Orf3a–Orf8 (E) complexes are shown as sticks. Control and mutant Orf3a are shown in violet, and S or Orf8 is shown in marine or cyan. The residues of Orf3a and docking proteins are colored in red and black, respectively. Asterisk (*) represents a mutated residue. N, nucleocapsid protein; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; S, spike protein
3.4. SARS‐CoV‐2 mutations were globally distributed with dynamic incidences over time
The changes in SARS‐CoV‐2 proteins caused by mutations can affect virus transmission, pathogenesis, and immunogenicity. And with the spread of the COVID‐19 pandemic, the incidence and lethality of SARS‐CoV‐2 infection varied from country to country. Hence, to link SARS‐CoV‐2 mutations and COVID‐19 prevalence, we analyzed the occurrence of these four types of mutations (Q57H and G251V in Orf3a, S194L and R203K/G204R in N) based on CNCB 2019nCoVR. To avoid deviation caused by insufficient sample size, we screened out the countries or regions where the number of SARS‐CoV‐2 genome sequences submitted was no more than 100, as of September 1st, 2020. The statistical results showed that SARS‐CoV‐2 mutations with high incidences were distributed nearly all over the world with dynamic occurrences over time (Figure 4). The frequency of all these mutations was relatively high in some countries (Figure 4). Among them, we noticed that the incidence of R203K/G204R in N was as high as 86.97% in Bangladesh; meanwhile, it was also over 50% in the other four countries, with a trend of escalation (Figure 4D). Analogously, the frequency of Q57H in Orf3a in Finland, Egypt, South Korea, and Denmark had shown high incidences, which were 69.4%, 67.26%, 59.72%, and 58.03%, respectively (Figure 4A).
Figure 4.
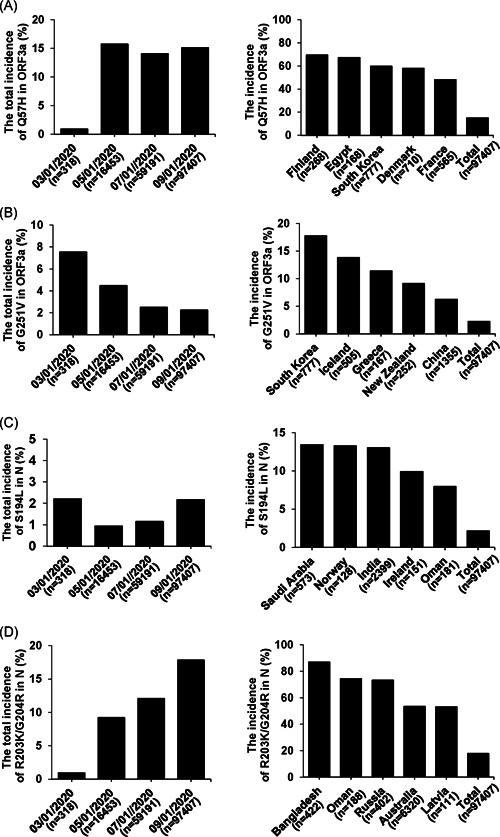
Temporal and spatial analyses of SARS‐CoV‐2 mutation incidences. Bars show the incidences of Q57H (A) and G251V (B) in Orf3a, S194L (C) and R203K/G204R (D) in N at different time periods or in the top five countries and globe (total), as of September 1st, 2020. N represents the all available nucleotide sequences of the country or globe submitted. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
4. DISCUSSION
The mutation is one of the major mechanisms of how viruses undergo continuous change as a result of genetic selection. Although most of the point mutations are neutral and do not change the protein that the gene encodes, yet there are always some occasionally favorable ones that can help viruses confer evolutionary advantages such as an abrupt epidemic outbreak. In addition, the single amino acid substitution would alter protein structure as well, and it has been reported that G104E mutation in SARS‐CoV nsp9 could prevent viral replication via changing protein structure and further destroying the helix–helix interface. 23 In the same vein, masses of single nucleotide polymorphisms (SNPs) have emerged in SARS‐CoV‐2 genomes. According to the data in CNCB 2019nCoVR, as of September 1st, 2020, we screened 20 SARS‐CoV‐2 nonsynonymous mutations with ≥1% incidence, which were categorized into 17 types of mutation combinations. After structural alignment, four protein mutants (Q57H Orf3a, G251V Orf3a, S194L N, and R203K/G204R N) were found displaying different structures from control ones, which corresponding mutations were located in the coding region of N and Orf3a known as essential proteins for coronavirus assembly 24 and cytotoxicity. 25 Remarkably, the incidences of Q57H in Orf3a and R203K/G204R in N were quite high (15.12% and 17.84%), which could be a sign that these mutations are correlated with enhanced virulence, evolvability, and traits considered beneficial for the virus. However, it is interesting that some mutations, such as P323L in nsp12 and D614G in S, with even much higher occurrence rates (43.21% and 43.27%) were found with no significant impact on protein structure, which indicates they maybe could affect virus characteristics via altering RNA second structure, 26 protein stability, 27 or partial structure, 28 instead of integral protein structure. For instance, a study reported that D614G in S could lead to increased virus infectivity by eliminating side‐chain hydrogen bond, which was only a tiny change in overall protein structure, 29 and our data also showed a slight structural difference between control and D614G mutant (RMSD = 2.33 Å). Therefore, more research about SARS‐CoV‐2 mutations are worth doing to determine whether these structure alterations of Orf3a or N could influence protein function and even virus infectivity.
Besides the protein structure alteration, the mutations in the SARS‐CoV‐2 genome could also affect protein–protein interactions. With molecular docking and ΔG value calculation, we found that protein‐binding affinity changed between each of all four above mutants and their intraviral docking proteins, and these protein interactions in SARS‐CoV have been confirmed to play indispensable mediating roles in virus replication and infected ability. 6 , 25 , 30 Moreover, the combination of SARS‐CoV Orf3a and S can prevent the release of premature viral RNA, 31 and Orf3a–M complex is localized in Golgi, 32 where virus particles are assembled by budding. 33 Our results showed stronger binding affinities in Q57H Orf3a–M and Q57H Orf3a–S complexes, but weaker affinities in G251V Orf3a–M and G251V Orf3a–S complexes. Meanwhile, considering the great disparity between the incidences of Q57H in Orf3a (15.12%) and G251V in Orf3a (2.25%), it could be speculated that the virus assembly and transmission would be disproportionately affected by different Orf3a mutations. Furthermore, some research indicated that the interaction between N and E had a function in the SARS‐CoV virus release, 21 , 34 , 35 and the N–M complex was necessary for the assembly of coronavirus. 22 Therefore, the enhanced interaction between each SARS‐CoV‐2 N mutant (S194L or R203K/G204R) and E might promote virus release, while decreased binding affinity of S194L N–M might attenuate virus assembly. Besides, SARS‐CoV N can bind to heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) of host cells and their interaction plays a regulatory role in the synthesis of SARS‐CoV RNA, 36 so it would be interesting to detect whether these N mutants would interfere with the combination of proteins in host cells. What is more, the hot spots that play critical roles in protein–protein interactions can be used as drugs target, 37 and our results demonstrated that all these selected SARS‐CoV‐2 mutations (Q57H and G251V in Orf3a, S194L and R203K/G204R in N) had great influences on hot spots within protein combinations, which the changes could have strong impacts on clinical treatment. Nevertheless, scientific experiments are indispensable to clarify the impacts of these mutations on virus characteristics, such as investigating mutated virus protein interaction with its partners and analyzing the infectivity of the mutated virus to host cells, which it is essential to confirm the interference of SARS‐CoV‐2 mutations during the development of vaccines or drugs. 38 Previous experimental research showed that Y195A mutation in SARS‐CoV M was found to disrupt its interaction with S, resulting in a declined ability of virus assembly. 39 Accordingly, mutations of the virus could alter intraviral protein–protein interactions and the characteristics of the virus, which illustrate that our results are valuable for providing important proofs for future studies.
In the last several months, populations in more than 180 countries/regions have been affected by SARS‐CoV‐2, and the emergence of mutations probably already makes important contributions to virus adaption to new environments and selective pressures, which, in turn, can impact transmissibility, pathogenesis, and immunogenicity of SARS‐CoV‐2. 40 It has been reported that variant SARS‐CoV‐2 genomes occurred in different areas. 41 , 42 , 43 , 44 , 45 Our results also showed that some mutations were widely distributed, and their occurrence rates showed different dynamic fluctuations. Take R203K/G204R in N as an example; this mutation combination with increased incidence was found mainly in Europe till March 2020, 46 , 47 while its high frequency also happened in Asian and African countries as of September 2020. Although there is no reliable evidence for any necessary link between SARS‐CoV‐2 mutations and epidemic outbreak in specific countries/regions, it is still particularly important to certify the relevance of geographically aggregated mutations to SARS‐CoV‐2 transmission and pathogenicity for effective containment of COVID‐19 outbreak.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Siqi Wu, Chang Tian, and Lijun Liu conceived the study. Siqi Wu, Chang Tian, and Panpan Liu collected and summarized the data of SARS‐CoV‐2 mutations information. Yang Zhang, Wei Zheng, Chang Tian, Siqi Wu, and Panpan Liu contributed to protein structure prediction. Xiaoqiang Huang, Chang Tian, Siqi Wu, and Panpan Liu contributed to molecular docking. Chang Tian, Siqi Wu and Panpan Liu predicted hot spots within the protein–protein interface. Dongjie Guo contributed to protein structures alignment and data visualizations. Siqi Wu, Chang Tian, and Lijun Liu wrote the paper. Lijun Liu directed the study.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We thank Xiaoting Li (Columbia University, USA) for helpful advice on modeling analysis. And we also would like to thank Dr. Qi Zhao (Northeastern University, China) for technical assistance on molecular docking. This study was supported by Northeastern University, PRC (N2020006), and Talent Project of Revitalizing Liaoning (XLYC1907052). The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Wu S, Tian C, Liu P, et al. Effects of SARS‐CoV‐2 mutations on protein structures and intraviral protein–protein interactions. J Med Virol. 2021;93:2132‐2140. 10.1002/jmv.26597
Siqi Wu and Chang Tian contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565‐574. 10.1016/s0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chan JFW, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221‐236. 10.1080/22221751.2020.1719902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490‐502. 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riojas MA, Frank AM, Puthuveetil NP, et al. A rare deletion in SARS‐CoV‐2 ORF6 dramatically alters the predicted three‐dimensional structure of the resultant protein. bioRxiv. 2020. 10.1101/2020.06.09.134460 [DOI] [Google Scholar]
- 5. Hu T, Zhang Y, Li L, et al. Two adjacent mutations on the dimer interface of SARS coronavirus 3C‐like protease cause different conformational changes in crystal structure. Virology. 2009;388:324‐334. 10.1016/j.virol.2009.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Siu YL, Teoh KT, Lo J, et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus‐like particles. J Virol. 2008;82:11318‐11330. 10.1128/jvi.01052-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Snijder EJ, Decroly E, Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv Virus Res. 2016;96:59‐126. 10.1016/bs.aivir.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim Y, Cheon S, Min CK, et al. Spread of mutant Middle East respiratory syndrome coronavirus with reduced affinity to human CD26 during the South Korean outbreak. mBio. 2016;7:e00019. 10.1128/mBio.00019-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang C, Zheng W, Huang X, Bell EW, Zhou X, Zhang Y. Protein structure and sequence reanalysis of 2019‐nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV‐1. J Proteome Res. 2020;19:1351‐1360. 10.1021/acs.jproteome.0c00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roy A, Kucukural A, Zhang Y. I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725‐738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Skolnick J. TM‐align: a protein structure alignment algorithm based on the TM‐score. Nucleic Acids Res. 2005;33:2302‐2309. 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kundrotas PJ, Vakser IA. Accuracy of protein–protein binding sites in high‐throughput template‐based modeling. PLOS Comput Biol. 2010;6:e1000727. 10.1371/journal.pcbi.1000727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Zundert GCP, Rodrigues JPGLM, Trellet M, et al. The HADDOCK2.2 Web Server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720‐725. 10.1016/j.jmb.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 14. Xue LC, Rodrigues JP, Kastritis PL, et al. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32:3676‐3678. 10.1093/bioinformatics/btw514 [DOI] [PubMed] [Google Scholar]
- 15. Kastritis PL, Bonvin AM. On the binding affinity of macromolecular interactions: daring to ask why proteins interact. J R Soc Interface. 2013;10:20120835. 10.1098/rsif.2012.0835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu X, Mitchell JC. KFC2: a knowledge‐based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins. 2011;79:2671‐2683. 10.1002/prot.23094 [DOI] [PubMed] [Google Scholar]
- 17. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng Q, Peng R, Yuan B, et al. Structural and biochemical characterization of the nsp12‐nsp7‐nsp8 core polymerase complex from SARS‐CoV‐2. Cell Rep. 2020;31:107774. 10.1016/j.celrep.2020.107774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von Brunn A, Teepe C, Simpson JC, et al. Analysis of intraviral protein–protein interactions of the SARS coronavirus ORFeome. PLOS One. 2007;2:e459. 10.1371/journal.pone.0000459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan YJ, Teng E, Shen S, et al. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J Virol. 2004;78:6723‐6734. 10.1128/jvi.78.13.6723-6734.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tseng YT, Wang SM, Huang KJ, Wang CT. SARS‐CoV envelope protein palmitoylation or nucleocapid association is not required for promoting virus‐like particle production. J Biomed Sci. 2014;21:34. 10.1186/1423-0127-21-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He R, Leeson A, Ballantine M, et al. Characterization of protein–protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus. Virus Res. 2004;105:121‐125. 10.1016/j.virusres.2004.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miknis ZJ, Donaldson EF, Umland TC, Rimmer RA, Baric RS, Schultz LW. Severe acute respiratory syndrome coronavirus nsp9 dimerization is essential for efficient viral growth. J Virol. 2009;83:3007‐3018. 10.1128/jvi.01505-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hurst KR, Ye R, Goebel SJ, Jayaraman P, Masters PS. An interaction between the nucleocapsid protein and a component of the replicase–transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J Virol. 2010;84:10276‐10288. 10.1128/jvi.01287-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Freundt EC, Yu L, Goldsmith CS, et al. The open reading frame 3a protein of severe acute respiratory syndrome‐associated coronavirus promotes membrane rearrangement and cell death. J Virol. 2010;84:1097‐1109. 10.1128/jvi.01662-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hosseini Rad A, McLellan Sm,AD. Implications of SARS‐CoV‐2 mutations for genomic RNA structure and host microRNA targeting. Int J Mol Sci. 2020;21:4807. 10.3390/ijms21134807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arévalo SJ, Sifuentes DZ, Robles CH, et al. Worldwide geographical and temporal analysis of SARS‐CoV‐2 haplotypes shows differential distribution patterns. bioRxiv. 2020:2020.2007.2012.199414. 10.1101/2020.07.12.199414 [DOI] [Google Scholar]
- 28. Liu S, Shen J, Fang S, et al. Genetic spectrum and distinct evolution patterns of SARS‐CoV‐2. medRxiv. 2020:2020.2006.2016.20132902. 10.1101/2020.06.16.20132902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS‐CoV‐2 spike: evidence that D614G increases infectivity of the COVID‐19 virus. Cell. 2020;182:812‐827. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McBride R, van Zyl M, Fielding BC. The coronavirus nucleocapsid is a multifunctional protein. Viruses. 2014;6:2991‐3018. 10.3390/v6082991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan YJ. The Severe Acute Respiratory Syndrome (SARS)‐coronavirus 3a protein may function as a modulator of the trafficking properties of the spike protein. Virol J. 2005;2:5. 10.1186/1743-422x-2-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yuan X, Li J, Shan Y, et al. Subcellular localization and membrane association of SARS‐CoV 3a protein. Virus Res. 2005;109:191‐202. 10.1016/j.virusres.2005.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Haan CA, Rottier PJ. Molecular interactions in the assembly of coronaviruses. Adv Virus Res. 2005;64:165‐230. 10.1016/s0065-3527(05)64006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boscarino JA, Logan HL, Lacny JJ, Gallagher TM. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J Virol. 2008;82:2989‐2999. 10.1128/jvi.01906-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopez S, Yao JS, Kuhn RJ, Strauss EG, Strauss JH. Nucleocapsid–glycoprotein interactions required for assembly of alphaviruses. J Virol. 1994;68:1316‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo H, Chen Q, Chen J, Chen K, Shen X, Jiang H. The nucleocapsid protein of SARS coronavirus has a high binding affinity to the human cellular heterogeneous nuclear ribonucleoprotein A1. FEBS Lett. 2005;579:2623‐2628. 10.1016/j.febslet.2005.03.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li X, Keskin O, Ma B, Nussinov R, Liang J. Protein–protein interactions: hot spots and structurally conserved residues often locate in complemented pockets that pre‐organized in the unbound states: implications for docking. J Mol Biol. 2004;344:781‐795. 10.1016/j.jmb.2004.09.051 [DOI] [PubMed] [Google Scholar]
- 38. Huang X, Pearce R, Zhang Y. De novo design of protein peptides to block association of the SARS‐CoV‐2 spike protein with human ACE2. Aging. 2020;12:11263‐11276. 10.18632/aging.103416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McBride CE, Machamer CE. A single tyrosine in the severe acute respiratory syndrome coronavirus membrane protein cytoplasmic tail is important for efficient interaction with spike protein. J Virol. 2010;84:1891‐1901. 10.1128/jvi.02458-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Opijnen T, Boerlijst MC, Berkhout B. Effects of random mutations in the human immunodeficiency virus type 1 transcriptional promoter on viral fitness in different host cell environments. J Virol. 2006;80:6678‐6685. 10.1128/jvi.02547-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yeh TY, Contreras GP. Emerging viral mutants in Australia suggest RNA recombination event in the SARS‐CoV‐2 genome. Med J Aust. 2020;213:44. 10.5694/mja2.50657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gong YN, Tsao KC, Hsiao MJ, et al. SARS‐CoV‐2 genomic surveillance in Taiwan revealed novel ORF8‐deletion mutant and clade possibly associated with infections in Middle East. Emerg Microbes Infect. 2020;9:1457‐1466. 10.1080/22221751.2020.1782271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castillo AE, Parra B, Tapia P, et al. Phylogenetic analysis of the first four SARS‐CoV‐2 cases in Chile. J Med Virol. 2020;92:1562‐1566. 10.1002/jmv.25797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poterico JA, Mestanza O. Genetic variants and source of introduction of SARS‐CoV‐2 in South America. J Med Virol. 2020;92:2139‐2145. 10.1002/jmv.26001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang M, Li M, Ren R, et al. International expansion of a novel SARS‐CoV‐2 mutant. J Virol. 2020;94:e00567‐20. 10.1128/jvi.00567-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pachetti M, Marini B, Benedetti F, et al. Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. J Transl Med. 2020;18:179. 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leary S, Gaudieri S, Chopra A, et al. Three adjacent nucleotide changes spanning two residues in SARS‐CoV‐2 nucleoprotein: possible homologous recombination from the transcription‐regulating sequence. bioRxiv. 2020:2020.2004.2010.029454. 10.1101/2020.04.10.029454 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.