

Abstract
The genomic sequences of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) worldwide are publicly available and are derived from studies due to the increase in the number of cases. The importance of study of mutations is related to the possible virulence and diagnosis of SARS‐CoV‐2. To identify circulating mutations present in SARS‐CoV‐2 genomic sequences in Mexico, Belize, and Guatemala to find out if the same strain spread to the south, and analyze the specificity of the primers used for diagnosis in these samples. Twenty three complete SARS‐CoV‐2 genomic sequences, available in the GISAID database from May 8 to September 11, 2020 were analyzed and aligned versus the genomic sequence reported in Wuhan, China (NC_045512.2), using Clustal Omega. Open reading frames were translated using the ExPASy Translate Tool and UCSF Chimera (v.1.12) for amino acid substitutions analysis. Finally, the sequences were aligned versus primers used in the diagnosis of COVID‐19. One hundred and eighty seven distinct variants were identified, of which 102 are missense, 66 synonymous and 19 noncoding. P4715L and P5828L substitutions in replicase polyprotein were found, as well as D614G in spike protein and L84S in ORF8 in Mexico, Belize, and Guatemala. The primers design by CDC of United States showed a positive E value. The genomic sequences of SARS‐CoV‐2 in Mexico, Belize, and Guatemala present similar mutations related to a virulent strain of greater infectivity, which could mean a greater capacity for inclusion in the host genome and be related to an increased spread of the virus in these countries, furthermore, its diagnosis would be affected.
Keywords: genetic variation, mutations, SARS coronavirus
1. INTRODUCTION
The first cases of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) were reported in December 2019 in Wuhan city, Hubei province, China 1 , 2 ; thus initiating the coronavirus pandemic (COVID‐19). 3 According to the information released from scientists around the world and the GISAID consortium, until September 15, 2020, SARS‐CoV‐2 has caused 29,445,572 cases worlwide and 931,454 deaths. 4 According to predictions, 5 the total number of deaths will increase to 2,778,330 by January 1, 2021.
To identify how the virus spread, cross‐sectional studies with phylogenetic analysis and markers that identified mutations were implemented. 6 It is known from epidemiological reports that the first cases started in Mexico from the East, particularly from the United States, Spain, France, Germany, Singapore, and especially from Bergamo, Italy. 7 , 8 In addition, we think that the dispersion went from Mexico to Belize and Guatemala, and therefore, there could be the same molecular characteristics, for this reason we included these three countries in our study.
SARS‐CoV‐2 is closely related to the SARS‐CoV and the Middle East respiratory syndrome coronavirus. 9 Its structure contains a single‐stranded RNA (ssRNA) genome with a length of 29,903 bp. It comprised of a 5ʹ‐untranslated region (5ʹ‐UTR), a conserved replicase domain (ORF1ab) cleaved into 16 nonstructural proteins (NSPs) that participate in virus transcription and genome replication, four structural proteins (S, E, M, and N), several accessory proteins (ORF3a, ORF6, ORF7a, ORF7b, ORF8, and ORF10), and a highly conserved 3ʹ‐UTR 1 , 10 , 11 (Table 1) among other coronaviruses. 12 , 13
Table 1.
Genomic structure of SARS‐CoV‐2
| 5ʹ‐UTR | Gene | 3ʹ‐UTR | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ORF1ab | ORF1a | S | ORF3a | E | M | ORF6a | ORF7a | ORF7b | ORF8 | N | ORF10 | |||
| CDS (bp) | 265 | 21291 | 13218 | 3822 | 828 | 228 | 669 | 186 | 366 | 132 | 366 | 1260 | 117 | 229 |
| Protein | Noncoding sequence | Replicase polyprotein 1ab | Replicase polyprotein 1a | Spike glycoprotein | Protein 3a | Envelope small membrane protein | Membrane protein | Nonstructural protein 6 | Protein 7a | Protein nonstructural 7b | Nonstructural proein8 | Nucleoprotein | ORF10 protein | Noncoding sequence |
| Amino acids | – | 7096 | 4405 | 1273 | 275 | 75 | 222 | 61 | 121 | 43 | 121 | 419 | 38 | – |
| Molecular weight (Da) | – | 794,058 | 489,989 | 141,178 | 31,123 | 8365 | 25,147 | 7273 | 13,744 | 5180 | 13,831 | 45,626 | 4449 | – |
| Functiona | Regulate the folding, processing of viral RNA | Transcription and replication of viral RNA, RNA‐directed RNA polymerase | Transcription and replication of viral, RNA‐binding and thiol protease | Host‐virus interaction and virulence | Modulate virus release | Virus morphogenesis and assembly and apoptosis | Host‐virus interaction and viral immunoreaction, interactions with other viral proteins | Indicator probable of virus virulence | Modulation of host cell cycle by virus | Transmembrane helix | Host‐virus interaction | Packages viral genome RNA into a helical ribonucleocapsid (RNP), assembly and interactions with genome and membrane protein M | Unknown function | Unknown function |
| IDa protein | YP_009724389.1 | YP_009725295.1 | YP_009724390.1 | YP_009724391.1 | YP_009724392.1 | YP_009724393.1 | YP_009724394.1 | YP_009724395.1 | YP_009725318.1 | YP_009724396.1 | YP_009724397.2 | YP_009724397.2 | ||
NCBI reference sequence: NC_045512.2.
The ORF1ab gene encodes for replicase polyprotein 1ab (pp1ab), which is constituted of NSPs (NSP1, NSP2…NSP16). Of these, NSP12 corresponds to RNA‐dependent RNA polymerase (RdRp) and is formed by 932 amino acids (4392–5324 residues). The spike (S) protein has been described as responsible for the interaction with the human receptor angiotensin‐converting enzyme 2 (hACE2) 14 ; it is constituted of two domains, the S1 domain, responsible for binding, and the S2 domain that mediates the fusion of the viral and cellular membrane. 15 Moreover, S1 has variations but S2 is highly conserved. 16
Nonsynonymous substitution changes the protein sequences, these have been reported in SARS‐CoV‐2 in the functional domains of ORF3a. 17 Issa et al. 17 reported that these substitutions are related to virulence, infectivity, ion channel formation, and virus release. ORF3a mutations have been found in other countries such as India. 18
We analyze and identify the characteristics of circulating SARS‐CoV‐2 mutations present in genomic sequences in Mexico, Belize, and Guatemala, to find out if they have the same molecular characteristics, we also evaluate how these mutations affect the primer design for reverse transcription‐polymerase chain reaction (RT‐PCR) from the Center for Disease Control and Prevention of the United States (CDC US), CDC China, Charité (Germany), Hong Kong University, and the National Institute of Infectious Diseases (Japan). The results indicated the presence of similar mutations in ORF1ab, S (S1, S2 or S2'), ORF 3a, ORF7a, ORF8, and N, as well as in the noncoding (5ʹ‐UTR and 3ʹ‐UTR) and intergenic regions (between ORF3a and E gene) in strains from Mexico, Belize, and Guatemala. Also, we found that primers from the Center for Disease Control and Prevention (CDC US) could present low specificity.
2. METHODS
We analyzed 457 SARS‐CoV‐2 genomic sequences from Mexico, Belize, and Guatemala available in the GISAID database (https://www.gisaid.org/) from May 8 until September 11, 2020. Of these, we only selected the complete sequences with approximately 29.800–29.900 base pairs (bp); 23 from Mexico (EPI_ISL_426362, EPI_ISL_424667, EPI_ISL_455456, EPI_ISL_426364, EPI_ISL_426363, EPI_ISL_424670, EPI_ISL_424673, EPI_ISL_516613, EPI_ISL_516620, EPI_ISL_454555, EPI_ISL_412972, EPI_ISL_452139, EPI_ISL_424672, EPI_ISL_516625, EPI_ISL_424626, EPI_ISL_455434, EPI_ISL_516609, EPI_ISL_496369, EPI_ISL_493348, EPI_ISL_493336, EPI_ISL_516611, EPI_ISL_455438, and EPI_ISL_496374), four from Belize (EPI_ISL_509713, EPI_ISL_509714, EPI_ISL_509712, and EPI_ISL_509711), and 10 from Guatemala (EPI_ISL_509710, EPI_ISL_509700, EPI_ISL_509699, EPI_ISL_509696, EPI_ISL_509695, EPI_ISL_509702, EPI_ISL_509703, EPI_ISL_509697, EPI_ISL_509698, and EPI_ISL_509701).
Genomic alignments were performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) versus the SARS‐CoV‐2 genomic sequence reported from Wuhan, China (NCBI accession number NC_045512.2) as reference. Open reading frames (ORFs) containing the identified variants were translated using the ExPASy Translate Tool (https://web.expasy.org/translate/) using standard code. Variants and their amino acids were used to create a table of variants (Table 2).
Table 2.
Nucleotide variants and amino acid substitutions in SARS‐CoV‐2 genomes in Mexico
| ID Mexico | Nucleotide change | Synonymous/nonsynonymous | Position genome | Amino acid substitution | Position protein | Gene | Product |
|---|---|---|---|---|---|---|---|
| EPI_ISL426362 | C>T | Synonymous | 8782 | S | 2839 | ORF1ab | NSP4 |
| T>A | Nonsynonymous | 9477 | F>Y | 3071 | ORF1ab | NSP4 | |
| C>T | Synonymous | 14805 | Y | 4847 | ORF1ab | NSP10 | |
| C>T | Synonymous | 23280 | I | 573 | S | S1 | |
| G>T | Nonsynonymous | 25979 | G>V | 196 | ORF3a | 3A protein | |
| C>T | Synonymous | 28657 | D | 128 | N | N | |
| C>T | Nonsynonymous | 28863 | S>L | 197 | N | N | |
| T>C | Nonsynonymous | 28144 | L>S | 84 | ORF8 | ORF8b protein | |
| T>C | ‐ | 26232 | ‐ | ‐ | Intergenic | ‐ | |
| EPI_ISL_424667 | C>T | Synonymous | 8782 | S | 2839 | ORF1ab | NSP4 |
| C>T | Synonymous | 17470 | L | 17470 | ORF1ab | Helicase | |
| C>T | Synonymous | 26088 | I | 232 | ORF3a | 3a Protein | |
| T>C | Nonsynonymous | 28144 | L>S | 84 | ORF8 | ORF8b protein | |
| EPI_ISL_455456 | C>T | Synonymous | 26088 | I | 232 | ORF3a | 3a Protein |
| T>C | Nonsynonymous | 28144 | L>S | 84 | ORF8 | ORF8b protein | |
| C>T | Synonymous | 8782 | S | 2839 | ORF1ab | NSP4 | |
| EPI_ISL_426364 | C>T | Synonymous | 8782 | S | 2839 | ORF1ab | NSP4 |
| C>T | Nonsynonymous | 17747 | P>L | 5828 | ORF1ab | Helicase | |
| A>G | Nonsynonymous | 17858 | Y>C | 5865 | ORF1ab | Helicase | |
| C>T | Synonymous | 18060 | L | 5932 | ORF1ab | 3ʹ–5ʹ Exonuclease | |
| C>T | Nonsynonymous | 21707 | H>Y | 49 | S | S1 | |
| C>T | Synonymous | 23422 | V | 620 | S | S1 | |
| A>T | Synonymous | 24694 | G | 1044 | S | S2' | |
| T>C | Nonsynonymous | 28144 | L>S | 84 | ORF8 | ORF8b protein | |
| EPI_ISL_426363 | C>T | Nonsynonymous | 21707 | H>Y | 49 | S | S1 |
| T>C | Nonsynonymous | 28144 | L>S | 84 | ORF8 | ORF8b protein | |
| C>T | Synonymous | 23422 | V | 620 | S | S1 | |
| A>T | Synonymous | 24694 | G | 1044 | S | S2' | |
| C>T | Synonymous | 8782 | S | 2839 | ORF1ab | NSP4 | |
| C>T | Nonsynonymous | 17747 | P>L | 5828 | ORF1ab | Helicase | |
| A>G | Nonsynonymous | 17858 | Y>C | 5865 | ORF1ab | Helicase | |
| C>T | Synonymous | 18060 | L | 5932 | ORF1ab | 3ʹ–5ʹ Exonuclease | |
| EPI_ISL_424670 | T>C | Nonsynonymous | 28144 | L>S | 84 | ORF8 | ORF8B protein |
| C>T | Synonymous | 26088 | I | 232 | ORF3a | 3A protein | |
| C>T | Synonymous | 8782 | S | 2839 | ORF1ab | NSP4 | |
| EPI_ISL_424673 | C>T | Nonsynonymous | 936 | T>I | 224 | ORF1ab | NSP2 |
| C>T | Synonymous | 8782 | S | 2839 | ORF1ab | NSP4 | |
| G>T | Nonsynonymous | 11083 | L>F | 3606 | ORF1ab | NSP6 | |
| C>T | Nonsynonymous | 17747 | P>L | 5828 | ORF1ab | Helicase | |
| A>G | Nonsynonymous | 17858 | Y>C | 5865 | ORF1ab | Helicase | |
| C>T | Synonymous | 18060 | L | 5932 | ORF1ab | 3ʹ–5ʹ Exonuclease | |
| A>T | Synonymous | 24694 | G | 1044 | S | S2' | |
| T>C | Nonsynonymous | 28144 | L>S | 84 | ORF8 | ORF8b protein | |
| G>T | Nonsynonymous | 28812 | S>I | 180 | N | N | |
| EPI_ISL_516613 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Nonsynonymous | 27964 | S>L | 24 | ORF8 | ORF8b protein | |
| C>T | Nonsynonymous | 28087 | A>V | 65 | ORF8 | ORF8b protein | |
| G>T | Nonsynonymous | 25563 | Q>H | 57 | ORF3a | 3a Protein | |
| C>T | Nonsynonymous | 28868 | P>S | 199 | N | N | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Nonsynonymous | 1059 | T>I | 265 | ORF1ab | NSP2 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 3768 | T>I | 1168 | ORF1ab | NSP3 | |
| G>T | Synonymous | 6421 | V | 2052 | ORF1ab | NSP3 | |
| T>A | Nonsynonymous | 6640 | H>Q | 2125 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 8739 | T>I | 2825 | ORF1ab | NSP4 | |
| C>T | Nonsynonymous | 10319 | L>F | 3352 | ORF1ab | 3c‐like proteinase | |
| C>T | Synonymous | 11575 | F | 3770 | ORF1ab | NSP6 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| C>T | Synonymous | 17503 | F | 5746 | ORF1ab | Helicase | |
| A>G | Synonymous | 20263 | L | 6666 | ORF1ab | 2'‐o‐ribose methyltransferase | |
| G>A | Nonsynonymous | 21306 | R>H | 7014 | ORF1ab | 2'‐o‐ribose methyltransferase | |
| EPI_ISL_516620 | C>T | ‐ | 106 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ | |
| G>T | Nonsynonymous | 2809 | R>S | 848 | ORF1ab | NSP3 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Synonymous | 5869 | Y | 1868 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| C>T | Synonymous | 18829 | V | 6188 | ORF1ab | 3ʹ–5ʹ Exonuclease | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Nonsynonymous | 26042 | T>I | 217 | ORF3a | 3a Protein | |
| G>A | Nonsynonymous | 28881 | R>K | 203 | N | N | |
| G>A | Nonsynonymous | 28882 | R>K | 203 | N | N | |
| G>C | Nonsynonymous | 28883 | G>R | 204 | N | N | |
| EPI_ISL_455455 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Nonsynonymous | 27046 | T>M | 175 | M | M | |
| G>A | Nonsynonymous | 28881 | R>K | 203 | N | N | |
| G>A | Nonsynonymous | 28882 | R>K | 203 | N | N | |
| G>C | Nonsynonymous | 28883 | G>R | 204 | N | N | |
| G>T | Nonsynonymous | 29224 | M>I | 317 | N | N | |
| EPI_ISL_412972 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| G>A | Nonsynonymous | 28881 | R>K | 203 | N | N | |
| G>A | Nonsynonymous | 28882 | R>K | 203 | N | N | |
| G>C | Nonsynonymous | 28883 | G>R | 204 | N | N | |
| EPI_ISL_452139 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| G>A | Nonsynonymous | 28881 | R>K | 203 | N | N | |
| G>A | Nonsynonymous | 28882 | R>K | 203 | N | N | |
| G>C | Nonsynonymous | 28883 | G>R | 204 | N | N | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| EPI_ISL_424672 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| A>G | Nonsynonymous | 1308 | N>S | 348 | ORF1ab | NSP2 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| C>T | Nonsynonymous | 17639 | S>L | 5792 | ORF1ab | Helicase | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| G>T | Nonsynonymous | 27506 | G>V | 38 | ORF7a | 7A protein | |
| C>T | Nonsynonymous | 28638 | P>L | 122 | N | N | |
| A>G | ‐ | 29700 | ‐ | ‐ | 3ʹ‐UTR | ‐ | |
| EPI_ISL_516625 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| G>T | Nonsynonymous | 25996 | V>L | 202 | ORF3a | 3a protein | |
| G>T | Nonsynonymous | 29477 | D>Y | 402 | N | N | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Synonymous | 3992 | A | 1043 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 6696 | P>L | 2144 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 7104 | T>I | 2280 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase | |
| EPI_ISL_424626 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Synonymous | 2940 | L | 893 | ORF1ab | NSP3 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| T>A | Nonsynonymous | 6842 | S>T | 2193 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| EPI_ISL_455434 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Nonsynonymous | 1059 | T>I | 265 | ORF1ab | NSP2 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>C | Nonsynonymous | 20756 | S>R | 6831 | ORF1ab | 2'‐O‐Ribose methyltransferase | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| G>T | Nonsynonymous | 25563 | Q>H | 57 | ORF3a | 3a protein | |
| EPI_ISL_516609 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| G>T | Nonsynonymous | 25690 | G>C | 100 | ORF3a | 3a protein | |
| C>T | Nonsynonymous | 28854 | S>L | 194 | N | N | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| T>C | Synonymous | 24076 | G | 838 | S | S2' | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Synonymous | 11074 | F | 3603 | ORF1ab | NSP6 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Nonsynonymous | 16052 | K>E | 5263 | ORF1ab | NSP10 | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase | |
| EPI_ISL_496369 | C>T | ‐ | 241 | ‐ | 5ʹ‐UTR | ‐ | |
| C>T | Nonsynonymous | 28854 | S>L | 194 | N | N | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| A>C | Nonsynonymous | 8805 | N>T | 2847 | ORF1ab | NSP4 | |
| C>T | Synonymous | 11575 | F | 3770 | ORF1ab | NSP6 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| C>T | Synonymous | 16888 | Y | 5541 | ORF1ab | Helicase | |
| C>T | Synonymous | 19030 | H | 6255 | ORF1ab | 3ʹ–5ʹ Exonuclease | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | ‐ | |
| EPI_ISL_493348 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| A>G | Nonsynonymous | 1558 | I>M | 431 | ORF1ab | NSP2 | |
| C>T | Nonsynonymous | 6573 | S>F | 2103 | ORF1ab | NSP3 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | Nsp10 | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase | |
| T>C | Synonymous | 22192 | I | 210 | S | S1 | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| EPI_ISL_493336 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Synonymous | 4582 | N | 1439 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 8175 | A>V | 2637 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase | |
| EPI_ISL_516611 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| G>A | Synonymous | 5668 | E | 1801 | ORF1ab | NSP3 | |
| C>T | Synonymous | 5884 | Y | 1873 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| A>T | Nonsynonymous | 23583 | Y>F | 674 | S | S2 | |
| C>T | Nonsynonymous | 28854 | S>L | 194 | N | N | |
| EPI_ISL_455438 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| G>A | Synonymous | 25183 | E | 1207 | S | S2' | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase | |
| EPI_ISL_496374 | C>T | ‐ | 241 | ‐ | ‐ | 5ʹ‐UTR | ‐ |
| C>T | Nonsynonymous | 28854 | S>L | 194 | N | N | |
| A>G | Nonsynonymous | 23403 | D>G | 614 | S | S1 | |
| C>T | Synonymous | 3037 | F | 924 | ORF1ab | NSP3 | |
| C>T | Nonsynonymous | 14408 | P>L | 4715 | ORF1ab | NSP10 | |
| A>G | Synonymous | 20268 | L | 6668 | ORF1ab | Endornase |
Note: Helicase: nsp13_ZBD, nsp13_TB, and nsp_HEL1core. 3ʹ–5ʹ exonuclease: nsp14A2_ExoN and nsp14B_NMT. endoRNAse: nsp15‐A1 and nsp15B‐NendoU. 2'‐O‐Ribose methyltransferase: nsp16_OMT. 3C‐like proteinase: nsp5A.
The amino acids corresponding to mutations D614G in the spike protein, P4715L in RdRp, and L84S in ORF8 protein were replaced by Visual Molecular Dynamics (VMD) v.1.9.1 (https://www.ks.uiuc.edu/) and visualized with Chimera v. 1.1.12 (https://www.cgl.ucsf.edu/chimera/), taking as templates structures from Protein the Data Bank (PDB) proposed by Zhang et al. 19 For the spike protein, we used 6VSB.pdb, which corresponds to the trimeric protein in open conformation that includes the S1 and S2 subdomains 20 while for the RdRp, we took chain A of 6M71.pdb reported by Gao et al. 21
As of September 09, 2020, the crystallographic structure of the ORF8 protein has not been reported, thus we take the one proposed by the Iterative Threading Assembly Refinement (I‐TASSER) server 22 (QHD43422.pdb). Also, in Mexico, the Berlin test with four oligonucleotides for the RdRp gene (GTGARATGGTCATGTGTGGCGG, FAM‐CAGGTGGAACCTCATCAGGAGATGC‐BBQ, FAM‐CCAGGTGGWACRTCATCMGGTGATGC‐BBQ, CARATGTTAAASACACTATTAGCATA), three for the E gene (ACAGGTACGTTAATAGTTAATAGCGT, FAM‐ACACTAGCCATCCTTACTGCGCTTCG‐BBQ, ATATTGCAGCAGTACGCACACA) and three for the N gene (CACATTGGCACCCGCAATC, FAM‐ACTTCCTCAAGGAACAACATTGCCA‐BBQ, GAGAACGAGAAGAGGCTTG) 23 are the reference in the Institute of Epidemiological Diagnosis and Reference “Dr. Manuel Martínez Báez” (InDRE) of the Secretary of Health of Mexico. In addition, the Institute has approved 53 molecular tests from different world companies for the detection of SARS‐CoV‐2. These tests detect different genes and use different primers with different analytical sensitivity (limit of detection; https://www.gob.mx/cms/uploads/attachment/file/576584/Listado_de_estuches_comerciales_utiles_para_el_diagn_stico_de_SARS-CoV-2.pdf).
On the other hand, the governments of Belize and Guatemala obtain diagnostic tests with different primers and with recommendations from the Pan American Health Organization and the World Health Organization (https://www.pressoffice.gov.bz/government-of-belize-to-procure-covid-19-test-kits-from-cayman-islands/; https://www.paho.org/es/documentos/directrices-laboratorio-para-deteccion-diagnostico-infeccion-con-virus-covid-19). The genomic sequences were aligned with primers used in the diagnosis of COVID‐19 by the RT‐PCR using the Sequence Manipulation Site: PCR Products tool v.2 (https://www.bioinformatics.org/sms2/pcr_products.html) and Primer3Plus v.2.4.2 (https://primer3plus.com/cgi-bin/dev/primer3plus.cgi), including the sequence NC_045512.2 as reference.
The expect‐value (E value) is a statistical parameter that describes the probability of the significance of an alignment. The lower the E‐value, the lower the alignment error; thus, the efficiency of the RT‐PCR amplification could show a higher concentration of product per cycle. We evaluated this and other characteristics of these tests, 24 such as the variations in specificity reported in the melting curve‐based multiplex quantitative RT‐PCR (RT‐qPCR) Assay for Human Coronaviruses 25 and secondary structures for other viruses, 26 for this reason, their specificity and E values were determinaed through the basic local alignment search tool (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) and the melting temperature (Tm) using Oligo Analyzer program development by Sigma‐Aldrich Co. (http://www.oligoevaluator.com/LoginServlet).
3. RESULTS
A total of 187 distinct variants were found in Mexico, 54 in Guatemala, and 39 in Belize. Alignment of genomic sequences of the three countries shared an approximately 99.9% identity. Of these variants, 161 correspond to transitions in Mexico, 50 to transitions in Guatemala and 31 to transitions in Belize (Figure 1) and the rest to transversions. The most common variant in all cases was C>T, which represents an average 52% of the variants that mainly correspond to ORF1ab (Figure 1). The translation revealed 46 amino acid substitutions and 28 synonyms. The substitutions in the noncoding regions included one in C106T and sixteen in C241T of 5ʹ‐UTR, one of 3ʹ‐UTR (A29700G), and also, one intergenic variant located between ORF3a and E (T26232C) in Mexico; while in Guatemala, we identified 13 amino acid substitutions and five synonyms.
Figure 1.
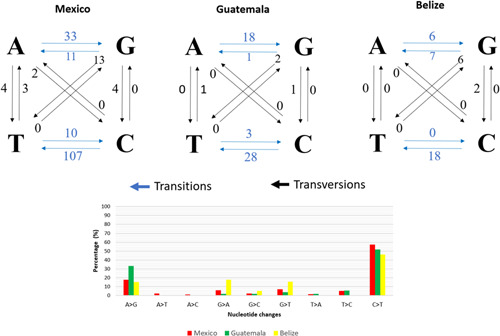
Nucleotide variations and distribution identified in the SARS‐CoV‐2 genomic sequences from Mexico, Guatemala, and Belize
The variants were distributed in six genes, four of which presented the highest number of mutations (almost 86%). The ORF1ab gene presented the maximum number of mutations, 15 amino acid substitutions, and 16 synonymous, followed by the S gene, with six substitutions and five synonymous. The third gene with the most mutations was the N gene, which contained six nonsynonymous amino acid substitutions and one synonym. And finally, in the ORF8 gene, six substitutions were observed. In the ORF7a gene, only one mutation (G38V) was found; while in the ORF3a gene, one substitution (G196V) and two synonymous (I232) were localized. Figure 1 shows the distribution of mutations in SARS‐CoV‐2 genomic sequences, including noncoding and intergenic regions. Furthermore, the distribution of the variants in the sequences, where the most common variants in all sequences were C>T and A>G. The variations observed in each genomic sequence are presented in Tables 2, 3, 4.
Table 3.
Nucleotide variants and amino acid substitutions in SARS‐CoV‐2 genomes in Guatemala

|
Note: In red: mutations exclusive to Guatemala and in green: mutations that coincide with Mexico.
Table 4.
Nucleotide variants and amino acid substitutions in SARS‐CoV‐2 genomes in Belize

|
Note: In red: mutations exclusive to BELIZE and in green: mutations that coincide with MEXICO.
We also localized NSP12 which corresponds to RNA‐dependent RNA polymerase (RdRp) and shows that the active sites are formed by the conserved amino acids D760 and D761 (blue spheres). Likewise, there are residues in the 5 Angstrom region (Å) that surround the active aspartates (in magenta bars, amino acids V763, C622, N695, Y619, E811 and F812); and residues H295, C301, C306, C310, H647, C487, C645 and C646 (in orange sticks) correspond to the binding sites of Zn+ ions. The mutation P4715L (red spheres) corresponds to amino acid 323 in RdRp and as it can be observed, do not affect, or influence active sites (Figure 2A). Also, Figure 2B shows an overall view of the trimeric spike protein. The D614G mutations found in Mexico, Belize and Guatemala were 29 out of a total of 37 strains.
Figure 2.
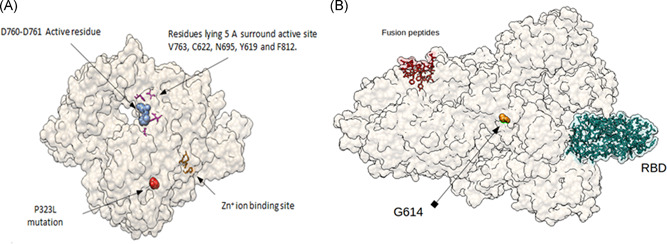
Mutations P4715L and G614G: (A) amino acid P4715 in red spheres, correspondent to residue 323 in RdRp protein of SARS‐CoV‐2. In blue spheres D760 and D761 active sites. In magenta sticks, V763, C622, N695, Y619, and F812 residues. These residues, lying 5 Å surrounding DD active site. In orange sticks, H295, C301, C306, and C310 residues (Zn+ ion binding site). Structure was downloaded from PDB (https://www.rcsb.org/) ID 6M71.pdb, and edited using UCSF Chimera, v.1.12. (B) Amino acid substitution D614G on spike (original model downloaded from rcsb.org, code 6VSB.pdb). G614 mutation is far away from important residues for attachment (RBD in green light spheres) and fusion (fusion peptides, red spheres, residues 788–806) with membrane. Visualization using Chimera UCSF, v.1.12
In Mexico, the genome sequences that we found had four sequences with lineage A5 and clade S; four sequences with linage A1 and clade S; three sequences with linage B1 and clade G; six sequences with linage B1.5 with clade G; one sequence with linage B1 and clade GH; two sequences with lineage B1.1 and clade GR; one sequence with lineage B1.2 and clade GH; one sequence with lineage B1 and clade GR and one sequence with lineage B1.75 with clade G. However, in Guatemala, we found four genome sequences with lineage B1.5 with clades O, four genome sequences with lineage B1.5 and clades G, one genome sequence with lineage B1 and clade H, and another with lineage A3 with clade S. In the genome sequences from Belize, we found two B1 lineages with H clades, and two other genomes with the B1.1 lineage and R clade.
A 3D model of ORF8 protein was obtained using the data of that proposed by Yang Zhang Lab, with its binding sites. ORF8 protein is 121 residues in length. 22 Figure 3 illustrates the spatial distribution of the L84S mutation along with R48, G50, L57, P56, Q72, and Y73 residues, which could be a glycerol binding site. S97 and L98 could be a region that binds to an Hg+ ion likewise, ORF8 could be related to pathogenesis. 14 , 27
Figure 3.
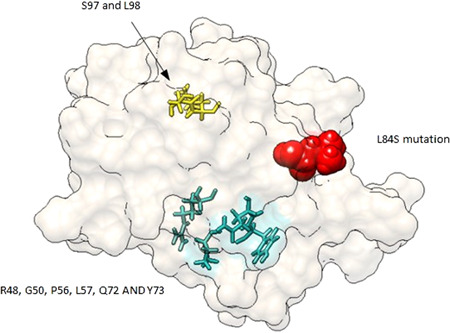
Spatial distribution of the mutation L84S in the ORF8 protein. L84P mutation is represented in red sticks and in wire light sea green color, R48, G50, L57, P56, Q72, and Y73. In yellow sticks, a probable Hg+ ion binding site
The in silico analysis of the primers is used in the RT‐qPCR for detection of SARS‐CoV‐2 (Table 5). The results reported by Yan et al. 28 and Udugama et al., 29 show that most of the primers contain 23%–58% of guanine‐cytosine content (GC). These generate a few dimers, as well as having a very weak or weak secondary structure, and possess a melting temperature (Tm) among 48.7°C–71.7°C. Also, we show the high sensitivity of the primers used because the E value is close to zero, except in the set designed by CDC from the United States where the E value is positive, indicating low sensitivity.
Table 5.
In silico analysis of primers used for diagnosis of COVID‐19
| Institution | Gene | Primers (5ʹ–3ʹ) | Estimated length (bp) | Position | Percentage of identitiesa (%) | Evaluea | Tmb (°C) | GCb (%) | Secondary structureb | Dimersb |
|---|---|---|---|---|---|---|---|---|---|---|
| Center for Disease Control and Prevention of the United States, CDC US 30 | N | GACCCCAAAATCAGCGAAAT | 72 | 28287–28358 | 100 | 3e‐07 | 64.9 | 45 | None | No |
| TCTGGTTACTGCCAGTTGAATCTG | 100 | 2e‐09 | 66.2 | 45.8 | Strong | No | ||||
| TTACAAACATTGGCCGCAAA | 67 | 29164–29230 | 100 | 3e‐07 | 66 | 40 | Very weak | No | ||
| GCGCGACATTCCGAAGAA | 100 | 3e‐06 | 67.5 | 55.6 | Very weak | No | ||||
| GGGAGCCTTGAATACACCAAAA | 72 | 28681–28752 | 100 | 2e‐08 | 65.8 | 45.5 | Very weak | No | ||
| TGTAGCACGATTGCAGCATTG | 100 | 7e‐08 | 67.1 | 47.6 | Weak | No | ||||
| RdRp | AGATTTGGACCTGCGAGCG | 160* | 14413–14572 | 52 | 3.6 | 67.7 | 57.9 | None | No | |
| GAGCGGCTGTCTCCACAAGT | 65 | 16 | 66.9 | 60 | Weak | No | ||||
| Chinese Center for Disease Control and Prevention, China CDC 31 | ORF1ab | CCCTGTGGGTTTTACACTTAA | 119 | 13342–13460 | 100 | 7e‐08 | 60.3 | 42.9 | Moderate | Yes |
| ACGATTGTGCATCAGCTGA | 100 | 1e‐06 | 63.3 | 47.4 | Very weak | No | ||||
| N | GGGGAACTTCTCCTGCTAGAAT | 99** | 28881–28979 | 86 | 2e‐08 | 63.6 | 50 | Weak | No | |
| CAGACATTTTGCTCTCAAGCTG | 100 | 2e‐08 | 63.9 | 45.5 | Weak | No | ||||
| Charité, Germany 29 | RdRp | GTGARATGGTCATGTGTGGCGG | 100* | 15431–15530 | 100 | 1e‐07 | 63.7 | 54.5 | Very weak | No |
| CARATGTTAAASACACTATTAGCATA | 80 | 1e‐07 | 48.7 | 23.1 | Very Weak | No | ||||
| E | ACAGGTACGTTAATAGTTAATAGCGT | 113 | 26269–26381 | 100 | 1e‐10 | 59.2 | 34.6 | Weak | No | |
| ATATTGCAGCAGTACGCACACA | 100 | 2e‐08 | 65.4 | 45.5 | Weak | No | ||||
| Hong Kong University 32 | ORF1b‐nsp14 | TGGGGYTTTACRGGTAACCT | 132 | 18778–18909 | 100 | 2e‐05 | 65.7 | 45.0 | None | No |
| AACRCGCTTAACAAAGCACTC | 95 | 5e‐07 | 60.1 | 42.9 | Moderate | No | ||||
| N | TAATCAGACAAGGAACTGATTA | 110 | 29145–29254 | 100 | 2e‐08 | 55.5 | 31.8 | Moderate | No | |
| CGAAGGTGTGACTTCCATG | 100 | 1e‐06 | 61.8 | 52.8 | Moderate | Yes | ||||
| National Institute of Infectious Diseases, Japan 33 | N | AAATTTTGGGGACCAGGAAC | 158* | 29125–29282 | 100 | 3e‐07 | 63.7 | 45 | Weak | No |
| TGGCAGCTGTGTAGGTCAAC | 95 | 7e‐05 | 64.0 | 55.0 | None | No | ||||
| ORF1a | TTCGGATGCTCGAACTGCACC | 413 | 484–896 | 100 | 7e‐08 | 71.7 | 57.1 | Moderate | No | |
| CTTTACCAGCACGTGCTAGAAGG | 100 | 6e‐09 | 65.8 | 52.2 | Weak | No | ||||
| CTCGAACTGCACCTCATGG | 346 | 492–837 | 100 | 1e‐06 | 64.7 | 57.9 | None | No | ||
| CAGAAGTTGTTATCGACATAGC | 100 | 2e‐08 | 58.0 | 40.9 | Very weak | No | ||||
| S | TTGGCAAAATTCAAGACTCACTTT | 547 | 24354–24900 | 100 | 2e‐09 | 64.6 | 33.3 | Very weak | No | |
| TGTGGTTCATAAAAATTCCTTTGTG | 100 | 4e‐10 | 64.5 | 32 | None | No | ||||
| CTCAAGACTCACTTTCTTCCAC | 494* | 24363–24856 | 95 | 8e‐08 | 59.6 | 45.5 | Weak | No | ||
| ATTTGAAACAAAGACACCTTCAC | 100 | 6e‐09 | 60.9 | 34.8 | Weak | No | ||||
| National Institute of Health, Thailand 34 | N | CGTTTGGTGGACCCTCAGAT | 57 | 28320–28376 | 100 | 3e‐07 | 66.2 | 55.0 | None | No |
| CCCCACTGCGTTCTCCATT | 100 | 1e‐06 | 67.3 | 57.9 | None | No |
Note: Forward and reverse.
Calculated by BLAST.
Calculated by Oligo Evaluator (Sigma‐Aldrich Co.).
Estimated by alignment using Clustal Omega.
No product in the EPI_ISL_412972 sequence.
Primers designed by the Chinese Center for Disease Control and Prevention (China CDC), Charité (Germany), and the National Institute of Infectious Diseases (Japan) for the RdRp, N and S genes give a product using Primer3Plus, and similarly, multiple alignments (Table 5). This indicates a high sensitivity to these primers, however, four sets of primers used by the Centers for Disease Control and Prevention (CDC US) present low sensitivity to RdRp, S, and N amplicons. They are misaligned with the primer forward that can recognize 11/19 base pairs causing low sensitivity.
4. DISCUSSION
The genomic sequences of first SARS‐CoV‐2 strain in Mexico (hCoV/Mexico/CDMX/InDRE_01/2020) show high identity with the sequence reported in China Wuhan‐Hu‐1 (NC_045512.2), it only differs in seven nucleotide substitutions. 8 This high sequence identity has been attributed to the recent spread of the virus in humans, suggesting a common lineage and source. 10 , 35 Our results show 84% more transitions than transversions. The effects of transition and transversion mutations have been studied in influenza (H1N1) and the human immunodeficiency virus virus, these studies conclude that it is likely that transversions cause radicals changes in amino acids 29 , 36 that could be involved in the high genomic conservation of the new coronavirus.
Different reports of variant analysis of SARS‐CoV‐2 genomes show similar results to those of Mexico, Belize, and Guatemala, for example, Koyama et al. 37 reported that C>T was the most common variant; they also identified mutations in C3037T (F924), C14408T (P4715L) in RdRp, ORF1ab and, A23403G (D614G) in the spike, this was reported mainly in Europe and the United States. 35 , 38 Additionally, D614G formed the largest phylogenic clade including C241T, F924, and P4715L, while the second largest clade was T28144C (L84S) present in ORF8, which was reported days after the outbreak in travelers from Wuhan. Among the L84S clade, the L84S/C17747T (P5828L) subclade was more frequent in the United States. 37 , 38 Regarding the P4715L mutation, it corresponds to amino acid residue 323 in the RdRp; however, it does not affect or influence the active sites (Figure 2A), according to Yang et al., 39 who predicted active site residues in the same region. Similarly, Khailany et al., 40 reported that C>T was the most frequent mutation observed and also found C8782T (S2839) mutations in ORF1ab and T28144C (L84S) in ORF8 genes. We did not identify the mutation in the C29095T (F274) N gene. 39
Moreover, current evidence of the mutation of an aspartate (D) at position 614 to glycine (G) in spike is possibly related to increased infectivity, 41 but also gives a more pathogenic strain. The G614G mutation alters the fusion of the cell membrane and the data reveals that it is located in a highly glycosylated region that also allows the identification of two viral clades. 39 , 42 The aspartate strain has been found in cases reported on the West Coast of United States, while the glycine strain has been reported on the East Coast. 43 In Mexico, our study revealed the presence of the D614 in samples, identified in a smaller number of cases at the moment of sequencing.
SARS‐CoV‐2 genomes have two major lineages with sublineages A (1, 2, 3, and 5) and B (1, 1.1, 2, 3, and 4). 44 In Mexico, Taboada et al. 45 found that the lineages changed from late February to March from A2 to B1. Considering that we selected only the complete sequences, we found a higher proportion of B lineages and clades G as in Guatemala and Belize.
Lineages have been associated with certain clinical manifestations. 35 Lineage A has sequences from Europe and conforms to a human coronavirus (HCoV‐OC43 and HCoV‐HKU1), may be associated with self‐limiting upper respiratory infections, and occasionally, with lower respiratory tract infections; while lineage B may cause severe lower respiratory tract infections with acute respiratory distress syndrome and extrapulmonary manifestations. 43
We found a mutation in the noncoding regions 5ʹ‐UTR (C241T), this type of mutation in UTRs of SARS‐Cov‐2 has been studied recently, suggesting that C241T in 5ʹ‐UTR appeared early during the outbreak, and could be key in virus replication and RNA folding, 46 affecting the steam‐loop 5b (SL5b) 47 , 48 and the host defense. 49 The intergenic mutation A29700G located between ORF3a and E genes might emerge through adenosine deaminase acting on RNA (ADAR) and could be important in the antiviral response 28 , 35 , 50 reducing the stability in the RNA fold. 29
Activation of the SARS‐CoV‐2 spike protein via sequential proteolytic cleavage can be at two distinct sites. For many CoVs, the spike protein is cleaved at the boundary between the S1 and S2 subunits (residues 685 and 686), which remain non covalently bound in the prefusion conformation while for all CoVs, the spike is further cleaved by host proteases at the so‐called S2 site located immediately upstream of the fusion peptide (residues 788–806). 51 Also, RBD is constituted by residues 333–527 and belongs to a region that attaches to hACE2, a highly conserved cryptic epitope in the receptor‐binding domains of SARS‐CoV‐2 and SARS‐CoV. 52 As we can see, the D614G mutation is not between important regions known, but recently has been associated with high prevalence, from <1% in January to 69% in March. The global spread of SARS‐CoV‐2 subtype with spike protein D614G mutation is shaped by Human Genomic Variations that regulate the expression of TMPRSS2 and MX1 genes, although the mechanism by which such a phenomenon occurs is not clear yet. 53
The ORF8 protein has 121 residues in length and very little is known about its function. Nevertheless, Zhang et al. 22 have proposed a 3D model along with its binding sites. The L84S mutation in genomic sequences in Mexico can indicate that the circulating strain shows a different characteristic, like the Wuhan strain. However, the number of analyzed samples is a limitation of guarantee. Due to several reports of low sensitivity in the RT‐PCR test, which is not considered the gold standard for diagnosis of COVID‐19, 54 , 55 we analyzed a predictive evaluation of the sensitivity of the primers used (Table 5).
The N gene has a high degree of conservation in coronaviruses, 56 however, in our study, the N gene is the third with the highest number of mutations, following the ORF1ab and S genes. The results suggest a high sensitivity of the primers. Nevertheless, that designed by CDC (US) for the RdRp gene, could generate low sensitivity of the forward and reverse primer related to the few complementary bases already reported. 57 Besides this, the mutations AAC (28881–28883) in the N gene could decrease sensitivity because they are part of the region where the primer is attached to the template strand. As a consequence, it is not recognized by Primer3Plus. We considered that not all primers possess high sensitivity for diagnosing of COVID‐19, and the mutations in the genomic sequences may decrease just the sensitivity. Recently in China, it has been reported that nonspecific primers may amplify high concentrations of human cathepsin C (CTSC) and messenger RNA in the tonsils. This could cause interference in diagnosing COVID‐19, 58 which could explain why RT‐PCR should not be considered the gold standard. Furthermore, the test presents other problems, such as those related to errors in swab tests, causing improper extraction of viral RNA. 59 A comprehensive review of the diagnosis of COVID‐19 can be found at Yan et al. 22 and Li and Ren. 60
As time passes, mutations in the genomic sequences of SARS‐CoV‐2 could appear in the highly conserved regions and the effectiveness of the diagnostic methods could be compromised. Factors, such as correct sampling, conservation and transport of the sample, extraction 61 quality and integrity of RNA, 37 calibration of the thermocycler, and optimal amplification conditions, may influence the results. Likewise, the design of primers in conserved regions is essential, and experimental studies are required for a wider understanding. Finally, several questions related to the mutations remain, a very important one is whether these mutations are related to the observed case‐fatality rate. Until September 13, 2020, Mexico has a very high case‐fatality rate of 10.6%, Belize 1.3%, and Guatemala 3.6%, in addition to a high number of people with comorbidities. 62
CONFLICT OF INTEREST
The authors declare that we have no conflicts of interest.
CONTRIBUTION STATEMENT
Formal analysis, investigation, methodology, data curation, software, writing: original draft, writing: review and editing: María T. Hernández‐Huerta and Laura P.‐C. Mayoral. Formal analysis, investigation and software: Carlos R. Díaz. Formal analysis, investigation and funding acquisition: Margarito Martínez Cruz. Formal analysis and investigation: Gabriel Mayoral‐Andrade. Resources and investigation: Luis M. S. Navarro. Investigation: María D. S. Pina Canseco, Ruth M. Cruz, Eduardo P.‐C. Mayoral, and Gabriela V. Martínez. Writing: review and editing: Eli C. Parada. Funding acquisition and investigation: Alma D. P. Santiago. Conceptualization, supervision, visualization, project administration, writing: review and editing: Eduardo Pérez‐Campos and Carlos A. Matias‐Cervantes.
ACKNOWLEDGEMENTS
The authors acknowledge all the people and laboratories responsible for the sequencing and submitting to the GISAID and NCBI databases for public consultation. Without them, this study could not have been carried out, likewise, we declare that the rights to the sequences belong to the people who participated in it, we have not participated in this. Additionally, we recognize the efforts of all the personnel involved in care of patients with COVID‐19. Also, the authors thank Charlotte Grundy for her assistance and the National Technology of Mexico (TecNM) project 8703.20‐P.
Hernández‐Huerta MT, Pérez‐Campos Mayoral L, Romero Díaz C, et al. Analysis of SARS‐CoV‐2 mutations in Mexico, Belize, and isolated regions of Guatemala and its implication in the diagnosis. J Med Virol. 2021;93:2099–2114. 10.1002/jmv.26591
María T. Hernández‐Huerta and Laura P.‐C. Mayoral contributed equally to this study.
Contributor Information
Eduardo Pérez‐Campos, Email: perezcampos@prodigy.net.mx.
Carlos Alberto Matias‐Cervantes, Email: carloscervantes.ox@outlook.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chan JFW, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person‐to‐person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elbe S, Buckland‐Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. 2017;1(1):33–46. 10.1002/gch2.1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Institute for Health Metrics and Evaluation . COVID‐19 projections. University of Washington. 2020. https://covid19.healthdata.org/global?view=total-deaths&tab=trend [Google Scholar]
- 6. Eybpoosh S, Haghdoost AA, Mostafavi E, Bahrampour A, Azadmanesh K, Zolala F. Molecular epidemiology of infectious diseases. Electron Physician. 2017;9(8):5149–5158. 10.19082/5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ornelas‐Aguirre JM. El nuevo coronavirus que llegó de Oriente: análisis de la epidemia inicial en México [The new coronavirus that came from the East: analysis of the initial epidemic in Mexico]. Gac Med Mex. 2020;156:209–217. 10.24875/GMM.20000165 [DOI] [PubMed] [Google Scholar]
- 8. Garcés‐Ayala F, Araiza‐Rodríguez A, Mendieta‐Condado E, et al. Full genome sequence of the first SARS‐CoV‐2 detected in Mexico. Arch Virol. 2020;165(9):2095–2098. 10.1007/s00705-020-04695-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181–192. 10.1038/s41579-018-0118-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan JFW, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–236. 10.1080/22221751.2020.1719902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. Accessory proteins of SARS‐CoV and other coronaviruses. Antiviral Res. 2014;109:97–109. 10.1016/j.antiviral.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baranov PV, Henderson CM, Anderson CB, Gesteland RF, Atkins JF, Howard MT. Programmed ribosomal frameshifting in decoding the SARS‐CoV genome. Virology. 2005;332(2):498–510. 10.1016/j.virol.2004.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang X, Pearce R, Zhang Y. De novo design of protein peptides to block association of the SARS‐CoV‐2 spike protein with human ACE2. Aging. 2020;12(12):11263–11276. 10.18632/aging.103416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He Y, Zhou Y, Liu S, et al. Receptor‐binding domain of SARS‐CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem Biophys Res Commun. 2004;324(2):773–781. 10.1016/j.bbrc.2004.09.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lokman SM, Rasheduzzaman M, Salauddin A, et al. Exploring the genomic and proteomic variations of SARS‐CoV‐2 spike glycoprotein: a computational biology approach. Infect Genet Evol. 2020;84:104389. 10.1016/j.meegid.2020.104389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Issa E, Merhi G, Panossian B, Salloum T. Tokajian S. SARS‐CoV‐2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems. 2020;5(3):e00266‐20. 10.1128/mSystems.00266-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hassan SS, Choudhury PP, Basu P, Jana SS. Molecular conservation and differential mutation on ORF3a gene in Indian SARS‐CoV2 genomes. Genomics. 2020;112(5):3226–3237. 10.1016/j.ygeno.2020.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Zhang Research Group. Zhang Lab. University of Michigan . https://zhanglab.ccmb.med.umich.edu/COVID-19/
- 20. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao Y, Yan L, Huang Y, et al. SARS‐Cov‐2 RNA‐dependent RNA polymerase in complex with cofactors. RCSB PBD. EMDataResource: EMD‐30127. 2020. 10.2210/pdb6M71/pdb [DOI] [Google Scholar]
- 22. Yang Zhang Research Group. Zhang Lab. University of Michigan. https://zhanglab.ccmb.med.umich.edu/
- 23. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Euro Surveill. 2020;25(3):2000045. 10.2807/1560-7917.ES.2020.25.3.2000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Houng HSH, Norwood D, Ludwig GV, Sun W, Lin M, Vaughn DW. Development and evaluation of an efficient 3'‐noncoding region based SARS coronavirus (SARS‐CoV) RT‐PCR assay for detection of SARS‐CoV infections. J Virol Methods. 2004;120(1):33–40. 10.1016/j.jviromet.2004.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wan Z, Zhang Y, He Z, et al. A melting curve‐based multiplex RT‐qPCR assay for simultaneous detection of four human coronaviruses. Int J Mol Sci. 2016;17(11):1880. 10.3390/ijms17111880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chou KX, Williams‐Hill DM. Improved TaqMan real‐time assays for detecting hepatitis A virus. J Virol Methods. 2018;254:46–50. 10.1016/j.jviromet.2018.01.014 [DOI] [PubMed] [Google Scholar]
- 27. Chen S, Zheng X, Zhu J, et al. Extended ORF8 gene region is valuable in the epidemiological investigation of severe acute respiratory syndrome‐similar coronavirus. J Infect Dis. 2020;222(2):223–233. 10.1093/infdis/jiaa278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan Y, Chang L, Wang L. Laboratory testing of SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 (2019‐nCoV): current status, challenges, and countermeasures. Rev Med Virol. 2020;30(3):e2106. 10.1002/rmv.2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Udugama B, Kadhiresan P, Kozlowski HN, et al. Diagnosing COVID‐19: the disease and tools for detection. ACS Nano. 2020;14(4):3822–3835. 10.1021/acsnano.0c02624 [DOI] [PubMed] [Google Scholar]
- 30. U.S. CDC . CDC 2019‐Novel coronavirus (2019‐nCoV) real‐time RT‐PCR diagnostic panel. Division of Viral Diseases, U.S. Centers for Disease Control and Prevention, Atlanta. 2020. [Google Scholar]
- 31. China CDC . Specific primers and probes for detection 2019 novel coronavirus. China National Institute For Viral Disease Control and Prevention, Beijing. 2020. [Google Scholar]
- 32. Hong Kong University . Detection of 2019 novel coronavirus (2019‐nCoV) in suspected human cases by RT‐PCR. School of Public Health. Hong Kong University, Hong Kong. 2020. [Google Scholar]
- 33. Naganori N, Shirato K, Katano H, et al. Detection of second case of 2019‐nCoV infection in Japan. Department of Virology III, National Institute of Infectious Diseases, Japan. 2020. [Google Scholar]
- 34. National Institute of Health . Diagnostic detection of novel coronavirus 2019 by real time RT‐PCR. Department of Medical Sciences, Ministry of Public Health, Thailand. 2020. [Google Scholar]
- 35. Gralinski LE, Menachery VD. Return of the coronavirus: 2019‐nCoV. Viruses. 2020;12(2):135. 10.3390/v12020135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lyons DM, Lauring AS. Evidence for the selective basis of transition‐to‐transversion substitution bias in two RNA viruses. Mol Biol Evol. 2017;34(12):3205–3215. 10.1093/molbev/msx251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koyama T, Platt D, Parida L. Variant analysis of COVID‐19 genomes. Bull World Health Organ. 2020;98(7):495–504. 10.2471/BLT.20.253591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mishra A, Pandey AK, Gupta P, et al. Mutation landscape of SARS‐CoV‐2 reveals three mutually exclusive clusters of leading and trailing single nucleotide substitutions. bioRxiv. 2020;2020:1–18. 10.1101/2020.05.07.082768 [DOI] [Google Scholar]
- 39. Yang J, Roy A, Zhang Y. Protein‐ligand binding site recognition using complementary binding‐specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29(20):2588–2595. 10.1093/bioinformatics/btt447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS‐CoV‐2. Gene Rep. 2020;19:100682. 10.1016/j.genrep.2020.100682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grubaugh ND, Hanage WP, Rasmussen AL. Making sense of mutation: what D614G means for the COVID‐19 pandemic remains unclear. Cell. 2020;182(4):794–795. 10.1016/j.cell.2020.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Becerra‐Flores M, Cardozo T. SARS‐CoV‐2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract. 2020;74:e13525. 10.1111/ijcp.13525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brufsky A. Distinct viral clades of SARS‐CoV‐2: implications for modeling of viral spread. J Med Virol. 2020;92:1386–1390. 10.1002/jmv.25902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rambaut A, Holmes EC, O′Toole Á, et al. A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5(11):1403–1407. 10.1038/s41564-020-0770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taboada B, Vazquez‐Perez JA, Muñoz‐Medina JE, et al. Genomic analysis of early SARS‐CoV‐2 variants introduced in Mexico. J Virol. 2020;94(18):e01056–20. 10.1128/JVI.01056-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rangan R, Zheludev IN, Hagey RJ, et al. RNA genome conservation and secondary structure in SARS‐CoV‐2 and SARS‐related viruses: a first look. RNA. 2020;26(8):937–959. 10.1261/rna.076141.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang C, Zheng W, Huang X, Bell EW, Zhou X, Zhang Y. Protein structure and sequence reanalysis of 2019‐nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV‐1. J Proteome Res. 2020;19(4):1351–1360. 10.1021/acs.jproteome.0c00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andrews RJ, Peterson JM, Haniff HS, et al. An in silico map of the SARS‐CoV‐2 RNA structurome. bioRxiv. 2020;2020:1–18. 10.1101/2020.04.17.045161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ryder SP. Analysis of rapidly emerging variants in structured regions of the SARS‐CoV‐2 genome. bioRxiv. 2020;2020:1–40. 10.1101/2020.05.27.120105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tomaselli S, Galeano F, Locatelli F, Gallo A. ADARs and the balance game between virus infection and innate immune cell response. Curr Issues Mol Biol. 2015;17:37–51. [PubMed] [Google Scholar]
- 51. Dramé M, Tabue Teguo M, Proye E, et al. Should RT‐PCR be considered a gold standard in the diagnosis of COVID‐19? J Med Virol. 2020;92:2312–2313. 10.1002/jmv.25996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yuan M, Wu NC, Zhu X, et al. A highly conserved cryptic epitope in the receptor binding domains of SARS‐CoV‐2 and SARS‐CoV. Science. 2020;368(6491):630–633. 10.1126/science.abb7269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bhattacharyya C, Das C, Ghosh A, Singh AK, et al. Global spread of SARS‐CoV‐2 subtype with spike protein mutation D614G is shaped by human genomic variations that regulate expression of TMPRSS2 and MX1 genes. bioRxiv. 2020;2020:1–30. 10.1101/2020.05.04.075911 [DOI] [Google Scholar]
- 54. Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci USA. 2009;106(14):5871–5876. 10.1073/pnas.0809524106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hernández‐Huerta MT, Pérez‐Campos Mayoral L, Sánchez Navarro LM, et al. Should RT‐PCR be considered a gold standard in the diagnosis of COVID‐19? J Med Virol. 2020;2020:26228. 10.1002/jmv.26228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johns Hopkins Center for Health Security . Fact sheet: comparison of national RT‐PCR primers, probes, and protocols for SARS‐CoV‐2 diagnostics. https://www.centerforhealthsecurity.org/resources/COVID-19/COVID-19-fact-sheets/200410-RT-PCR.pdf
- 57. Vogels CBF, Brito AF, Wyllie AL, et al. Analytical sensitivity and efficiency comparisons of SARS‐CoV‐2 RT–qPCR primer–probe sets. Nat Microbiol. 2020;5:1299–1305. 10.1038/s41564-020-0761-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu W. Non‐specific primers reveal false‐negative risk in detection of COVID‐19 infections. medRxiv. 2020;2020:1–13. 10.1101/2020.04.07.20056804 [DOI] [Google Scholar]
- 59. Xie X, Zhong Z, Zhao W, Zheng C, Wang F, Liu J. Chest CT for typical coronavirus disease 2019 (COVID‐19) pneumonia: relationship to negative RT‐PCR testing. Radiology. 2020;296(2):E41–E45. 10.1148/radiol.2020200343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li C, Ren L. Recent progress on the diagnosis of 2019 novel coronavirus. Transbound Emerg Dis. 2020;67(4):1485–1491. 10.1111/tbed.13620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grant PR, Turner MA, Shin GY, Nastouli E, Levett LJ. Extraction‐free COVID‐19 (SARS‐CoV‐2) diagnosis by RTPCR PCR to increase capacity for national testing programme during a pandemic. BioRxiv. 2020;2020:1–6. 10.1101/2020.04.06.028316 [DOI] [Google Scholar]
- 62. Johns Hopkins University . Mortality analyses, maps & trends. 2020. https://coronavirus.jhu.edu/data/mortality
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.