

Abstract
The novel betacoronavirus severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) emerged at the end of 2019 and caused the coronavirus disease 19 (COVID‐19) pandemic due to its high transmissibility and early immunosuppression. Previous studies on other betacoronaviruses suggested that betacoronavirus infection is associated with the host autophagy pathway. However, it is unclear whether any components of autophagy or virophagy can be therapeutic targets for COVID‐19 treatment. In this report, we examined the antiviral effect of four well‐characterized small molecule inhibitors that target the key cellular factors involved in key steps of the autophagy pathway. They include small molecules targeting the ULK1/Atg1 complex involved in the induction stage of autophagy (ULK1 inhibitor SBI0206965), the ATG14/Beclin1/VPS34 complex involved in the nucleation step of autophagy (class III PI3‐kinase inhibitor VPS34‐IN1), and a widely‐used autophagy inhibitor that persistently inhibits class I and temporary inhibits class III PI3‐kinase (3‐MA) and a clinically approved autophagy inhibitor that suppresses autophagy by inhibiting lysosomal acidification and prevents the formation of autophagolysosome (HCQ). Surprisingly, not all the tested autophagy inhibitors suppressed SARS‐CoV‐2 infection. We showed that inhibition of class III PI3‐kinase involved in the initiation step of both canonical and noncanonical autophagy potently suppressed SARS‐CoV‐2 at a nano‐molar level. In addition, this specific kinase inhibitor VPS34‐IN1, and its bioavailable analogue VVPS34‐IN1, potently inhibited SARS‐CoV‐2 infection in ex vivo human lung tissues. Taken together, class III PI3‐kinase may be a possible target for COVID‐19 therapeutic development.
Keywords: class III PI3‐K, COVID‐19, ex vivo human lung tissues, SARS‐CoV‐2, Vps34
1. INTRODUCTION
Coronavirus is an enveloped positive‐strand RNA virus belonging to the family of Coronaviridae, under the order of Nidovirudae. It is classified into four genera, namely the alpha‐, beta‐, gamma‐ and delta‐coronavirus. There have been seven human coronaviruses (HCoVs) identified by far. Four of them, including the alphacoronaviruses HCoV‐229E and HCoV‐NL63, and betacoronaviruses HCoV‐HKU1 and HCoV‐OC43, are among the major causes of the common cold. The rest of them, including severe acute respiratory syndrome coronavirus (SARS‐CoV), novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), and middle east respiratory syndrome coronavirus (MERS‐CoV), are all highly pathogenic betacoronaviruses. SARS‐CoV and SARS‐CoV‐2 belong to the subgenus Sarbecovirus, while MERS‐CoV belongs to the subgenus Merbecovirus. 1 , 2 It is alarming that SARS‐CoV‐2 that causes the coronavirus disease 19 (COVID‐19) pandemic is already the third highly pathogenic human coronavirus since the first appearance of SARS‐CoV in 2003. More understanding of the virology of coronaviruses is urgently needed for combating the COVID‐19 pandemic and the possible upcoming coronavirus diseases.
Autophagy is an essential cellular pathway important for the clearance and recycling of intracellular materials. The activation of autophagy is induced by metabolic stress including nutrient deprivation and hypoxia, which causes inhibition of the autophagy negative regulator mammalian target of rapamycin (mTOR). 3 , 4 Suppression of mTOR results in activation of a key molecule Unc‐51‐like autophagy activating kinase 1 (ULK1), which translocates to the endoplasmic reticulum (ER) for the initiation of autophagy. ULK1 further recruits the class III phosphoinositide 3‐kinase (PI3‐K) complex consisting of Beclin‐1, VPS34, and ATG14, leading to phagophore nucleation and formation of a crescent‐shaped double‐membrane structure known as omegasome budded out from the ER. The class III PI3‐K complex mediates accumulation of phosphatidylinositol 3‐phosphate (PIP3) on the surface of the omegasome, leading to the recruitment of LC3, conjugation of phosphatidylethanolamine (PE) onto LC3‐I to form LC3‐II and membrane elongation of the phagophore. The phagophore eventually seals to form the autophagosome, which then fuses with the lysosome for breaking down the enclosed content. It is noteworthy that autophagy, apart from the recycling of useful materials, is also an important mechanism for the elimination of intracellular pathogens including viruses, a process termed virophagy. 5 , 6
Although autophagy is involved in the clearance of invading pathogens, some viruses are able to evade, and even benefit from autophagy. 5 , 6 , 7 , 8 , 9 It has been speculated that coronavirus replication is associated with autophagy, although the exact interaction is still poorly understood. Coronavirus infection causes the formation of numerous membranous structures including double‐membrane vesicles (DMVs), which is coincidentally a hallmark of autophagy. 10 , 11 , 12 Evidence, although still debatable, showed that coronavirus nsp6 can mediate the formation of DMVs. 13 , 14 , 15 , 16 Moreover, SARS‐CoV nsp6 has been reported to partially co‐localize with LC3 in DMVs. 17 In view of the implied relationship between autophagy and coronavirus, previous studies have attempted to elucidate the possibility of inhibiting coronavirus infection by manipulating the autophagy pathway. 18 Nonetheless, contrasting results were observed across studies using various gain‐of‐function and loss‐of‐function approaches. 18 , 19 On one hand, this might be due to discrepancies between the systems used in these studies, it is also possible that coronaviruses may take advantage of only particular component(s) of the autophagy pathway instead of the entire autophagy machinery. It is therefore worthwhile to dissect the molecular interactions between coronavirus and autophagy.
In view of the association between autophagy and coronavirus infection, we speculated whether suppression of coronavirus replication could be attained by interrupting the autophagy pathway. Four well‐characterized commonly used autophagy inhibitors targeting various steps of autophagy were selected for testing their inhibitory effect on SARS‐CoV‐2 replication. These are small molecules targeting (1) ULK1 (a key protein in the ULK1/Atg1 complex involved in the induction stage of autophagy, (2) VPS34 (a class III PI3‐kinase in the ATG14/Beclin1/VPS34 complex involved in the nucleation step of autophagy), and (3) a widely‐used autophagy inhibitor (3‐methyladenine [3‐MA]) that persistently inhibits class I PI3‐K and temporarily inhibits class III PI3‐K, and (4) a clinically approved autophagy inhibitor (hydroxychloroquine [HCQ]) that suppresses the autophagy by inhibiting lysosomal acidification and prevents the degradation of autophagolysosome. Interestingly, inhibition of class III PI3‐K by VPS34‐IN1, but not the upstream ULK1, resulted in potent suppression of SARS‐CoV‐2 infection in a highly susceptible VeroE6 cell‐line at submicromolar concentration. Most importantly, this specific class III PI3‐kinase inhibitor VPS34‐IN1, and its bioavailable analogue VVPS34‐IN1, potently inhibited SARS‐CoV‐2 infection in ex vivo human lung tissues culture. The low half‐maximal effective dose of VPS34‐IN1 and the presence of the bioavailable analogue with known pharmacokinetic properties support further examination on the in vivo inhibition of class III PI3‐K as COVID‐19 therapeutics. Taken together, further investigation on how coronaviruses exploit host autophagy machinery will reveal a new avenue for the development of effective COVID‐19 antivirals.
2. MATERIALS AND METHODS
2.1. Cells, viruses, and small molecule inhibitors
VeroE6 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Gibco) at 37°C with 5% CO2. SARS‐CoV‐2 HKU‐001a (GenBank accession number: MT230904) originally derived from a nasopharyngeal aspirate of a COVID‐19 patient was propagated in VeroE6 cells and titered by plaque assay using VeroE6 cells. All SARS‐CoV‐2 infection experiments were performed in a Biosafety level 3 laboratory at the Department of Microbiology, HKU. Autophagy inhibitors SBI‐0206965 (Selleckchem), Vps34‐IN1 (Selleckchem), VVps34‐IN1 (Selleckchem), 3‐MA (Cayman Chemical), and HCQ (Cayman Chemical) were used at the indicated conditions.
2.2. Immunofluorescence staining
Cells seeded on chamber slides were fixed with 4% paraformaldehyde, NP‐40 permeabilized, and blocked with 5% normal donkey serum (Jackson ImmunoResearch). Viral NP proteins were stained using our in‐house mouse anti‐NP antibody, and then Alexa Flour‐488 conjugated donkey anti‐mouse immunoglobulin G (Abcam). The immunofluorescence signals were detected using a Carl Zeiss LSM 880 confocal microscope (Zeiss).
2.3. Cytotoxicity detection assay
Cytotoxicity was assayed by an LDH‐Glo Cytotoxicity Assay kit (Promega). Culture supernatant diluted 1:100 with lactate dehydrogenase (LDH) storage buffer was mixed and incubated with an equal volume of LDH detection reagent. After 60 min of incubation at room temperature, luciferase activity was measured using a multi‐well plate reader (Beckman Coulter).
2.4. Human ex vivo lung tissue culture
Human lung tissues were obtained with written consent from patients undergoing surgical operations at the Queen Mary Hospital, Hong Kong. This study has been approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (UW13‐364). The culture method has been described previously. 20 Briefly, freshly obtained tissues were dissected into small cubes and maintained in a basal medium of Advanced DMEM:F12 (Gibco) supplemented with 2 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) (Gibco), 1× GlutaMAX (Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μg/ml ciprofloxacin, 20 μg/ml vancomycin, 50 μg/ml amikacin, and 50 μg/ml nystatin at 37°C with 5% CO2. Tissues were treated with the indicated drugs overnight, followed by SARS‐CoV‐2 infection at 2 × 106 PFU/well. Six hours postinoculation, the inoculum was removed, and the tissue cubes were thoroughly washed with phosphate‐buffered saline. Infected tissues were cultured in a basal medium for 48 h and supernatant was collected for plaque assay.
3. RESULTS
3.1. Suppression of class III PI3‐K, but not ULK1, inhibited SARS‐CoV‐2 infection
Autophagy is a multi‐step mechanism for clearing and recycling of cellular materials (Figure 1A). Although it has been implicated that autophagy plays an intimate role with coronavirus infection, the interplay between autophagy and SARS‐CoV‐2 infection is poorly understood. 18 , 19 To dissect the involvement of autophagy during SARS‐CoV‐2 infection, we examined the effect of four autophagy inhibitors targeting different steps of autophagy on SARS‐CoV‐2 infection in vitro. VeroE6 cells supportive to SARS‐CoV‐2 infection were pretreated with either 20 μM of SBI‐0206965 (ULK1 inhibitor), 10 μM of Vps34‐IN1 (Vps34 inhibitor), 5 mM of 3‐MA (class I PI3‐K inhibitor), or 100 μM of HCQ (autophagolysosome inhibitor) for 6 h, followed by SARS‐CoV‐2 infection. As expected, treatment with the four inhibitors resulted in various degrees of influence on SARS‐CoV‐2 infection. Inhibition of ULK1 promoted SARS‐CoV‐2 infection as indicated by an increased amount of viral transcripts detected in culture supernatant at both 24‐ and 48‐h postinfection (Figure 1B). This is in line with the idea that autophagy, or virophagy, is a scavenger mechanism that promotes pathogen clearance. Interestingly, selective inhibition of class III PI3‐kinase Vps34 downstream of ULK1, in contrast to inhibition of ULK1, caused a significant decrease in SARS‐CoV‐2 replication (Figure 1C). 3‐MA, which mainly inhibits class I PI3‐kinase, caused minimal inhibition (Figure 1D). HCQ, consistent with previous reports, 21 , 22 , 23 potently inhibited SARS‐CoV‐2 viral replication (Figure 1E). The results suggested that the early stage of autophagy induction or its components may be involved in SARS‐CoV‐2 replication at a step downstream of ULK1. The presence of functional class III PI3‐kinase Vps34 is required for efficient SARS‐CoV‐2 replication.
Figure 1.
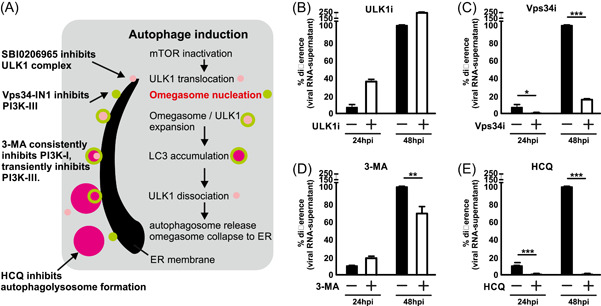
Suppression of class III PI3‐K, but not ULK1, inhibited SARS‐CoV‐2 infection. (A) Schematic diagram of autophagy induction, autophagosome, and autophagolysosome formation. (B–E) Inhibition of SARS‐CoV‐2 infection by various inhibitors targeting different steps of autophagy. VeroE6 cells were pretreated with ULK1 inhibitor SBI‐0206965 (20 µM), Vps34 inhibitor Vps34‐IN1 (10 µM), 3‐methyladenine (3‐MA) (5 mM), or hydroxychloroquine (HCQ) (100 µM) for 6 h, followed by SARS‐CoV‐2 infection at MOI 0.05 for 24 and 48 h. Viral RNA in the culture supernatant was extracted and quantitated using RT‐qPCR. Percentage difference with reference to 48 h mock‐treated sample was calculated. Data were statistically analyzed by unpaired Student's t test and presented as mean ± SD. *p < .05; **p < .01; ***p < .001. MOI, multiplicity of infection; RT‐qPCR, quantitative reverse‐transcription polymerase chain reaction
3.2. Vps34‐IN1 effectively inhibited SARS‐CoV‐2 infection with low cytotoxicity
The inhibitory effect of Vps34‐IN1 on SARS‐CoV‐2 infection was further confirmed by the quantitation of infectious virions. The prophylactic and therapeutic effect of Vps34‐IN1 was tested by pretreatment or posttreatment of the drug upon infection, and the titer of infectious virions in the culture supernatant was quantitated by plaque assay at 48 h postinfection with HCQ treatment serving as a positive control (Figures 2A and 2C). As expected, both pretreatment and posttreatment of VPS34‐IN1 inhibited SARS‐CoV‐2 replication, with the former being more effective. The viral replication was also examined by confocal microscopy for staining of viral NP protein 24 h postinfection (Figures 2B and 2D). In line with data presented in Figures 2A and 2C, Vps34‐IN1 efficiently inhibited viral replication in a dose‐dependent manner. A sample of 10 µM Vps34‐IN1 completely inhibited viral NP protein expression as potently as 100 µM HCQ treatment. The effective concentration and cytotoxicity of Vps34‐IN1 were also determined in comparison with HCQ (Figure 2E,F). Although the half‐maximal effective concentration (EC50) of HCQ was 19 µM, that of Vps34‐IN1 was 0.82 µM, which is more than 20 times lower than HCQ. Moreover, the drug did not show significant cytotoxicity within the tested soluble range of concentration.
Figure 2.
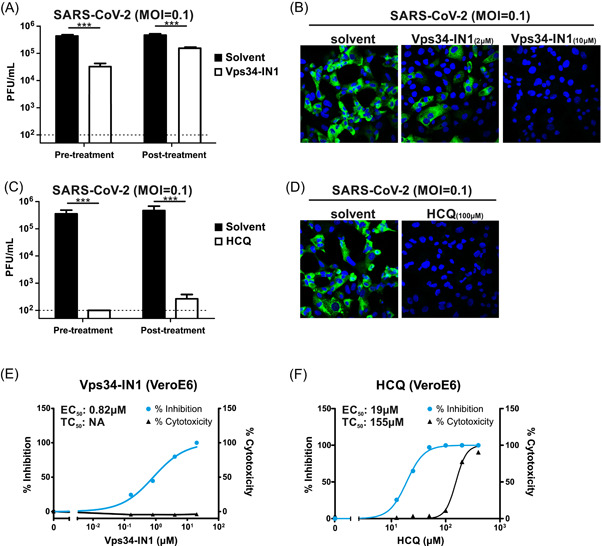
Inhibition of Vps34 quelled SARS‐CoV‐2 infection. (A, B) Treatment with Vps34‐specific inhibitor Vps34‐IN1 reduced SARS‐CoV‐2 replication. (A) VeroE6 cells were either pretreated or posttreated for 6 h with Vps34‐IN1 (10 µM) or control solvent DMSO. Cells were then infected by SARS‐CoV‐2 at MOI 0.1 and maintained for 48 h in the presence of drugs. Culture supernatant was harvested 48 h postinfection for quantitation of infectious virions by plaque assay. (B) VeroE6 cells pretreated with either 2 or 10 µM Vps34‐IN1 were infected at MOI 0.1 for 24 h. Cells were then fixed and stained by the anti‐NP antibody for confocal microscopy. Green, NP. Blue, DAPI nucleus counterstain. (C, D) HCQ (100 µM) treatment inhibited SARS‐CoV‐2 replication. Conditions were the same as A and B respectively. (E, F) Determination of effective concentration and toxicity of Vps34‐IN1 and HCQ posttreatment in VeroE6 cells. Inhibition of viral replication at an increasing dose of drugs was measured by RT‐qPCR of supernatant viral transcripts. Toxicity of drugs was measured by LDH‐based luciferase assay. Data were statistically analysed by unpaired Student's t test and presented as mean ± SD. *p < .05; **p < .01; ***p < .001. DAPI, 4′,6‐diamidino‐2‐phenylindole; DMSO, dimethyl sulfoxide; EC50, 50% of maximal effective concentration; HCQ, hydroxychloroquine; LDH, lactate dehydrogenase; MOI, multiplicity of infection; PFU, plaque‐forming unit; RT‐qPCR, quantitative reverse‐transcription polymerase chain reaction; TC50, 50% of maximal toxicity concentration
3.3. Inhibition of class III PI3‐K suppressed SARS‐CoV‐2 infection in ex vivo human lung tissue culture
Many cellular pathways, including autophagy, have been altered in cancer cell lines, leading to possible misinterpretation in in vitro settings. We have previously demonstrated the use of an ex vivo human lung tissue culture model for studying SARS‐CoV‐2 infection. 20 We, therefore, sought to examine the inhibitory effect of Vps34‐IN1 in noncancerous lung tissues freshly isolated from human patients (Figure 3A,B). After dissection, the lung tissues were pretreated overnight with Vps34‐IN1, followed by SARS‐CoV‐2 infection for 48 h. Consistent with our observation in cell‐lines, Vps34‐IN1 potently inhibited SARS‐CoV‐2 viral replication in normal ex vivo human lung tissue culture in a dose‐dependent manner (Figure 3C). Vps34‐IN1 is an in vitro selective inhibitor for the class III PI3‐kinase Vps34. A modified variant VVps34‐IN1 that showed reasonable bioavailability in animals has been developed for in vivo studies. 24 Here, we further showed that VVps34‐IN1 was also able to impede SARS‐CoV‐2 infection in our ex‐vivo human lung tissue culture model (Figure 3D). Taken together, we demonstrated in this study that the class III PI3‐kinase Vps34 might be a host target hijacked by SARS‐CoV‐2. Inhibition of Vps34 by the specific inhibitor Vps34‐IN1 or VVps34‐IN1 could efficiently inhibit SARS‐CoV‐2 replication.
Figure 3.
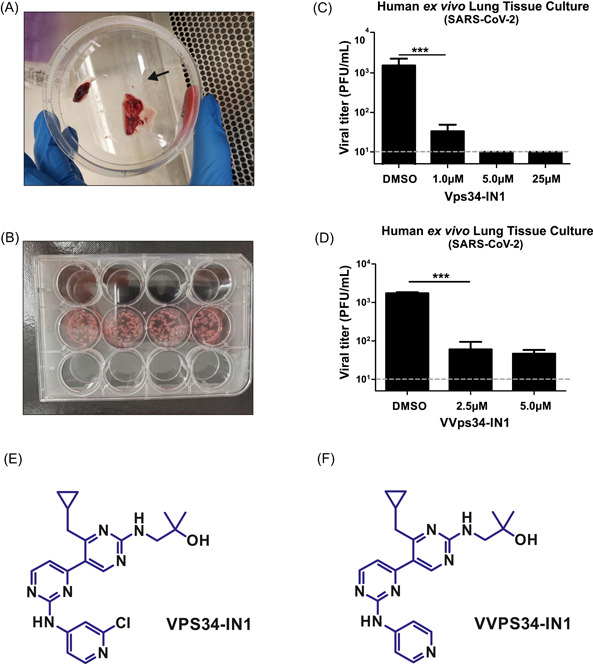
Inhibition of SARS‐CoV‐2 infection by Vps34‐specific inhibitor in ex vivo human lung tissue culture. (A) Human normal lung tissues were collected from the patient under surgical removal of its adjacent lung cancerous tissues. (B) Fresh human lung tissues were dissected into small pieces and evenly distributed into a culture plate. (C, D) After overnight pretreatment with mock, and either Vps34‐IN1 or VVps34‐IN1 at the indicated concentrations, the lung tissues were infected with SARS‐CoV‐2 at 2 × 106 PFU/well. The culture supernatant was collected at 48 h postinfection and the infectious virion in the supernatant was quantitated by plaque‐forming assay. Arrow, human lung tissue; dotted line, detection limit of the plaque‐forming assay at 1 × 101 PFU/ml. Data were statistically analyzed by unpaired Student's t test and presented as mean ± SD. ***p < .001. PFU, plaque‐forming unit. (E, F) Chemical structures of Vps34‐IN1 and VVPS34‐IN1
4. DISCUSSION
It has long been speculated that coronaviruses might have exploited host autophagy machinery for their efficient replication. However, previous studies examining the effect of various autophagy inducers and inhibitors and silencing of particular autophagy components have reported contradicting observations. 18 , 19 The lack of knowledge about the manipulation of autophagy by coronaviruses makes it difficult for designing anti‐coronavirals specifically targeting autophagy. In this study, we selected four well‐characterized small‐molecule autophagy inhibitors targeting ULK1, class III PI3‐K, class I PI3‐K, and autophagosome‐lysosome fusion, and found that inhibition of class III PI3‐K significantly inhibited SARS‐CoV‐2 replication.
Vps34‐IN1 is a potent inhibitor of class III PI3‐K Vps34 with high specificity. It was first discovered by Alessi's group at the University of Dundee and has been clearly demonstrated no significant nonspecific inhibition on 340 protein kinases and 25 lipid kinases including all class I and class II PI3Ks. 25 It is noted that the Vps34‐IN1 has been well‐characterized not only in in vitro assays 25 but also in a subsequent in vivo animal study. 26 This potent and specific Vps34‐IN1 inhibitor has also been applied for specific inhibition of Vps34 in numerous studies. 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 Therefore, the potential off‐target effect of class III PI3‐K inhibitor, Vps34‐IN1, used in this study will be minimal. In addition, another group studying the chemistry optimization of the Vps34‐IN1 (Figure 3E) found that its derivative VVps34‐IN1 (also known as compound 19; Figure 3F) maintains the high selectivity and in vitro potency. 24 The compound 19 was further characterized in C57BL/6 and nude mice, demonstrating its good bioavailability and in vivo inhibition of autophagy. However, no direct comparison of Vps34‐IN1 and VVps34‐IN1 was included in that report. 24 As we found that SARS‐CoV‐2 inhibition by Vps34‐IN1 was slightly better than that of VVps34‐IN1 in ex vivo human lung tissues (Figure 3C,D), it will be of great interest to determine the pharmacokinetic profile of Vps34‐IN1 in animal models such as hACE2‐transgenic mice and hamsters.
Next, the inhibition of SARS‐CoV‐2 infection was further confirmed using our previously established ex vivo human lung tissue culture model, 20 which helped minimize the possible artifacts arising from in vitro studies using cancer cell‐lines with altered signaling pathways. As Vps34‐IN1 treatment has been tested in various mouse models and no toxicity has been reported, we did not include the toxicity test in primary human lung tissues. In contrast to the treatment of Vps34‐IN1, inhibition of ULK1, the upstream activator of class III PI3‐K, did not suppress SARS‐CoV‐2 replication. This may imply that SARS‐CoV‐2 might have hijacked the autophagy machinery by exploiting the class III PI3‐K complex, or its related proteins, for its infection. It also agrees with the notion that coronaviruses might hijack specific component(s) of autophagy instead of the entire autophagy pathway. It is noteworthy that the class III PI3‐K complex is an essential component for the nucleation of the omegasome, which originated from the membrane of the ER. 38 Coincidentally, SARS‐CoV and MERS‐CoV encoded nonstructural proteins nsp3, nsp4 and nsp6 have been proposed to initiate the formation of DMVs for viral replication. 13 , 14 , 16 , 39 The DMVs are also shown to originate from and interconnect with the membrane of the ER. It is therefore of great interest to determine whether class III PI3‐K plays a crucial role in initiating DMV formation, through a similar mechanism as host omegasome nucleation.
Currently, there are still no defined medications that can cure or prevent COVID‐19 infection. Therefore, the search for effective therapeutics is vigorously undergoing. Besides the development of antivirals targeting viral components such as polymerase and proteases, temporal modulation of host machinery also serves as a plausible direction for COVID‐19‐specific drug design. In this study, we examined the antiviral property of four potent autophagy inhibitors and found that the class III PI3‐K inhibitors, Vps34‐IN1 and VVps34‐IN1, potently inhibited SARS‐CoV‐2 in ex vivo human lung tissue culture. Orally available VVps34‐IN1 is an analogue of Vps34‐IN1, characterized by improved pharmacokinetic properties such as good oral bioavailability but retaining high selectivity and potency. 24 The peak serum level of VVps34‐IN1 is 2.99 µM in mice following a single oral dose. This opens the possibility of using Vps34‐IN1 or its analogue for the treatment of SARS‐CoV‐2 infection. In addition, we observed that the inhibitory effect of Vps34‐IN1 on SARS‐CoV‐2 infection is more potent than that of VVps34‐IN1 at a concentration of 5.0 μM. Further in vivo pharmacokinetic study of Vps34‐IN1 and in vivo evaluation of its antiviral activity in animal infection models will facilitate the development of COVID‐19 therapeutics.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Contributed equally to this study: Chun‐Kit Yuen and Wan‐Man Wong. Conceptualized the project: Kin‐Hang Kok. Performed the experiments with assistance and/or reagents from Xiaohui Wang, Hin Chu, and Kwok‐Yung Yuen: Chun‐Kit Yuen, Wan‐Man Wong, and Long‐Fung Mak. Wrote the manuscript: Chun‐Kit Yuen and Kin‐Hang Kok.
ACKNOWLEDGMENTS
This study was supported by the Theme‐Based Research Scheme (T11/707/15‐R) and General Research Fund (17123420) of the Research Grants Council, Hong Kong Special Administrative Region.
Yuen C‐K, Wong W‐M, Mak L‐F, et al. Suppression of SARS‐CoV‐2 infection in ex‐vivo human lung tissues by targeting class III phosphoinositide 3‐kinase. J Med Virol. 2021;93:2076‐2083. 10.1002/jmv.26583
Chun‐Kit Yuen and Wan‐Man Wong contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Coronaviridae Study Group of the International Committee on Taxonomy of V . The species severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5(4):536‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Decaro N, Lorusso A. Novel human coronavirus (SARS‐CoV‐2): a lesson from animal coronaviruses. Vet Microbiol. 2020;244:108693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281(40):30299‐30304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Munz C. Macroautophagy—friend or foe of viral replication? EMBO Rep. 2013;14(6):483‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong X, Levine B. Autophagy and viruses: adversaries or allies? J Innate Immun. 2013;5(5):480‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shibutani ST, Saitoh T, Nowag H, Munz C, Yoshimori T. Autophagy and autophagy‐related proteins in the immune system. Nat Immunol. 2015;16(10):1014‐1024. [DOI] [PubMed] [Google Scholar]
- 8. Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle. 2010;9(7):1295‐1307. [DOI] [PubMed] [Google Scholar]
- 9. Munz C. The autophagic machinery in viral exocytosis. Front Microbiol. 2017;8:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection—a double‐edged sword. Nat Rev Microbiol. 2018;16(6):341‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mao J, Lin E, He L, Yu J, Tan P, Zhou Y. Autophagy and viral infection. Adv Exp Med Biol. 2019;1209:55‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong HH, Sanyal S. Manipulation of autophagy by (+) RNA viruses. Semin Cell Dev Biol. 2020;101:3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Hemert MJ, van den Worm SH, Knoops K, Mommaas AM, Gorbalenya AE, Snijder EJ. SARS‐coronavirus replication/transcription complexes are membrane‐protected and need a host factor for activity in vitro. PLOS Pathog. 2008;4(5):e1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knoops K, Kikkert M, Worm SHE, et al. SARS‐coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLOS Biol. 2008;6(9):e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baliji S, Cammer SA, Sobral B, Baker SC. Detection of nonstructural protein 6 in murine coronavirus‐infected cells and analysis of the transmembrane topology by using bioinformatics and molecular approaches. J Virol. 2009;83(13):6957‐6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oudshoorn D, Rijs K, Limpens RWAL, et al. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3‐4 polyprotein induce the formation of double‐membrane vesicles that mimic those associated with coronaviral RNA replication. mBio. 2017;8(6):e01658‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cottam EM, Maier HJ, Manifava M, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7(11):1335‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bello‐Perez M, Sola I, Novoa B, Klionsky DJ, Falco A. Canonical and noncanonical autophagy as potential targets for COVID‐19. Cells. 2020;9(7):1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang N, Shen HM. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID‐19. Int J Biol Sci. 2020;16(10):1724‐1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chu H, Chan JFW, Wang Y, et al. Comparative replication and immune activation profiles of SARS‐CoV‐2 and SARS‐CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID‐19. Clin Infect Dis. 2020;71:1400‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonam SR, Muller S, Bayry J, Klionsky DJ. Autophagy as an emerging target for COVID‐19: lessons from an old friend, chloroquine. Autophagy. 2020;24:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30(3):269‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu J, Cao R, Xu M, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS‐CoV‐2 infection in vitro. Cell Discov. 2020;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Honda A, Harrington E, Cornella‐Taracido I, et al. Potent, selective, and orally bioavailable inhibitors of VPS34 provide chemical tools to modulate autophagy in vivo. ACS Med Chem Lett. 2016;7(1):72‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bago R, Malik N, Munson MJ, et al. Characterization of VPS34‐IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3‐phosphate‐binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3‐kinase. Biochem J. 2014;463(3):413‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bilanges B, Alliouachene S, Pearce W, et al. Vps34 PI 3‐kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat Commun. 2017;8(1):1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pemberton JG, Kim YJ, Humpolickova J, et al. Defining the subcellular distribution and metabolic channeling of phosphatidylinositol. J Cell Biol. 2020;219(3):e201906130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu F, Wu X, Qian Y, Jiang X, Wang Y, Gao J. PIK3C3 regulates the expansion of liver CSCs and PIK3C3 inhibition counteracts liver cancer stem cell activity induced by PI3K inhibitor. Cell Death Dis. 2020;11(6):427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zachari M, Rainard JM, Pandarakalam GC, et al. The identification and characterisation of autophagy inhibitors from the published kinase inhibitor sets. Biochem J. 2020;477(4):801‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lystad AH, Carlsson SR, de la Ballina LR, et al. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy‐related processes. Nat Cell Biol. 2019;21(3):372‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miao G, Zhang Y, Chen D, Zhang H. The ER‐localized transmembrane protein TMEM39A/SUSR2 regulates autophagy by controlling the trafficking of the PtdIns(4)P phosphatase SAC1. Mol Cell. 2020;77(3):618‐632 e615. [DOI] [PubMed] [Google Scholar]
- 32. Singla A, Fedoseienko A, Giridharan SSP, et al. Endosomal PI(3)P regulation by the COMMD/CCDC22/CCDC93 (CCC) complex controls membrane protein recycling. Nat Commun. 2019;10(1):4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baba T, Toth DJ, Sengupta N, Kim YJ, Balla T. Phosphatidylinositol 4,5‐bisphosphate controls Rab7 and PLEKHM1 membrane cycling during autophagosome‐lysosome fusion. EMBO J. 2019;38(8):e100312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H, Huang F, Zhang Z, et al. Feedback activation of SGK3 and AKT contributes to rapamycin resistance by reactivating mTORC1/4EBP1 axis via TSC2 in breast cancer. Int J Biol Sci. 2019;15(5):929‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Janser F, Adams O, Bütler V, et al. Her2‐targeted therapy induces autophagy in esophageal adenocarcinoma cells. Int J Mol Sci. 2018;19(10):3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Daste F, Walrant A, Holst MR, et al. Control of actin polymerization via the coincidence of phosphoinositides and high membrane curvature. J Cell Biol. 2017;216(11):3745‐3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Filippakis H, Alesi N, Ogorek B, et al. Lysosomal regulation of cholesterol homeostasis in tuberous sclerosis complex is mediated via NPC1 and LDL‐R. Oncotarget. 2017;8(24):38099‐38112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Proikas‐Cezanne T, Takacs Z, Donnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128(2):207‐217. [DOI] [PubMed] [Google Scholar]
- 39. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double‐membrane vesicles. mBio. 2013;4(4):e00524‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.