

Abstract
Bone homeostasis is intimately relied on the balance between osteoblasts (OBs) and osteoclasts (OCs). Our previous studies have revealed that regulator of G protein signaling protein 12 (Rgs12), the largest protein in the Rgs super family, is essential for osteoclastogenesis from hematopoietic cells and OC precursors. However, how Rgs12 regulates OB differentiation and function is still unknown. To understand that, we generated an OB-targeted Rgs12 conditional knockout (CKO) mice model by crossing Rgs12fl/fl mice with Osterix (Osx)-Cre transgenic mice. We found that Rgs12 was highly expressed in both OB precursor cells (OPCs) and OBs of Wide Type (WT) mice, and gradually increased during OB differentiation, whereas Rgs12-CKO mice (Osx; Rgs12fl/fl) exhibited a dramatic decrease in cancellous bone mass, evidenced by reduced percentage of trabecular bone volume (BV/TV, 48%), trabecular number (Tb.N, 64%), trabecular thickness (Tb.Th, 76%), bone formation rate (BFR, 59%) and increased trabecular space (Tb.Sp, 171%). Loss of Rgs12 in OPCs in vitro significantly inhibited OB differentiation and the expression of OB marker genes, resulted in suppression of OB maturation and mineralization. Further mechanism study showed, deletion of Rgs12 in OPCs significantly inhibited GTPase activity and impaired Ca2+ oscillations via restraints of major Ca2+ entry sources (extracellular Ca2+ influx and intracellular ER Ca2+ release), partially contributed by the blockage of L-type Ca2+ channel mediated Ca2+ influx. Downstream mediator ERK was also inactive in Rgs12-deficient OPCs, while application of pertussis toxin (PTX) or overexpression of Rgs12 could partially rescue the defective OB differentiation and function via activation of ERK. Our findings reveal that Rgs12 is an important regulator in OB differentiation and function, and highlight Rgs12 as a potential therapeutic target for bone disorders.
Keywords: Osteoblasts, Rgs12, Calcium channel / oscillation, Gαi signaling
Introduction
Bone homeostasis is intimately relied on the balance between bone-forming osteoblasts (OBs) and bone-degrading osteoclasts (OCs) (1, 2). Any dysfunctional bone formation relative to bone resorption leads to defective skeletal integrity (3, 4). In contrast to anti-resorptive treatment for bone diseases due to excessive bone resorption, bone disorders such as skeletal dysplasias or osteogenic bone tumors (too much or disorganized bone formation), and chronic kidney disease (CKD) or aging (reduced bone formation) are more challenging to treat due to the limited number of reliable drugs promoting osteogenesis and activity (5–8). Therefore, uncovering potential therapeutic targets that regulate OB differentiation and function during skeletal development and remodeling remains important objectives for skeletal restoration (7).
OBs are mononucleated cells derived from mesenchymal stem cell (MSC) lineage in the bone marrow and are uniquely responsible for bone formation (2, 9). The speed and effectiveness of OB precursor cells (OPCs) differentiating into mature OBs determines the rate of bone formation and is dependent on the expression of OB-specific transcription factors like runt related transcription factor 2 (Runx2) and osterix (Osx), followed by the expression of alkaline phosphatase (ALP) and secretion of extracellular matrix containing type1 collagen (Col1α1) and non-collagenous proteins, leading to the deposition and mineralization of bone matrix (2, 5, 10, 11). Calcium (Ca2+) oscillations play an indispensible role during these processes through controlling gene expression and affecting OB differentiation and proliferation via Ca2+ signalling (9, 12–14). The excitability of Ca2+ signalling is determined by the contribution of extracellular Ca2+ influx, the effect of inositol 1, 4, 5-trisphosphate (IP3) to release Ca2+ from the endoplasmic reticulum (ER) and the capacity of removing Ca2+ from the cytosol by sarco/endoplasmic reticulum Ca2+ ATPase pump (SERCA) and plasma membrane Ca2+ ATPase (PMCA) (13, 15, 16). Ca2+ channels such as voltage-sensitive Ca2+ channels (VSCCs) are involved in these Ca2+ signalling activities (16–19). Some studies have showed that elevated intracellular Ca2+ by Ca2+ influx through the L-type Ca2+ channel (LTCC), one of the VSCCs, will eventually lead to the activation of the extracellular signal-related protein kinase (ERK) signaling which is critical for OB differentiation and survival (11, 19–21). Nevertheless, it remains largely unknown which factor(s) trigger and maintain Ca2+ oscillations in OBs and how Ca2+ signalling regulates OB differentiation via Ca2+ channels.
Regulators of G-protein signalling (Rgs) 12 (Rgs12) is the largest protein in the Rgs family with its multi-domain architecture (9, 22). It possesses a PDZ (PSD-95/Dlg/ZO-1) domain and a phosphotyrosine-binding (PTB) domain that are capable of binding the C terminus of G protein–coupled receptors (GPCRs) and interacts with GPCR chemokine receptors respectively, a conserved Rgs domain that is responsible for the GTPase accelerating protein (GAP) activity, a pair of Ras-binding domains (RBDs) that allows Rgs12 to integrate both heterotrimeric and monomeric G proteins signalling, and a GoLoco motif that carries guanine nucleotide dissociation inhibitor (GDI) activity toward Gαi subunits (9, 22, 23). This multi-domain structure provides Rgs12 with the potential to modulate multiple signalling pathways (9, 24, 25), including the signaling mediated by Gi-coupled GPCR, which has been reported to lead to osteopenia phenotype through the transgenic expression of a constitutively active Gi-coupled GPCR in OB (26). Our previous studies have revealed that Rgs12 plays an essential role in osteoclastogenesis through the regulation of PLCγ-Ca2+ channel-Ca2+ oscillations-nuclear factor of activated T cells 2 (NFAT2) pathway (27–29). Loss of Rgs12 impaired Ca2+ oscillations and reduced NFAT2 expression in OC, leading to the failure in OC differentiation and function (27, 29). However, the role of Rgs12 in OB differentiation and function, and how Ca2+ oscillations and signaling regulate these processes is poorly understood.
In this study, we generated OB-specific knockout of Rgs12 in mice and found that Rgs12 influences cancellous bone mass, leading to an osteopenia phenotype, and targeted deletion of Rgs12 in OPCs impaired OB differentiation and function through the disruption of Ca2+ channel/ Gαi-Ca2+ oscillations-ERK activation. Further, the defective OB differentiation and function could partially be rescued by the application of pertussis toxin (PTX), a Gi specific inhibitor, or overexpression of Rgs12 in Rgs12 deficient OPCs. We concluded that Rgs12 is required for OB differentiation and function, and highlight Rgs12 as a potential therapeutic target for bone disorders.
Materials and Methods
Detail of all reagents and antibodies are listed in supplemental materials and methods
Generation of Rgs12 conditional knockout (CKO) mice
Methodologies for generation of homozygous Rgs12fl/fl and Osx-Cre mice on C57Bl/6 backgrounds have been previously described (27, 30). To specifically inactivate Rgs12 in the OB lineage, we crossed Rgs12fl/fl mice with Osx-Cre transgenic mice to generate Osx; Rgs12fl/+ progeny, which were used for subsequent mating to produce homozygous Osx; Rgs12fl/fl mice. Because the phenotype in Rgs12fl/fl mice were indistinguishable from that in Osx-Cre mice after the age of 12 weeks, Rgs12fl/fl mice from the same litter of Osx; Rgs12fl/fl were used as control (31–34). All mice were maintained in C57BL/6J background and housed maximally at 5 per cage under a standard 12-hour light/12-hour dark cycle condition and free access to water and rodent diet. Once reached 12 weeks old, qualified mice (average body shape and bodyweight) were randomized in each group according to their genotyping (see supplemental materials and methods). Both genders were used to include potential variation related to gender. All experiments using mice were performed following protocols and approved by the Institutional Animal Care and Use Committee (IACUC) of the State University of New York (Buffalo) and the University of Pennsylvania.
Primary OPCs culture, virus infection and differentiation
Primary OPCs were isolated from the calvarial bone of Rgs12fl/fl mice at postnatal day 4 (4 pups of either gender) according to a serial digestion method as previously performed (30). In brief, calvarial bone was dissected, fragmented and subjected to sequential digestions in collagenase type I and 0.25% trypsin. Cells from this digestion were plated in complete culture medium (α-MEM, 10% FBS, penicillin and streptomycin) at 37°C in a humidified 5% CO2 atmosphere. Upon reaching 90% of confluence, cells were trypsinized, reseeded, and cultured for further experiments. OPCs from Rgs12fl/fl mice were infected with adenovirus (Ad-Cre, Ad-Null or Ad-GFP). Ad-Cre infection yielded an approximate 85% deletion of Rgs12 in Rgs12fl/fl OPCs, which were labeled as Rgs12d/d. Ad-Null or Ad-GFP treated cells were used as control and marked as Rgs12f/f and Ad-Null treated cells were specially used in Ca2+ imaging experiments. For osteogenic differentiation, OPCs were treated with osteogenic medium (OS medium), composed of complete culture medium with addition of β-glycerophosphate, ascorbic acid and dexamethasone. For experiments evaluating Gα signalling, PTX (100 ng/ml, Thermo Fisher Scientific) was also added to the OS medium (35, 36).
MTS assay
OPCs after adenovirus infection were seeded at 5000 cells per well in 96-well plate and performed according to manufacturer’s instructions (CellTiter 96 AQueous kit, Promega, Madison, WI, USA).
Gene transfer
Plasmid pCMV-Flag-Rgs12 vector was constructed by inserting the full length mouse Rgs12 cDNA into the plasmid of p3xFlag-myc-CMV-26 (Sigma). For transfection of 5 x 105 cells, 2 μg DNA mixed with 6 μl Fugene HD transfection reagent (Promega) was used. After adding serum-free medium for 4 h, cells were changed into 2% FBS medium and incubated overnight, and replenished with complete culture medium containing G418 (400 μg/ml, Gibco) for 2 weeks.
ALP activity assay
ALP activity was measured at day 7 of osteogenic differentiation as previously described (30). Briefly, the cells were washed twice with PBS then harvested with harvest buffer (see supplemental materials and methods), and sonicated at low power, followed by centrifugation and assay buffer adding, then, incubated at 37ºC until color change. NaOH solution was added to stop the reaction, and recorded the time. The optical density of ALP was measured at 405nm in a 96-well plate using microplate reader.
Measurement of bone nodule formation
Extracellular matrix calcium deposits were captured by Von Kossa staining and Alizarin red staining at day 14, day 21 or day 28 of osteogenesis as described previously (30). In brief, cells were fixed in 4% (w/v) paraformaldehyde (Sigma) and stained with 5% silver nitrate (Sigma) solution then exposed under ultraviolet (UV) light, or stained with 40 mM Alizarin red S (Sigma) solution (pH 4.4) at room temperature avoid light. Stained cells were scanned for image acquisition. To quantify Alizarin Red staining, 10% (w/v) cetylpyridinium chloride (Sigma) in 10 mM sodium phosphate (pH 7.0) was used to detain cells and measured using microplate reader (see supplemental materials and methods).
Quantitative real-time PCR (qPCR)
QPCR was carried out as described previously, with slight modifications (30, 37) (see supplemental materials and methods). In brief, total RNA was extracted and quantified. cDNA was synthesized from 2 μg of total RNA, and PCR amplifications were performed in triplicate with SYBR Green qPCR Master Mix (Bimake) on CFX96 real-time PCR machine (Bio-Rad) using the following steps: 95°C for 5 min, 40 cycles of 95°C for 15 s, and 60°C for 30 s. Gene expression were calculated using 2−ΔΔCT method and normalized to housekeeping gene GAPDH. Primer sequences were listed in supplementary Table 1.
Western blot (WB)
WB was performed as described previously (27, 30, 37). Briefly, cells and tissues were homogenized in RIPA buffer (see supplemental materials and methods) and protein concentrations were measured using BCA protein assay (Pierce). A total of 20 μg of protein was subjected to 10% SDS-PAGE gels, then transferred to a PVDF membrane (Millipore), followed by 30 min 5% milk blocking and overnight primary antibodies incubation at 4°C. After horseradish peroxidase–conjugated secondary antibodies incubation, visualization was performed using enhanced chemiluminescence (ECL) detection kit (Bio-Rad) on a ChemiDoc™ touch imaging system (Bio-Rad) and analyzed using the Image J software (National Institutes of Health, Bethesda, MD). Primary antibodies directed against Rgs12 (1:500, Abcam), Col1 (1:1000, Abcam), OCN (1:500, Abcam), phospho-p44/42 ERK (1:1000, CST), p44/42 ERK (1:1000, CST), β-actin (1:3000, Santa Cruz) and GAPDH (1:2000, Genscript) were used. GAPDH or β-actin was used as the internal control.
Bone Micro-CT analysis
A quantitative analysis of the femur morphology and microarchitecture was performed with the Micro-CT system (USDA Grand Forks Human Nutrition Research Center, Grand Forks, ND, USA). Femurs from 12 weeks old Rgs12fl/fl and Osx; Rgs12fl/fl mice of either gender (n=3 per gender, n=6 in total of each group) were fixed with 4% (w/v) paraformaldehyde for 24 h, rinsed with PBS, scanned and reconstituted as three-dimensional (3D) images. The trabecular and cortical bone architecture were assessed at the distal femoral metaphysis and midshaft. About 150 slices (1.5 mm) of bone were evaluated to determine percentage of bone volume (BV/TV, %), trabecular number (Tb.N, mm−1), trabecular thickness (Tb.Th, mm) and trabecular spacing (Tb.Sp, mm) as previously descried (27, 28, 30).
Bone histomorphometric analysis
To measure dynamic bone formation, mice were intraperitoneally injected with calcein (25 mg/kg) at postnatal day 90 and day 96. Mice were euthanized under CO2 condition and harvested 2 days after last injection. Tibias were fixed in 4% (w/v) paraformaldehyde and longitudinal sections (8 μm) of proximal tibias were cut (see supplemental materials and methods). Bone formation rate per bone surface (BFR/BS, μm3/um−2 per day), mineral apposition rate (MAR, μm per day), OB surface per bone surface (Ob.S/BS, %), OB number per bone perimeter (N.Ob/B.Pm, mm−1) and OC number per bone perimeter (N.Oc/B.Pm, mm−1) were measured.
Bone histological analysis
Tibia from 12 weeks old mice were fixed in 4% (w/v) paraformaldehyde for 24 h and subsequently decalcified at 4°C in 10% EDTA for 3-4 weeks. The samples were dehydrated in gradients of ethanol, followed by 100% xylene, and then embedded in paraffin. After deparaffinization, sectioned slides in 5 μm were stained with haemotoxylin and eosin (H&E) and imaged using a Leica inverted microscope (DMI6000B, Leica, Germany)
IF staining
IF staining was performed based on protocols described previously with slight modifications (27, 30, 38). In brief, sectioned slides prepared above were permeabilized with 0.05% Triton X-100, blocked in 10% goat serum (Vector Laboratories, Burlingame, CA, USA) and incubated with anti-Rgs12 antibody (1:500, Abcam) at 4°C overnight. Primary antibody was coupled with Alexa Fluor 488-conjugated anti-Chicken secondary antibody (1:500, Abcam), followed by counterstained for nuclei using DAPI. Slides were mounted and images were acquired using the Leica fluorescent microscope (DMI6000B, Leica, Germany).
Ca2+ imaging
Measurements of Ca2+ oscillation were performed as previously described with slight modification (27, 39). OPCs after Ad-Null or Ad-Cre infection were reseeded at a density of 2 × 105 cells/ 35 mm-dish and induced with OS medium for 3 days. Then, cells were loaded with 5 μM fluo-4 AM (Thermo Fisher Scientific) for 50 min in complete culture medium containing 0.05% pluronic F127 (Sigma), washed and incubated in Hank’s Balanced Salt Solution (HBSS). Change of intracellular Ca2+ concentration was accessed by a Leica fluorescent microscope (DMI6000B, Leica, Germany) with excitation wavelength at 488 nm and emission wavelength at 505–530 nm. Signals were recorded simultaneously at 5-s intervals for 30 min, and fluorescence intensity was analyzed using Leica Application Suite X (Leica, Germany) software and plotted over time.
To measure intracellular Ca2+ concentration change during extracellular Ca2+ influx or release from ER in OPCs, Cells were washed and incubated a in Ca2+ free isotonic solution (ISO) (see supplemental materials and methods) after fluo-4 AM loading. Change of intracellular Ca2+ concentration was accessed and recorded for 45 min as mentioned above. Once the cells reached a stable baseline level of cytosolic Ca2+, the Ca2+ free ISO was exchanged for a Ca2+ free ISO containing 1μM TG (Sigma) to trigger Ca2+ release from the ER, or a normal ISO containing 1.3 mM CaCl2 to induce extracellular Ca2+ influx, or a normal ISO containing 10μM Bay K8644 (Sigma) to induce L-type Ca2+ channel amplified influx (19, 39). Changes of intracellular Ca2+ intensity was normalized by the initial Ca2+ level of cell itself.
GTPase assay
GTPase activity was performed according to manufacturer’s instructions (602-0120, Innova Biosciences, San Diego, CA, USA) (40). Colorimetric measurements were read at the wavelength of 635 nm and GTPase activities were assessed on the basis of inorganic complex solutions.
Statistical Analysis
Animal number in each group was calculated to detect a 50% difference at alpha = 0.05 and power = 0.8 using Graphpad StatMate 2.0 software (GraphPad Software, San Diego, USA). All data are presented as mean ± standard deviation (SD) (n >3) and analyzed by using unpaired, two-tailed Student’s t-test for the comparison between two groups or one-way ANOVA followed by Sidak’s multiple comparison test for grouped samples. p<0.05 were considered to be significant. Graphpad Prism 6.0 software (GraphPad Software, San Diego, USA) was used to perform statistical analysis.
Results
Rgs12 was expressed in murine OBs and gradually increased throughout osteogenesis
To evaluate Rgs12 expression is different tissues, samples from brain, lung, kidney and bone were isolated from WT mice. WB showed Rgs12 protein highly expressed in brain and lung, with a lower expression level in bone (Fig. 1A). Further, to monitor Rgs12 expression pattern during osteogenesis, OPCs derived from calvarial bone of WT mice were stimulated with OS medium for 0, 3, 7 and 14 days. The mRNA level of Rgs12 was gradually increased and a significant increase occurred after day 7 of osteogenesis (Fig. 1B). The result was further confirmed at the protein level (Fig. 1C).
Fig. 1. Rgs12 is expressed in murine bone tissues and osteoblasts (OBs), with expression increases during osteogenesis.
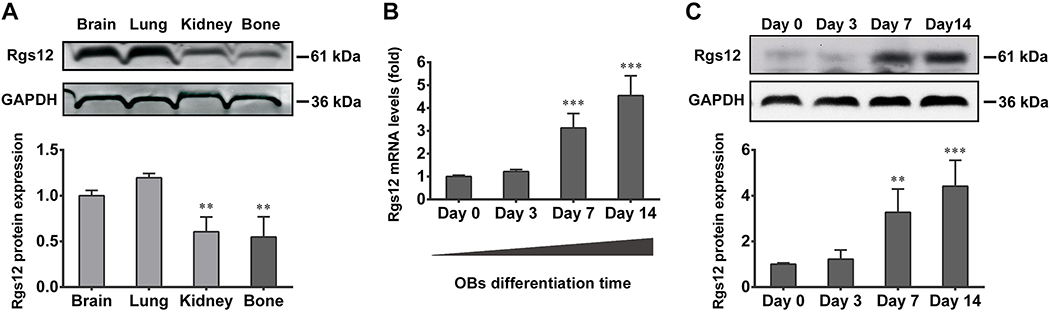
(A) WB showed Rgs12 protein expression in brain, lung, kidney and bone tissue of WT mice. **p <0.01 versus (vs) brain, n=4. (B) qPCR results showed Rgs12 mRNA expressed in primary OB precursors (OPCs) derived from WT calvarial bone, and increased with osteogenic medium (OS medium) stimulation days. (C) qPCR results were confirmed at protein level. All quantitative data were normalized to GAPDH and presented as mean ± SD, **p <0.01, ***p <0.001 vs day 0, n=4.
Targeted deletion of Rgs12 in OPCs reduced cancellous bone mass with decreased OB number
To better understand the role of Rgs12 during osteogenesis in vivo, we generated Rgs12 CKO mice (Osx; Rgs12fl/fl mice) described previously. Osx; Rgs12fl/fl mice were born with expected Mendelian ratios. Deletion of Rgs12 was examined using genomic DNA from calvarial bone (Supplemental Fig. 1A). Low levels of Rgs12 mRNA (Supplemental Fig. 1B) and significantly diminished expression of Rgs12 protein were found in calvarial bone of Osx; Rgs12fl/fl mice (Supplemental Fig. 1C). In addition, expression of Rgs12 was significantly reduced in femur sample of Osx; Rgs12fl/fl mice (Supplemental Fig. 1D), indicating a successful generation of knockout model.
To assess whether deletion of Rgs12 affects bone physiology, femurs of 12 weeks old mice were evaluated by Micro-CT analysis. With a marked reduction in cancellous bone mass (Fig. 2A), the percentage of BV/TV, Tb.N, and Tb.Th in Osx; Rgs12fl/fl mice were 48%, 64% and 76% of those in Rgs12fl/fl mice, whereas Tb.Sp increased to 171% (Fig. 2B), indicating an osteopenia phenotype. H&E staining on tibias also exhibited a decreased trabecular bone in Osx; Rgs12fl/fl mice (Fig. 2C). Dynamic histomorphometric analysis of 12 weeks old Osx; Rgs12fl/fl mice further demonstrated a significant reduction in BFR/BS, MAR, Ob.S/BS and N.Ob/B.Pm (Fig. 2D, E), and no change was found in N.Oc/B.Pm between Osx; Rgs12fl/fl mice and control littermates (Fig. 2E). These data suggested an osteopenia phenotype caused by impaired OB differentiation after Rgs12 deletion.
Fig. 2. Deletion of Rgs12 in OPCs (OSX; Rgs12fl/fl) causes significant osteopenia with reduced OB formation.
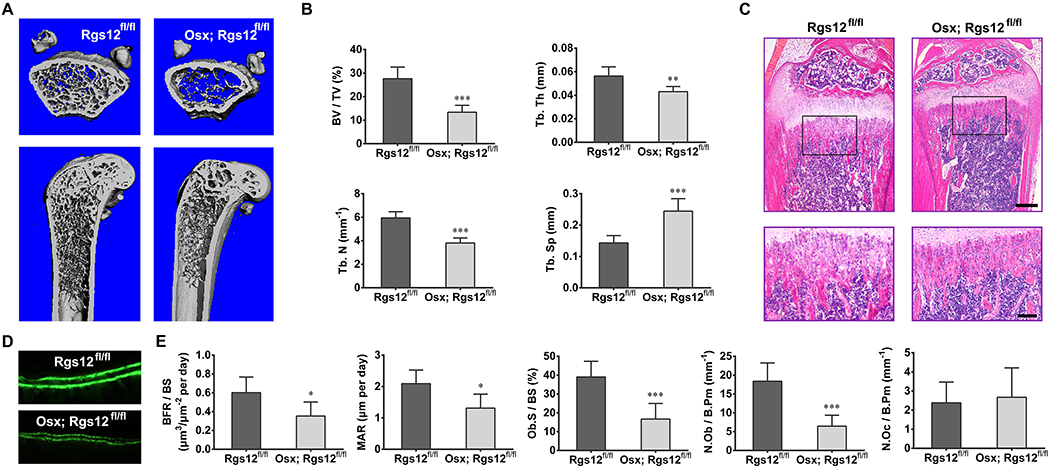
(A) Micro-CT analysis of the femurs from 12 weeks old Rgs12fl/fl and OSX; Rgs12fl/fl mice (upper panel, axial view of the metaphyseal region; lower panel, longitudinal view). An apparent reduce in the cancellous bone mass was observed in OSX; Rgs12fl/fl mice. (B) Quantitative analysis of the percentage of bone volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th) and trabecular spacing (Tb.Sp) of samples shown in (A). *p <0.05, ***p <0.001 vs Rgs12fl/fl mice, n=6. (C) H&E staining of the proximal tibia metaphyseal regions of 12 weeks old mice. Lower panel: higher magnification. Scale bars, 500μm (upper) or 50μm (lower). (D) Dynamic histomorphometry of tibia after double calcein labeling. (E) Quantitative analysis of bone formation rate per bone surface (BFR/BS), mineral apposition rate (MAR), OB surface per bone surface (Ob.S/BS), OB number per bone perimeter (N.Ob/B.Pm) and OC number per bone perimeter (N.Oc/B.Pm) from double calcein labeling. *p <0.05, ***p <0.001 vs Rgs12fl/fl mice, n=6. Quantitative data were presented as mean ± SD.
Deletion of Rgs12 impairs OB differentiation
To further determine whether the decreased bone mass in Osx; Rgs12fl/fl mice resulted from impaired OBs, we investigated the role of Rgs12 in OB differentiation and function in vitro. OPCs were isolated from Rgs12fl/fl mice and infected with different adenovirus as previously described. QPCR result showed Ad-Cre infection yielded an approximate 85% reduction of Rgs12 in Rgs12fl/fl OPCs, while Ad-GFP and Ad-Null infection had no reduced effects of Rgs12 in OPCs (Fig. 3A). Deletion of Rgs12 in OPCs apparently impaired OB differentiation, evidenced by lower expression level of OB differentiation markers including ALP, Col1α1, OCN and Runx2 after 7 days of osteogenic induction (Fig. 3B). These impairments were also confirmed by reduced expression of extracellular matrix proteins, Col1 and OCN (Fig. 3C). Blockage of OPCs differentiation and mineralization were further detected by exhibiting less ALP activity (Fig. 3D) and reduced bone nodule formation (Fig. 3D, E) in Rgs12d/d OBs. All differentiation-related experiments were performed under cell confluency condition to eliminate the effect of proliferation on cell differentiation (Supplemental Fig. 2).
Fig. 3. Deletion of Rgs12 impairs OB differentiation.
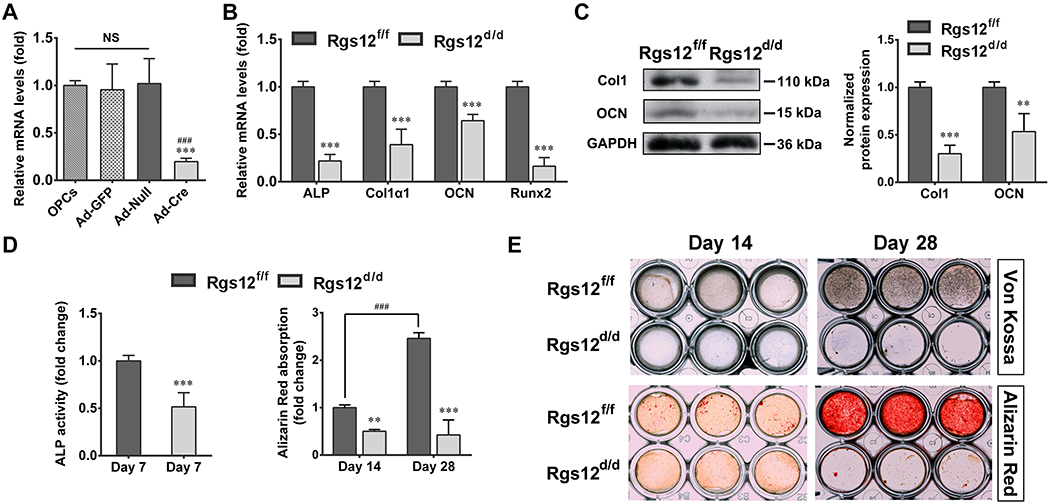
(A) Rgs12 mRNA level in OPCs after different adenovirus infection, normalized to GAPDH (n=6, triplicates per group). ***p <0.001 vs Ad-GFP; ###p <0.001 vs Ad-Null; NS, not statistical significant between groups. (B) qPCR analysis of OB marker genes ALP, collagen type I alpha 1 (Col1α1), osteocalcin (OCN) and Runx2 in OPCs after 7 days of OS medium culture. OPCs from Rgs12fl/fl mice that infected with Ad-GFP were marked as Rgs12f/f, while infected with Ad-Cre were labelled as Rgs12d/d. ***p <0.001 vs control, n=4, triplicates per group. (C) WB analysis of extracellular matrix proteins expression at day 7 of osteogenic induction. **p <0.01, ***p <0.001 vs control, n=4. (D) ALP activity and Alizarin Red absorption of OPCs at indicated days of osteogenic induction. **p < 0.01, ***p < 0.001 vs Rgs12f/f cells within groups; ###p < 0.001 vs Rgs12f/f cells at day 14 of osteogenic induction; n=4, triplicates per group. (E) Representative images of Von Kossa and Alizarin Red staining (triplicates per group). Quantitative data were presented as mean ± SD.
Deletion of Rgs12 inhibited GTPase activity and Ca2+ oscillations, blocking ERK activation in OPCs, and application of PTX partially rescued defective OB differentiation and function.
Studies show that signaling mediated by Gi-coupled GPCR served as a negative regulator of OB differentiation (9, 26, 41), while Rgs12 acts as an antagonist of this signaling via enhancing GTPase activity towards Gαi subunit (9, 22). Therefore, we compared the differences of GTPase activities between Rgs12f/f and Rgs12d/d OPCs and our result showed that Pi released in Rgs12d/d OPCs was 53% of that in Rgs12f/f OPCs (Fig. 4A), indicating a possible enhanced Gαi-mediated signaling in Rgs12d/d OPCs. Accumulating studies show that Gαi-mediated signaling can crosstalk with Ca2+ signaling (42–44), which also plays a critical role during OB differentiation and function (9, 22), and our previous study demonstrated that Rgs12 regulated Ca2+ oscillations during OC differentiation (27). Thus, we hypothesized Rgs12 is also required for Ca2+ oscillations during OB differentiation. By monitoring intracellular Ca2+ change in OPCs after OS medium stimulation for 3 days, we found a sustained frequency of Ca2+ oscillations at approximately two minutes per interval in the Rgs12f/f cells (Fig. 4B, left panel), nevertheless, in Rgs12d/d cells, Ca2+ oscillations were barely detectable (Fig. 4B, right panel). To further confirm the involvement of Rgs12 in these two pathways, phosphorylated ERK (p-ERK), the downstream mediator of both pathways, was examined (20–22, 45). Our results showed the expression level of p-ERK was significantly lower in Rgs12d/d cells after OS medium stimulation (Fig. 4C).
Fig. 4. Deletion of Rgs12 inhibits GTPase activity, Ca2+ oscillations and sequentially blocks ERK activation; and PTX treatment partially rescued defective OB differentiation and function.
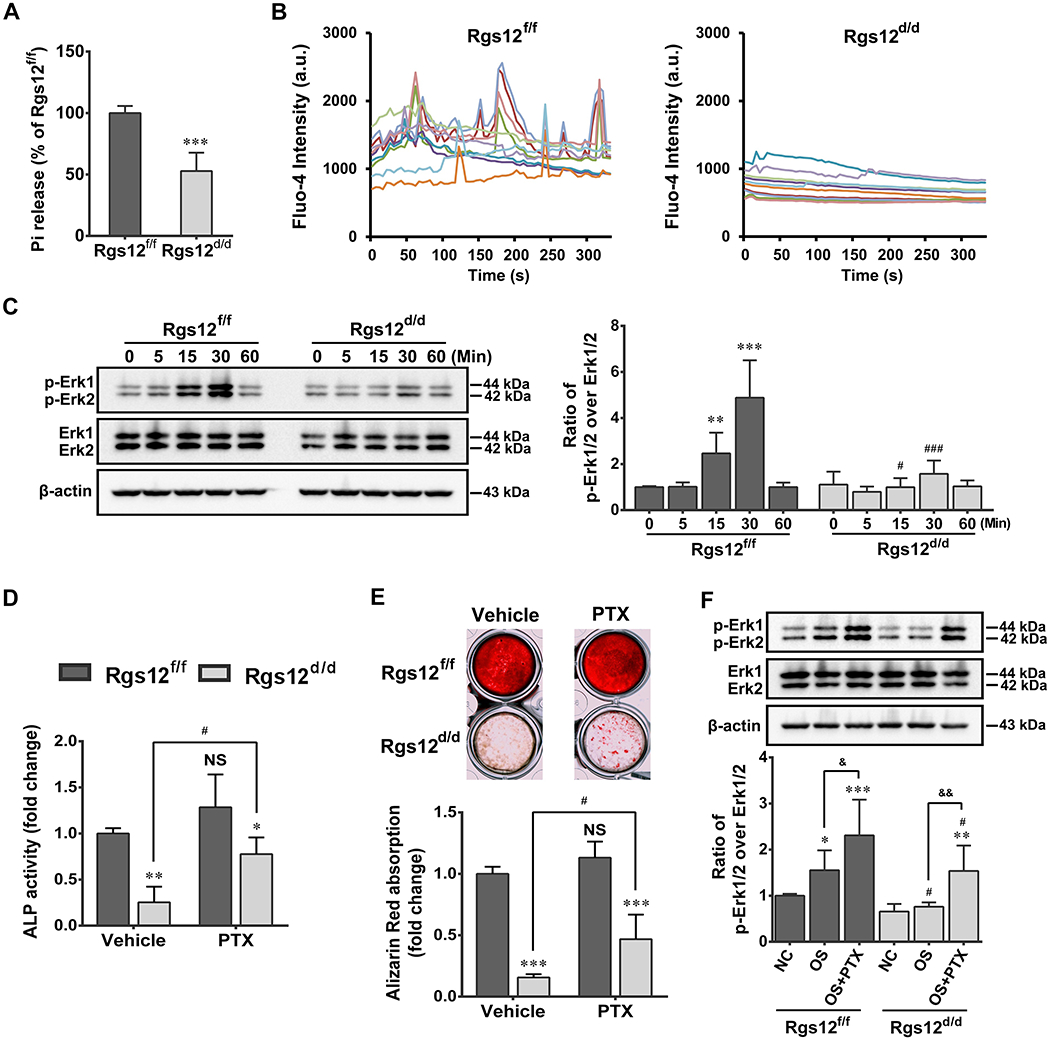
(A) GTPase assay showed inorganic phosphate (Pi) release level in OPCs. ***p <0.001 vs Rgs12f/f cells; n=4. (B) Analysis of Ca2+ oscillations in OPCs after 3 days of OS medium stimulation. Cells were traced, calculated, and plotted over time using different colors (n=10 cells of each group). (C) WB showed phosphorylated ERK (p-ERK) expression in OPCs. Cells were starved for 6h prior to OS medium stimulation, p-ERK were normalized to total ERK. **p <0.01, ***p <0.001 vs 0 min; #p <0.05, ###p < 0.001 vs same time point in Rgs12f/f cells; n=4. (D) ALP activity of OPCs with or without PTX treatment at day 7 of osteogenic induction. (E) Alizarin Red absorption and staining of OPCs with or without PTX treatment at day 28 of osteogenic induction. *p <0.05, **p <0.01, ***p <0.001 vs Rgs12f/f cells within groups; NS, not statistical significant or #p <0.05 vs same cells without PTX treatment; n=4. (F) WB analysis of p-ERK in OPCs with or without PTX treatment. Cells in PTX group were pretreated with PTX (100ng/ml) overnight in complete culture medium, followed by 6h starvation and then OS medium stimulation combine PTX for 15 min. NC and OS group were only stimulated with OS medium for 0 or 15 min. *p <0.05, **p <0.01, ***p <0.001 vs NC within groups; #p <0.05 vs same subgroup in Rgs12f/f; &p <0.05, &&p <0.01 vs OS within groups; n=4. Quantitative data were presented as mean ± SD.
PTX was applied to purposely feedback the role of Rgs12 in Gαi-mediated signaling during osteogenesis. As expected, application of PTX displayed a partial rescue of ALP activity and bone nodule formation in Rgs12d/d OBs (Fig. 4D, E). Further results showed p-ERK was significantly upregulated in PTX-treated Rgs12d/d cells (Fig. 4F). These findings supported the regulator role of Rgs12 in OB differentiation and function through Gαi-mediated signaling.
Deletion of Rgs12 restrained the entry sources of cytosolic free Ca2+ in OPCs
Given that Ca2+ oscillations arise from periodic Ca2+ influx and repetitive release and reuptake of free Ca2+ from intracellular stores (46, 47), we investigated whether Rgs12 deletion affects Ca2+ entry from these two main sources. First, we determined the influence of Rgs12 on Ca2+ influx. The fluorescence intensity increased gradually in Rgs12f/f cells after normal ISO application, whereas elevating amplitude in Rgs12d/d cells was dramatically decreased (Fig. 5A, B; Supplemental Video 1 and 2). Significant differences between Rgs12f/f and Rgs12d/d cells occurred after 3 min of ISO application, and reached maximum after 13:10 min (Fig. 5C), indicating a restraint on Ca2+ entry to cytosol from extracellular environment. Next, we determined whether deletion of Rgs12 also affects Ca2+ release from ER. To achieve that, cells were treated with 1μM TG to inhibit SERCA and unmask an endogenous leak for Ca2+ (39, 46). Similar to Ca2+ influx, elevating amplitude of ER Ca2+ release in Rgs12d/d cells were dramatically decreased (Fig. 5D, E; Supplemental Video 3 and 4), and significant differences were captured after 15 s of TG application, and reached maximum level after 90 s (Fig. 5F). Collectively, these data suggested deletion of Rgs12 restrained both extracellular and intracellular entry sources of cytosolic free Ca2+, leading to inhibition on Ca2+ oscillations in OPCs.
Fig. 5. Deletion of Rgs12 restrains Ca2+ entry from extracellular environment and intracellular ER store to the cytosol.
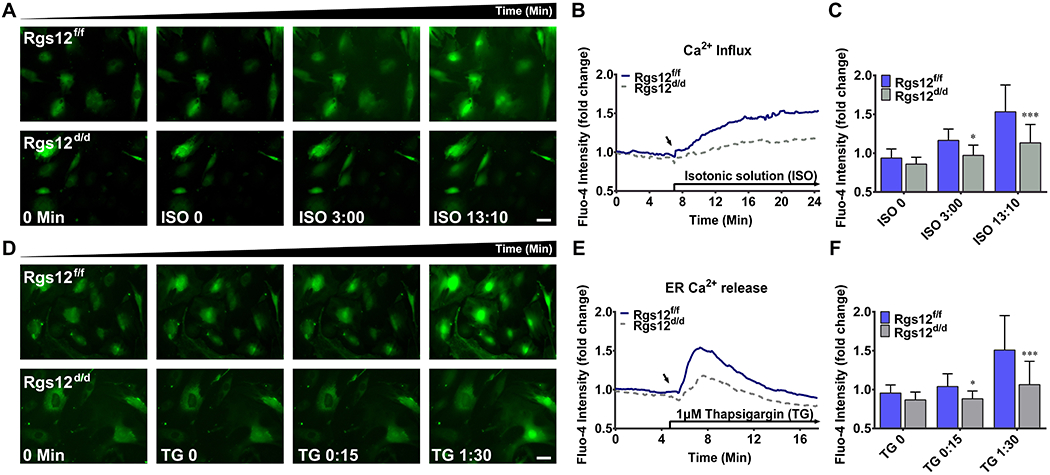
(A), (B) and (C): Ca2+ influx in Rgs12f/f or Rgs12d/d OPCs. (A) Representative images of intracellular Ca2+ in OPCs before (0 min) and after isotonic solution (ISO, started from ISO 0 min) stimulation. OPCs were incubated in Ca2+ free ISO to reach a stable baseline level of cytosolic Ca2+, and then changed into normal ISO, which contains 1.3 mM CaCl2 to induce extracellular Ca2+ influx. Upper panel, Rgs12f/f OPCs; lower panel, Rgs12d/d OPCs. (B) Overlay of traces from OPCs. Arrow indicated the time point when extracellular Ca2+ presented. Data was expressed as a fold change of fluo-4 intensity, accomplished by normalized each cell’s fluorescence intensity to its initial intensity (n=39 cells per group). (C) Quantitative analysis of fluo-4 intensity at indicated time point. *p <0.05, ***p <0.001 vs Rgs12f/f cells, n=39 cells per group. (D), (E) and (F): ER Ca2+ release in Rgs12f/f or Rgs12d/d OPCs. (D) Representative images of intracellular Ca2+ in OPCs before (0 min) and after stimulation of 1μM thapsigargin (TG, started from TG 0 min). OPCs were incubated in Ca2+ free ISO as previous, and treated with TG in Ca2+ free ISO to trigger Ca2+ release form the ER. Upper panel, Rgs12f/f OPCs; lower panel, Rgs12d/d OPCs. (E) Overlay of traces from OPCs. Arrow indicated the time point of TG application (n=47 cells per group). (F) Quantitative analysis of fluo-4 intensity at indicated time point. *p <0.05, ***p <0.001 vs Rgs12f/f cells, n=47 cells per group. Scale bars, 50μm. Quantitative data were presented as mean ± SD.
Deletion of Rgs12 attenuated the BayK8644-amplified Ca2+ influx
LTCCs, via Ca2+ influx, play key role in regulating intracellular Ca2+ homeostasis of OB (19, 20). Thus, we use BayK8644, an agonist of LTCCs, to determine whether LTCCs partly contribute to the restrain of Ca2+ influx after Rgs12 deletion (48). First, Rgs12f/f OPCs were treated with or without 10μM BayK8644 in normal ISO, to ensure an amplified Ca2+ influx induced by BayK8644. Our results showed the fluorescence intensity of BayK8644-treated cells increased immediately after BayK8644 application (Fig. 6A, B; Supplemental Video 5 and 6) and reached significant differences after 30 s of application (Fig. 6C), demonstrating an amplified Ca2+ influx via LTCCs. Next, we compared the effect of BayK8644 on Rgs12f/f and Rgs12d/d cells, and no obvious increasing signals were detected in Rgs12d/d cells after a limited and transient fluorescence peak, induced by BayK8644 application (Fig. 6D, E; Supplemental Video 7 and 8). Significant differences between Rgs12f/f and Rgs12d/d cells were even captured at logarithmic phase of fluorescence signals (Fig. 6F). These results confirmed the involvement of LTCCs in restraint of Ca2+ influx caused by Rgs12 deletion.
Fig. 6. Deletion of Rgs12 attenuates the BayK8644-amplified Ca2+ influx through L-type Ca2+ channel.
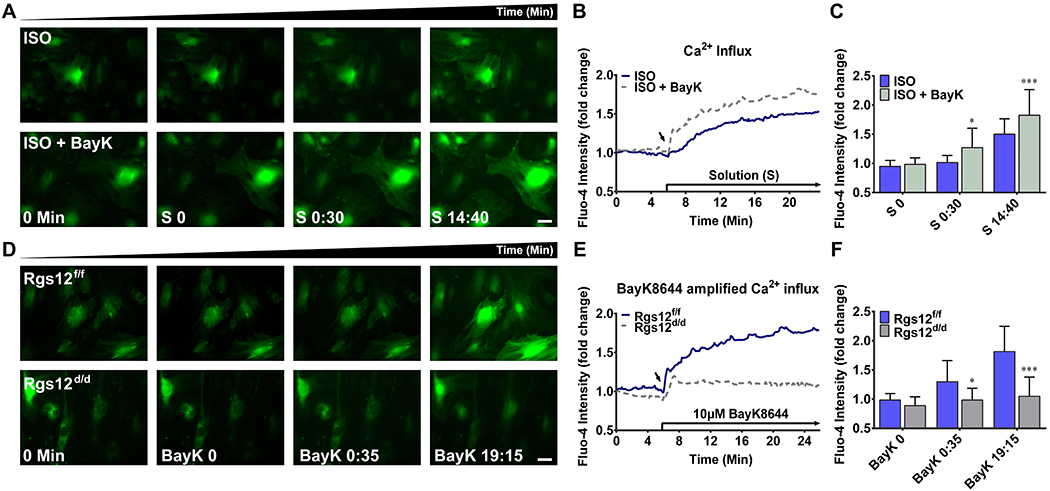
(A), (B) and (C): Effect of BayK8644 on intracellular Ca2+ in Rgs12f/f OPCs. (A) Representative images of intracellular Ca2+ in Rgs12f/f OPCs in the presence or absence of 10μM BayK8644 stimulation. OPCs were incubated in Ca2+ free ISO as previous described, and treated with BayK8644 in ISO (lower panel) or ISO alone (upper panel). (B) Overlay of traces from OPCs. Arrow indicated the time point when different solutions applied (S, started from S 0 min). Data was expressed as a fold change as previous described (n=28 cells per group). (C) Quantitative analysis of fluo-4 intensity at indicated time point. *p <0.05, ***p <0.001 vs cells without BayK8644, n=28 cells per group. (D), (E) and (F): Effect of BayK8644 on Rgs12f/f or Rgs12d/d OPCs. (D) Representative images of intracellular Ca2+ for OPCs before (0 min) and after stimulation of 10μM BayK8644 (BayK, started from BayK 0 min). OPCs were incubated in Ca2+ free ISO as previous, and treated with BayK8644 in ISO. Upper panel, Rgs12f/f OPCs; lower panel, Rgs12d/d OPCs. (E) Overlay of traces from OPCs. Arrow indicated the time point of BayK8644 application. Data was expressed as a fold change (n=28 cells per group). (F) Quantitative analysis of fluo-4 intensity at indicated time point. *p <0.05, ***p <0.001 vs Rgs12f/f cells, n=28 cells per group. Scale bars, 50μm. Quantitative data were presented as mean ± SD.
Ectopic expression of Rgs12 partially rescued defective OB differentiation in Rgs12d/d cells
To test potential therapeutic effect of Rgs12 on defective OB, we ectopically expressed Rgs12 in Rgs12d/d OPCs. Successful Rgs12 overexpression was confirmed by WB (Fig. 7A). Overexpression of Rgs12 in Rgs12fl/fl OPCs exhibited an enhanced effect on OB differentiation, while differentiation in Rgs12d/d OBs was also partially rescued, with a significant improvement in both ALP activity and bone nodule formation (Fig. 7B, C), supporting the regulator role of Rgs12 during osteogenesis (Fig. 7D).
Fig. 7. Ectopic expression of Rgs12 partially rescues defective OB differentiation and function.
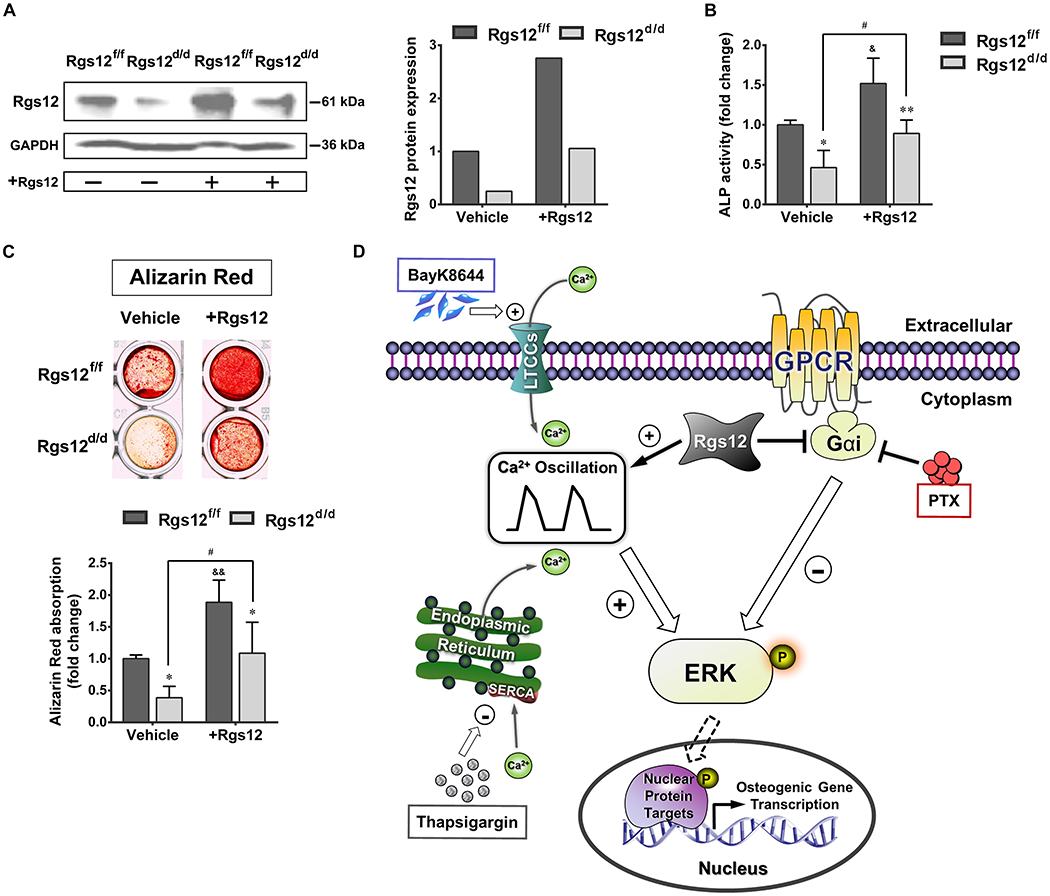
(A) WB showed Rgs12 protein expression in Rgs12f/f and Rgs12d/d OPCs transfected with or without pCMV-Flag-Rgs12 plasmid, normalized to GAPDH. (B) ALP activity of OPCs at day 7 of osteogenic induction. (C) Alizarin Red staining and absorption of OPCs at day 21 of osteogenic induction. *p <0.05, **p <0.01 vs Rgs12f/f cells within groups; &p <0.05, &&p <0.01 or #p <0.05 vs same cells without Rgs12 overexpression; n=3, triplicates per group. Quantitative data were presented as mean ± SD. (D) Proposed role of Rgs12-regulated Gαi and Ca2+ signaling during OB differentiation.
Discussion
Rgs proteins are broadly involved in bone development and remodeling via their influences on OC and OB differentiation and function individually or mutually (9, 22, 49). Our previous study demonstrated that Rgs12 promotes osteoclastogenesis during bone remodeling (27). Here, we provide the first evidence to demonstrate a close dependency of osteogenesis on the expression of Rgs12, and to reveal the mechanism underlying how Rgs12, through both Ca2+ channel-Ca2+ oscillation-ERK signaling and traditional Gαi-ERK signaling, promotes OB differentiation and function during osteogenesis. The facilitating role of Rgs12 in bone formation was defined by obvious osteopenia phenotypes in Rgs12 conditional knockout mice (Osx; Rgs12fl/fl) which specially inactivated Rgs12 on OB lineage (Fig. 2), and also supported by OPCs which underwent in vitro Rgs12 deletion, exhibited a striking inability to differentiate into mature and functional OBs (Fig. 3). Additionally, ectopic expression of Rgs12 in Rgs12 defective OPCs (Rgs12 d/d) demonstrated a partial restoration of differentiation ability and surprisingly showed enhanced differentiation ability when overexpressed in normal OPCs (Rgs12 f/f) (Fig. 7B, C). Therefore, our findings indicate that Rgs12 plays an indispensable role during osteogenesis.
We identify Gαi-ERK signaling as one of the pathway that Rgs12 exerts its regulatory role during osteogenesis. Rgs12 is a GTPase-activating protein that enhances the intrinsic GTPase activity of Gαi subunit, thereby inactivate downstream signal transduction (9, 22, 23). It was previously noted that hyperactivation of the Gαi-mediated signalling served as a negative regulator of OB differentiation through inhibition of AC, and eventually lead to a reduction in trabecular bone formation (26, 41), while the opposite, Gαs-AC-cAMP signalling is required for OB differentiation (50, 51). In such a way, it would be rational to postulate that the deletion of Rgs12 shrinks GTPase activity towards Gαi subunit and subsequently eases the inhibitory effect on Gαi-mediated signalling, leading to an osteopenia phenotype. We found that Pi release reduced approximately 53% in Rgs12d/d OPCs compare with control OPCs, indicating a dramatic shrink of GTPase activity towards Gαi subunit after Rgs12 deletion (Fig. 4A). Several studies showed that ERK activation serves as downstream effector of Gαi-mediated signalling via membrane recruitment of rap1GAPII and reduction of GTP-bound Rap1 (41, 51–53), and is essential for OB differentiation and function (11, 45, 54). Our results are in agreement with these findings and further showed a significantly lower expression of p-ERK in Rgs12d/d cells induced by OS medium (Fig. 4C), suggesting an inactivation of ERK in OPCs after Rgs12 deletion. We also applied PTX on Rgs12d/d cells to identify whether the effects followed by Rgs12 deletion could be reverse. Our results exhibit partial rescued effects on OB differentiation and maturation, and to some extent, were similar to the outcomes after ectopic expression of Rgs12 in defective OPCs (Fig. 4D, E, F), further confirming the involvement of Gαi-mediated ERK signaling pathway in Rgs12 regulated OB differentiation and function.
We also identify Ca2+ channel-Ca2+ oscillation-ERK signaling as another critical pathway that contributes to the regulatory role of Rgs12 during osteogenesis (Fig. 7D). Ca2+, as an intracellular second messenger, impacts nearly every aspect of biological processes through enormous versatility of Ca2+ signaling (17, 55, 56). Components of Ca2+ signaling, including Ca2+ influx from extracellular environment through Ca2+ channel and Ca2+ release from intracellular Ca2+ store, regulate the frequency and amplitude of Ca2+ oscillations (13, 22, 57), which is well known to play an critical role in promoting osteogenesis (9, 14, 58). In our work, we found that Ca2+ oscillations were barely detectable in Rgs12d/d OPCs (Fig. 4B), indicating an impairment on Ca2+ oscillations caused by Rgs12 deletion. We further determined whether this impairment on Ca2+ oscillations was attributed to the impact on major Ca2+ entry sources after Rgs12 deletion. Surprisingly, our results suggested that both extracellular Ca2+ influx and Ca2+ release from ER store were restrained after Rgs12 deletion in OPCs (Fig. 5). Several studies had conducted that these two major Ca2+ sources of Ca2+ oscillations played fundamental roles in regulating OB proliferation, differentiation and function individually (20, 22, 46, 47). Therefore, our work establishes the role of Rgs12 as a key regulator that affects Ca2+ oscillations during osteogenesis. Moreover, Ca2+ enters cytosol through a variety of Ca2+ channels, among them, LTCCs have been listed as critical regulators of intracellular Ca2+ homeostasis in OB and are closely associated with the regulation of OB functions (19, 47, 48). Our current data corroborates with those ideas and further suggested that LTCCs involve in the restraint of Ca2+ influx caused by Rgs12 deletion. Our results clearly showed steadily increased fluorescence signals in control OPCs after BayK8644 treatment, indicating a successful induction of amplified Ca2+ influx via LTCCs (Fig. 6A, B, C). Based on this, we further found that the long-lasting increasing signals of cytosolic free Ca2+ in control cells were unable to be detected in Rgs12d/d cells, indicating a negative impact on LTCCs function after Rgs12 deletion (Fig. 6D, E, F). ERK also serves as downstream effectors of Ca2+ signaling, and the frequencies of Ca2+ oscillations was reported to influence Ca2+-mediated activation of Ras and signaling through the ERK/mitogen-activated protein kinase (MAPK) cascade (21, 59, 60). The inactivation of ERK after Rgs12 deletion in OPCs that showed previously could also due to the decreased Ca2+ signaling originating from LTCCs (Fig. 4C, 7D).
A crosstalk between Gαi and Ca2+ signaling has long been reported (42–44), as both these pathways could share same terminal signals from cAMP signaling through Ca2+ sensor and Ca2+ oscillations. It was noted that Ca2+ sensor plays dual role on cAMP signaling, exerting through intracellular Ca2+ and PTX-sensitive Gαi pathways (42). On the other hand, it is well known that Gαi subunits dampen the cAMP production via inhibiting AC, whereas Gαq subunits increase intracellular Ca2+ via phospholipase C (PLC) activation (45, 61). Emerging evidence demonstrated that Gαi3 mutants with defective GTP binding and displayed lower GTPase activity, can block the Gαq-PLC-Ca2+ signaling pathway by forming an unproductive complex (52, 62). In our work, we also found lower GTPase activity towards Gαi after Rgs12 deletion.
An interesting phenomenon from our present and previous studies is that Rgs12 deletion impaired Ca2+ oscillations in both OBs and OCs, and RANKL–Ca2+ oscillations–NFAT2 signaling pathway had been blocked in Rgs12 defective OCs (27). A further study with more focus on bone remodeling using global Rgs12-knock out model is therefore suggested, and the deletion effects on Ca2+ channels and their subunits. Taken together, our results reveal a crucial role of Rgs12 in OB differentiation and bone formation via controlling Ca2+ channel/Gαi –Ca2+ oscillation-ERK activity, and highlight Rgs12 as a potential therapeutic target for skeletal abnormalities.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
Research reported in this publication was supported by the National Institute on Aging, National institute of Arthritis and Musculoskeletal and Skin Diseases, and the National Institute of Dental and Craniofacial Research, part of the National Institutes of Health, under Award Numbers AG048388, AR066101, and DE023105 to S.Y.
Footnotes
Supplemental methods and data have been included with the submission.
Disclosures
All authors state that they have no conflicts of interest.
Reference
- 1.Zaidi M Skeletal remodeling in health and disease. Nat Med. 2007; 13: 791–801. [DOI] [PubMed] [Google Scholar]
- 2.Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: Now and the future. Lancet. 2011; 377: 1276–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seeman E Pathogenesis of bone fragility in women and men. Lancet. 2002; 359: 1841–1850. [DOI] [PubMed] [Google Scholar]
- 4.Khosla S, Riggs BL. Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin North Am. 2005; 34: 1015–1030. [DOI] [PubMed] [Google Scholar]
- 5.Marie PJ. Osteoblast dysfunctions in bone diseases: From cellular and molecular mechanisms to therapeutic strategies. Cell Mol Life Sci. 2015; 72: 1347–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riggs BL, Parfitt AM. Drugs Used to Treat Osteoporosis: The Critical Need for a Uniform Nomenclature Based on Their Action on Bone Remodeling. J Bone Miner Res. 2004; 20: 177–184. [DOI] [PubMed] [Google Scholar]
- 7.Kawai M, Mödder UI, Khosla S, Rosen CJ. Emerging therapeutic opportunities for skeletal restoration. Nat Rev Drug Discov. 2011; 10: 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green JT, Mills AM. Osteogenic tumors of bone. Semin Diagn Pathol. 2014; 31: 21–29. [DOI] [PubMed] [Google Scholar]
- 9.Jules J, Yang S, Chen W, Li Y-P. Role of Regulators of G Protein Signaling Proteins in Bone Physiology and Pathophysiology. Prog Mol Biol Transl Sci. 2015; 133: 47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdallah BM, Jafari A, Zaher W, Qiu W, Kassem M. Skeletal (stromal) stem cells: An update on intracellular signaling pathways controlling osteoblast differentiation. Bone. 2015; 70: 28–36. [DOI] [PubMed] [Google Scholar]
- 11.Raucci A, Bellosta P, Grassi R, Basilico C, Mansukhani A. Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. J Cell Physiol. 2008; 215: 442–451. [DOI] [PubMed] [Google Scholar]
- 12.Hanna H, Andre FM, Mir LM. Electrical control of calcium oscillations in mesenchymal stem cells using microsecond pulsed electric fields. Stem Cell Res Ther. 2017; 8: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki T, Notomi T, Miyajima D, et al. Osteoblastic differentiation enhances expression of TRPV4 that is required for calcium oscillation induced by mechanical force. Bone. 2013; 54: 172–178. [DOI] [PubMed] [Google Scholar]
- 14.Sun S, Liu Y, Lipsky S, Cho M. Physical manipulation of calcium oscillations facilitates osteodifferentiation of human mesenchymal stem cells. FASEB J. 2007; 21: 1472–1480. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Huang G, Luo X, Penninger JM, Muallem S. Role of regulator of G protein signaling 2 (RGS2) in Ca2+ oscillations and adaptation of Ca2+ signaling to reduce excitability of RGS2−/− cells. J Biol Chem. 2004; 279: 41642–41649. [DOI] [PubMed] [Google Scholar]
- 16.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011; 3: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015; 95: 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Putney JW, Steinckwich-Besançon N, Numaga-Tomita T, et al. The functions of store-operated calcium channels. Biochim Biophys Acta - Mol Cell Res. 2017; 1864: 900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Z, Cao X, Zhang Z, et al. Simulated microgravity inhibits L-type calcium channel currents partially by the up-regulation of miR-103 in MC3T3-E1 osteoblasts. Sci Rep. 2015; 5: 8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zayzafoon M Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem. 2006; 97: 56–70. [DOI] [PubMed] [Google Scholar]
- 21.Mulvaney JM, Zhang T, Fewtrell C, Roberson MS. Calcium Influx through L-type Channels Is Required for Selective Activation of Extracellular Signal-regulated Kinase by Gonadotropin-releasing Hormone*. J Biol Chem. 1999; 274: 29796–29804. [DOI] [PubMed] [Google Scholar]
- 22.Keinan D, Yang S, Cohen RE, Yuan X, Liu T, Li Y-P. Role of regulator of G protein signaling proteins in bone. Front Biosci. 2014; 19: 634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snow BE, Hall RA, Krumins AM, et al. GTPase activating specificity of RGS12 and binding specificity of an alternatively spliced PDZ (PSD-95/Dlg]ZO-1) domain. J Biol Chem. 1998; 273: 17749–17755. [DOI] [PubMed] [Google Scholar]
- 24.Sambi BS, Hains MD, Waters CM, et al. The effect of RGS12 on PDGFbeta receptor signalling to p42/p44 mitogen activated protein kinase in mammalian cells. Cell Signal. 2006; 18: 971–81. [DOI] [PubMed] [Google Scholar]
- 25.Willard M, Willard F, Li X, Cappell S. Selective role for RGS12 as a Ras/Raf/MEK scaffold in nerve growth factor-mediated differentiation. EMBO J. 2007; 26: 2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng J, Bencsik M, Louie A, et al. Conditional expression of a Gi-coupled receptor in osteoblasts results in trabecular osteopenia. Endocrinology. 2008; 149: 1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan X, Cao J, Liu T, et al. Regulators of G protein signaling 12 promotes osteoclastogenesis in bone remodeling and pathological bone loss. Cell Death Differ. 2015; 22: 2046–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang S, Li Y-P, Liu T, et al. Mx1-Cre mediated Rgs12 conditional knockout mice exhibit increased bone mass phenotype. Genesis. 2013; 51: 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S, Li Y-P. RGS12 Is Essential for RANKL-Evoked Signaling for Terminal Differentiation of Osteoclasts In Vitro. J Bone Miner Res. 2006; 22: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan X, Cao J, He X, et al. Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nat Commun. 2016; 7: 11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha KM, Zhou X. Genetic and molecular control of osterix in skeletal formation. J Cell Biochem. 2013; 114: 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davey RA, Clarke MV, Sastra S, et al. Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res. 2012; 21: 885–893. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Mishina Y, Liu F. Osterix-Cre Transgene Causes Craniofacial Bone Development Defect. Calcif Tissue Int. 2014; 96: 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Fang F, Yuan H, et al. Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J Bone Min Res. 2013; 28: 2414–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russo C, Lazzaro V, Gazzaruso C, et al. Proinsulin C-peptide modulates the expression of ERK1/2, type I collagen and RANKL in human osteoblast-like cells (Saos-2). Mol Cell Endocrinol. 2017; 442: 134–141. [DOI] [PubMed] [Google Scholar]
- 36.Khoshniat S, Bourgine A, Julien M, et al. Phosphate-dependent stimulation of MGP and OPN expression in osteoblasts via the ERK1/2 pathway is modulated by calcium. Bone. 2011; 48: 894–902. [DOI] [PubMed] [Google Scholar]
- 37.Li Z, Li C, Zhou Y, et al. Advanced glycation end products biphasically modulate bone resorption in osteoclast-like cells. Am J Physiol - Endocrinol Metab. 2016; 310: E355–E366. [DOI] [PubMed] [Google Scholar]
- 38.Ping S, Liu S, Zhou Y, et al. Protein disulfide isomerase-mediated apoptosis and proliferation of vascular smooth muscle cells induced by mechanical stress and advanced glycosylation end products result in diabetic mouse vein graft atherosclerosis. Cell Death Dis. 2017; 8: e2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gómez NM, Lu W, Lim JC, et al. Robust lysosomal calcium signaling through channel TRPML1 is impaired by lysosomal lipid accumulation. FASEB J. 2017; : fj.201700220RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin S-J, Chiang M-C, Shih H-Y, et al. Regulator of G protein signaling 2 (Rgs2) regulates neural crest development through Pparδ-Sox10 cascade. Biochim Biophys Acta - Mol Cell Res. 2017; 1864: 463–474. [DOI] [PubMed] [Google Scholar]
- 41.Hsiao EC. G s /G i Regulation of Bone Cell Differentiation: Review and Insights from Engineered Receptors. Gs/Gi Regul Bone … Horm Metab Res Horm Metab Res. 2016; 48: 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerbino A, Ruder WC, Curci S, Pozzan T, Zaccolo M, Hofer AM. Termination of cAMP signals by Ca2+ and Gαi via extracellular Ca2+ sensors: A link to intracellular Ca2+ oscillations. J Cell Biol. 2005; 171: 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruce JIE, Straub S V, Yule DI Crosstalk between cAMP and CA2+ signaling in non-excitable cells. Cell Calcium. 2003; 34: 431–444. [DOI] [PubMed] [Google Scholar]
- 44.Cooper DM, Mons N, Karpen JW. Adenylyl cyclases and the interaction between calcium and cAMP signalling. Nature. 1995; 374: 421–424. [DOI] [PubMed] [Google Scholar]
- 45.Caverzasio J, Palmer G, Suzuki a, Bonjour JP. Evidence for the involvement of two pathways in activation of extracellular signal-regulated kinase (Erk) and cell proliferation by Gi and Gq protein-coupled receptors in osteoblast-like cells. J Bone Miner Res. 2000; 15: 1697–1706. [DOI] [PubMed] [Google Scholar]
- 46.Dolmetsch RE, Lewis RS. Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J Gen Physiol. 1994; 103: 365–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freisinger CM, Schneider I, Westfall TA, Slusarski DC. Calcium dynamics integrated into signalling pathways that influence vertebrate axial patterning. Philos Trans R Soc Lond B Biol Sci. 2008; 363: 1377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park R, Ji JD. Calcium channels: the potential therapeutic targets for inflammatory bone destruction of rheumatoid arthritis. Inflamm Res. 2016; 65: 347–354. [DOI] [PubMed] [Google Scholar]
- 49.Sims NA, Martin TJ. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 2014; 3: 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakamoto A, Chen M, Nakamura T, Xie T, Karsenty G, Weinstein LS. Deficiency of the G-protein α-subunit Gsα in osteoblasts leads to differential effects on trabecular and cortical bone. J Biol Chem. 2005; 280: 21369–21375. [DOI] [PubMed] [Google Scholar]
- 51.Luttrell LM. Reviews in molecular biology and biotechnology: Transmembrane signaling by G protein-coupled receptors. Mol Biotechnol. 2008; 39: 239–264. [DOI] [PubMed] [Google Scholar]
- 52.Blaukat A, Barac A, Cross MJ, Offermanns S, Dikic I. G protein-coupled receptor-mediated mitogen-activated protein kinase activation through cooperation of Galpha(q) and Galpha(i) signals. Mol Cell Biol. 2000; 20: 6837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mochizuki N, Ohba Y, Kiyokawa E, et al. Activation of the ERK/MAPK pathway by an isoform of rap1GAP associated with G alpha(i). Nature. 1999; 400: 891–894. [DOI] [PubMed] [Google Scholar]
- 54.Zanotti S, Smerdel-Ramoya A, Stadmeyer L, Canalis E. Activation of the ERK pathway in osteoblastic cells, role of gremlin and BMP-2. J Cell Biochem. 2008; 104: 1421–1426. [DOI] [PubMed] [Google Scholar]
- 55.Clapham DE. Calcium Signaling. Cell. 2007; 131: 1047–1058. [DOI] [PubMed] [Google Scholar]
- 56.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000; 1: 11–21. [DOI] [PubMed] [Google Scholar]
- 57.Kiselyov K, Shin DM, Muallem S. Signalling specificity in GPCR-dependent Ca2+ signalling. Cell Signal. 2003; 15: 243–253. [DOI] [PubMed] [Google Scholar]
- 58.Li YJ, Batra NN, You L, et al. Oscillatory fluid flow affects human marrow stromal cell proliferation and differentiation. J Orthop Res. 2004; 22: 1283–1289. [DOI] [PubMed] [Google Scholar]
- 59.Kupzig S, Walker S a, Cullen PJ. The frequencies of calcium oscillations are optimized for efficient calcium-mediated activation of Ras and the ERK/MAPK cascade. Proc Natl Acad Sci U S A. 2005; 102: 7577–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morita M, Higuchi C, Moto T, et al. Dual regulation of calcium oscillation in astrocytes by growth factors and pro-inflammatory cytokines via the mitogen-activated protein kinase cascade. J Neurosci. 2003; 23: 10944–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Werry TD, Christopoulos A, Sexton PM. Mechanisms of ERK1 / 2 Regulation by Seven-Transmembrane-Domain Receptors. Curr Pharm Des. 2006; : 1683–1702. [DOI] [PubMed] [Google Scholar]
- 62.Marivin A, Leyme A, Parag-Sharma K, et al. Dominant-negative Gα subunits are a mechanism of dysregulated heterotrimeric G protein signaling in human disease. Sci Signal. 2016; 9. doi: 10.1126/scisignal.aad2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.