

Abstract
Background
BTBD7_hsa_circ_0000563, which is located on chromosome 14, contains conserved binding sites with miR‐155/130a and RNA‐binding proteins according to bioinformatic prediction. We investigated the association of BTBD7_hsa_circ_0000563 expression in coronary artery segments with atherosclerotic stenosis and identified the proteome‐wide BTBD7_hsa_circ_0000563‐regulated proteins in human coronary artery.
Methods
The atherosclerotic grade and extent in coronary artery segments were determined by hematoxylin and eosin staining. BTBD7_hsa_circ_0000563 expression in eight coronary artery segments from one patient was quantified by RT‐qPCR assay. A proteomic approach was adopted to reveal significant differences in protein expression between among four groups differing in their BTBD7_hsa_circ_0000563 expression levels.
Results
The RT‐qPCR assay revealed that coronary artery segments with severe atherosclerotic stenosis had significantly low BTBD7_hsa_circ_0000563 levels. The proteomic analysis identified 49 differentially expressed proteins among the segment groups with different BTBD7_hsa_circ_0000563 expression levels, of which 10 were downregulated and 39 were upregulated with increases in the BTBD7_hsa_circ_0000563 level. The 10 downregulated proteins were P61626 (LYSC_HUMAN), P02760 (AMBP_HUMAN), Q02985 (FHR3_HUMAN), P01701 (LV151_HUMAN), P06312(KV401_HUMAN), P01624 (KV315_HUMAN), P13671 (CO6_HUMAN), P01700(LV147_HUMAN), Q9Y287(ITM2B_HUMAN), and A0A075B6I0 (LV861_HUMAN). The top 10 upregulated proteins were Q92552 (RT27_HUMAN), Q9UJY1(HSPB8_HUMAN), Q9Y235(ABEC2_HUMAN), P19022 (CADH2_HUMAN), O43837(IDH3B_HUMAN), Q9H479(FN3K_HUMAN), Q9UM22(EPDR1_HUMAN), P48681(NEST_HUMAN), Q9NRP0(OSTC_HUMAN), and Q15628(TRADD_HUMAN).
Conclusion
BTBD7_hsa_circ_0000563 is involved in the atherosclerotic changes in human coronary artery segments. Verification, mechanistic, and function studies are needed to confirm whether patients with coronary artery disease would benefit from such personalized medicine in the future.
Keywords: BTBD7_hsa_circ_0000563, circRNA, coronary artery segment, coronary heart disease
We investigated BTBD7_hsa_circ_0000563 expression level in coronary artery segments with atherosclerotic stenosis and identified 49 differentially BTBD7_hsa_circ_0000563‐regulated proteins in human coronary artery which were mainly located in the mitochondria and involved in the citrate cycle (TCA cycle) pathway. It provided new insights into ATP synthase important role in the regulatory network of BTBD7_hsa_circ_0000563 in the coronary artery.
1. INTRODUCTION
Cardiovascular diseases, especially coronary artery disease (CAD) arising from atherosclerosis, are the leading causes of human morbidity and mortality worldwide. 1 Although imaging modalities and serological indicators are available for estimating the extent of atherosclerosis in affected individuals, the established methods used for the monitoring of CAD are concentrated on measurements at the late symptomatic phases. Therefore, there remains an urgent need for the exploration of biomarkers for the early detection and timely therapeutic intervention of CAD.
Increasing evidence has implicated circular RNAs (circRNAs) in the pathogenesis of CAD. 2 , 3 We had previously discovered through hierarchical clustering analysis that coronary artery segments with severe atherosclerosis showed distinct differences in circRNA profiles to those of their non‐atherosclerotic counterparts. These circRNAs may play pathological roles in coronary artery atherosclerosis. Among them, BTBD7_hsa_circ_0000563 stands out as a clinically relevant tissue‐associated circRNA, 4 as it is involved in various human diseases and participates in many biological processes. 5 , 6 The BTBD7_hsa_circ_0000563 coding sequence starts from position 93 760 203 of chromosome 14 and ends at position 93 762 503 (https://circinteractome.nia.nih.gov/bin/circsearch?circrna=hsa_circ_0000563). Furthermore, bioinformatic prediction has indicated that BTBD7_hsa_circ_0000563 contains conserved binding sites with miR‐155/130a and RNA‐binding proteins. In this present study, reverse transcription (RT) followed by the real‐time quantitative polymerase chain reaction (RT‐qPCR) detection of BTBD7_hsa_circ_0000563 was employed to uncover and validate candidate biomarkers of CAD in the coronary artery, using samples from patients with CAD. Additionally, quantitative proteomics‐based strategies were employed to identify BTBD7_hsa_circ_0000563‐regulated proteins in the human coronary artery.
2. MATERIALS AND METHODS
2.1. Study subjects
Coronary artery samples from two deceased individuals were obtained by autopsy at the Department of Human Anatomy at Nanjing Medical University. Informed consent for the research use only of the samples was obtained from the bereaved families, and the autopsy was conducted according to the guidelines of the university. All experimental protocols used were performed in accordance with established guidelines and approved by the ethics committee of Nanjing Medical University and the First Affiliated Hospital of Nanjing Medical University. At the time of death, the two individuals were 64 and 68 years old, respectively, and the female:male ratio was 0:2. The postmortem delay varied between 1 and 2 days. For each autopsy, the epicardial coronary artery was removed from the heart and divided into 10 segments, namely the proximal, middle, and distal segments of the left anterior descending, left circumflex, and right coronary artery, respectively, and the left main trunk. Each of the 10 coronary artery segments was subjected to RNA, protein, and pathological analyses. The segment samples used for the RNA and protein analyses were snap‐frozen in liquid nitrogen and stored at –80°C for RT‐qPCR analysis. For the pathological study, the segment samples were fixed overnight in 10% formalin and embedded in paraffin for histological analysis. The datasets used and/or analyzed during this study are available from the corresponding author upon request.
2.2. Pathological analysis
First, the coronary artery segment samples were decalcified with EDTA decalcification fluid for approximately 2 weeks. Then, the samples were fixed overnight in 10% formalin and processed for paraffin embedment. Thereafter, consecutive 5‐µm‐thick longitudinal sections were obtained using a rotary microtome (Leica RM2235, Leica Biosystems Nussloch GmbH) and stained with hematoxylin and eosin (H&E) for morphometric analysis. Each slide was examined under a stereomicroscope 10× (Leica DM2500, Leica) at 5× to 20× original magnification, and the image was digitized using an image analysis system (Leica LAS). The grading and extent of the atherosclerosis of the segments were determined independently by pathologists who assessed the histological grading according to the American Heart Association classification guidelines. 7
2.3. Selection of circular RNAs
In our previous study, significant differences were found between patients with and those without CAD with respect to their circulating levels of miR‐221 (P = .001), miR‐155 (P = .049), and miR‐130a (P = .001). 8 Therefore, we selected hsa‐miR‐221‐3p, hsa‐miR‐155‐5p, and hsa‐miR‐130a‐3p, which were validated by qPCR detection, for a joint analysis of the miRNA and circRNA profiling data. We used miRNA target predictions based on Starbase v2.0 (http://starbase.sysu.edu.cn/mirCircRNA.php) and found interactions between BTBD7_hsa_circ_0000563 and hsa‐miR‐155‐5p and hsa‐miR‐130a‐3p, respectively. Therefore, BTBD7_hsa_circ_0000563 was selected as the target circRNA in the present study.
2.4. RNA extraction and RT‐qPCR
Total RNA was extracted from the coronary artery segments using TRIzol reagent (15596018, Invitrogen) according to the manufacturer's instructions. The RNA integrity number was determined using the Agilent Bioanalyzer 2100 system (Agilent Technologies).
Taking BTBD7.22 2F (5′‐ATGCTTGCACAAGAAATGGA‐3′) and BTBD7.22 2R (5′‐GAACATGAATGAGGATAATTAG‐3′) as primers, total RNA (1 µg) was used as a template to prepare cDNA for the PCR analysis, as reported by Pan et al 9 The BTBD7_hsa_circ_0000563 expression level was quantified using the SYBR Green Real‐time PCR Master Mix (QPK‐212, TOYOBO) on an ABI 7900HT sequence detection system (7900HT, Applied Biosystems). 10
2.5. Protein extraction, LC‐MS/MS analysis
For the quantification of the protein, 500 μg of total protein was extracted using a multistep L3 lysis buffer/M2 lysis buffer protocol, as reported by Zhou et al 11 in 2020. First, the Bradford method was used to quantify the protein. Then, 100 μg of protein was dissolved to 1 μg/μL using Μ2 lysis buffer (without thiourea). The sample was then diluted 6‐fold by the adding addition of 5 volumes of 100 mmol/L tetraethylammonium bicarbonate before shaking by centrifugation. The protein samples were then digested overnight at 37°C by the addition of 2 μg of trypsin. Next, 10 mg of C18 resin powder was weighed for each 100 µg of peptide sample activated with 1 mL of methanol. After centrifugation of the C18 column and discarding of the supernatant, the sample was desalted by washing the column twice with 0.1% formic acid (FA) + 3% acetonitrile and then eluted with 1 mL of 0.1% FA + 80% ACN. The eluted peptides were dried with a vacuum concentrator. The dried sample was diluted to 1 μg/μL and then analyzed by liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) on a Triple TOF 5600 + LC/MS system (AB SCIEX, Redwood City, CA, USA). The scanning mode was set to 5 μL and 120 min, and peptides in the sample with a mass‐to‐charge (m/z) ratio of 350‐1500 were scanned. For information‐dependent acquisition of the data, the first‐order mass spectrum was scanned with an ion accumulation time of 250 ms, and the secondary mass spectrum of 30 precursor ions was acquired with an ion accumulation time of 50 ms The MS1 spectrum was acquired in the 350‐1200 m/z range, and the MS2 spectrum was acquired in the 100‐1500 m/z range. The dynamic exclusion time for the precursor ion was set to 15 s.
2.6. Data analysis
Protein identification and quantification were performed using ProteinPilot software (https://sciex.com.cn/products/software/proteinpilot‐software, v4.5, AB SCIEX). The MS/MS spectral data were searched against the UniProt‐Homo_sapiens protein database and identified by the Paragon algorithm. The parameters were set as follows: cysteine modified with iodoacetamide; and biological modifications selected as the ID focus. For calculation of the false discovery rate (FDR), an automatic decoy database search strategy was employed where the value was estimated using the Proteomics System Performance Evaluation Pipeline (PSPEP) Software, algorithm integrated with the ProteinPilot software package. Only unique peptides with global FDR values of less than 1% were considered for further analysis. Skyline v4.2 software was applied for the MS1 filtering and ion chromatogram extractions for label‐free peptide quantification, with the parameter settings for the Skyline MS1 filtering being the same as. The mean ratio for each group, as determined from the Skyline quantification, was used to calculate the fold change.
For the final analysis, eight segment samples (viz., the left main trunk; proximal, middle, and distal segments of the left anterior descending; proximal, middle, and distal segments of the left circumflex; and proximal segment of the right coronary artery) were divided into four groups according to ascending order of BTBD7_hsa_circ_0000563 expression levels, functioning as a comparative basis for further analysis of the proteomic data. The median quantitative value of the samples in each group was taken as the expression value of a protein in that group, and thus, a new expression value of BTBD7_hsa_circ_0000563 was obtained for each group. Next, the fold change between groups was also compared with the new expression value of each group. To test whether the difference between groups was significant, a t test was carried out and the P‐value was obtained. The screening criteria for proteins with significant expression differences were as follows: significantly differentially expressed, with a fold change threshold of over 1.5 or less than 0.667 between groups; P < .05 for the comparison between groups; and of any three groups being compared, at least one of them satisfies the above criteria with an increasing or a decreasing ratio trend among all three groups following the increase in BTBD7_hsa_circ_0000563 expression level.
2.7. Bioinformatics and annotations
To determine the biological and functional properties of all identified proteins, Gene Ontology (GO) terms (http://geneontology.org/) were used to describe the properties of the genes and gene products in organisms. To this end, homology searches were first performed on all identified sequences, using the local NCBI blastp program for the NCBInr animal database. The e‐value was set at less than 1e‐5, and the best hit for each query sequence took into account the GO term match. Functional annotation of the genes from the new genome and studies of the genome evolution were carried out using the Clusters of Orthologous Groups (COG) database (http://www.ncbi.nlm.nih.gov/COG/), which covers the orthologous protein system population. Additionally, using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, 12 pathway analysis was carried out to identify the most important metabolic and signal transduction pathways in which the proteins were involved. To identify candidate biomarkers, hypergeometric testing for GO enrichment and KEGG pathway enrichment was used. The protein‐protein interaction (PPI) network of the BTBD7_hsa_circ_0000563‐regulated proteins was generated using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database v10.0 13 with default settings and visualized using Cytoscape v3.2.1 (http://www.cytoscape.org).
3. RESULTS
3.1. Natural history and histological classification of atherosclerotic lesions in the coronary artery samples
The natural history and histological classification of atherosclerotic lesions in the coronary artery samples were determined by H&E staining. Although atherosclerotic lesions were found in the coronary artery segments of both individuals, the grade and extent of the lesions were more severe in patient 1. All of the examined segment samples from patient 1 showed atherosclerotic changes in the tunica intima, ranging from lesions classifiable as atherosclerotic tunica intima to those classified as secondary affection tunica intima, with the coronary stenosis ranging from 51% to 100%. The atherosclerotic lesions grading and staging in coronary artery samples of patient 1 were shown by means of H&E staining. LM, LAD‐M, and LCX‐D were classified as stenosis from 51% to 75% while LAD‐P, LAD‐D, LCX‐P, LCX‐M, RCA‐P, RCA‐M, and RCA‐D were defined as stenosis from 76% to 100%. LAD‐P, LAD‐M, LAD‐D, LCX‐M, LCX‐D, RCA‐M, and RCA‐D were atherosclerotic tunica intima; LM, LCX‐P, and RCA‐P were secondary affection tunica intima. By contrast, only eight of the coronary artery segments from patient 2 had atherosclerotic changes in the normal tunica intima, namely fatty streak tunica intima, fibrous plaque tunica intima, and atherosclerotic tunica intima, with the coronary stenosis ranging from 0% to 75%. The results of atherosclerotic lesions grading and staging in coronary artery samples of patient 2 were observed as follows: LAD‐D, LCX‐P, LCX‐M, LCX‐D, RCA‐P, and RCA‐D were determined as stenosis from 0% to 25%; LM, LAD‐P, LAD‐M, and RCA‐M were stenosis from 51% to 75%. With regard to the staging of these samples, LAD‐D and LCX‐P were normal tunica intima. LCX‐M, LCX‐D, and RCA‐D were fatty streak tunica intima; RCA‐P was defined as fibrous plaque tunica intima; LM, LAD‐P, LAD‐M, and RCA‐M were divided as atherosclerotic tunica intima.
3.2. BTBD7_hsa_circ_0000563 expression in the coronary artery segments
Owing to RNA degradation, the purity and concentration of the total RNA in all the coronary artery segments from patient 1 and in the middle and distal segments of the right coronary artery from patient 2 failed to meet the standards for RT‐qPCR quantification. Therefore, only the BTBD7_hsa_circ_0000563 expression levels in the coronary artery samples from patient 2 (except for the middle and distal segments of the right coronary artery) were obtained by RT‐qPCR. The results of Sanger sequencing from PCR products confirmed the cyclization site and the results were shown in Figure 1.
FIGURE 1.
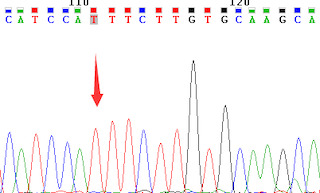
Cyclization site of the PCR sequencing confirmed by Sanger sequencing
The results revealed that the BTBD7_hsa_circ_0000563 expression levels were significantly higher in the normal coronary artery segments than in their severely atherosclerotic counterparts (Table 1 and Figure 2). In the distal segment of the left anterior descending with a normal tunica intima and 0‐25% stenosis, the BTBD7_hsa_circ_0000563 expression level was significantly higher (by 20‐fold) than that in the left main trunk with an atherosclerotic tunica intima and 51‐75% stenosis (P = .037). Compared to the distal segment of the left anterior descending functioning as control group, significant differences in the BTBD7_hsa_circ_0000563 expression level were also observed in the distal segment of the left circumflex (Stage 1: fatty streak tunica intima) and the proximal segment of the right coronary artery (Stage 2: fibrous plaques tunica intima; P ≤ .05).
TABLE 1.
BTBD7_hsa_circ_0000563 expression in coronary artery segments
| Segment | h‐actin | ct | △ct | △△ct | 2−△△ct | Expression level |
|---|---|---|---|---|---|---|
| LM | 20.15 | 33.85 | 13.7 | 2.08 | 0.236514412 | 0.1705 |
| 20.16 | 35.04 | 14.88 | 3.26 | 0.10438599 | ||
| 20.18 | 31.87 | 11.69 | 0.07 | 0.952637998 | ||
| LAD‐P | 20.06 | 31.41 | 11.35 | ‐0.27 | 1.205807828 | 0.013726 |
| 19.85 | 37.38 | 17.53 | 5.91 | 0.016630784 | ||
| 20.06 | 38.21 | 18.15 | 6.53 | 0.010821168 | ||
| LAD‐M | 20.17 | 37.29 | 17.12 | 5.5 | 0.022097087 | 1.325879 |
| 20.17 | 31.46 | 11.29 | ‐0.33 | 1.257013375 | ||
| 20.19 | 31.33 | 11.14 | ‐0.48 | 1.394743666 | ||
| LAD‐D | 19.14 | 30.86 | 11.72 | 0 | 1 | 1 |
| 19.17 | 29.8 | 10.63 | 0 | 1 | ||
| 18.91 | 31.42 | 12.51 | 0 | 1 | ||
| LCX‐P | 18.85 | 28.19 | 9.34 | −2.28 | 4.856779538 | 3.99756 |
| 18.98 | 30.86 | 11.88 | 0.26 | 0.835087919 | ||
| 19.07 | 29.04 | 9.97 | −1.65 | 3.138336392 | ||
| LCX‐D | 20.18 | 29.35 | 9.17 | −2.45 | 5.464161027 | 5.31411 |
| 20.11 | 30.09 | 9.98 | −1.64 | 3.116658319 | ||
| 20.26 | 29 | 8.74 | −2.88 | 7.361501205 | ||
| RCA‐P | 20.11 | 30.08 | 9.97 | −1.65 | 3.138336392 | 2.28259 |
| 19.94 | 30.5 | 10.56 | −1.06 | 2.084931522 | ||
| 20.02 | 30.94 | 10.92 | −0.7 | 1.624504793 | ||
| LCX‐M | 20.39 | 34.04 | 13.65 | 2.03 | 0.244855074 | 2.15609 |
| 20.38 | 31.45 | 11.07 | −0.55 | 1.464085696 | ||
| 20.22 | 30.33 | 10.11 | −1.51 | 2.848100391 |
Abbreviations: LAD‐D, distal segment of the left anterior descending; LAD‐M, middle segment of the left anterior descending; LAD‐P, proximal segment of the left anterior descending; LCX‐D, distal segment of the left circumflex; LCX‐M, middle segment of the left circumflex; LCX‐P, proximal segment of the left circumflex; LM, left main trunk; RCA‐P, proximal segment of the right coronary artery.
FIGURE 2.
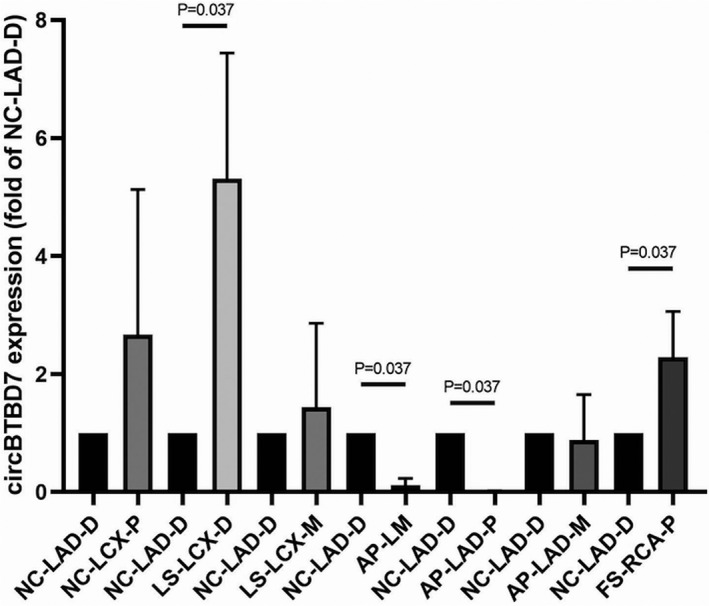
BTBD7_hsa_circ_0000563 expression levels in coronary artery segments. LAD‐P, proximal segment of the left anterior descending; AP, atherosclerotic plaques; FS, fatty streak; LAD‐M, middle segment of the left anterior descending; LAD‐D, distal segment of the left anterior descending; LCX‐P, proximal segment of the left circumflex; LCX‐M, middle segment of the left circumflex; LCX‐D, distal segment of the left circumflex; LS, lipid strain; NC, normal tunica intima; RCA‐P, proximal segment of the right coronary artery
3.3. Screening of proteins with significant expression differences according to the BTBD7_hsa_circ_0000563 expression level
Table 2 lists the proteins with significant expression differences according to the BTBD7_hsa_circ_0000563 expression levels. The proteome‐wide analysis revealed that 49 proteins were differentially expressed among the various groups with different BTBD7_hsa_circ_0000563 expression levels. Of these 49 proteins, 10 were found to be downregulated and 39 were upregulated with the increases in the BTBD7_hsa_circ_0000563 expression level.
TABLE 2.
Screening of proteins with significant expression differences according to the BTBD7_hsa_circ_0000563 expression level
| ID | Log2 (Mean Ratio) | Expression Trend | ID | Log2 (Mean Ratio) | Expression Trend |
|---|---|---|---|---|---|
| sp|P00367|DHE3_HUMAN | 0.396890153 | UP | sp|P07954|FUMH_HUMAN | 1.751035177 | UP |
| sp|Q92552|RT27_HUMAN | 9.964340868 | UP | sp|Q02985|FHR3_HUMAN | ‐0.776783417 | DOWN |
| sp|P13861|KAP2_HUMAN | 0.86710573 | UP | sp|Q9HA77|SYCM_HUMAN | 1.357458168 | UP |
| sp|O75746|CMC1_HUMAN | 2.949846161 | UP | sp|O75208|COQ9_HUMAN | 4.345230767 | UP |
| sp|Q9UJY1|HSPB8_HUMAN | 9.964340868 | UP | sp|Q9UM22|EPDR1_HUMAN | 9.964340868 | UP |
| sp|P48735|IDHP_HUMAN | 2.575070247 | UP | sp|Q9Y6W5|WASF2_HUMAN | 0.672953291 | UP |
| sp|O96000|NDUBA_HUMAN | 2.471621028 | UP | sp|P09622|DLDH_HUMAN | 1.333042018 | UP |
| sp|P13671|CO6_HUMAN | ‐1.037019189 | DOWN | sp|P48681|NEST_HUMAN | 9.964340868 | UP |
| sp|Q99733|NP1L4_HUMAN | 1.285106251 | UP | sp|Q16836|HCDH_HUMAN | 0.98112199 | UP |
| sp|P01624|KV315_HUMAN | ‐0.928694966 | DOWN | sp|P01700|LV147_HUMAN | ‐1.088244513 | DOWN |
| sp|Q9Y235|ABEC2_HUMAN | 9.964340868 | UP | sp|P36542|ATPG_HUMAN | 1.609124551 | UP |
| sp|P16615|AT2A2_HUMAN | 2.409979209 | UP | sp|P06312|KV401_HUMAN | ‐0.824528626 | DOWN |
| sp|P02760|AMBP_HUMAN | ‐0.522840789 | DOWN | sp|Q9NRP0|OSTC_HUMAN | 9.964340868 | UP |
| sp|P19022|CADH2_HUMAN | 9.964340868 | UP | sp|P01701|LV151_HUMAN | ‐0.784217877 | DOWN |
| sp|P61626|LYSC_HUMAN | ‐0.387726146 | DOWN | sp|P02511|CRYAB_HUMAN | 1.893750502 | UP |
| sp|O43837|IDH3B_HUMAN | 9.964340868 | UP | sp|Q15628|TRADD_HUMAN | 9.964340868 | UP |
| sp|Q9H479|FN3K_HUMAN | 9.964340868 | UP | sp|P23786|CPT2_HUMAN | 9.964340868 | UP |
| sp|O95831|AIFM1_HUMAN | 2.471881105 | UP | sp|P00846|ATP6_HUMAN | 9.964340868 | UP |
| sp|P17540|KCRS_HUMAN | 6.466926155 | UP | sp|O75367|H2AY_HUMAN | 0.732052073 | UP |
| sp|P21912|SDHB_HUMAN | 2.522976396 | UP | sp|P12235|ADT1_HUMAN | 3.202051973 | UP |
| sp|P50213|IDH3A_HUMAN | 1.393965276 | UP | sp|P08559|ODPA_HUMAN | 3.087349684 | UP |
| sp|P26440|IVD_HUMAN | 1.047538389 | UP | sp|P42765|THIM_HUMAN | 1.299244658 | UP |
| sp|Q9Y287|ITM2B_HUMAN | ‐1.146669649 | DOWN | sp|P30046|DOPD_HUMAN | 0.498761466 | UP |
| sp|Q99798|ACON_HUMAN | 2.022545365 | UP | sp|P46777|RL5_HUMAN | 0.738983955 | UP |
| sp|A0A075B6I0|LV861_HUMAN | ‐1.546526319 | DOWN |
UP, upregulated; DOWN, downregulated.
3.4. Gene Ontology annotation of the differentially expressed proteins
To further determine the biological and functional properties of all differentially expressed proteins, the 49 proteins were submitted to GO annotation (Figure 3). The results suggested that in the cellular component category, these proteins were predominantly derived from the mitochondrial matrix (GO:0 005 759) and mitochondrial inner membrane (GO:0 005 743) (Figure 3A). Moreover, in the molecular function category, the proteins were annotated primarily to NAD or NADH binding; 3 iron, 4 sulfur cluster binding; FAD binding; NAD binding; and isocitrate dehydrogenase (NAD+) activity (Figure 3B). In the biological process category, the proteins participated predominantly in the respiratory electron transport chain (GO: 0 022 904); tricarboxylic acid cycle (GO:0 006 099); and mitochondrial electron transport, NADH to ubiquinone (GO:0 006 120) (Figure 3C).
FIGURE 3.
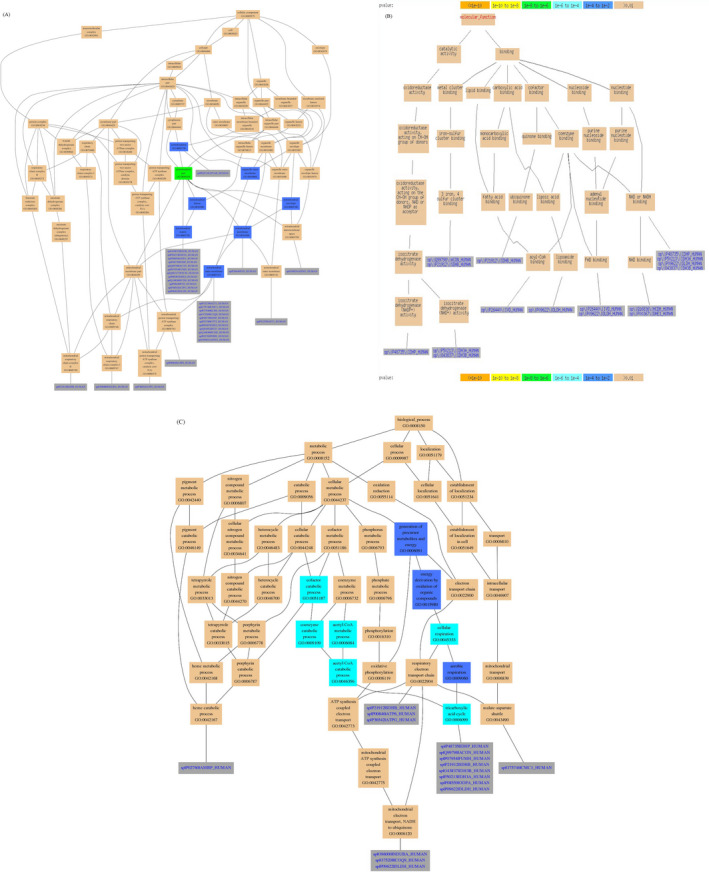
Gene Ontology annotation of the differentially expressed proteins. A, Cellular compartment annotation results for the differentially expressed proteins; B, Molecular function annotation results for the differentially expressed proteins; C, Biological process annotation results for the differentially expressed proteins
3.5. KEGG enrichment of the differentially expressed proteins
According to the KEGG pathway enrichment analysis, the BTBD7_hsa_circ_0000563‐related proteins were associated with the following pathways: Parkinson's disease; Alzheimer's disease; fatty acid metabolism; primary immunodeficiency; oxidative phosphorylation; Huntington's disease; d‐glutamine and d‐glutamate metabolism; valine, leucine, and isoleucine degradation; fatty acid elongation in mitochondria; metabolic pathways; apoptosis; microbial metabolism in diverse environments; and the citrate cycle (TCA cycle; Figure 4).
FIGURE 4.
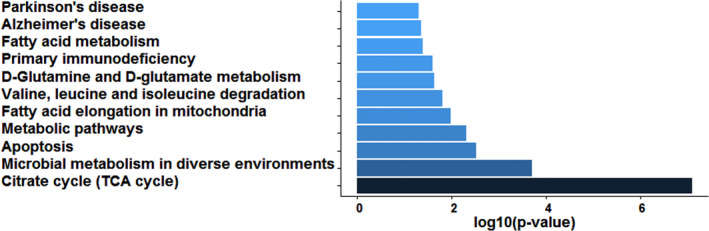
KEGG enrichment of the differentially expressed proteins
3.6. Protein‐protein interactions of the differentially expressed proteins
The PPI network of the BTBD7_hsa_circ_0000563‐regulated proteins was generated using the STRING database. Of the 49 differentially expressed proteins, 25 were engaged in the PPI network, which contained 30 nodes and 86 edges (Figure 5A). These 25 proteins were categorized into several functional groups. Therefore, the PPI network indicated that the BTBD7_hsa_circ_0000563‐regulated proteins had the potential to interact with one another and to work in unison to exercise their functions.
FIGURE 5.
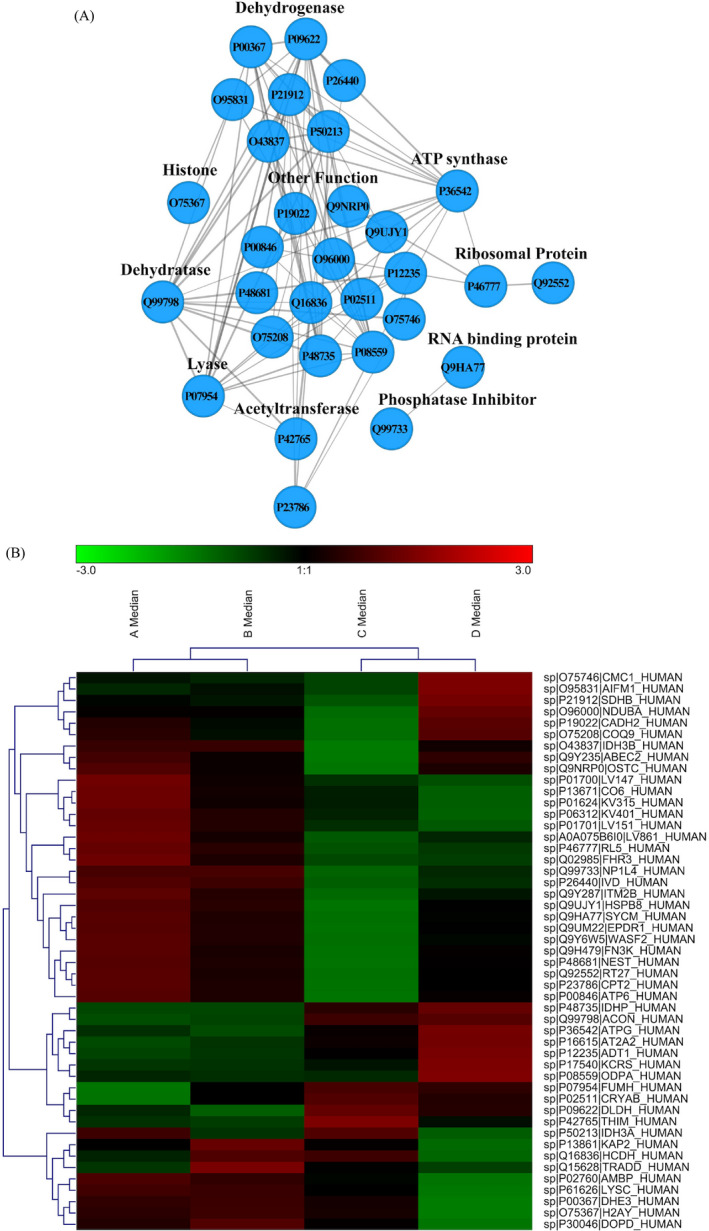
A, Protein‐protein interaction network of the differentially expressed proteins; B, Heat map of the differentially expressed proteins
Moreover, to evaluate the classification of the BTBD7_hsa_circ_0000563‐regulated proteins, we used the classification system to sort out the protein. The 49 proteins hit by the classification system were divided into several classes (Figure 5B). Taking these results together, the BTBD7_hsa_circ_0000563‐regulated proteins may make a difference to various biological processes, particularly in the regulation of dehydrogenases and ATP synthase.
4. DISCUSSION
In this study, we found that the circRNA BTBD7_hsa_circ_0000563 was involved in atherosclerotic changes in human coronary artery segments. Specifically, its expression levels were higher in relatively normal coronary artery segments than in those with severe atherosclerosis. In addition, by means of a proteomic approach, 49 proteins in the human coronary artery segments were identified to be regulated by enhanced BTBD7_hsa_circ_0000563 expression, thereby providing an abundant resource for future functional studies of BTBD7_hsa_circ_0000563 in the coronary artery.
Having being first reported by Memczak et al, 5 BTBD7_hsa_circ_0000563 is located on chromosome 14, with the gene code starting from position 93 760 203 and ending at position 93 762 503. Significant differences in the expression of BTBD7_hsa_circ_0000563 have since been found in human cells (H1‐hESC, NHEK, and AG04450), 14 human brain regions, 6 and human endothelial progenitor cells 4 by RNA sequencing analysis. However, our present study demonstrated that coronary artery segments with severe atherosclerotic stenosis had significantly lower levels of BTBD7_hsa_circ_0000563 than normal coronary artery segments. Furthermore, it was previously predicted that hsa‐miR‐155‐5p and hsa‐miR‐130a‐3p are targets of BTBD7_hsa_circ_0000563. In this present study, proteome‐wide analysis identified 49 proteins regulated by BTBD7_hsa_circ_0000563 overexpression in human coronary artery segments. By means of Gene Ontology annotation and KEGG enrichment of the differentially expressed proteins, we discovered that a large number of the differentially expressed proteins were located in the mitochondria and involved in the citrate cycle (TCA cycle) pathway. Furthermore, the PPI network of the 49 differentially expressed proteins regulated by BTBD7_hsa_circ_0000563 was generated using the STRING database. Interestingly, we found that 25 were engaged in the PPI network, which contained 30 nodes and 86 edges. Combined with these findings, we concluded that ATP synthase may play an important role in the regulatory network of BTBD7_hsa_circ_0000563 in the coronary artery. These results lay an epigenetic foundation for the future study of the underlying mechanisms of coronary artery atherosclerosis development.
As complex and dynamic organelles, the mitochondria play numerous functions pertaining to cellular metabolism and homeostasis, with the hallmark function being cellular energy generation by means of oxidative phosphorylation. 15 , 16 The mitochondrial DNA encodes only 13 oxidative phosphorylation proteins; the approximately 1500 other proteins from the mitochondrial proteome are encoded by the nuclear genome and transferred to the mitochondria. 17 In addition, several converging metabolic pathways, including those of folate metabolism, the TCA cycle, and sulfur metabolism, exist in whole or partial components of the mitochondria. 18
CAD has a complex etiology, with mitochondrial dysfunction being one of the factors that can have an effect on various CAD‐related cellular aberrations, including energy deficits, the deregulation of autophagy, metabolic abnormalities, the excessive production of reactive species, endoplasmic reticulum stress, and the activation of apoptosis. Impairments of ATP synthesis and the respiratory chain have been considered key to mitochondrial dysfunction. Therefore, the recovery of mitochondrial function, including the respiratory activity and ATP‐producing capacity, has been considered as a primary therapeutic target for improving the prognosis of patients with CAD. 19 Results from a recent study have suggested that the serum concentration of mitochondrial ATP synthase inhibitory factor 1 is independently negatively associated with all‐cause and cardiovascular mortality in patients with CAD. 20 Another study suggested that ectopic ATP synthase on endothelial cells could be a potential and novel therapeutic target for CAD. 21
This study had several limitations. First, the sample size was too small, given that the BTBD7_hsa_circ_0000563 expression levels based on the RT‐qPCR assay and the proteome‐wide protein identification based on the LC‐MS/MS analysis were conducted on only eight coronary artery segments from one of the patients. Second, only the association of BTBD7_hsa_circ_0000563 with the proteins that it regulates in the coronary artery segments was explored, whereas the possible mechanisms underlying these associations were not studied. Finally, the therapeutic value of the present findings cannot as yet be applied to clinical practice owing to the absence of function and mechanistic studies of BTBD7_hsa_circ_0000563. Therefore, further verification, mechanistic, and function studies are needed to determine whether patients with CAD can benefit from such personalized medicine in the future.
In conclusion, the most relevant finding of this study was that coronary artery segments with severe atherosclerotic stenosis showed significantly lower levels of BTBD7_hsa_circ_0000563 expression than normal coronary artery segments. Additionally, by means of a proteomic approach, 49 proteins were found to be regulated by enhanced BTBD7_hsa_circ_0000563 expression in human coronary artery segments. Bioinformatic analysis revealed that a large number of the differentially expressed proteins were located in the mitochondria and were involved in the citrate cycle (TCA cycle) pathway. Combined with the PPI network of the 49 differentially expressed proteins regulated by BTBD7_hsa_circ_0000563, ATP synthase may play an important role in the regulatory network of BTBD7_hsa_circ_0000563 in the coronary artery.
CONFLICT OF INTEREST
There are no conflicts of interest associated with this manuscript.
AUTHORS’ CONTRIBUTIONS
As a guarantor, Enzhi Jia conceived the study. Jia‐Xin Chen and Lei Hua, who contributed equally to this paper, designed the study and wrote the draft. Yong‐Jie Zhang, Jian‐Liang Jin, Mu‐Feng Gu, Zhi‐Yuan Mao, and Hai‐Jian Sun enrolled the participants and collected data under the supervision of Chen‐Hui Zhao and Qiao‐Wei Jia. Jing Zhang, Jin‐Xia Yuan, Lian‐Sheng Wang, and Wen‐Zhu Ma coordinated the study.
ETHICAL APPROVAL
Informed consent for the research use only of the samples was obtained from the bereaved families. The autopsy was conducted according to the guidelines of the university. All experimental protocols used were performed in accordance with the Declaration of Helsinki and were approved by the ethics committee of the Nanjing Medical University and the First Affiliated Hospital of Nanjing Medical University.
ACKNOWLEDGEMENTS
This study received support from the National Natural Science Foundation of China (grants 81170180, 30400173, 30971257, and 81970302) and the Priority Academic Program Development of Jiangsu Higher Education Institutions. We are grateful to Dr En‐Zhi Jia, Assistant Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine, for acting as a guarantor and conceiving this study.
Chen J‐X, Hua L, Zhao C‐H, et al. Quantitative proteomics reveals the regulatory networks of circular RNA BTBD7_hsa_circ_0000563 in human coronary artery. J Clin Lab Anal. 2020;34:e23495 10.1002/jcla.23495
Chen and Hua contributed equally to this paper.
Funding information
This study received support from the National Natural Science Foundation of China (Grants 81170180, 30400173, 30971257, and 81970302) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
REFERENCES
- 1. Mendis S, Davis S, Norrving B. Organizational update: the World Health Organization global status report on noncommunicable diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke. 2015;46(5):e121‐e122. [DOI] [PubMed] [Google Scholar]
- 2. Zhou Q, Zhang Z, Bei Y, Li G, Wang T. Circular RNAs as novel biomarkers for cardiovascular diseases In: X. J, ed. Circular RNAs: Biogenesis and Functions. Vol. 1087 of the book series Advances in Experimental Medicine and Biology. New York: Springer Singapore; 2018:159‐170. [DOI] [PubMed] [Google Scholar]
- 3. Altesha MA, Ni T, Khan A, Liu K, Zheng X. Circular RNA in cardiovascular disease. J Cell Physiol. 2019;234(5):5588‐5600. [DOI] [PubMed] [Google Scholar]
- 4. Maass PG, Glažar P, Memczak S, et al. A map of human circular RNAs in clinically relevant tissues. J Mol Med. 2017;95(11):1179‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333‐338. [DOI] [PubMed] [Google Scholar]
- 6. Rybak‐Wolf A, Stottmeister C, Glažar P, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870‐885. [DOI] [PubMed] [Google Scholar]
- 7. Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20(5):1177‐1178. [DOI] [PubMed] [Google Scholar]
- 8. Jia Q‐W, Chen Z‐H, Ding X‐Q, et al. Predictive effects of circulating miR‐221, miR‐130a and miR‐155 for coronary heart disease: a multi‐ethnic study in china. Cell Physiol Biochem. 2017;42(2):808‐823. [DOI] [PubMed] [Google Scholar]
- 9. Pan RY, Zhao CH, Yuan JX, et al. Circular RNA profile in coronary artery disease. Am J Transl Res. 2019;11(11):7115‐7125. [PMC free article] [PubMed] [Google Scholar]
- 10. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101‐1108. [DOI] [PubMed] [Google Scholar]
- 11. Zhou YQ, Yuan JX, Fan YW, et al. Proteomic landscape of human coronary artery atherosclerosis. Int J Mol Med. 2020;371‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanehisa M. Inferring antimicrobial resistance from pathogen genomes in KEGG. Methods Mol Biol. 2018;1807:225‐239. [DOI] [PubMed] [Google Scholar]
- 13. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447‐D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell‐type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. 2018;19(2):77‐92. [DOI] [PubMed] [Google Scholar]
- 16. Gorman GS, Chinnery PF, DiMauro S, et al. Mitochondrial diseases. Nature Rev Disease Prim. 2016;2(1):16080. [DOI] [PubMed] [Google Scholar]
- 17. Calvo SE, Clauser KR, Mootha VK. MitoCarta2. 0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251‐D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. 2018;50(2):121‐127. [DOI] [PubMed] [Google Scholar]
- 20. Maierean S, Serban MC, Rizzo M, Lippi G, Sahebkar A, Banach M. The potential role of mitochondrial ATP synthase inhibitory factor 1 (IF1) in coronary heart disease: a literature review. Lipids Health Dis. 2017;16(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu Y, Zhu Y. Ectopic ATP synthase in endothelial cells: a novel cardiovascular therapeutic target. Curr Pharm Des. 2010;16(37):4074‐4079. [DOI] [PubMed] [Google Scholar]