

Conspectus
In nature, light is harvested by photoactive proteins to drive a range of biological processes, including photosynthesis, phototaxis, vision, and ultimately life. Bacteriorhodopsin, for example, is a protein embedded within archaeal cell membranes that binds the chromophore retinal within its hydrophobic pocket. Exposure to light triggers regioselective photoisomerization of the confined retinal, which in turn initiates a cascade of conformational changes within the protein, triggering proton flux against the concentration gradient, providing the microorganisms with the energy to live. We are inspired by these functions in nature to harness light energy using synthetic photoswitches under confinement. Like retinal, synthetic photoswitches require some degree of conformational flexibility to isomerize. In nature, the conformational change associated with retinal isomerization is accommodated by the structural flexibility of the opsin host, yet it results in steric communication between the chromophore and the protein. Similarly, we strive to design systems wherein isomerization of confined photoswitches results in steric communication between a photoswitch and its confining environment. To achieve this aim, a balance must be struck between molecular crowding and conformational freedom under confinement: too much crowding prevents switching, whereas too much freedom resembles switching of isolated molecules in solution, preventing communication.
In this Account, we discuss five classes of synthetic light-switchable compounds—diarylethenes, anthracenes, azobenzenes, spiropyrans, and donor–acceptor Stenhouse adducts—comparing their behaviors under confinement and in solution. The environments employed to confine these photoswitches are diverse, ranging from planar surfaces to nanosized cavities within coordination cages, nanoporous frameworks, and nanoparticle aggregates. The trends that emerge are primarily dependent on the nature of the photoswitch and not on the material used for confinement. In general, we find that photoswitches requiring less conformational freedom for switching are, as expected, more straightforward to isomerize reversibly under confinement. Because these compounds undergo only small structural changes upon isomerization, however, switching does not propagate into communication with their environment. Conversely, photoswitches that require more conformational freedom are more challenging to switch under confinement but also can influence system-wide behavior.
Although we are primarily interested in the effects of geometric constraints on photoswitching under confinement, additional effects inevitably emerge when a compound is removed from solution and placed within a new, more crowded environment. For instance, we have found that compounds that convert to zwitterionic isomers upon light irradiation often experience stabilization of these forms under confinement. This effect results from the mutual stabilization of zwitterions that are brought into close proximity on surfaces or within cavities. Furthermore, photoswitches can experience preorganization under confinement, influencing the selectivity and efficiency of their photoreactions. Because intermolecular interactions arising from confinement cannot be considered independently from the effects of geometric constraints, we describe all confinement effects concurrently throughout this Account.
Key References
Samanta D.; Gemen J.; Chu Z.; Diskin-Posner Y.; Shimon L. J. W.; Klajn R.. Reversible photoswitching of encapsulated azobenzenes in water. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 9379–9384. A flexible Pd6L4coordination cage encapsulates structurally simple azobenzenes in a 2:1 ratio. Isomerization from trans- to cis-azobenzene results in a steric clash and expulsion of one guest from the cavity, offering an opportunity to translate light energy into system-wide communication.1
Kundu P. K.; Olsen G. L.; Kiss V.; Klajn R.. Nanoporous frameworks exhibiting multiple stimuli responsiveness. Nat. Commun. 2014, 5, 3588.. The switching behavior of spiropyran within nanoporous frameworks is dependent on the density of the photochromic units. In less crowded frameworks, spiropyran switches reversibly as in solution; in more crowded frameworks, spiropyran converts spontaneously to merocyanine, driven by mutual stabilization of neighboring zwitterions.2
Chu Z.; Han Y.; Bian T.; De S.; Král P.; Klajn R.. Supramolecular Control of Azobenzene Switching on Nanoparticles. J. Am. Chem. Soc. 2019, 141, 1949–1960. Confinement of azobenzene on the surface of nanoparticles enables preorganization of these switches adjacent to background ligands that facilitate their isomerization. We also observe cooperative switching of neighboring azobenzenes within small aggregates on the nanoparticle surface.3
Zhao H.; Sen S.; Udayabhaskararao T.; Sawczyk M.; Kučanda K.; Manna D.; Kundu P. K.; Lee J.-W.; Král P.; Klajn R.. Reversible trapping and reaction acceleration within dynamically self-assembling nanoflasks. Nat. Nanotechnol. 2016, 11, 82–88. Reversible assembly of nanoparticles leads to the creation and destruction of confined environments, or “nanoflasks”, located interstitially within the aggregate. Preorganization of anthracenes within these nanoflasks results in reaction acceleration and regioselective photodimerization to the syn product.4
1. Introduction
Photoisomerization of retinal drives diverse processes that are critical to life in all organisms, ranging from ion transport across bacterial membranes to vision in animals. The proteins responsible for harnessing the isomerization of retinal are called opsins. Bacteriorhodopsin, for example, is a seven-transmembrane-helix proton pump that binds retinal within its hydrophobic cavity, wherein the chromophore forms an imine bond with the protein’s Lys216 residue (Figure 1).5 Illumination of bacteriorhodopsin triggers trans→cis isomerization of the bound all-trans retinal selectively at the 13-position. This selectivity is remarkable considering that retinal contains several photoisomerizable C=C bonds—in fact, photoirradiation of all-trans retinal in solution results in an ill-defined mixture of 9-cis-, 11-cis-, and 13-cis-retinal. Furthermore, photoswitching of retinal within the confinement of the protein proceeds with a quantum yield of up to 67%, whereas each of the solution products forms in a poor yield of a few percent. Other protein scaffolds can guide the same process with entirely different selectivities. For example, proteins called photoisomerases can similarly bind all-trans-retinal and direct its switching precisely at the 11-position.6
Figure 1.
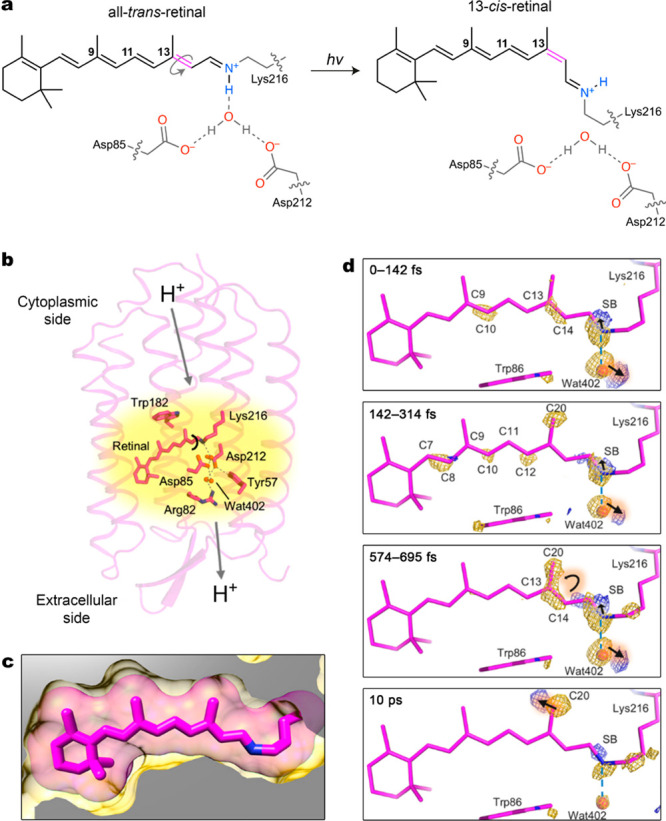
(a) Light-induced regioselective isomerization of retinal within the binding pocket of bacteriorhodopsin. (b) Structure of bacteriorhodopsin with retinal buried inside its hydrophobic cavity. The arrows indicate the direction of light-induced proton transfer. (c) Binding of retinal (pink sticks with van der Waals radii shown as a transparent halo) encased within the hydrophobic cavity of bacteriorhodopsin (yellow). (d) Structural dynamics of retinal and its immediate surroundings captured by a femtosecond X-ray laser. The transition from trans-retinal to cis-retinal is mapped onto a dark-state model based on the difference Fourier electron density (Fobslight – Fobs) contoured at 4σ (yellow, negative; blue, positive). Adapted with permission from ref (5). Copyright 2018 American Association for the Advancement of Science.
The trans→cis isomerization of the bound retinal is accompanied by a large conformational change, which initiates a sequence of structural changes within the bacteriorhodopsin host, ultimately resulting in unidirectional transport of protons outside the cell.7 While achieving the level of photoswitching precision found in nature is challenging, we are inspired by these processes to harness molecular photoswitches for the creation of functional molecular machines and materials.
Photoswitchable molecules have been studied extensively in solution and have been used as building blocks for synthetic molecular machines, which have the potential to convert light energy into mechanical work. The vast majority of known photoswitches require some degree of conformational freedom for reversible switching. Just as isomerization of retinal within opsins initiates a signaling cascade, isomerization of synthetic photoswitches can enable their communication with the environment. Switching of free (noninteracting) molecules in solution, however, while highly efficient, does not entail communication with other components of the system. To facilitate better communication between photoswitches and their environment, we are thus investigating the behavior of these molecules under confinement. Molecular confinement generally arises when a species is physically trapped (as opposed to covalently bound) within a partially or completely enclosed environment.8 As such, their diffusion is limited and their movement is constrained, thereby influencing their chemical reactivity. For the purpose of this Account, we also consider species that are tethered to a surface or framework to be confined, as they likewise experience a partially enclosed environment and their movement is restricted.
Successful switching and communication under confinement require achieving a delicate balance between conformational freedom and molecular crowding. Historically, the term molecular crowding has been used to describe cellular environments wherein the majority of the volume is physically occupied by large (i.e., not solvent) molecules.9 In nature, this crowded environment influences the way biomolecules diffuse through the cell, which in turn affects their structure, interactions, and functions.10 Similarly, a very crowded molecular environment prohibitively restricts the freedom available for a photoswitch; unsuccessful switching has been observed within single crystals,11,12 densely packed monolayers,13,14 and cavities of rigid coordination cages.15 Confined systems that offer too much conformational freedom and not enough molecular crowding, however, constitute environments that are functionally similar to solution-state systems. As in solution, photoswitches communicate weakly with these loosely confined systems.
In pursuit of this balance, a range of different strategies have been developed to accommodate the conformational changes associated with photoswitching under confinement.8 These strategies include diluting densely packed monolayers of photoswitches with shorter molecules,16,17 decorating photoswitches with spacer groups,18,19 and confining them on curved substrates,20 within porous solids,21,22 within the spaces between aggregated nanoparticles,4 or within the cavities of flexible coordination cages.23 To compare the behaviors of photoswitchable molecules within different confined environments, we have organized this Account according to the type of photoswitch, roughly in order of increasing demand for conformational freedom. Furthermore, we have included switches that undergo unimolecular photoisomerization or photodimerization, as these two types of molecules show similar behavioral responses under confinement. We begin by discussing diarylethenes, including dihydropyrene switches, which require only a small degree of freedom for efficient isomerization. We then transition to anthracenes, which undergo cyclodimerization between two neighboring molecules and require a greater degree of conformational freedom. Next, we discuss the behavior of confined azo switches, including azobenzenes and arylazopyrazoles, which require a high degree of conformational freedom for switching. Uniquely, spiropyran and donor–acceptor Stenhouse adducts form zwitterionic isomers upon switching, and we show in the final section how intermolecular interactions between zwitterions often govern the behavior of these switches under confinement.
2. Confined Diarylethenes
In this section, we discuss bridged diarylethenes (DAEs), whose switching entails 6π electrocyclization (Figure 2a) and thus requires a minimal degree of conformational freedom. As they isomerize, these molecules reversibly switch between an open, nonconjugated form and a closed, conjugated form. In general, switching of these molecules does not result in steric clashes with their environment, and reversible switching can be readily achieved when these molecules are confined on solid surfaces or within small cavities. The small degree of conformational freedom required for DAE isomerization is best manifested by the observation of photoswitching in the crystalline state, which occurs efficiently while being accompanied by small changes in the overall shape of the single crystal (Figure 2b).24
Figure 2.
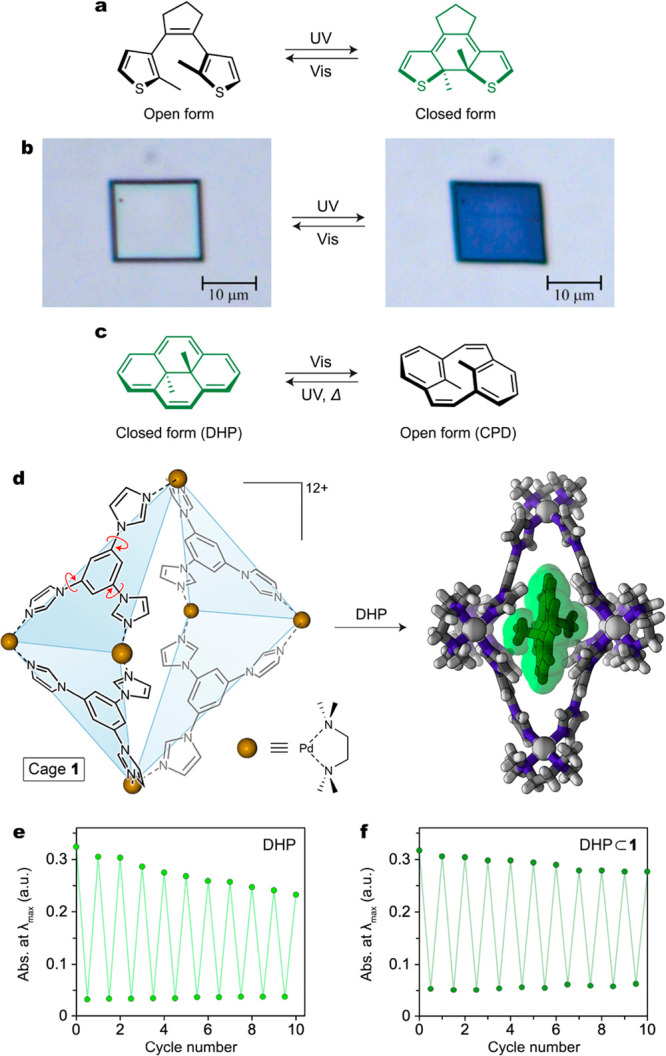
(a) Reversible photoisomerization of a diarylethene. The colored isomer featuring extensive conjugation of π electrons (here, the closed form) is shown in green. (b) Reversible light-induced deformation of a single crystal of a simple diarylethene (here, 1,2-bis(2-ethyl-5-phenyl-3-thienyl)perfluorocyclopentene). (c) Reversible photoisomerization between dihydropyrene (DHP) and cyclophanediene (CPD). (d) Structural formula of coordination cage 1 used to investigate the behavior of photoswitchable molecules under confinement (left) and crystal structure of an inclusion complex of DHP inside cage 1 (right). (e) Gradual decomposition of DHP in pentane solution over 10 switching cycles. (f) Improved fatigue resistance of DHP⊂1 over 10 cycles under the same irradiation conditions. (b) Adapted with permission from ref (24). Copyright 2007 Springer Nature. (e, f) Adapted from ref (31). Copyright 2020 American Chemical Society.
Because the electrocyclic ring closing of DAE establishes efficient π-electron conjugation throughout the molecule, interest in using these photoswitches to modulate the electronic properties of materials25 has motivated the study of their behavior under confinement. In pioneering studies, mixed self-assembled monolayers of a thiolated DAE and dodecanethiol were prepared on planar gold surfaces. Dodecanethiol ligands are electrically insulating and similar in length to the DAE ligands, creating a crowded molecular environment around the photoswitches.26,27 By scanning tunneling microscopy (STM), the confined DAE units were nevertheless observed to switch reversibly and with high efficiency, comparable to their behavior in solution. These results suggest that confinement of DAEs within densely packed monolayers does not restrict the small conformational changes associated with isomerization. Similar observations were recorded upon confinement of DAE switches within an amorphous polymer matrix.28 Within more rigid environments, such as highly ordered polymer matrices, some differentiation between the two isomers of DAE can be achieved: while the closed form is planar and can pack closely with other closed-ring DAEs, the open form is more bulky and requires slightly more space. The closed form is thus favored within this environment, hindering switching to the open form.28 Reversible switching of DAEs under confinement has also been achieved within networks of carbon nanotubes29 and organic thin films.30
Uniquely among DAE photoswitches, dihydropyrene (DHP) exhibits negative photochromism: upon irradiation with visible light, the closed, colored form undergoes a ring-opening reaction to form cyclophanediene (CPD), and the back reaction can be performed either upon irradiation with UV light or thermally in the dark (Figure 2c). To accommodate the conformational freedom required for DHP isomerization, we encapsulated DHP within flexible coordination cage 1 (Figure 2d).31 This pseudo-octahedral cage self-assembles from six Pd2+ cations, four triimidazole ligands, and six tetramethylethylenediamine (TMEDA) ligands (the latter occupy the two external coordination sites on each metal center).32 The inherent flexibility of cage 1 is derived from torsion around the bonds between the imidazole groups and the central benzene ring within each ligand. Encapsulation of DHP within cage 1 is accompanied by a red shift in the absorption bands associated with DHP in the UV/vis spectrum and an upfield shift of the signals corresponding to DHP in the 1H NMR spectrum. By following the forward reaction and the thermal back reaction over time using UV/vis spectrophotometry, we found that the rates of DHP switching under confinement within the cage cavity and in solution are similar. Specifically, isomerization of DHP to CPD proceeds approximately 3 times slower under confinement than in solution, but encapsulation does not influence the thermal back reaction. Furthermore, molecular dynamics (MD) simulations indicate that cage 1 deforms similarly upon encapsulation of either DHP or CPD.31
Considering these experimental and computational results concurrently, we surmise that cage 1 can easily adapt in response to the small structural changes that accompany DHP isomerization. Interestingly, although encapsulation does not significantly alter the switching behavior of DHP, we determined that confinement within cage 1 serves to protect the unstable biradical intermediate33 associated with this reaction. While only 70% of free DHP remains intact after 10 switching cycles, >90% of encapsulated DHP is maintained under the same irradiation conditions (Figure 2e,f, respectively). This degree of stabilization, which we attribute primarily to distancing of reactive radical species from one another, is remarkable given the large open panels within cage 1 (Figure 2d, right).
3. Confined Anthracenes
Most of the photoswitches highlighted in this Account undergo unimolecular photoisomerization; in contrast, anthracenes (and similar compounds, such as coumarins) undergo a dimerization reaction upon irradiation with light. While the mechanisms for unimolecular photoisomerization and photodimerization are very different, both reactions are triggered upon irradiation with light of a certain wavelength, and both can be reversed thermally and/or upon irradiation with light of another wavelength. More specifically, irradiation of anthracenes with near-UV (∼350 nm) light triggers a [4+4] photodimerization reaction (Figure 3a). The dimer can then revert back to anthracene upon exposure to higher-energy (∼250 nm) UV light or upon heating above 100 °C. The dimerization reaction requires a relatively small degree of conformational freedom, and in this respect, the behavior of confined anthracenes is similar to that of DAE photoswitches described in the previous section. While dimerization of anthracene under confinement rarely results in steric clashes with the environment, confinement effects can nevertheless influence the reactivity of these species.
Figure 3.

(a) Reversible photodimerization of anthracene. (b) Proposed photoreaction of anthracene inside the cavity of ZIF-8. (c) Crystal structure of a ZIF-8 cavity encapsulating four molecules of anthracene. (d) UV/vis absorption spectra accompanying UV irradiation of ZIF-8 encapsulating anthracene. (e) Schematic illustration of light-induced trapping and increased reactivity of small molecules within colloidal crystals (“dynamically self-assembling nanoflasks”). (f) Electron micrographs (at different magnifications) of colloidal crystals prepared by exposing azobenzene-coated gold nanoparticles to UV light. (g) Accelerated photodimerization of 9-anthracenemethanol in the presence of photoresponsive nanoparticles. (h) Stereoselectivity in the dimerization of 9-anthracenemethanol in the presence and absence of photoresponsive nanoparticles. (i) Schematic illustration of the photoreaction of 9-(4-mercaptophenylethynyl)anthracene on a Au(111) surface. (j) Dependence of the anthracene dimerization yield on the curvature of the underlying nanoparticle. (c, d) Adapted with permission from ref (34). Copyright 2019 Wiley-VCH. (f–h) Adapted with permission from ref (4). Copyright 2016 Springer Nature. (i) Adapted with permission from ref (37). Copyright 2011 American Association for the Advancement of Science.
In confined spaces, there is a delicate balance between having enough conformational freedom for anthracene switching and having too much molecular crowding. This balance is beautifully illustrated in a recent report wherein four anthracene molecules were encapsulated within a spherical cavity of a metal–organic framework (ZIF-8).34 Results from X-ray crystallography and UV/vis spectrophotometry suggested that irradiation with UV light leads to selective photodimerization of one pair of anthracene molecules, while the remaining two anthracenes remain as monomers (Figure 3b–d). The initial (preirradiation) state within the cavity in this system provides enough conformational freedom for two anthracene molecules to dimerize; however, this reaction results in an environment that is too sterically hindered for switching of the second anthracene pair.
Anthracenes can also be switched within cavities formed upon aggregation of nanoparticles (NPs), which we have termed “nanoflasks” (Figure 3e,f).4 By following changes in the characteristic absorbance of photodimerization products around 350–370 nm, we found that when 9-anthracenemethanol molecules were entrapped within these nanoflasks, the photodimerization reaction was accelerated by 2 orders of magnitude compared with the same reaction in solution (Figure 3g). This effect is primarily attributed to the increased local concentration of anthracene molecules under confinement. Furthermore, hydrogen bonding between 9-anthracenemethanol and ligands on the NPs results in preorganization of the trapped molecules within the cavity. In addition to contributing to the observed increase in dimerization rate, this effect significantly modulates the regioselectivity of this photoreaction (Figure 3h). We used 1H NMR spectroscopy to identify the anti and syn isomers on the basis of the chemical shifts of the tertiary protons adjacent to the hydroxymethyl groups, which appear at 4.1 and 4.4 ppm, respectively. The photodimerization of 9-anthracenemethanol in solution yields the thermodynamically favored anti isomer, with the hydroxyl groups pointing in opposite directions within dianthracene. Interactions between anthracene molecules and NP ligands within these nanoflasks, however, preorganize the molecules around the “edge” of the nanoflask, resulting in preferential (>80%) formation of the syn isomer.
While typical anthracenes undergo [4+4] photocycloaddition upon irradiation with UV light, installing an ethynyl moiety at the 9-position can also induce a [4+2] Diels–Alder addition.35 Confining these 9-ethynylanthracenes, however, can induce either of these reactions to proceed with high selectivity. For example, confinement within supramolecular gels induces the [4+2] reaction selectively.36 In contrast, preorganization of thiolated 9-ethynylanthracene on a planar gold surface prevents the mutual orientation of molecules required for the [4+2] Diels–Alder reaction, thus resulting in the [4+4] photodimerization (Figure 3i).37 Similar regioselectivity was observed upon preorganizing the same ligands on a curved surface, but the reaction proceeded with lower efficiency.38 This modulation in reactivity is attributed to the difference in curvature of planar and curved surfaces. On the planar surface, the terminal anthracene units are in close proximity, facilitating the [4+4] photodimerization. On the curved surface, however, the distance between anthracene moieties is larger because the angle between adjacent ligands is greater, hindering the reaction between neighboring switches. Changing the surface curvature thus modulates the photostationary state of this reaction.
To further examine preorganization on curved surfaces, we appended the same 9-ethynylanthracene ligands to gold NPs of different sizes (Figure 3j).39 Importantly, the curved surfaces of NPs offer a convenient method of modulating the reactivity of anthracenes by adjusting the surface curvature. On 2.5 nm gold NPs, no photoreaction was observed (as concluded from the fact that there were no changes in absorption at ∼400 nm, which is diagnostic of unreacted anthracene). We repeated the same experiment using 5.5 and 7.5 nm gold NPs, which have less curvature. Upon irradiation with UV light, the reaction yields were determined as 9% and 26% on 5.5 and 7.5 nm NPs, respectively. These results suggest that curvature plays an important role in modulating the switching efficiency of ethynylanthracenes. Compared with 5.5 and 7.5 nm NPs, the 2.5 nm particles have more curvature and thus longer distances between the ligands. This geometry positions neighboring anthracene headgroups too distant from one another to undergo [4+4] photodimerization. In contrast, the distance between ligands is shorter on larger NPs, and neighboring anthracene units are positioned close enough to react. This effect was verified by the trend relating increasing NP size to increasing [4+4] reaction yield.39
4. Confined Azo Compounds
In this section, we compare the behaviors of azo photoswitches, including azobenzenes and arylazopyrazoles, in the presence and absence of confinement. Upon irradiation with UV light, the planar trans isomer of azobenzene undergoes a large conformational change to the nonplanar cis form (Figure 4a). Because azo compounds require a high degree of conformational freedom to achieve switching, there are a plethora of examples wherein confinement of azo switches leads to a marked decrease in their switchability.11,13−15 Although the environment around confined azo switches must be carefully designed, these photoswitches are perhaps the most promising candidates for harnessing the power of light to drive more complex systems.40,41 Crucially, the high degree of conformational freedom required by azo switches enables them to interact with their environment, facilitating the emergence of cooperative effects and communication throughout a system.42−44
Figure 4.
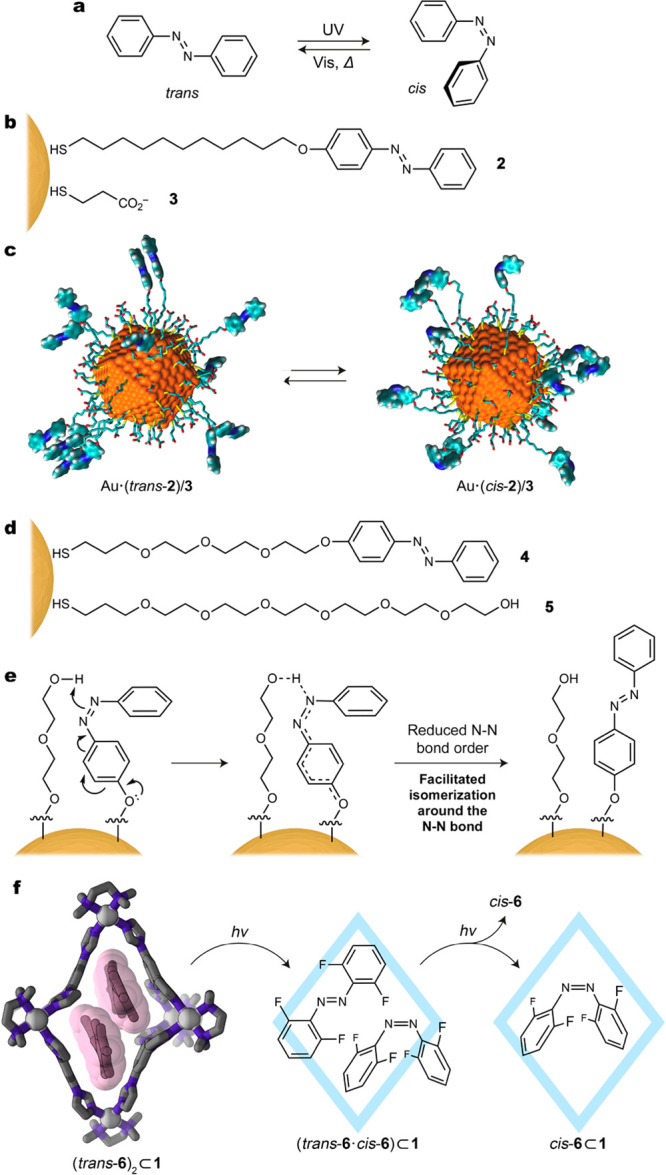
(a) Reversible photoisomerization of azobenzene. (b) Structural formulas of azobenzene 2 and background ligand 3. (c) Snapshots from molecular dynamics simulations of a 2/3-coated gold nanoparticle in the trans and cis states of azobenzene. (d) Structural formulas of azobenzene 4 and background ligand 5.3 (e) Proposed mechanism for accelerated azobenzene isomerization on a nanoparticle surface upon coadsorption with a hydroxy-terminated background ligand.3 (f) Crystal structure of an inclusion complex comprising cage 1 and two molecules of tetra-o-fluoroazobenzene (6) (left) and the stepwise mechanism underlying the photoisomerization of 6 within the cavity of 1 (right). (c) Adapted from ref (3). Copyright 2019 American Chemical Society.
As in previous sections, our early investigations into the behavior of azobenzene molecules under confinement focused on elucidating the impact of molecular crowding on switching efficiency.3,16,45 By using gold NPs of different sizes, we tuned surface curvature and thus the distance between terminal azobenzene groups on the ligands. Larger NPs, for instance, have less curvature and shorter ligand–ligand distances, creating a more crowded environment. We measured the rates of azobenzene isomerization on the surface of these nanoparticles by following the evolution of their UV/vis absorption spectra over time under irradiation with light. Upon comparing the behaviors of azobenzene confined on 2.6, 4.4, 6.2, and 7.8 nm gold NPs, we observed that decreasing the distance between azobenzene units decreases photoswitching efficiency.16 Similarly, steric effects have also been reported to suppress azobenzene switching on planar gold surfaces.46−48 On these surfaces, chromophore density can be modulated by preparing mixed self-assembled monolayers of azobenzene and alkanethiolate spacer ligands.16 Decreasing chromophore density in these systems can thus be accomplished by increasing the percentage of spacer ligands; as observed on curved surfaces,20 less molecular crowding around the azobenzene ligands resulted in higher rates of photoswitching.16 To follow photoswitching on planar surfaces, we used UV/vis differential reflectance spectroscopy to show that less molecular crowding is also correlated with more complete conversion from trans to cis. We observed a similar effect upon increasing the percentage of spacer ligands on gold NPs: a higher percentage of the cis isomer can be obtained with greater chromophore dilution.3
Having achieved successful photoswitching of azobenzenes on gold NPs, we investigated intermolecular interactions between ligands within these environments and their effects on the rate of azobenzene switching. To this end, we synthesized NPs functionalized with mixed monolayers of different azobenzenes and different background ligands, systematically varying the length of the thiolate tether and the terminal functional group on the background ligand.3 MD simulations suggested that when azobenzene 2 is combined with significantly shorter background ligand 3 (Figure 4b), the hydrophobic azobenzene units bundle together in water, minimizing their exposure to the polar solvent (Figure 4c). These bundles displayed remarkable photoswitching behavior; instead of inefficient switching, as one might expect from a crowded environment, aggregation was observed to accelerate isomerization of azobenzenes. As inferred from similar behavior exhibited by azobenzene on planar surfaces16,42 and within single crystals,49 this result suggests a cooperative switching mechanism whereby isomerization of one azobenzene molecule within the bundle results in communication with a neighboring azobenzene, facilitating its isomerization.
To further investigate the effect of intermolecular interactions on azobenzene switching, we synthesized NPs cofunctionalized with azobenzene ligand 4 and hydroxy-terminated thiol 5 (Figure 4d).3 Upon confinement on the NP surface, the hydroxyl group of the background ligand can form a hydrogen bond with azobenzene’s nitrogen atoms, decreasing the double-bond character of the azo bond and facilitating the trans ⇄ cis switching in both directions (Figure 4e). Compared with an analogous system with methyl-terminated background ligands, azobenzene back-isomerization in this system proceeded up to 500 times faster.
In addition to investigating azobenzene isomerization on surfaces, we have probed the behavior of azo photoswitches within the cavity of a coordination cage. Although photoisomerization of azo compounds requires a high degree of conformational freedom and does not proceed in the cavity of rigid coordination cages,15 we anticipated that switching could be accommodated by the inherent flexibility of cage 1 (Figure 2d). Upon encapsulation of azo analogues, 1:1 or 2:1 inclusion complexes were observed to form depending on the substitution pattern on the azobenzene core.1 The formation of these complexes was accompanied by an upfield shift of the guest signals in the 1H NMR spectra. Furthermore, we obtained X-ray crystal structures that illustrate the adaptivity of cage 1, showing significant distortion of the cage upon conforming to the geometry of the encapsulated guest molecules (Figure 4f, left). Recently, Pavan et al. used atomistic MD and metadynamics simulations to map out the conformational free energy landscape of the empty cage in solution.50 According to these simulations, the structural rearrangement of the cage upon encapsulation of azo guests incurs a relatively large penalty (e.g., ∼8 kcal/mol for cage deformation caused by binding of two molecules of trans-azobenzene). This energetic cost is offset by favorable host–guest interactions and release of water molecules from the hydrophobic cavity.
Upon irradiation with UV light, all of the encapsulated trans-azo compounds were observed (by UV/vis and 1H NMR spectroscopies) to convert to their corresponding cis isomers.1,51 For complexes encapsulating two trans-azo guests, isomerization within the cavity was followed by expulsion of one guest, while the second guest remained within the cavity. Additional simulations provided insight into the mechanism of isomerization and guest release. Upon irradiation of the system with light, one of the two trans isomers converts to the cis form under confinement (Figure 4f). The resulting complex, in which trans and cis isomers are coencapsulated, is highly unstable, leading to preferential expulsion of the cis isomer.50
5. Confined Spiropyrans and DASA Switches
Within this section, we examine the effects governing the switching behavior of spiropyran/merocyanine and donor–acceptor Stenhouse adduct (DASA) switches under confinement. Spiropyran is composed of two heterocyclic rings bound through a single (spiro) carbon atom; upon light-induced ring-opening followed by Z–E isomerization, spiropyran is converted to merocyanine,52 the colloquial name for the open form of this photoswitch (Figure 5a). By comparison, DASAs first undergo photoinduced Z–E isomerization, followed by a 4π electrocyclization (Figure 5b).53 Preference for the open versus closed form of these two switches is dependent on environmental conditions such as solvent polarity, and switching to the metastable form can be accomplished upon irradiation with light of a particular wavelength. As for azo compounds, the ring-opening/closing reactions experienced by spiropyrans and DASAs require high degrees of conformational freedom (Figure 5b). Furthermore, both types of switches form zwitterionic compounds upon isomerization. The effects that emerge upon confinement of zwitterions are strong and are often observed to dominate the behavior of these photoswitches.
Figure 5.

(a, b) Reversible photoisomerization of (a) spiropyran and (b) DASA. (c) Comparison of the kinetics of merocyanine→spiropyran back-isomerization in solution and on the surface of 2.6 nm gold nanoparticles (χ = 0.33). (d) Dependence of the kinetics of disassembly of gold nanoparticle aggregates on the spiropyran coverage. (e, f) Spontaneous bleaching of the open form of DASA on the surfaces of (e) 4.2 and (f) 8.6 nm magnetite nanoparticles. (g) TEM image of a spiropyran-incorporating framework. The arrows indicate individual nanopores. (h) Encapsulation of spiropyran 7 within cage 1 and the resulting crystal structure (hydrogen atoms of the cage have been omitted for clarity). (i) Mechanism of the light-induced decoloration of 7⊂1. (c, d) From ref (55). CC BY 3.0. (e, f) Adapted with permission from ref (57). Copyright 2017 Wiley-VCH. (g) From ref (2). CC BY NC ND 3.0.
Because spiropyran molecules require a high degree of conformational freedom to achieve switching, one may anticipate that highly crowded environments should hinder isomerization. We began our investigation within this section by appending spiropyran ligands onto Au25 nanoclusters.54 These nanoclusters have a high degree of curvature, resulting in long distances between individual ligands. Furthermore, we incorporated shorter background ligands onto the nanoclusters such that only approximately four spiropyrans (vs ∼14 background ligands) were present on each particle. Because of the combined effects of surface curvature and chromophore dilution, the extent of crowding around each spiropyran in these systems was minimal. Upon sequential irradiation with UV and visible light, reversible isomerization between surface-confined spiropyran and merocyanine was observed by following the merocyanine absorption peak at λmax = 587 nm; we detected no significant difference in photoswitching behavior compared with spiropyran in solution.54
To further investigate the effect of surface curvature on the switching behavior, we confined spiropyran ligands onto larger gold NPs.55 We functionalized 2.6 nm NPs with a mixture of spiropyran and alkyl (spacer) ligands. At relatively high chromophore dilution (χ = 0.33, meaning 33% of the ligands were terminated with a spiropyran unit), isomerization of spiropyran to merocyanine was readily achieved upon irradiation with UV light, as confirmed by a decrease in absorbance at 800 nm. Interestingly, thermal relaxation back to spiropyran proceeded several times slower on these NPs than in solution (Figure 5c), which was attributed to the mutual stabilization of the zwitterionic merocyanine units on the NPs.52,56 At higher chromophore densities, isomerization from spiropyran to merocyanine drove NP aggregation. When the mole fraction of spiropyran was increased from χ = 0.6 to χ = 0.9, the half-life of the resulting aggregates lengthened from 9 to 72 s. In these systems, stabilization occurs not only between merocyanine units on the same NP but also between different NPs within aggregates, slowing their disassembly (Figure 5d).55
We observed a similar effect upon coating the surface of magnetite NPs with a monolayer of DASA ligands.57 We used UV/vis spectrophotometry to confirm adsorption of the ligands onto the NPs and to monitor their photoswitching behavior. Upon attachment to NPs, the otherwise-stable open form of these switches spontaneously and irreversibly isomerized to the closed, zwitterionic form. In other words, the photostationary state of this switch was significantly perturbed under confinement. As for merocyanine confined on gold NPs, this effect was attributed to mutual stabilization between the zwitterionic form of DASA under confinement on the NP surface. Crucially, the rate and degree of stabilization were dependent on the curvature of the NPs: spontaneous conversion to the zwitterionic form was faster and proceeded to a larger extent on larger NPs, which have less curvature and thus support stabilization between the neighboring DASA zwitterions more efficiently (Figure 5e,f).
Furthermore, upon covalent incorporation of spiropyran within a porous aromatic framework (PAF), we observed a similar stabilization effect using electron microscopies (SEM and TEM) and UV/vis spectrophotometry.2 Upon desolvation of this material, the solid grains were observed to shrink and change in color from faint yellow to deep blue (λmax ≈ 550 nm), indicating spontaneous conversion from spiropyran to merocyanine. As on the surface of NPs, this effect is due to interactions between zwitterionic merocyanine units, which experience mutual stabilization under confinement. When the PAF was resolvated, the material returned to its original structure, and merocyanine was converted back to spiropyran. By using other building blocks, a more rigid photoresponsive framework was obtained (Figure 4g).2 The rigidity of this PAF prevented collapse upon desolvation, and spontaneous conversion to merocyanine was not observed. Instead, we successfully drove reversible switching between spiropyran and merocyanine using light of two different wavelengths. Efficient reversible photoisomerization within nanoporous materials has also been accomplished for other spiropyran derivatives58 as well as azobenzenes59,60 and overcrowded alkenes.22,61
In addition to confining spiropyrans on surfaces and within porous solids, we also probed the behavior of these photoswitches within the cavity of a coordination cage using UV/vis spectrophotometry and 1H NMR spectroscopy. Upon encapsulation of spiropyran 7SP within cage 1, spontaneous conversion to zwitterionic merocyanine 7MC was observed (Figure 5h); this process was accompanied by a downfield shift of the acidic imidazole protons of the cage (8.8–9.3 ppm) and an increase in the absorbance at 592 nm (due to the MC isomer).23 Unlike previous examples within this section, wherein mutual stabilization of zwitterions drove the formation of the zwitterionic form, only one merocyanine unit can be encapsulated within cage 1. In this case, stabilization of the merocyanine form is primarily due to π–π stacking interactions with aromatic panels from the cage framework.62 Furthermore, we observed a dynamic equilibrium between deprotonated and protonated (7MCH) merocyanine within the cage (Figure 5i). While both merocyanine species are stabilized by π–π stacking with the cage, 7MC is stabilized by additional electrostatic interactions with the positively charged cage framework, which renders it non-photoresponsive. Upon irradiation with light corresponding to the absorbance maximum of the minor species 7MCH, however, efficient ring closing to form 7SP proceeded, gradually shifting the 7MC ⥃ 7MCH equilibrium to the right and ultimately achieving a system-wide conversion to spiropyran (Figure 5i). Although the merocyanine and spiropyran forms feature very different conformations, the conformational freedom required for switching was readily accommodated by the flexible structure of the cage.
6. Conclusion
Communication between molecular photoswitches and their environment relies on achieving a balance between conformational freedom and molecular crowding. In nature, this balance is showcased by the isomerization of retinal within bacteriorhodopsin, which constitutes the initial step of the cascade leading to transmembrane proton flux against the concentration gradient. In this Account, we have discussed the behaviors of different classes of synthetic photoswitches within different types of confined environments and compared their behaviors in the presence and absence of confinement. We observe a trend wherein photoswitches that require less conformational freedom, such as diarylethenes and anthracenes, require less stringent design conditions to achieve successful switching under confinement. Confinement of these molecules often results in switching rates similar to those in solution, and no significant communication between these switches and their environment is observed. Even in these cases, however, confinement influences the behavior of photoactive species, e.g., by improving fatigue resistance of dihydropyrene switching or by modulating the selectivity of anthracene cycloaddition. Conversely, photoswitches that require a high degree of conformational freedom, such as azobenzenes, can present a significant design challenge in confined systems. While there are many examples in which azo compounds do not switch within crowded environments, successful photoisomerization of these molecules can lead to dramatically different behaviors under confinement compared with in solution. Effective communication between these switches and their environment can lead to effects such as modulation of the isomerization rate or guest release. In particular, we anticipate that isomerization-triggered guest release could ultimately be harnessed for chemical signaling, similar to the way retinal isomerization initiates phototransduction in nature. Photoswitches that form zwitterionic compounds upon isomerization, such as spiropyrans and DASA compounds, constitute an interesting exception to the trends described above. Upon switching to their zwitterionic forms, these photoswitches often experience a stabilization effect under confinement, either because of zwitterion–zwitterion interactions or because of interactions with their immediate environment.
In the future, we plan to explore the behavior of molecular photoswitches within an even wider range of confined environments, including densely packed63,64 and non-close-packed65 nanoparticle arrays and binding pockets within biomolecules.66 Already we have begun to harness the behavior of photoswitches under confinement to perform system-wide functions. Switching of azobenzenes confined on the surface of NPs, for instance, can drive their reversible aggregation.67−71 By systematically varying the degree of molecular crowding within confined environments, we will continue to develop our understanding of the trade-off between crowding and conformational freedom, tuning the balance required for using photoswitches within functional systems and materials.
Acknowledgments
This work was supported by the European Research Council (Grant 336080). A.B.G. and L.M.L. acknowledge funding from the Zuckerman STEM Leadership Program.
Biographies
Angela B. Grommet received a Bachelor of Science with Honors from Kansas State University in 2013. She then moved to the University of Cambridge and conducted her doctoral research under the supervision of Jonathan Nitschke, employing coordination cages for the separation and transport of molecular cargo. Upon earning her Ph.D. in 2018, she was awarded a Zuckerman STEM Leadership Postdoctoral Fellowship and joined the group of Rafal Klajn at the Weizmann Institute of Science. She now focuses on utilizing confinement to modulate chemical reactivity.
Lucia M. Lee received a B.Sc. with Honors in 2011 from McMaster University, where she stayed as a Natural Sciences and Engineering Research Council (NSERC) doctoral student to study organochalcogen supramolecular building blocks under the mentorship of Ignacio Vargas-Baca. At the completion of her Ph.D. in 2017, she joined the group of Stefan Matile at the University of Geneva as a postdoctoral fellow to pursue studies on transmembrane anion transport using main-group supramolecular interactions. In 2019, she joined the Klajn group as a Zuckerman STEM Leadership Postdoctoral Fellow. Her current work involves using σ-hole interactions to guide the assembly of nanoparticles.
Rafal Klajn completed his Ph.D. at Northwestern University in 2009 and joined the Department of Organic Chemistry at the Weizmann Institute of Science, where he is currently an Associate Professor. The interests of his research group revolve around supramolecular chemistry, photochromism, molecular switches, nanoscale self-assembly and reactivity, and the development of new stimuli-responsive and dissipative materials.
The authors declare no competing financial interest.
References
- Samanta D.; Gemen J.; Chu Z.; Diskin-Posner Y.; Shimon L. J. W.; Klajn R. Reversible photoswitching of encapsulated azobenzenes in water. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9379–9384. 10.1073/pnas.1712787115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu P. K.; Olsen G. L.; Kiss V.; Klajn R. Nanoporous frameworks exhibiting multiple stimuli responsiveness. Nat. Commun. 2014, 5, 3588. 10.1038/ncomms4588. [DOI] [PubMed] [Google Scholar]
- Chu Z.; Han Y.; Bian T.; De S.; Král P.; Klajn R. Supramolecular Control of Azobenzene Switching on Nanoparticles. J. Am. Chem. Soc. 2019, 141, 1949–1960. 10.1021/jacs.8b09638. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Sen S.; Udayabhaskararao T.; Sawczyk M.; Kučanda K.; Manna D.; Kundu P. K.; Lee J.-W.; Král P.; Klajn R. Reversible trapping and reaction acceleration within dynamically self-assembling nanoflasks. Nat. Nanotechnol. 2016, 11, 82–88. 10.1038/nnano.2015.256. [DOI] [PubMed] [Google Scholar]
- Nogly P.; Weinert T.; James D.; Carbajo S.; Ozerov D.; Furrer A.; Gashi D.; Borin V.; Skopintsev P.; Jaeger K.; Nass K.; Båth P.; Bosman R.; Koglin J.; Seaberg M.; Lane T.; Kekilli D.; Brünle S.; Tanaka T.; Wu W.; Milne C.; White T.; Barty A.; Weierstall U.; Panneels V.; Nango E.; Iwata S.; Hunter M.; Schapiro I.; Schertler G.; Neutze R.; Standfuss J. Retinal isomerization in bacteriorhodopsin captured by a femtosecond X-ray laser. Science 2018, 361, eaat0094. 10.1126/science.aat0094. [DOI] [PubMed] [Google Scholar]
- Shichida Y.; Matsuyama T. Evolution of opsins and phototransduction. Philos. Trans. R. Soc., B 2009, 364, 2881–2895. 10.1098/rstb.2009.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T.; Skopintsev P.; James D.; Dworkowski F.; Panepucci E.; Kekilli D.; Furrer A.; Brünle S.; Mous S.; Ozerov D.; Nogly P.; Wang M. T.; Standfuss J. Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Science 2019, 365, 61–65. 10.1126/science.aaw8634. [DOI] [PubMed] [Google Scholar]
- Grommet A. B.; Feller M.; Klajn R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 2020, 15, 256–271. 10.1038/s41565-020-0652-2. [DOI] [PubMed] [Google Scholar]
- Nakano S.; Miyoshi D.; Sugimoto N. Effects of Molecular Crowding on the Structures, Interactions, and Functions of Nucleic Acids. Chem. Rev. 2014, 114, 2733–2758. 10.1021/cr400113m. [DOI] [PubMed] [Google Scholar]
- Banks D. S.; Fradin C. Anomalous Diffusion of Proteins Due to Molecular Crowding. Biophys. J. 2005, 89, 2960–2971. 10.1529/biophysj.104.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A.; Kengmana E. S.; Fonseca M. V.; Han G. G. D. Solid-state photoswitching molecules: structural design for isomerization in condensed phase. Materials Today Adv. 2020, 6, 100058. 10.1016/j.mtadv.2020.100058. [DOI] [Google Scholar]
- Cohen M. D.; Ron I.; Schmidt G. M. J.; Thomas J. M. Photochemical Decoration of Dislocations inside Crystals of Acenaphthyne. Nature 1969, 224, 167–168. 10.1038/224167a0. [DOI] [Google Scholar]
- Wang R.; Iyoda T.; Jiang L.; Tryk D. A.; Hashimoto K.; Fujishima A. Structural investigation of azobenzene-containing self-assembled monolayer films. J. Electroanal. Chem. 1997, 438, 213–219. 10.1016/S0022-0728(96)05031-0. [DOI] [Google Scholar]
- Klajn R. Immobilized azobenzenes for the construction of photoresponsive materials. Pure Appl. Chem. 2010, 82, 2247–2279. 10.1351/PAC-CON-10-09-04. [DOI] [Google Scholar]
- Kusukawa T.; Fujita M. Ship-in-a-Bottle” Formation of Stable Hydrophobic Dimers of cis-Azobenzene and -Stilbene Derivatives in a Self-Assembled Coordination Nanocage. J. Am. Chem. Soc. 1999, 121, 1397–1398. 10.1021/ja9837295. [DOI] [Google Scholar]
- Moldt T.; Brete D.; Przyrembel D.; Das S.; Goldman J. R.; Kundu P. K.; Gahl C.; Klajn R.; Weinelt M. Tailoring the Properties of Surface-Immobilized Azobenzenes by Monolayer Dilution and Surface Curvature. Langmuir 2015, 31, 1048–1057. 10.1021/la504291n. [DOI] [PubMed] [Google Scholar]
- Moldt T.; Przyrembel D.; Schulze M.; Bronsch W.; Boie L.; Brete D.; Gahl C.; Klajn R.; Tegeder P.; Weinelt M. Differing Isomerization Kinetics of Azobenzene-Functionalized Self-Assembled Monolayers in Ambient Air and in Vacuum. Langmuir 2016, 32, 10795–10801. 10.1021/acs.langmuir.6b01690. [DOI] [PubMed] [Google Scholar]
- Han M.; Ishikawa D.; Honda T.; Ito E.; Hara M. Light-driven molecular switches in azobenzene self-assembled monolayers: effect of molecular structure on reversible photoisomerization and stable cis state. Chem. Commun. 2010, 46, 3598–3600. 10.1039/b921801g. [DOI] [PubMed] [Google Scholar]
- Santos Hurtado C.; Bastien G.; Mašát M.; Štoček J. R.; Dračínský M.; Rončević I.; Císařová I.; Rogers C. T.; Kaleta J. Regular Two-Dimensional Arrays of Surface-Mounted Molecular Switches: Switching Monitored by UV–vis and NMR Spectroscopy. J. Am. Chem. Soc. 2020, 142, 9337–9351. 10.1021/jacs.0c01753. [DOI] [PubMed] [Google Scholar]
- Negishi Y.; Kamimura U.; Ide M.; Hirayama M. A photoresponsive Au25 nanocluster protected by azobenzene derivative thiolates. Nanoscale 2012, 4, 4263–4268. 10.1039/c2nr30830d. [DOI] [PubMed] [Google Scholar]
- Dolgopolova E. A.; Berseneva A. A.; Faillace M. S.; Ejegbavwo O. A.; Leith G. A.; Choi S. W.; Gregory H. N.; Rice A. M.; Smith M. D.; Chruszcz M.; Garashchuk S.; Mythreye K.; Shustova N. B. Confinement-Driven Photophysics in Cages, Covalent–Organic Frameworks, Metal–Organic Frameworks, and DNA. J. Am. Chem. Soc. 2020, 142, 4769–4783. 10.1021/jacs.9b13505. [DOI] [PubMed] [Google Scholar]
- Danowski W.; van Leeuwen T.; Abdolahzadeh S.; Roke D.; Browne W. R.; Wezenberg S.; Feringa B. L. Unidirectional rotary motion in a metal–organic framework. Nat. Nanotechnol. 2019, 14, 488–494. 10.1038/s41565-019-0401-6. [DOI] [PubMed] [Google Scholar]
- Samanta D.; Galaktionova D.; Gemen J.; Shimon L. J. W.; Diskin-Posner Y.; Avram L.; Král P.; Klajn R. Reversible chromism of spiropyran in the cavity of a flexible coordination cage. Nat. Commun. 2018, 9, 641. 10.1038/s41467-017-02715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobatake S.; Takami S.; Muto H.; Ishikawa T.; Irie M. Rapid and reversible shape changes of molecular crystals on photoirradiation. Nature 2007, 446, 778–781. 10.1038/nature05669. [DOI] [PubMed] [Google Scholar]
- Jia C.; Migliore A.; Xin N.; Huang S.; Wang J.; Yang Q.; Wang S.; Chen H.; Wang D.; Feng B.; Liu Z.; Zhang G.; Qu D.-H.; Tian H.; Ratner M. A.; Xu H. Q.; Nitzan A.; Guo X. Covalently bonded single-molecule junctions with stable and reversible photoswitched conductivity. Science 2016, 352, 1443–1445. 10.1126/science.aaf6298. [DOI] [PubMed] [Google Scholar]
- Katsonis N.; Kudernac T.; Walko M.; van der Molen S. J.; van Wees B. J.; Feringa B. L. Reversible Conductance Switching of Single Diarylethenes on a Gold Surface. Adv. Mater. 2006, 18, 1397–1400. 10.1002/adma.200600210. [DOI] [Google Scholar]
- Arramel; Pijper T. C.; Kudernac T.; Katsonis N.; van der Maas M.; Feringa B. L.; van Wees B. J. Reversible light induced conductance switching of asymmetric diarylethenes on gold: surface and electronic studies. Nanoscale 2013, 5, 9277–9282. 10.1039/c3nr00832k. [DOI] [PubMed] [Google Scholar]
- Hou L.; Leydecker T.; Zhang X.; Rekab W.; Herder M.; Cendra C.; Hecht S.; McCulloch I.; Salleo A.; Orgiu E.; Samorì P. Engineering Optically Switchable Transistors with Improved Performance by Controlling Interactions of Diarylethenes in Polymer Matrices. J. Am. Chem. Soc. 2020, 142, 11050–11059. 10.1021/jacs.0c02961. [DOI] [PubMed] [Google Scholar]
- Sciascia C.; Castagna R.; Dekermenjian M.; Martel R.; Srimath Kandada A. R.; Di Fonzo F.; Bianco A.; Bertarelli C.; Meneghetti M.; Lanzani G. Light-Controlled Resistance Modulation in a Photochromic Diarylethene–Carbon Nanotube Blend. J. Phys. Chem. C 2012, 116, 19483–19489. 10.1021/jp212231j. [DOI] [Google Scholar]
- El Gemayel M.; Börjesson K.; Herder M.; Duong D. T.; Hutchison J. A.; Ruzié C.; Schweicher G.; Salleo A.; Geerts Y.; Hecht S.; Orgiu E.; Samorì P. Optically switchable transistors by simple incorporation of photochromic systems into small-molecule semiconducting matrices. Nat. Commun. 2015, 6, 6330. 10.1038/ncomms7330. [DOI] [PubMed] [Google Scholar]
- Canton M.; Grommet A. B.; Pesce L.; Gemen J.; Li S.; Diskin-Posner Y.; Credi A.; Pavan G. M.; Andréasson J.; Klajn R. Improving Fatigue Resistance of Dihydropyrene by Encapsulating within a Coordination Cage. J. Am. Chem. Soc. 2020, 142, 14557–14565. 10.1021/jacs.0c06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D.; Mukherjee S.; Patil Y. P.; Mukherjee P. S. Self-Assembled Pd6 Open Cage with Triimidazole Walls and the Use of Its Confined Nanospace for Catalytic Knoevenagel- and Diels–Alder Reactions in Aqueous Medium. Chem. - Eur. J. 2012, 18, 12322–12329. 10.1002/chem.201201679. [DOI] [PubMed] [Google Scholar]
- Sheepwash M. A. L.; Mitchell R. H.; Bohne C. Mechanistic Insights into the Photochromism of trans-10b,10c-Dimethyl-10b,10c-dihydropyrene Derivatives. J. Am. Chem. Soc. 2002, 124, 4693–4700. 10.1021/ja017229e. [DOI] [PubMed] [Google Scholar]
- Tu M.; Reinsch H.; Rodríguez-Hermida S.; Verbeke R.; Stassin T.; Egger W.; Dickmann M.; Dieu B.; Hofkens J.; Vankelecom I. F. J.; Stock N.; Ameloot R. Reversible Optical Writing and Data Storage in an Anthracene-Loaded Metal–Organic Framework. Angew. Chem., Int. Ed. 2019, 58, 2423–2427. 10.1002/anie.201813996. [DOI] [PubMed] [Google Scholar]
- Becker H. D.; Andersson K. Photochemical Diels–Alder dimerization of 9-phenylethylnylanthracene. J. Photochem. 1984, 26, 75–77. 10.1016/0047-2670(84)85029-7. [DOI] [Google Scholar]
- Das S.; Okamura N.; Yagi S.; Ajayaghosh A. Supramolecular Gel Phase Controlled [4 + 2] Diels–Alder Photocycloaddition for Electroplex Mediated White Electroluminescence. J. Am. Chem. Soc. 2019, 141, 5635–5639. 10.1021/jacs.9b00955. [DOI] [PubMed] [Google Scholar]
- Kim M.; Hohman J. N.; Cao Y.; Houk K. N.; Ma H.; Jen A. K.-Y.; Weiss P. S. Creating Favorable Geometries for Directing Organic Photoreactions in Alkanethiolate Monolayers. Science 2011, 331, 1312–1315. 10.1126/science.1200830. [DOI] [PubMed] [Google Scholar]
- Zheng Y. B.; Payton J. L.; Song T.-B.; Pathem B. K.; Zhao Y.; Ma H.; Yang Y.; Jensen L.; Jen A. K.-Y.; Weiss P. S. Surface-Enhanced Raman Spectroscopy To Probe Photoreaction Pathways and Kinetics of Isolated Reactants on Surfaces: Flat versus Curved Substrates. Nano Lett. 2012, 12, 5362–5368. 10.1021/nl302750d. [DOI] [PubMed] [Google Scholar]
- Zdobinsky T.; Sankar Maiti P.; Klajn R. Support Curvature and Conformational Freedom Control Chemical Reactivity of Immobilized Species. J. Am. Chem. Soc. 2014, 136, 2711–2714. 10.1021/ja411573a. [DOI] [PubMed] [Google Scholar]
- Muraoka T.; Kinbara K.; Aida T. Mechanical twisting of a guest by a photoresponsive host. Nature 2006, 440, 512–515. 10.1038/nature04635. [DOI] [PubMed] [Google Scholar]
- Coskun A.; Friedman D. C.; Li H.; Patel K.; Khatib H. A.; Stoddart J. F. A Light-Gated STOP–GO Molecular Shuttle. J. Am. Chem. Soc. 2009, 131, 2493–2495. 10.1021/ja809225e. [DOI] [PubMed] [Google Scholar]
- Pace G.; Ferri V.; Grave C.; Elbing M.; von Hänisch C.; Zharnikov M.; Mayor M.; Rampi M. A.; Samorì P. Cooperative light-induced molecular movements of highly ordered azobenzene self-assembled monolayers. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 9937–9942. 10.1073/pnas.0703748104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredy J. W.; Méndez-Ardoy A.; Kwangmettatam S.; Bochicchio D.; Matt B.; Stuart M. C. A.; Huskens J.; Katsonis N.; Pavan G. M.; Kudernac T. Molecular photoswitches mediating the strain-driven disassembly of supramolecular tubules. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11850–11855. 10.1073/pnas.1711184114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochicchio D.; Kwangmettatam S.; Kudernac T.; Pavan G. M. How Defects Control the Out-of-Equilibrium Dissipative Evolution of a Supramolecular Tubule. ACS Nano 2019, 13, 4322–4334. 10.1021/acsnano.8b09523. [DOI] [PubMed] [Google Scholar]
- Chu Z.; Klajn R. Polysilsesquioxane Nanowire Networks as an “Artificial Solvent” for Reversible Operation of Photochromic Molecules. Nano Lett. 2019, 19, 7106–7111. 10.1021/acs.nanolett.9b02642. [DOI] [PubMed] [Google Scholar]
- Akiyama H.; Tamada K.; Nagasawa J.; Abe K.; Tamaki T. Photoreactivity in Self-Assembled Monolayers Formed from Asymmetric Disulfides Having para Substituted Azobenzenes. J. Phys. Chem. B 2003, 107, 130–135. 10.1021/jp026103g. [DOI] [Google Scholar]
- Titov E.; Granucci G.; Götze J. P.; Persico M.; Saalfrank P. Dynamics of Azobenzene Dimer Photoisomerization: Electronic and Steric Effects. J. Phys. Chem. Lett. 2016, 7, 3591–3596. 10.1021/acs.jpclett.6b01401. [DOI] [PubMed] [Google Scholar]
- Cantatore V.; Granucci G.; Rousseau G.; Padula G.; Persico M. Photoisomerization of Self-Assembled Monolayers of Azobiphenyls: Simulations Highlight the Role of Packing and Defects. J. Phys. Chem. Lett. 2016, 7, 4027–4031. 10.1021/acs.jpclett.6b02018. [DOI] [PubMed] [Google Scholar]
- Lai C.-Y.; Raj G.; Liepuoniute I.; Chiesa M.; Naumov P. Direct Observation of Photoinduced trans–cis Isomerization on Azobenzene Single Crystal. Cryst. Growth Des. 2017, 17, 3306–3312. 10.1021/acs.cgd.7b00288. [DOI] [Google Scholar]
- Pesce L.; Perego C.; Grommet A. B.; Klajn R.; Pavan G. M. Molecular Factors Controlling the Isomerization of Azobenzenes in the Cavity of a Flexible Coordination Cage. J. Am. Chem. Soc. 2020, 142, 9792–9802. 10.1021/jacs.0c03444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanopolskyi A. I.; De S.; Białek M. J.; Diskin-Posner Y.; Avram L.; Feller M.; Klajn R. Reversible switching of arylazopyrazole within a metal–organic cage. Beilstein J. Org. Chem. 2019, 15, 2398–2407. 10.3762/bjoc.15.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klajn R. Spiropyran-based dynamic materials. Chem. Soc. Rev. 2014, 43, 148–184. 10.1039/C3CS60181A. [DOI] [PubMed] [Google Scholar]
- Lerch M. M.; Wezenberg S. J.; Szymanski W.; Feringa B. L. Unraveling the Photoswitching Mechanism in Donor–Acceptor Stenhouse Adducts. J. Am. Chem. Soc. 2016, 138, 6344–6347. 10.1021/jacs.6b01722. [DOI] [PubMed] [Google Scholar]
- Udayabhaskararao T.; Kundu P. K.; Ahrens J.; Klajn R. Reversible Photoisomerization of Spiropyran on the Surfaces of Au25 Nanoclusters. ChemPhysChem 2016, 17, 1805–1809. 10.1002/cphc.201500897. [DOI] [PubMed] [Google Scholar]
- Kundu P. K.; Das S.; Ahrens J.; Klajn R. Controlling the lifetimes of dynamic nanoparticle aggregates by spiropyran functionalization. Nanoscale 2016, 8, 19280–19286. 10.1039/C6NR05959G. [DOI] [PubMed] [Google Scholar]
- Cabrera I.; Krongauz V. Dynamic ordering of aggregated mesomorphic macromolecules. Nature 1987, 326, 582–585. 10.1038/326582a0. [DOI] [Google Scholar]
- Ahrens J.; Bian T.; Vexler T.; Klajn R. Irreversible Bleaching of Donor–Acceptor Stenhouse Adducts on the Surfaces of Magnetite Nanoparticles. ChemPhotoChem 2017, 1, 230–236. 10.1002/cptc.201700009. [DOI] [Google Scholar]
- Williams D. E.; Martin C. R.; Dolgopolova E. A.; Swifton A.; Godfrey D. C.; Ejegbavwo O. A.; Pellechia P. J.; Smith M. D.; Shustova N. B. Flipping the Switch: Fast Photoisomerization in a Confined Environment. J. Am. Chem. Soc. 2018, 140, 7611–7622. 10.1021/jacs.8b02994. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Müller K.; Valášek M.; Grosjean S.; Bräse S.; Wöll C.; Mayor M.; Heinke L. Series of Photoswitchable Azobenzene-Containing Metal–Organic Frameworks with Variable Adsorption Switching Effect. J. Phys. Chem. C 2018, 122, 19044–19050. 10.1021/acs.jpcc.8b05843. [DOI] [Google Scholar]
- Müller K.; Helfferich J.; Zhao F. L.; Verma R.; Kanj A. B.; Meded V.; Bléger D.; Wenzel W.; Heinke L. Switching the Proton Conduction in Nanoporous, Crystalline Materials by Light. Adv. Mater. 2018, 30, 1706551. 10.1002/adma.201706551. [DOI] [PubMed] [Google Scholar]
- Castiglioni F.; Danowski W.; Perego J.; Leung F. K.-C.; Sozzani P.; Bracco S.; Wezenberg S. J.; Comotti A.; Feringa B. L. Modulation of porosity in a solid material enabled by bulk photoisomerization of an overcrowded alkene. Nat. Chem. 2020, 12, 595–602. 10.1038/s41557-020-0493-5. [DOI] [PubMed] [Google Scholar]
- Saha R.; Devaraj A.; Bhattacharyya S.; Das S.; Zangrando E.; Mukherjee P. S. Unusual Behavior of Donor–Acceptor Stenhouse Adducts in Confined Space of a Water-Soluble PdII8 Molecular Vessel. J. Am. Chem. Soc. 2019, 141, 8638–8645. 10.1021/jacs.9b03924. [DOI] [PubMed] [Google Scholar]
- Singh G.; Chan H.; Baskin A.; Gelman E.; Repnin N.; Král P.; Klajn R. Self-assembly of magnetite nanocubes into helical superstructures. Science 2014, 345, 1149–1153. 10.1126/science.1254132. [DOI] [PubMed] [Google Scholar]
- Sawczyk M.; Klajn R. Out-of-Equilibrium Aggregates and Coatings during Seeded Growth of Metallic Nanoparticles. J. Am. Chem. Soc. 2017, 139, 17973–17978. 10.1021/jacs.7b09111. [DOI] [PubMed] [Google Scholar]
- Udayabhaskararao T.; Altantzis T.; Houben L.; Coronado-Puchau M.; Langer J.; Popovitz-Biro R.; Liz-Marzán L. M.; Vuković L.; Král P.; Bals S.; Klajn R. Tunable porous nanoallotropes prepared by post-assembly etching of binary nanoparticle superlattices. Science 2017, 358, 514–518. 10.1126/science.aan6046. [DOI] [PubMed] [Google Scholar]
- Amdursky N.; Kundu P. K.; Ahrens J.; Huppert D.; Klajn R. Noncovalent Interactions with Proteins Modify the Physicochemical Properties of a Molecular Switch. ChemPlusChem 2016, 81, 44–48. 10.1002/cplu.201500417. [DOI] [PubMed] [Google Scholar]
- Bian T.; Chu Z.; Klajn R. The Many Ways to Assemble Nanoparticles Using Light. Adv. Mater. 2020, 32, 1905866. 10.1002/adma.201905866. [DOI] [PubMed] [Google Scholar]
- Manna D.; Udayabhaskararao T.; Zhao H.; Klajn R. Orthogonal Light-Induced Self-Assembly of Nanoparticles using Differently Substituted Azobenzenes. Angew. Chem., Int. Ed. 2015, 54, 12394–12397. 10.1002/anie.201502419. [DOI] [PubMed] [Google Scholar]
- Das S.; Ranjan P.; Maiti P. S.; Singh G.; Leitus G.; Klajn R. Dual-Responsive Nanoparticles and their Self-Assembly. Adv. Mater. 2013, 25, 422–426. 10.1002/adma.201201734. [DOI] [PubMed] [Google Scholar]
- Chovnik O.; Balgley R.; Goldman J. R.; Klajn R. Dynamically Self-Assembling Carriers Enable Guiding of Diamagnetic Particles by Weak Magnets. J. Am. Chem. Soc. 2012, 134, 19564–19567. 10.1021/ja309633v. [DOI] [PubMed] [Google Scholar]
- Lee J.-W.; Klajn R. Dual-responsive nanoparticles that aggregate under the simultaneous action of light and CO2. Chem. Commun. 2015, 51, 2036–2039. 10.1039/C4CC08541H. [DOI] [PubMed] [Google Scholar]