

Abstract
Neoagarobiose (NA2) is the repeating disaccharide unit of agarose and possesses various promising biological activities. To identify an efficient exolytic β-agarase required for NA2 production from agarose, the GH50A β-agarase gene from agar-degrading Cellvibrio sp. KY-GH-1 was overexpressed as a recombinant His-tagged protein using the Escherichia coli expression system. GH50A β-agarase that consists of 797 amino acids was able to produce predominantly NA2 from agarose at an optimal temperature and pH of 35 °C and 7.5, respectively. The enzyme was stable up to 35 °C and within a pH range of 7.0–9.0. The Km, Vmax, Kcat, and Kcat/Km values of the enzyme were 26.5 mg/mL, 16.9 U/mg, 25.2 s–1, and 1.2 × 105 s–1 M–1, respectively. The copresence of 5 mM MnSO4 and 10 mM tris(2-carboxyethyl)phosphine (TCEP) resulted in a 2.5-fold enhancement of the enzyme activity. For NA2 production, neoagaro-oligosaccharides (NAOSs) containing NA4–NA18 were preferred over agarose or agaro-oligosaccharides (AOSs) as substrates. NA2 was produced along with minor amounts of agarotriose (A3) after treatment of AOS with the enzyme, indicating that the exolytic digestion of AOS by the enzyme was initiated by releasing A3 from nonreducing ends. Enzymatic hydrolysis of 0.4% agarose (100 mL) using GH50A β-agarase (20 μg/mL) for 4 h under optimal reaction conditions (5 mM MnSO4, 10 mM TCEP, 35 °C, 20 mM Tris–HCl, and pH 7.5) and purification of NA2 from hydrolysis products by Bio-Gel P-2 column chromatography resulted in the recovery of 216 mg of NA2 (∼54% yield from agarose). Altogether, these results suggest that the recombinant GH50A β-agarase is useful to convert agarose to NA2.
Introduction
Agar, a well-known marine red algae cell wall polysaccharide, consists of a mixture of two different components: neutral agarose and negatively charged agaropectin. Agarose is composed of alternating residues of α-1,3-linked 3,6-anhydro-l-galactose (l-AHG) and β-1,4-linked d-galactose. Agaropectin consists of the same repeating units but with partial replacement of AHG residues with l-galactose sulfate and partial replacement of the d-galactose residues with pyruvic acid acetal 4,6-O-(1-carboxyethylidene)-d-galactose.1 Agar is widely used as a gelling agent in food industries and for microbial culture media because it is resistant to microbial degradation. At least 15 genera of marine bacteria and seven genera of nonmarine bacteria have been reported to produce agarases that can degrade agar into its monomers d-galactose and l-AHG.2−6
Agarases are divided into two types based on their mode of cleavage: α-agarase (EC 3.2.1.158), which cleaves the α-1,3-glycosidic linkage, and β-agarase (EC 3.2.1.81), which cleaves the β-1,4-glycosidic linkage.2,3 Reported agarases are mostly agar-liquefying endo-β-agarases that produce neoagaro-oligosaccharides (NAOSs). Fewer reports are available on α-agarases that generate agaro-oligosaccharides (AOSs). Comparing amino acid sequence similarities among bacterial agarases, the Carbohydrate-Active Enzymes (CAZyme) database classifies agarases into different glycoside hydrolase (GH) families: GH16, GH50, GH86, and GH118 for β-agarases,7−9 as well as GH96 and GH117 for α-agarases.10,11 GH16, GH86, and GH118 β-agarases are endolytic and generate neoagarotetraose (NA4)/neoagarohexaose (NA6), NA6/neoagarooctaose (NA8), and NA8/neoagarodecaose (NA10). GH50 β-agarases produce neoagarobiose (NA2), NA4, or NA2/NA4 via their exolytic and endolytic activities.2,12−15 GH96 α-agarases produce agarobiose from agarose, and GH117 α-neoagarobiose hydrolase (α-NABH) catalyzes the hydrolytic degradation of NA2 to l-AHG and d-galactose.11
NAOS and AOS display various biological activities, including antioxidant,16 antitumor,17,18 prebiotic,19 anti-inflammatory,20 and antidiabetic and anti-obesity activities.21 In addition, NA2 and l-AHG demonstrate skin-moisturizing and whitening, anti-inflammatory, and anticarcinogenic effects.22−24 The biological activities of agar degradation products are likely associated with the presence of AHG, yet, in vivo studies providing direct evidence for the presumed l-AHG biological functions remain largely unavailable. This situation is largely attributable to the difficulty of mass-producing commercially available l-AHG. Recently, an enzymatic process to produce l-AHG from agarose has been developed, which involves a cotreatment of GH50 β-agarase and GH117 α-NABH to degrade agarose into NA2 and then into l-AHG and d-galactose.25,26 However, this process can still be improved for better enzymatic saccharification of agarose. Further exploration of the GH50 family β-agarase that can efficiently produce NA2 from agarose is still needed.
In a previous study, we sequenced the entire genome of Cellvibrio sp. KY-GH-1 (KCTC13629BP) to explore genetic information encoding agarases that hydrolyze agarose into l-AHG and d-galactose.27 The KY-GH-1 strain possesses three exolytic GH50 β-agarases in an agarase gene cluster spanning approximately 77 kb. However, which of these three isozymes possesses the most prominent exolytic GH50 β-agarase activity for the production of NA2 from agarose remains unknown.
In this study, we obtained three recombinant His-tagged GH50 family β-agarases (GH50A, GH50B, and GH50C) from Cellvibrio sp. KY-GH-1 using the Escherichia coli expression system with the pET-30a vector and compared their enzyme activities. We identified that GH50A β-agarase exhibits the highest exolytic β-agarase activity among the three isozymes. Finally, we further examined the enzymatic properties of GH50A β-agarase and assessed its efficiency for NA2 production from agarose.
Results and Discussion
Production of Individual GH50 β-Agarases from Cellvibrio sp. KY-GH-1 as C-Terminal His-Tagged Proteins Using E. coli Expression System with pET-30a Vector
Our previous sequence analysis of the Cellvibrio sp. KY-GH-1 genome demonstrated the presence of three GH50 β-agarase genes (GH50A, GH50B, and GH50C).27 Based on these data, we examined which recombinant His-tagged β-agarases expressed in the E. coli had the most prominent exolytic activity toward agar degradation.
After E. coli transformants were subjected to isopropyl β-d-1-thiogalactopyranoside (IPTG) induction, cells were harvested and prepared for fractionation into total, soluble, and insoluble portions. All three recombinant β-agarases were detected in cellular soluble fractions (Figure 1A). The levels of individual enzymes detected in the insoluble inclusion body fraction were insignificant. Molecular weights (MWs) of GH50A, GH50B, and GH50C β-agarases were calculated from the amino acid composition, including the 6× His-tag. These estimated values were 90.3, 88.7, and 88.5 kDa for GH50A, GH50B, and GH50C β-agarases, respectively. However, the estimated MWs of the recombinant His-tagged proteins using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) suggested 89.0, 90.1, and 94.0 kDa, respectively.
Figure 1.
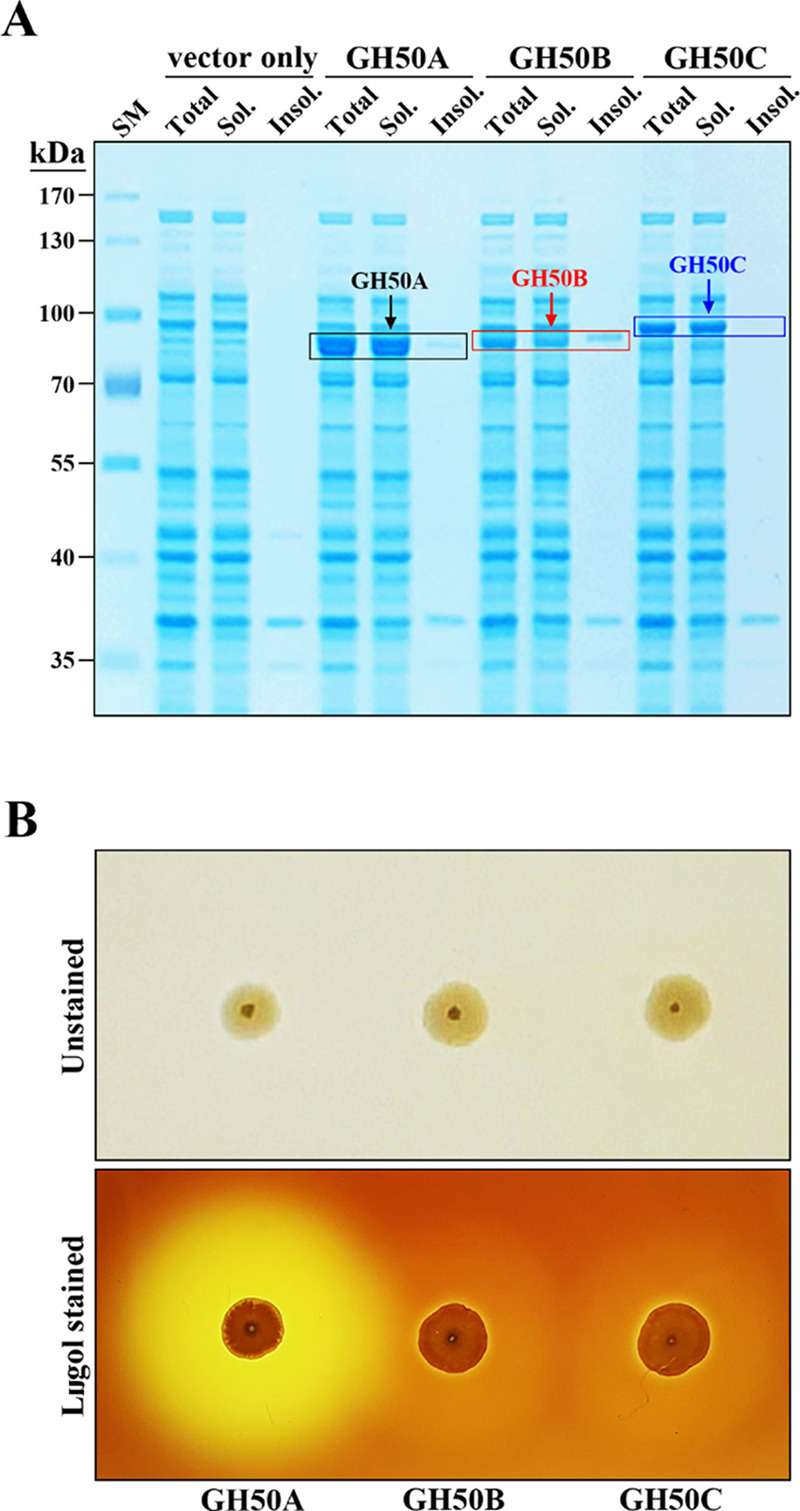
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of recombinant His-tagged GH50A, GH50B, and GH50C β-agarases expressed in E. coli and detection of extracellular agarase activity on agarose plates using Lugol’s iodine solution. (A) Total, soluble, and insoluble cellular portions were isolated from E. coli BL21 (DE3) transformants cultured in the presence of 0.075 mM IPTG. An equivalent amount of each portion was electrophoresed on SDS-polyacrylamide gel. (B) Transformants expressing recombinant His-tagged GH50A, GH50B, or GH50C β-agarase were spotted onto Luria broth (LB) agarose plates containing kanamycin and IPTG and incubated for 2 days at 30 °C. Plates were stained with Lugol’s iodine solution at RT. Representative results are shown; two additional experiments yielded similar results.
Transformants expressing GH50A, GH50B, or GH50C β-agarase were cultured on LB agarose plates containing 0.05 mM IPTG for 2 days, followed by staining with Lugol’s iodine solution. Agarose-degrading activity, which is reflected by a bright clear zone around the colonies, was the highest for the GH50A β-agarase-expressing transformant (Figure 1B). These data suggest that GH50A β-agarase is the major exolytic β-agarase required for the agarose-degrading enzyme machinery in Cellvibrio sp. KY-GH-1. Therefore, the recombinant GH50A β-agarase may be useful for enzymatic production of NA2 from agarose. The functional roles of GH50B and GH50C β-agarases in the degradation of agarose remain to be elucidated.
Comparison of GH50A β-Agarase Amino Acid Sequences with Related Enzymes from Other Sources
The amino acid sequence of GH50A β-agarase was aligned with other β-agarase sequences using the CLUSTALW program (Figure 2A). The overall sequence of Cellvibrio sp. KY-GH-1 GH50A β-agarase shares 97.6% similarity with Alteromonas sp. E-1 β-agarase (GenBank Accession No. BAE97587.1),28 as well as 96.9, 96.4, 95.6, and 91.7% similarity with Cellvibrio sp. OA-2007 (GenBank Accession No. WP_062065002.1),29Cellvibrio sp. pealriver (GenBank Accession No. WP_049629422.1),30Cellvibrio sp. KY-YJ-3 (GenBank Accession No. WP_151059417.1), and Cellvibrio sp. BR (GenBank Accession No. WP_157152532.1), respectively. This observation indicates that there is a high degree of homology among Cellvibrio GH50 β-agarases. GH50A β-agarase also shares similar identity (66.9, 64.3, 63.2, and 62.7%) with Catenovulum sp. RQJ05 (GenBank Accession No. WP_111979256.1), Catenovulum sediminis (GenBank Accession No. WP_143873817.1), Catenovulum agarivorans (GenBank Accession No. WP_035016017.1),31 and Saccharophagus degradans (GenBank Accession No. WP_143710996.1).32 Although these β-agarases found in the NCBI GenBank database (http://www.ncbi.nlm.nih.gov/) possess high levels of amino acid sequence homology (62.7–96.9%), none of their enzymatic properties have been tested to date.
Figure 2.

Multiple amino acid sequence alignment comparing GH50A β-agarase with nine GH50 family β-agarases and their phylogenetic relationship. (A) Amino acids are displayed in single-letter abbreviations after alignment of open-reading frames using Clustal X.33 Identical residues in all sequences are indicated by asterisks under the column; conserved substitutions are indicated by colons, semi-conserved substitutions are indicated by dots, and conserved residues are highlighted using the Clustal X color scheme. Deletions are indicated by dashes. (B) Unrooted tree was constructed using UPGMA.34 Numbers at nodes are levels of bootstrap support.
The unrooted phylogenetic relationships of individual β-agarases show that Cellvibrio sp. KY-GH-1 GH50A β-agarase is more closely related to β-agarases from Alteromonas sp. E-1, Cellvibrio sp. OA-2007, Cellvibrio sp. pealriver, Cellvibrio sp. KY-YJ-3, and Cellvibrio sp. BR isolated from nonmarine sources than to the enzymes from Catenovulum sp. RQJ05, C. sediminis, C. agarivorans, and S. degradans isolated from marine sources (Figure 2B).
Several GH50 family β-agarases were previously reported to cleave agarose by either exolytic activity alone or by a combination of exolytic and endolytic activities to produce NA2, NA4, or NA2/NA4.2,12−15 Since GH50A homologues were present in all of the agar-degrading bacteria aligned, we reasoned that GH50A β-agarase was most likely to be associated with degradation of agarose into NA2 and/or NA4 and is an essential component of the agar-degrading enzyme machinery in Cellvibrio sp. KY-GH-1.
Enzymatic Characteristics of GH50A β-Agarase
To examine recombinant GH50A β-agarase for mass production of NA2 from agarose, we investigated various enzymatic characteristics. As shown in Figure 3, the soluble β-agarase band was approximately 32% of the total soluble cellular proteins when compared to a standard curve, suggesting its successful overexpression in a soluble form in the E. coli expression system. The purified GH50A β-agarase was detected as a single band on an 8% SDS-PAGE gel without remarkable contaminants, demonstrating the high purity of the enzyme.
Figure 3.
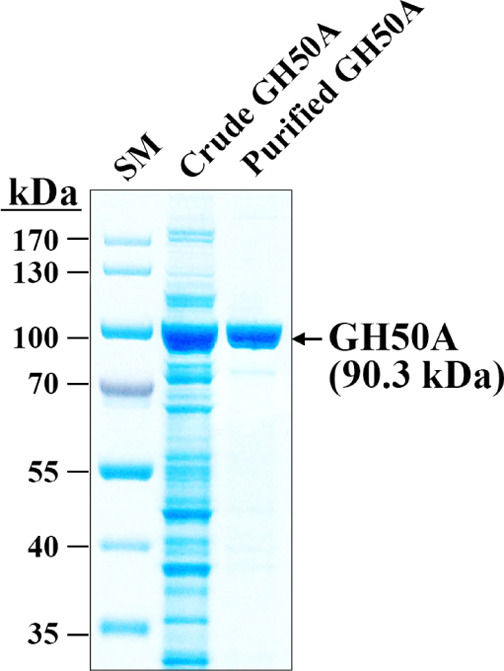
SDS-PAGE of purified recombinant His-tagged GH50A β-agarase. Lane: SM, prestained size markers; Crude GH50A, the soluble cell lysate (10 μg) obtained from E. coli transformant expressing GH50A β-agarase; and Purified GH50A, the purified recombinant His-tagged GH50A from the soluble cell lysate using the NI-NTA Purification System.
The highest enzymatic activity was found at 35 °C when the purified GH50A β-agarase was measured at various temperatures (Figure 4A). More than 80% of the maximum activity was retained throughout the temperature range of 20–50 °C. The enzyme exhibited maximum activity in a broad pH range of 6.0–10.0 (Figure 4B). The enzyme was stable up to 35 °C (Figure 4C). However, GH50A β-agarase retained only 60% of the maximum activity after treatment at 40 °C for 4 h and completely lost all its enzyme activity after incubating at 50 °C for 4 h. GH50A β-agarase was stable at the pH range of 7.0–9.0 but rapidly lost its activity in acidic pHs as it retained 30% of the maximum activity after incubating at pH 6.0 for 4 h (Figure 4D). This suggests that GH50A β-agarase is more stable at alkaline pH. The Km, Vmax, Kcat, and Kcat/Km values of the enzyme were 26.5 mg/mL, 16.9 U/mg, 25.2 s–1, and 1.2 × 105 s–1 M–1, respectively (Figure 4E).
Figure 4.

Biochemical characteristics of GH50A β-agarase. (A, B) Effects of temperature and pH on the enzyme activity were measured using 0.4% agarose and 20 μg/mL purified GH50A β-agarase. (C, D) Temperature and pH stability of the GH50A β-agarase were investigated following 4 h incubation at individual pH or temperature. (E) Lineweaver–Burk plot to obtain Km, Vmax, Kcat, and Kcat/Km values of GH50A β-agarase was investigated using indicated concentrations of the enzyme and agarose. Each value is expressed as the mean ± standard deviation (SD) (n = 3; three replicates per independent experiment). Representative results are shown; two additional experiments yielded similar results.
GH50A β-agarase activity was enhanced 1.7–1.8-fold in the presence of 5 mM MnCl2 or 5 mM MnSO4, suggesting that enzyme activity may rely on Mn2+ (Figure 5A). However, the enzyme activity was not affected by 5 mM ethylenediaminetetraacetic acid (EDTA). Interestingly, reported marine β-agarases are generally dependent on the presence of Mg2+ and Na+.14,35,36 However, GH50A β-agarase activity in this study was not affected by MgCl2 or NaCl. This demonstrates that the Mn2+-dependent enhancement of GH50A activity is a unique enzymatic property. The presence of 5 mM TCEP also enhanced the enzyme activity by 1.2-fold.
Figure 5.
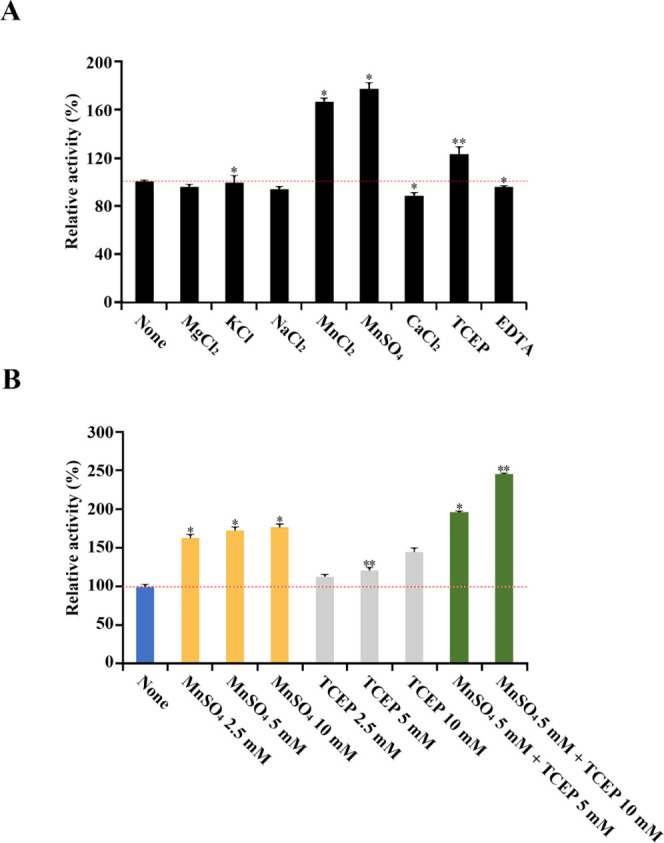
Effects of metal ions, reducing agent, and chelating agent on GH50A β-agarase activity. Individual effects of metal ions, a sulfhydryl reductant TCEP, and a chelator EDTA at a concentration of 5 mM (A), and co-treatment effect of MnSO4 (5 mM) and TCEP (5 mM or 10 mM) on the activity of GH50A β-agarase activity (B) were measured by mixing purified enzyme (20 μg/mL) with 0.4% agarose in 20 mM Tris-HCl buffer (pH 7.5) and then incubated at 35°C for 30 min. Enzyme activity measured without treatment with various ions and reagents was defined as 100%. Each value is expressed as the mean ± SD (n = 3; three replicates per independent experiment). *P < 0.05 and **P < 0.01 compared with non-treated control.
The enhancing effects by Mn2+ and TCEP on GH50A β-agarase activity were in a dose-dependent manner; 10 mM MnSO4 and 10 mM TCEP increased the enzyme activity to a maximum of 1.8-fold and 1.4-fold, respectively (Figure 5B). Also, GH50A β-agarase activity was elevated 2.5-fold in the copresence of 5 mM MnSO4 and 10 mM TCEP, suggesting a synergistic effect with Mn2+ and TCEP on GH50A β-agarase activity. However, the copresence of Mn2+ and 1,4-dithiothreitol (DTT) failed to demonstrate synergy between their individual positive effects on enzyme activity (data not shown). Although TCEP contributed to promoting the enzyme activity presumably by preventing oxidation of sulfhydryl groups of the enzyme, the DTT promoting effect was not compatible with the Mn2+ enhancing effect on the enzyme activity. This is possibly due to a suppressive effect on ionizing manganese salts.37 Therefore, GH50A β-agarase might require manganese ions and a sulfhydryl reductant TCEP for full enzyme activity. The manganese-binding affinity of GH50A β-agarase might be strong enough to resist inhibition by the chelating action of EDTA.
Individual effects of metal ions, a sulfhydryl reductant TCEP, and a chelator EDTA at a concentration of 5 mM (A) and cotreatment effects of MnSO4 (5 mM) and TCEP (5 or 10 mM) on the GH50A β-agarase activity (B) were measured by mixing purified enzyme (20 μg/mL) with 0.4% agarose in 20 mM Tris–HCl buffer (pH 7.5) and then incubating at 35 °C for 30 min. Enzyme activity measured without treatment with various ions and reagents was defined as 100%. Each value is expressed as the mean ± SD (n = 3; three replicates per independent experiment). *P < 0.05 and **P <0.01 compared with non-treated control.
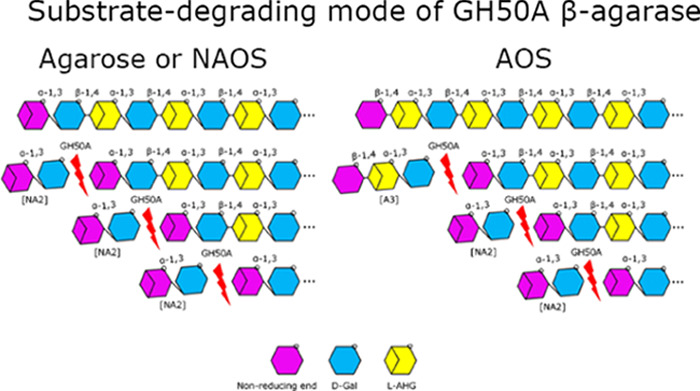
Substrate Degradation Mode of GH50A β-Agarase
In the literature, GH16, GH86, and GH118 β-agarases are endo-type enzymes that degrade agarose into NAOS with different DPs of 4–10, whereas GH50 β-agarases degrade agarose into NA2, NA4, or NA2/NA4 as predominant end products through their exolytic and endolytic activities.2,12−15
We performed thin-layer chromatography (TLC) analysis of the hydrolysis products from a time-kinetic treatment of the substrate (agarose or NAOS mixture of NA2–NA18) with GH50A β-agarase to determine the substrate-degrading mode. GH50A β-agarase-catalyzed time-kinetic hydrolysis patterns showed exo-typic hydrolysis of agarose to yield NA2 from the initiation of the reaction, and the amount of NA2 consistently increased as the primary product during the reaction period of 60 min (Figure 6A). In addition, NA4 and some NAOS with higher DPs were detected to a much lesser extent. TLC analysis of the hydrolysates of an NAOS mixture containing NA2–NA18 revealed that NAOSs as large as or larger than NA6 were preferred substrates for the enzyme reaction to produce NA2 (Figure 6B). TLC analysis data also revealed that GH50A β-agarase could degrade an AOS mixture with various DPs to produce NA2 as the major product. However, this process also produced minor amounts of A3 that was presumably generated at the first step of the enzyme-catalyzed digestion of AOS (Figure 6C).
Figure 6.

Time-kinetic analysis of GH50A β-agarase-catalyzed hydrolysates of agarose, NAOS, or AOS by TLC. For the hydrolysis of each substrate, 0.4% agarose (A), 1.0% NAOS (B), or 1.0% AOS (C) was incubated with 10 μg/mL enzyme in 20 mM Tris–HCl (pH 7.5) at 35 °C for the indicated time periods. Standard neoagaro-oligosaccharides (STD NAOSs) and agaro-oligosaccharides (AOSs) were prepared as described in Experimental Section. Equivalent amounts of each reaction mixture were analyzed by TLC. Saccharides were visualized with ethanol solution containing 0.2% (w/v) naphthoresorcinol and 10% (v/v) H2SO4 and then heating at 80 °C. Representative results are shown; two additional experiments yielded similar results.
To confirm the substrate preference of GH50A β-agarase for NA6–NA8 over NA4, NAOS hydrolysis products treated with GH50A β-agarase for the indicated time periods were analyzed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). Consistent with the TLC data, LC-MS/MS data showed that the enzyme-catalyzed hydrolysis of NA6–NA8 occurred prior to that of NA4, confirming that NAOSs larger than NA4 are preferred substrates for GH50A β-agarase to produce NA2 (Figure 7). In addition, LC-MS/MS analysis of the AOS enzyme-catalyzed hydrolysates showed that the major product NA2 and the minor product A3 were produced in a time-dependent manner (Figure 8). These results demonstrate that the AOS exolytic digestion by GH50A β-agarase to produce NA2 is initiated after releasing A3 from the nonreducing end of AOS.
Figure 7.

Time-kinetic analysis of GH50A β-agarase-catalyzed hydrolysates of NAOS by LC-MS/MS. For NAOS hydrolysis, 1% NAOS was incubated with 10 μg/mL enzyme in 20 mM Tris–HCl (pH 7.5) at 35°C for individual time periods. LC-MS/MS analysis was performed as described in the Experimental Section.
Figure 8.

Time-kinetic analysis of GH50A β-agarase-catalyzed AOS hydrolysates by LC-MS/MS. For hydrolysis of AOS, 1% AOS was incubated with 10 μg/mL enzyme in 20 mM Tris–HCl (pH 7.5) at 35 °C for individual time periods. LC-MS/MS analysis was performed as described in the Experimental Section.
To the best of our knowledge, there are no reported GH50 family β-agarases that produce NA2 in an exo-splitting manner after releasing A3 from the nonreducing end of AOS. Therefore, this novel GH50A β-agarase reported in this study is likely to be the first discovered enzyme capable of catalyzing such reactions.
In addition, although most of the GH50 β-agarases were reported to produce NA4 or NA2/NA4 from agarose by their exolytic and endolytic activities,2,12−15 GH50 β-agarase that produces NA2 directly from agarose by exolytic action is relatively rare. In these regards, GH50A β-agarase from a freshwater bacterium Cellvibrio sp. KY-GH-1 is a unique GH50 family β-agarase that can exolytically digest its substrates (agarose, NAOS, or AOS) to produce NA2 as the major product (Figure 9).
Figure 9.

Model for GH50A β-agarase-catalyzed hydrolysis of agarose, NAOS, or AOS. GH50A β-agarase produces NA2 directly from agarose or NAOS from the nonreducing end by exolytic activity. GH50A β-agarase can produce NA2 from AOS in the same manner after releasing A3 at the nonreducing end of AOS.
Previously, we reported that Cellvibrio sp. KY-GH-1 possesses genes encoding four endolytic GH16 family β-agarases, two endolytic GH86 family β-agarases, and three exolytic GH50 family β-agarases.27 Agarose degradation into NA2 by combined treatment with endolytic GH16/GH86 β-agarases and exolytic GH50A β-agarase or with acid pretreatment and GH50A β-agarase treatment may be more efficient than single treatment with GH50A β-agarase.
Yield of NA2 from Agarose or AOS Treatment with GH50A β-Agarase under Optimal Reaction Conditions
To examine whether GH50A β-agarase may be an efficient enzyme for NA2 production from agarose, we examined the yield of NA2 from 0.4% agarose under optimum reaction conditions (5 mM MnSO4, 10 mM TCEP, 20 mM Tris–HCl, pH 7.5, 35 °C). The concentration of GH50A β-agarase required for maximal production of reducing sugars from 0.4% agarose was measured following a 4 h treatment under optimal reaction conditions. The optimal concentration was 20 μg/mL (Figure 10A). When treated for 12 h under the same conditions, the concentration of the enzyme required for maximum production was 5 μg/mL (data not shown). Following the treatment of 0.4% agarose (100 mL) with 20 μg/mL GH50A β-agarase for 4 h under optimal conditions, the reaction mixture was lyophilized. The dried sample was dissolved in DI water and subjected to size-exclusion chromatography using a Bio-Gel P-2 column. The TLC analysis showed that NA2 was identified in fractions 18–19, demonstrating that NA2 could be purified from the enzyme reaction products by size-exclusion chromatography (Figure 10B). Lyophilization of fractions 18–19 recovered NA2 as a powder. When the enzyme hydrolysate of agarose (400 mg) was fractionated by three rounds of column chromatography, a total of 216 mg of NA2 was obtained, indicating that the yield of NA2 was ∼54% from agarose.
Figure 10.
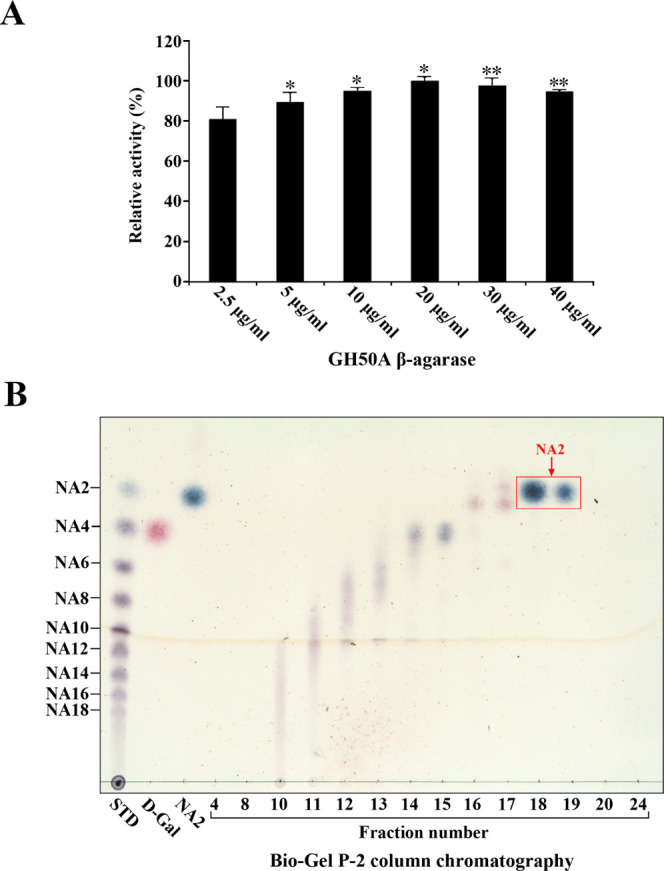
Dosage effect of GH50A β-agarase activity and TLC analysis of each fraction obtained by Bio-Gel P-2 column chromatography of GH50A β-agarase-catalyzed hydrolysate of agarose. (A) Enzyme reaction was performed by incubating 0.4% agarose with individual amounts of enzyme for 4 h under optimal conditions. Each value is expressed as the mean ± SD (n = 3; three replicates per independent experiment). *P < 0.05 and **P < 0.01 compared with non-treated control. (B) For the preparation of the TLC sample, 0.4% agarose was treated with the enzyme (20 μg/mL) for 4 h under optimal conditions and then lyophilized. The lyophilized sample was subjected to Bio-Gel P-2 column chromatography with DI water as the mobile phase. Individual fractions were analyzed by TLC. Saccharides were visualized as described in the legend of Figure 6. Representative results are shown; two additional experiments yielded similar results.
To examine whether GH50A β-agarase may be an efficient enzyme for NA2 production from AOS, we examined the yield of NA2 from AOS. After 1.0% AOS (30 mL) was treated with GH50A β-agarase (20 μg/mL) for 4 h under optimum reaction conditions (5 mM MnSO4, 10 mM TCEP, 20 mM Tris–HCl, pH 7.5, 35 °C), the reaction mixture was lyophilized. The dried sample was dissolved in DI water and subjected to size-exclusion chromatography using a Sephadex G-10 column. The TLC analysis of individual fractions obtained from the column chromatography showed that NA2 was identified in fractions 21–23 along with A3 in fractions 18–19 (Figure 11A). In addition, NA2 and A3 in individual fractions were further confirmed with an LC-MS/MS analysis (Figure 11B). This indicates that NA2 and A3 could be purified from the enzyme reaction products by Sephadex G-10 column chromatography. Lyophilization of fractions 21–23 resulted in recovery of NA2 powder (184 mg), indicating ∼61% yield of NA2 from AOS.
Figure 11.
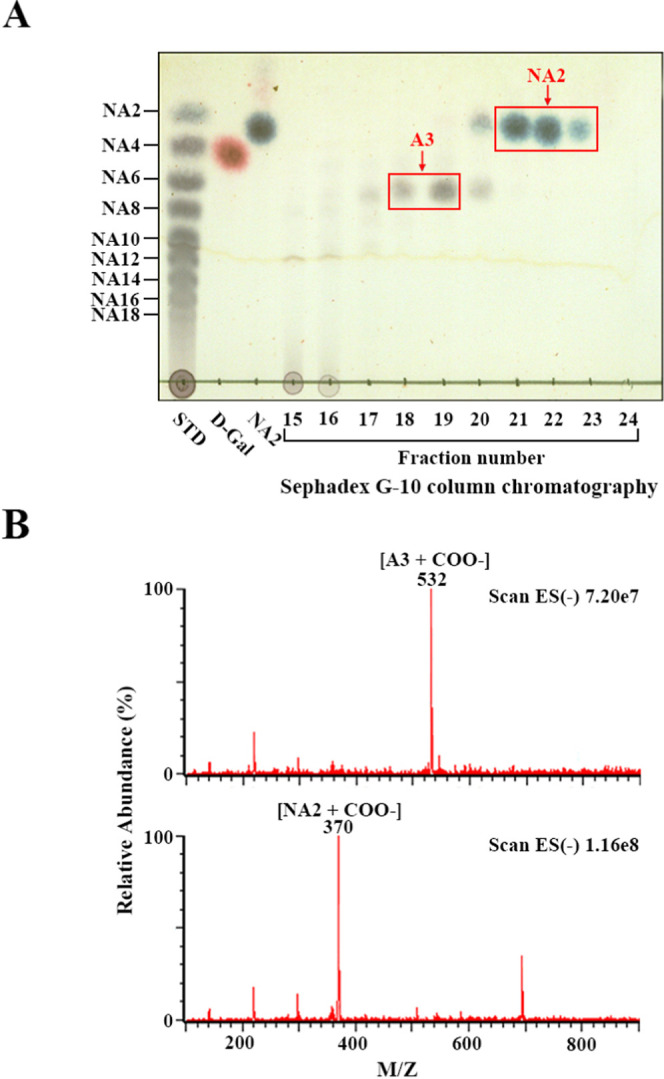
TLC analysis of each fraction obtained by Sephadex G-10 column chromatography of GH50A β-agarase-catalyzed hydrolysate of AOS and LC-MS/MS analysis of NA2 and A3 obtained from column chromatography. (A) After 1.0% AOS was treated with 20 μg/mL enzyme for 4 h under optimal conditions, the reaction mixture was lyophilized. The lyophilized sample was subjected to Sephadex G-10 column chromatography with DI water as the mobile phase. Individual fractions were analyzed by TLC. Saccharides were visualized as described in the legend of Figure 6. (B) Confirmation of NA2 and A3 by LC-MS/MS as described in the Experimental Section.
Conclusions
In summary, a recombinant His-tagged GH50A β-agarase derived from a freshwater agar-degrading Cellvibrio sp. KY-GH-1 produces neoagarobiose (NA2) as the primary product from agarose. Enzyme activity exhibits more than 80% of maximum activity at pH 6.0–10.0 and temperature 20–50 °C and is enhanced 2.5-fold due to the copresence of MnSO4 and TCEP, but it is not affected by 5 mM EDTA. GH50A β-agarase hydrolyzes NAOSs irrespective of their DPs to produce NA2 but with a preference for NA6–NA18. An NAOS mixture (NA6–NA18) prepared by endolytic β-agarase treatment of agarose was a preferred substrate over either agarose itself or an AOS mixture prepared by acetic acid treatment of agarose. The GH50A β-agarase enzymatic properties suggest that it may be useful as a one-step process to convert agarose to NA2 with ∼54% yield or as a cotreatment process with an endolytic β-agarase (GH16/GH86/GH118 β-agarases) to maximize agarose conversion to NA2. Further, GH50A β-agarase will be useful for saccharification of agarose biomass to produce monomers l-AHG and d-galactose, via cotreatment with GH117 family α-NABH. Also, since none of the nine β-agarases possessing high-level homology (62.7–96.9%) in their amino acid sequences were examined for enzymatic properties to date, current data for GH50A β-agarase reported in this study may provide insight into β-agarases that exhibit significant homology to GH50A β-agarase.
Experimental Section
Cloning and Expression of GH50 β-Agarase Genes Using the E. coli Expression System with pET-30a Vector
Three Cellvibrio sp. KY-GH-1 β-agarase genes (GH50A, GH50B, and GH50C) were amplified from genomic DNA by PCR using NdeI-forward primers (5′-CCCGCATATGATGTGTTCGAGTTATAA-GCTTG-3′ for GH50A and 5′-GCGGCATATGAAAAACTCACAACATCTTAATC-3′ for GH50B), BamH1-forward primer (5′-GGCGGGATCCATGAAAAAAAATCAACTACATTTA-3′ for GH50C), and Xho1-reverse primers (5′-CGGGCTCGAGTTTTTTCGCGCGGCGAGTA-3′ for GH50A, 5′-GCGCTCGAGTTTT-CCAAAACGCTTAATGTAAAGG-3′ for GH50B, and 5′-GAATCTCGAGTTGGATGGGAGGAATTTTA-3′ for GH50C). The PCR products were purified and digested by NdeI/XhoI for both GH50A and GH50B genes or BamHI/XhoI for the GH50C gene. The digested PCR fragments were ligated into the pET-30a expression vector (Novagen, Madison, WI) using T4 ligase (Roche, Basel, Switzerland) to express each enzyme as a C-terminal 6× His-tagged protein. The recombinant pET-30a plasmids were transformed into E. coli BL21 (DE3) (Novagen). Transformants harboring individual GH50 β-agarase genes were cultured overnight at 30 °C in a Luria broth (LB) plate with 50 μg/mL kanamycin (Sigma-Aldrich, St. Louis, MO).
Expression of individual β-agarase genes in the E. coli transformants was induced as previously described.38 Transformants were cultured in LB media containing 50 μg/mL kanamycin at 25 °C until an OD600 nm of 0.5–0.6 was reached. For induction of GH50 β-agarases, 0.075 mM isopropyl β-d-1-thiogalactopyranoside (IPTG, Sigma-Aldrich, St. Louis, MO) was added and cells were further cultured for an additional 5 h at 25 °C.
Sequence Analysis and Phylogenetic Tree Construction
The amino acid sequences of nine GH50 family β-agarases were obtained by BLAST searches using the NCBI database (http://www.ncbi.nlm.nih.gov). The amino acid sequences of individual open-reading frames were aligned using Clustal X.33 The conserved residues are highlighted using the Clustal X color scheme. The unrooted phylogenetic tree containing individual GH50 family members was constructed based on the amino acid sequence similarities using UPGMA.34
Zymogram of Agarase Activity on LB Agarose Plates
The E. coli transformants harboring the empty pET-30a plasmid or the recombinant β-agarase pET-30a plasmids (GH50A, GH50B, or GH50C) were spotted on LB plates containing 1.5% agarose (Invitrogen, Life Technology, Grand Island, NY), 50 μg/mL kanamycin, and 0.05 mM IPTG and incubated for 2 days at 30 °C. The plates were stained with Lugol’s iodine solution at room temperature (RT).
Cell Lysate Preparation, Protein Quantification, and Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
Transformant cells were suspended in 40 mM Tris–HCl buffer (pH 8.0), disrupted by sonication with 20 short bursts of 10 s, extracted at 4 °C for 30 min, and fractionated into three portions (total, soluble, and insoluble inclusion) as previously described.39 Cell lysate protein quantification was performed using a Micro BCA kit (Pierce, Rockford, IL). An equivalent amount of cell lysate (10 μg) was electrophoresed on an 8% SDS-polyacrylamide gel using the Laemmli method.40 The gels were stained with Coomassie brilliant blue (CBB) R-250. To quantify the concentration of the soluble recombinant protein in the cell lysate, its arbitrary densitometric unit was compared to a serial diluted bovine serum albumin standard curve as previously described.41 Densitometry was performed using ImageQuant TL software (Amersham, Arlington Heights, IL).
Purification of the Recombinant His-Tagged GH50A β-Agarase
The soluble portion from cell lysates containing the recombinant His-tagged GH50A β-agarase protein was subjected to immobilized metal ion affinity chromatography (IMAC) using Ni-NTA resin (ThermoFisher Scientific, Rockford, IL).42
β-Agarase Activity Assay
Quantification of GH50 β-agarase activity was performed using the 3,5-dinitrosalicylic acid (DNS) method,43 which detects reducing sugars released from agarose. The enzyme solution (100 μL) was mixed with an equal volume of 0.8% agarose dissolved in 20 mM Tris–HCl buffer (pH 7.5). After incubation at 35 °C for 30 min, reducing sugars formed in the reaction mixture were colorimetrically measured using the DNS reagent. One unit of the enzyme activity was defined as the amount of the enzyme that produced reducing power equivalent to 1 μmol of d-galactose (Sigma-Aldrich) per minute.
Thin-Layer Chromatography (TLC)
TLC analysis for GH50A β-agarase-catalyzed agarose hydrolysates, NAOS, or AOS was performed using silica gel 60 aluminum plates (F254 Merck, Darmstadt, Germany) and developed with n-butanol–ethanol–H2O (3:2:2 (v/v)). Saccharides were detected using a visualization solution containing 0.2% (w/v) naphthoresorcinol (Sigma-Aldrich) and 10% (v/v) H2SO4 in ethanol, followed by heating at 80 °C. d-Galactose and the NAOS standard mixture containing NA2–NA18 (prepared as described44 and gifted by Dr. Sang-Hyeon Lee) were used as standards. An AOS mixture was prepared by mild acid hydrolysis of 1% agarose in 3 M acetic acid treated at 80 °C for 3 h, followed by adjusting pH to ∼7.0 using 5 M NaOH. After lyophilization, the solid sample was washed with 95% (v/v) cold ethanol three times and then dried using a rotary evaporator to obtain the AOS powder.
Size-Exclusion Chromatography Using Bio-Gel P-2 or Sephadex G-10 Column
NA2-containing hydrolysis products were prepared by treating agarose with GH50A β-agarase under optimal reaction conditions, followed by lyophilization. The lyophilized products were dissolved in deionized (DI) water and subjected to size-exclusion chromatography using a Bio-Gel P-2 column (I.D. 1.3 cm × 60 cm, Bio-Rad Laboratories, Hercules, CA) equilibrated with DI water. Each 3 mL fraction was obtained by elution with DI water as the mobile phase, and fractions containing NA2 only were determined by TLC and collected.
NA2 and agarotriose (A3) containing enzymatic reaction products were prepared by treating 1% AOS with GH50A β-agarase (20 μg/mL) for 4 h under optimal reaction conditions, followed by lyophilization. Purification of NA2 and A3 was performed by size-exclusion chromatography using a Sephadex G-10 column (I.D. 1.3 cm × 60 cm, GE Healthcare Bio-Sciences AB, Uppsala, Sweden) equilibrated with DI water. Each 2 mL fraction was obtained by elution with DI water as the mobile phase, and fractions containing NA2 or A3 were determined by TLC and collected.
Identification of Enzyme Reaction Products by Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS)
The LC-MS/MS analysis was performed using a Xevo TQ-S micro mass spectrometer with an electrospray ionization (ESI) ion source equipped with an Acquity ultra-performance liquid chromatography (UPLC) H-Class core system (Waters Corporation, Milford, MA) previously described.45,46 The sample elution was performed using a Waters Acquity UPLC Spherisorb amino column (2 mm × 100 mm, 3 μm particle size) held at 40 °C with a solvent flow rate of 200 μL/min. The LC system consisted of (A) 0.1% aqueous formic acid and (B) 0.1% acetonitrile. NA2–NA8 in enzymatic hydrolysates from NAOS were identified by chromatography initiated using a binary gradient solvent system of 35:65 of A/B for 5 min, which gradually changed to 65:35 of A/B at min 12. Finally, the gradient was switched to 35:65 of A/B at min 13 and continued until the chromatography stopped at min 20. NA2 and A3 in enzymatic hydrolysates from AOS were identified by chromatography initiated by a binary gradient solvent system of 95:5 of A/B for 3 min, which was gradually changed to 75:25 of A/B until min 13. The flow was maintained at 75:25 of A/B until min 15. The initial condition of 95:5 of A/B was then attained at min 16 and continued until the chromatography stopped at min 30. The injection volume was 5 μL, and the sample manager temperature was set at 5 °C. The mass spectrometer detector conditions were set as follows: capillary voltage, 2.0 kV; cone voltage, 20 V; source temperature, 150 °C; desolvation temperature, 250 °C; desolvation gas flow, 550 m/h; cone gas flow, 5 L/h; and mass range, 100–1500.
Statistical Analyses
Unless otherwise indicated, data are representative of at least three independent experiments. All data are expressed as the mean ± standard deviation (SD, for each group n ≤ 3). Statistical analyses were performed using Student’s t test to evaluate the significance of differences between two groups and one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test for comparing three or more groups. P values <0.05 were considered significant. Statistical analysis was conducted using SPSS Statistics version 23 (IBM, Armonk, NY).
Acknowledgments
This work was supported by a grant from Kyungpook National University, Daegu 41566, Republic of Korea, 2019.
Glossary
Abbreviations used
- AHG
3,6-anhydro-l-galactose
- AOS
agaro-oligosaccharide
- DNS
3,5-dinitrosalicylic acid
- DP
degree of polymerization
- GH
glycoside hydrolase
- IMAC
immobilized metal ion affinity chromatography
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- NA2
neoagarobiose
- NAOS
neoagaro-oligosaccharide
- PCR
polymerase chain reaction
- SD
standard deviation
- SDS
sodium dodecyl sulfate
- TCEP
tris (2-carboxyethyl) phosphine
- TLC
thin-layer chromatography
The authors declare no competing financial interest.
References
- Duckworth M.; Yaphe W. The structure of agar. Part I. Fractionation of a complex mixture of Polysaccharides. Carbohydr. Res. 1971, 16, 189–197. 10.1016/S0008-6215(00)86113-3. [DOI] [Google Scholar]
- Fu X. T.; Kim S. M. Agarase: review of major sources, categories, purification method, enzyme characteristics and applications. Mar. Drugs 2010, 8, 200–218. 10.3390/md8010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi S. T.; Barzkar N. Future direction in marine bacterial agarases for industrial applications. Appl. Microbiol. Biotechnol. 2018, 102, 6847–6863. 10.1007/s00253-018-9156-5. [DOI] [PubMed] [Google Scholar]
- Rhee Y. J.; Han C. R.; Kim W. C.; Jun D. Y.; Rhee I. K.; Kim Y. H. Isolation of a novel freshwater agarolytic Cellvibrio sp. KY-YJ-3 and characterization of its extracellular β-agarase. J. Microbiol. Biotechnol. 2010, 20, 1378–1385. 10.4014/jmb.1007.07010. [DOI] [PubMed] [Google Scholar]
- Agbo J. A. C.; Moss M. O. The Isolation and characterization of agarolytic bacteria from a lowland river. J. Gen. Microbiol. 1979, 115, 355–368. 10.1099/00221287-115-2-355. [DOI] [Google Scholar]
- Bannikova G. E.; Lopatin S. A.; Varlamov V. P.; Kuznetsov B. B.; Kozina I. V.; Miroshnichenko M. L.; Chernykh N. A.; Turova T. P.; Bonch-Osmolovskaia E. A. The thermophilic bacteria hydrolyzing agar: Characterization of thermostable agarose. Appl. Biochem. Microbiol. 2008, 44, 366–371. 10.1134/S0003683808040054. [DOI] [PubMed] [Google Scholar]
- Cantarel B. L.; Coutinho P. M.; Rancurel C.; Bernard T.; Lombard V.; Henrissat B. The carbohydrate-active EnZyme database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi W. J.; Chang Y. K.; Hong S. K. Agar degradation by microorganisms and agar-degrading enzymes. Appl. Microbiol. Biotechnol. 2012, 94, 917–930. 10.1007/s00253-012-4023-2. [DOI] [PubMed] [Google Scholar]
- Michel G.; Nyval-Collen P.; Barbeyron T.; Czjzek M.; Helbert W. Bioconversion of red seaweed galactans. A focus on bacterial agarases and carrageenases. Appl. Microbiol. Biotechnol. 2006, 71, 23–33. 10.1007/s00253-006-0377-7. [DOI] [PubMed] [Google Scholar]
- Flament D.; Barbeyron T.; Jam M.; Potin P.; Czjzek M.; Kloareg B.; Michel G. α-Agarases define a new family of glycoside hydrolases, distinct from β-agarase families. Appl. Environ. Microbiol. 2007, 73, 4691–4694. 10.1128/AEM.00496-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S. C.; Lee S.; Lee J.; Kim H. T.; Ko H. J.; Kim K. H.; Choi I. G. Crystal structure of a key enzyme in the agarolytic pathway, α-neoagarobiose hydrolase from Saccharophagus degradans 2-40. Biochem. Biophys. Res. Commun. 2011, 412, 238–244. 10.1016/j.bbrc.2011.07.073. [DOI] [PubMed] [Google Scholar]
- Fu X. T.; Lin H.; Kim S. M. Purification and characterization of a novel β-agarase, AgaA34, from Agarivorans albus YKW-34. Appl. Microbiol. Biotechnol. 2008, 78, 265–273. 10.1007/s00253-007-1303-3. [DOI] [PubMed] [Google Scholar]
- Li G.; Sun M.; Wu J.; Ye M.; Ge X.; Wei W.; Li H.; Hu F. Identification and biochemical characterization of a novel endo-type β-agarase AgaW from Cohnella sp. strain LGH. Appl. Microbiol. Biotechnol. 2015, 99, 10019–10029. 10.1007/s00253-015-6869-6. [DOI] [PubMed] [Google Scholar]
- Han W.; Cheng Y.; Wang D.; Wang S.; Liu H.; Gu J.; Wu Z.; Li F. Biochemical characteristics and substrate degradation pattern of a novel exo-type β-agarase from the polysaccharide-degrading marine bacterium Flammeovirga sp. strain MY04. Appl. Environ. Microbiol. 2016, 82, 4944–4954. 10.1128/AEM.00393-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. W.; Lin H. J.; Huang W. C.; Hsuan S. L.; Lin J. H.; Wang J. P. Molecular cloning, expression, and functional characterization of the β-agarase AgaB-4 from Paenibacillus agarexedens. AMB Express 2018, 8, 49 10.1186/s13568-018-0581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. M.; Yan X. J. Antioxidant activities of agaro-oligosaccharides with different degrees of polymerization in cell-based system. Biochim. Biophys. Acta, Gen. Subj. 2005, 1722, 103–111. 10.1016/j.bbagen.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Enoki T.; Tominaga T.; Takashima F.; Ohnogi H.; Sagawa H.; Kato I. Anti-tumor-promoting activities of agaro-oligosaccharides on two-stage mouse skin carcinogenesis. Biol. Pharm. Bull. 2012, 35, 1145–1149. 10.1248/bpb.b12-00188. [DOI] [PubMed] [Google Scholar]
- Lee M. H.; Jang J. H.; Yoon G. Y.; Lee S. J.; Lee M. G.; Kang T. H.; Park Y. M.; et al. Neoagarohexaose-mediated activation of dendritic cells via Toll-like receptor 4 leads to stimulation of natural killer cells and enhancement of antitumor immunity. BMB Rep. 2017, 50, 263–268. 10.5483/BMBRep.2017.50.5.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Gong Q.; Wang Y.; Ma Y.; Li J.; Yu W. Prebiotic effects of neoagaro-oligosaccharides prepared by enzymatic hydrolysis of agarose. Anaerobe 2006, 12, 260–266. 10.1016/j.anaerobe.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Wang W.; Liu P.; Hao C.; Wu L.; Wan W.; Mao X. Neoagaro-oligosaccharide monomers inhibit inflammation in LPS-stimulated macrophages through suppression of MAPK and NF-κB pathways. Sci. Rep. 2017, 7, 44252 10.1038/srep44252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. J.; Lee J. H.; Kim E. J.; Yang H. J.; Park J. S.; Hong S. K. Anti-obesity and anti-diabetic effect of neoagarooligosaccharides on high-fat diet-Induced obesity in mice. Mar. Drugs 2017, 15, 90. 10.3390/md15040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi R.; Takisada M.; Suzuki T.; Kirimura K.; Usami S. Neoagarobiose as a novel moisturizer with whitening effect. Biosci., Biotechnol., Biochem. 1997, 61, 162–163. 10.1271/bbb.61.162. [DOI] [PubMed] [Google Scholar]
- Yun E. J.; Lee A. R.; Kim J. H.; Cho K. M.; Kim K. H. 3,6-Anhydro-l-galactose, a rare sugar from agar, a new anticariogenic sugar to replace xylitol. Food Chem. 2017, 221, 976–983. 10.1016/j.foodchem.2016.11.066. [DOI] [PubMed] [Google Scholar]
- Yun E. J.; Lee S.; Kim J. H.; Kim B. B.; Kim H. T.; Lee S. H.; Pelton J. G.; Kang N. J.; Choi I. G.; Kim K. H. Enzymatic production of 3,6-anhydro-L-galactose from agarose and its purification and in vitro skin whitening and anti-inflammatory activities. Appl. Microbiol. Biotechnol. 2013, 97, 2961–2970. 10.1007/s00253-012-4184-z. [DOI] [PubMed] [Google Scholar]
- Alkotaini B.; Han N. S.; Kim B. S. Fusion of agarase and neoagarobiose hydrolase for mono-sugar production from agar. Appl. Microbiol. Biotechnol. 2017, 101, 1573–1580. 10.1007/s00253-016-8011-9. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Sun J.; Liu Z.; Huang W.; Xue C.; Mao X. Coimmobilization of β-agarase and α-neoagarobiose hydrolase for enhancing the production of 3,6-anhydro-l-galactose. J. Agric. Food Chem. 2018, 66, 7087–7095. 10.1021/acs.jafc.8b01974. [DOI] [PubMed] [Google Scholar]
- Kwon G. H.; Kwon M. J.; Park J. E.; Kim Y. H. Whole genome sequence of a freshwater agar-degrading bacterium Cellvibrio sp. KY-GH-1. Biotechnol. Rep. 2019, 23, e00346 10.1016/j.btre.2019.e00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirimura K.; Masuda N.; Iwasaki Y.; Nakagawa H.; Kobayashi R.; Usami S. Purification and characterization of a novel beta-agarase from an alkalophilic bacterium. Alteromonas sp. E-1. J. Biosci. Bioeng. 1999, 87, 436–441. 10.1016/S1389-1723(99)80091-7. [DOI] [PubMed] [Google Scholar]
- Syazni; Yanagisawa M.; Kasuu M.; Nakasaki K.; Ariga O. Draft genome sequence of the nonmarine agarolytic bacterium Cellvibrio sp. OA-2007. Genome Announce. 2015, 3, e00468-15 10.1128/genomeA.00468-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z.; Lin W.; Luo J. Comparative phenotype and genome analysis of Cellvibrio sp. PR1, a xylanolytic and agarolytic bacterium from the Pearl River. BioMed Res. Int. 2017, 2017, 6304248 10.1155/2017/6304248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan D.; Li X.; Gu Z.; Wei G.; Gao Z.; Shao Z. Draft genome sequence of the agar-degrading bacterium Catenovulum sp. strain DS-2, isolated from intestines of Haliotis diversicolor. Genome Announce. 2014, 2, e00144-14 10.1128/genomeA.00144-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekborg N. A.; Taylor L. E.; Longmire A. G.; Henrissat B.; Weiner R. M.; Hutcheson S. W. Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2-40. Appl. Environ. Microbiol. 2006, 72, 3396–3405. 10.1128/AEM.72.5.3396-3405.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneath P. H. A.; Sokal R. R.. Numerical Taxonomy: The Principles and Practice of Numerical Classification; W.H. Freeman and Company: San Francisco, 1973; pp 1–573. [Google Scholar]
- An K.; Shi X.; Cui F.; Cheng J.; Liu N.; Zhao X.; Zhang X. H. Characterization and overexpression of a glycosyl hydrolase family 16 β-agarase YM01-1 from marine bacterium Catenovulum agarivorans YM01T. Protein Expression Purif. 2018, 143, 1–8. 10.1016/j.pep.2017.10.002. [DOI] [PubMed] [Google Scholar]
- Xie W.; Lin B.; Zhou Z.; Lu G.; Lun J.; Xia C.; Li S.; Hu Z. Characterization of a novel β-agarase from an agar-degrading bacterium Catenovulum sp. X3. Appl. Microbiol. Biotechnol. 2013, 97, 4907–4915. 10.1007/s00253-012-4385-5. [DOI] [PubMed] [Google Scholar]
- Charrier J. G.; Anastasio C. On dithiothreitol (DTT) as a measure of oxidative potential for ambient particles: evidence for the importance of soluble transition metals. Atmos. Chem. Phys. Discuss. 2012, 12, 11317–11350. 10.5194/acpd-12-11317-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W.; Rosenberg A. H.; Dunn J. J.; Dubendorff J. W. Use of T7 RNA Polymerase to Direct Expression of Cloned Genes. Methods Enzymol. 1990, 185, 60–89. 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Larkin M. A.; Blackshields G.; Brown N. P.; Chenna R.; McGettigan P. A.; McWilliam H.; Valentin F.; Wallace I. M.; Wilm A.; Lopez R.; Thompson J. D.; Gibson T. J.; Higgins D. G. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Jun D. Y.; Kim M. K.; Kim Y. H. Rabbit antibody against murine cyclin D3 protein overexpressed in bacterial system. J. Micobiol. Biotechnol. 1996, 6, 474–481. [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Syrový I.; Hodný Z. Staining and quantification of proteins separated by polyacrylamide gel electrophoresis. J. Chromatogr. B: Biomed. Sci. Appl. 1991, 569, 175–196. 10.1016/0378-4347(91)80229-6. [DOI] [PubMed] [Google Scholar]
- Spriestersbach A.; Kubicek J.; Schafer F.; Block H.; Maertens B. Purification of his-tagged proteins. Methods Enzymol. 2015, 559, 1–15. 10.1016/bs.mie.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Miller G. L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 1959, 31, 426–428. 10.1021/ac60147a030. [DOI] [Google Scholar]
- Lee D. G.; Jang M. K.; Lee O. H.; Kim N. Y.; Ju S. A.; Lee S. H. Over-production of a glycoside hydrolase family 50 β-agarase from Agarivorans sp. JA-1 in Bacillus subtilis and the whitening effect of its product. Biotechnol. Lett. 2008, 30, 911–918. 10.1007/s10529-008-9634-4. [DOI] [PubMed] [Google Scholar]
- Zeng C.; Zhang L.; Miao S.; Zhang Y.; Zeng S.; Zheng B. Preliminary characterization of a novel β-agarase from Thalassospira profundimonas. SpringerPlus 2016, 5, 1086 10.1186/s40064-016-2748-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koti B. A.; Shinde M.; Lalitha J. Repeated batch production of agar-oligosaccharides from agarose by an amberlite IRA-900 immobilized agarase system. Biotechnol. Bioprocess Eng. 2013, 18, 333–341. 10.1007/s12257-012-0237-5. [DOI] [Google Scholar]