

Abstract
Deuterium is one of the few stable isotopes that have the capacity to significantly alter a compound’s chemical and biological properties. The addition of a single neutron to a protium atom results in the near doubling of its mass, which gives rise to deuterium’s characteristic isotope effects. Since the incorporation of deuterium into organic substrates is known to alter enzyme/protein–substrate interactions, we tested the extent to which deuterium enrichment would modify fungal secondary metabolite production. Several fungal cultures were tested, and in all cases their secondary metabolomes were marked by changes in natural product production. Workup of one Aspergillus sp. grown under deuterium-enrichment conditions resulted in the production of several secondary metabolites not previously detected from the fungus. Bioassay testing revealed that in comparison to the inactive crude fungal extract derived from growing the fungus under non-deuterium-enriched conditions, an extract derived from the same isolate cultured in a deuterium-enriched medium inhibited methicillin-resistant Staphylococcus aureus. Using an assortment of NMR and mass spectrometry experiments, we were able to identify the bacterial inhibitor as an isotope-labeled version of pigmentosin A (6). Five additional isotopically labeled metabolites were also obtained from the fungus including brevianamide F (1), stephacidin A (2), notoamide D (3), notoamide L (4), and notoamide C (5). Given the assorted changes observed in the secondary metabolite profiles of this and other fungi grown in deuterium-enriched environments, as well as the fact that 1 and 3–6 had not been previously observed from the Aspergillus sp. isolate used in this study, we propose that deuterium enrichment might offer an effective method for further expanding a fungus’s chemical diversity potential.
Graphical Abstract
Stable isotopes occur throughout the natural world and are woven into the chemical fabric of life. In most cases, their assimilation into biomolecules affords negligible effects on chemical substances since the addition of one or more neutrons in an atom typically imparts trivial deviations in a compound’s physical and chemical properties. However, deuterium (2H or D) and tritium (3H or T) represent exceptions to this rule since the gain of one and two neutrons, respectively, results in the near doubling (2H) and tripling (3H) of protium’s (1H or H) atomic mass. The chemical consequences of replacing protium with deuterium are considerable, resulting in marked isotope effects. Specifically, the lower vibrational frequency of deuterium (lower zero-point energy of vibration) increases the bond dissociation energy of C–D bonds by ~5 kJ·mol−1 compared to their C−H counterparts,1 and these effects are even further amplified for C−T bonds.2 Thus, the shorter and stronger C–D and C–T bonds exhibit appreciable differences in their physical and chemical properties compared to C–H bonds.
Although both deuterium and tritium have the potential to significantly alter the chemical and biological properties of compounds, both isotopes occur in relatively low abundances on Earth and, therefore, have limited practical effects on life’s chemical and biochemical processes. In the case of tritium, the amount present in the biosphere fluctuates at very low levels,3 while deuterium’s abundance has remained small on a geologic time scale relative to the proportion of available protium (D/H = (149 ± 3) × 10−6).4,5 Consequently, the considerably more abundant protium atoms have had a far greater influence on the evolutionary development of life’s chemical processes compared to its isotopic counterparts.
Earth’s natural ecosystems do not afford conditions in which the relative abundance of hydrogen isotopes is vastly skewed. However, that has not dissuaded researchers from testing the consequences of how organisms might respond to synthetic environments containing elevated quantities of deuterium or tritium. Many studies have evaluated the effects of tritium-containing substances on cells and whole organisms due mainly to concerns associated with the isotope’s radiological decay.6,7 In comparison, deuterium is nonradioactive, which has enabled researchers to obtain a better understanding of the isotope’s bond-strength-related toxicity. A majority of these studies have focused on the toxicity of D2O, which generally causes death or the arrest of cell growth/function at elevated concentrations.8,9 Intriguingly, some microorganisms can be acclimated to environments containing very high and otherwise inhibitory levels of D2O.10,11 On the basis of these observations, we were curious about how a deuterium-enriched environment might impact secondary metabolism in fungi. We hypothesized that secondary metabolism would be altered as a result of both the direct (e.g., efficiency of biosynthetic enzymes to utilize deuterated substrates) and indirect (e.g., modulation of primary metabolic and intracellular cell signaling pathways) effects of deuterium’s presence. In this investigation, we tested the impact of a deuterium-enriched environment on a panel of fungi whose natural product compositions had been previously examined in our lab. Herein, we describe the effects of deuterium-enrichment on secondary metabolite production by these fungi.

RESULTS AND DISCUSSION
Impact of Deuterium Enrichment on the Fungal Secondary Metabolome.
Eight fungi that had been subject to previous chemical studies in our lab were selected for a screening experiment.12–16 Published work had indicated that acclimation is an important consideration for enabling microorganisms to grow on a deuterium-enriched medium.10,11 Accordingly, each isolate was cultured on a medium containing malt extract agar made with D2O to create deuterium-acclimated fungal strains. In all cases, fungi transferred to plates containing the D2O-based medium initially exhibited slowed growth, but recovered to near normal rates of mycelium expansion after one or more weeks of incubation. Both D2O-acclimated and nonacclimated control fungal cultures were used to seed flasks containing modified versions of Czapek broth prepared with deuterium-enriched (treatment) and non-deuterium-enriched (control) components. After 4 weeks of static incubation, fungal cultures were partitioned against EtOAc, and their secondary metabolites were recovered from the organic layers. We observed that the acclimation process had little effect on the appearances or relative amounts of mycelial biomass produced by the fungi (Figure S1), which indicated that either acclimation was unnecessary or our acclimation process was ineffective at priming fungi for the switch to the deuterium-enriched Czapek broth. Comparisons of the metabolomes by LC-ESIMS of fungi grown in deuterium-enriched versus non-deuterium-enriched environments revealed that in many cases deuterium enrichment was effective at altering their metabolite profiles (Figures S2–S9). These changes appeared to be nonpermanent since switching the fungi back from a deuterium-enriched medium to a non-deuterium-enriched medium was sufficient for restoring each fungus’s original metabolite profile. On the basis of this screening, we deduced that deuterium enrichment induces an assortment of quantitative and qualitative changes in a fungus’s natural product output.
Preparation and Structure Determination of Deuterium-Enriched Metabolites.
To explore in detail how deuterium enrichment specifically impacted the secondary metabolome of a fungus, we picked one of the fungi from our screening experiments for further investigation. The selected isolate was a soil-derived Aspergillus sp. that had been obtained from Waikiki Beach (Honolulu, HI, USA).12 This isolate had been subjected to extensive prior chemical investigation, and our new observations were highly suggestive that the fungus was now producing a qualitatively different assortment of secondary metabolites that had not been previously encountered in our lab.
Small (~125 mm3) pieces of deuterium-acclimated fungal mycelium and its attached agar were used to inoculate 1 L Erlenmeyer flasks containing 25 mL of the modified, deuterium-enriched Czapek broth. The flasks were capped with foil and incubated at room temperature for 8 weeks with a 12 h light and 12 h dark cycle. Although the modified Czapek medium was prepared using only D2O and deuterium-enriched organic and inorganic components (>99% deuterium), the autoclaving process and maintenance of the foil-sealed flasks under atmospheric conditions for 8 weeks resulted in a substantial amount of protium in the form of water entering the flasks. Since we expected limited yields of metabolites from the fungus grown in the deuterium-enriched culture broth, we utilized glucose that was fully labeled with 2H and 13C as the major carbon source; however, the medium’s deuterated acetate was not augmented by 13C labeling. Our initial experiments showed that the 13C labeling of the glucose was sufficient to provide the enhanced carbon sensitivity needed for 2D NMR experiments.
The treated (deuterium-enriched modified Czapek broth) and control (nonenriched modified Czapek broth) Aspergillus sp. cultures were pooled separately and partitioned three times against EtOAc (1:1, vol/vol). The organic residue obtained from the EtOAc layer from the treated cultures exhibited inhibition (>85%) of methicillin-resistant Staphylococcus aureus cell proliferation at 25 μg/mL, whereas the sample derived from the control culture did not. Subsequent purification performed on the EtOAc-soluble components (0.87 g) provided the putative bioactive substance and five additional secondary metabolites. Since all chromatography procedures were performed using nondeuterated solvents, the exchangeable deuterium atoms were lost during the Sephadex LH-20, preparative C18 HPLC, and semipreparative C18 HPLC purification steps.
While the use of dual-labeled glucose did provide increased capacity for NMR-based carbon detection, it also added certain challenges for the structure determination experiments. The mixed incorporation of hydrogen (1H and 2H) isotopes and the high degree of 13C labeling achieved under our experimental conditions meant that routine dereplication and structure determination efforts become challenging processes. For example, the HRESIMS data for all products exhibited Gaussian distributions (Figure 1A) that hindered the straightforward deduction of secondary metabolite molecular weights. We also noted that the presence of significant quantities of three (1H, 2H, and 13C) NMR-active nuclei in the metabolites resulted in problematic multinuclear couplings that were not easily suppressed. This resulted in extensive signal overlap (Figure 1B), as well as spurious long-range couplings that manifested in most spectral data (Figure 1C).
Figure 1.

HRESIMS and NMR data for compound 1 illustrating the effects of deuterium enrichment and 13C incorporation: (A) expansion of an HRESIMS ion cluster showing the Gaussian distribution resulting from isotope enrichment; (B) 1H NMR spectrum showing the relatively low intensity nonexchangeable 1H signals with large 1H−13C couplings; (C) 13C−13C INADEQUATE NMR spectrum revealing a combination of one-bond and multiple-bond correlations.
Compound 1 was obtained as a white, amorphous solid. HRESIMS displayed clusters of isotopic ions exhibiting Gaussian distributions in a range of m/z 324.2189 to 337.2688 under positive mode, as well as m/z 300.2215 to 313.2720 under negative mode (Figures S10–S13), which were informative for establishing a range of putative molecular weights for the metabolite. The 1H NMR spectrum of 1 (Figure S14) exhibited two amide protons (δH 7.69, 10.85) that integrated as singlets, which were many times more intense than the compound’s other proton signals. This enabled us to estimate that roughly 85–95% of the nonexchangeable hydrogens in 1 were deuterium atoms, which was further confirmed by BilevelDec 1H NMR. While 2H NMR spectra were collected (data not shown), we found that for practical reasons (e.g., 2H signal broadening and relatively weak 3JD−D couplings) the residual protium in 1 made 1H NMR and 1H-based multinuclear detection strategies more feasible for most experiments. All of the 1H signals displayed large couplings (1JCH ≈ 140–160 Hz), and the HSQC NMR (Figure S17) exhibited the expected 13C satellite signals. Upon closer inspection, the 13C NMR signals (1H-decoupled 13C NMR, Figure S16) were found to be composed of clusters of peaks in Gaussian distributions, which confirmed the metabolite’s substantial 2H and 13C isotope labeling. The 13C and HSQC NMR results (Table S1) suggested the presence of four methylene, seven methine (including five olefinic), and five quaternary (including three olefinic and two carbonyl) carbons. Taking these NMR data into consideration, we returned to the MS results and concluded that the [M + Na]+ ion at m/z 337.2688 indicated a molecular formula of 13C16H2D15N3O2Na (calcd 337.2697, Δ 2.67 ppm) for compound 1. INADEQUATE (Figure S18) correlations supported the presence of phenylalanine and proline residues. These observations enabled us to reengage in a more informed dereplication search, which, when used in combination with the UV, IR, and optical rotation values obtained for 1, led us to identify the metabolite as isotope-labeled brevianamide F.17,18
Similarly, compound 2 was obtained as a white, amorphous solid. Like 1, the HRESIMS data for compound 2 exhibited a cluster of ions with a Gaussian distribution in the range of m/z 483.3558 to 506.4615. 1H, 13C, and HSQC NMR (Figures S21, S23, S25; Table S1) confirmed that a majority of the carbons and hydrogens, except for two exchangeable amide protons (δH 8.62, 10.39), were isotopically labeled. The 13C NMR spectrum was recognized as identical to that of stephacidin A (Figure S24), which had been previously purified by our group. On the basis of INADEQUATE correlation data (Figure S26) and comparisons of the metabolite’s UV, IR, and optical rotation values with published values, compound 2 was confirmed to be an isotope-labeled version of stephacidin A.19
Compound 3 was purified as a white, amorphous solid with HRESIMS data that appeared as a cluster of ions in a Gaussian distribution over the range of m/z 507.4074 to 525.4807. 1H, 13C, and HSQC NMR results (Figures S29, S31, and S33) suggested the presence of one amide proton (δH 5.42). The 13C and HSQC NMR data (Table S1) indicated the presence of four methyl, five methylene (including one olefinic carbon), seven methine (including five olefinic carbons), and 10 quaternary (including two carbons attached to oxygen, four olefinic carbons, and two carbonyl carbons) carbons. A comparison of the 13C NMR signals for C-11–C-18, C-4–C-9, and C-25–C-29 in 3 revealed they were comparable to data obtained for compound 2 (Figure S32). This pointed toward the presence of a 2,5-diketopiperazine incorporating a proline residue and a 5,6-substituted-2,2-dimethyl-2H-chromene moiety. INADEQUATE correlations between C-22–C-21, C-21–C-20, and C-20–C-23/C-24/C-2 (Figure S34) were informative for revealing the presence of an isoprenyl group. In contrast to compound 2, quaternary carbons C-11 and C-17 were replaced by methine carbons in 3, which indicated that the bicyclo[2.2.2]diazaoctane system formed by the cyclization of the isoprenyl unit in 2 was open in 3. Further observations derived from the INADEQUATE data for 3, along with comparisons of its 1D NMR, UV, IR, and optical rotation results to published values, indicated that the metabolite was an isotopically isotope-labeled version of notoamide D.20
Compound 4, which appeared as a white, amorphous solid, was analyzed by HRESIMS, leading to the production of a cluster of ions with a Gaussian distribution ranging from m/z 489.3724 to 508.4468. The 1H, 13C, and HSQC NMR data (Figures S37, S39, and S41) suggested the presence of amide (δH 8.21, 1H) and amine (δH 7.30, 2H) protons. The 13C and HSQC NMR data (Table S1) revealed that compound 4 contained three methyl, six methylene (including an olefinic carbon), five methine (including four olefinic carbons), 11 quaternary (including one attached to an oxygen, five olefinic, and three carbonyl carbons) carbons. A comparison of the 13C NMR spectra for compounds 4 and 2 indicated the presence of bicyclo[2.2.2]diazaoctane and 5,6-substituted-2,2-dimethyl-2H-chromene moieties, as well as an additional ketone in the compound (Figure S40). INADEQUATE correlations from C-22 to C-21, C-23, and C-24 indicated the presence of an isopropenyl unit at C-21. The correlations from C-3 to C-9 and C-10 were used to assign the position of the ketone between C-9 and C-10 (Figure S42). Further observations from the INADEQUATE correlation data, as well as comparisons of its 1D NMR, UV, IR, and optical rotation values with published data, confirmed that 4 was an isotope-labeled form of notoamide L.21
Compound 5 appeared as a white, amorphous solid. The HRESIMS data exhibited a cluster of ions with a Gaussian distribution from m/z 506.4045 to 527.4128. The 1H, 13C, and HSQC NMR data (Figures S45, S47, and S49) suggested the presence of two amide protons (δH 6.20, 10.64). The 1H, HSQC, and 13C NMR data (Table S1) for compound 5 were similar to those of compound 3, except that C-2 in 3 showed a downfield shift from δC 91.3 to 180.6, while C-3 moved upfield from δC 87.4 to 55.3 (Table S1, Figure S48). Additionally, the presence of an amide proton on N-19 indicated that the ring system to which it belonged in 3 was now open. The HMBC correlations (Figure S50) from H2-22, H3-23, and H3-24 to C-20 and from H3-23 and H3-24 to C-21 provided evidence for an isoprenyl unit. HMBC correlations from H3-23 and H3-24 to C-9 supported the connection between the isoprenyl unit and C-9. Additional observations derived from HMBC correlation results, as well as comparisons of its 1D NMR, UV, IR, and optical rotation data with published values, confirmed 5 was isotope-labeled notoamide C.20
Compound 6 was obtained as a yellow, amorphous solid. HRESIMS analysis provided a cluster of ions with a Gaussian distribution in the range of m/z 572.2839 to 591.3526. On the basis of the 1H, 13C, and HSQC NMR data for 6 (Figures S53–S55), two exchangeable protons were detected along with two methyl (including a methoxy carbon), one methylene, three methine (including two olefinic carbons and one attached to an oxygen atom), and nine quaternary (including eight olefinic and a carbonyl carbon) carbons. This accounted for less than half of the compound’s presumed mass, which suggested that 6 was a dimer. 13C–13C TOCSY correlations (Figure S57) from C-2 to C-1, C-3, C-4, C-11, C-13, and C-14, as well as from C-10 to C-1, C-2, C-4, C-11, C-12, and C-13, supported the presence of two fused aromatic rings. HMBC correlations (Figure S56) from H2-8 to C-9, as well as 13C–13C TOCSY correlations from C-5 to C-4, C-8, and C9 and from C-8 to C-7, indicated the presence of a lactone-containing ring adjacent to the fused aromatic ring system. Further observation derived from HMBC correlations, as well as a comparison of its 1D NMR data with published values, confirmed that the planar structure of 6 was identical to pigmentosin A.22 Since 6 started to degrade when stored in DMSO and its yield was limited (0.5 mg), the absolute configuration of 6 was not determined.
Determination of Deuterium Content in Aspergillus sp. Metabolites.
Given that all the metabolites contained one or more exchangeable protons, we surmised that these could be used as internal references to determine the relative proportion of protium/deuterium in each compound. This approach, while simple, was not entirely satisfactory since the 13C satellites (one-bond coupling of 13C–1H) resulted in the overlap of many proton signals. Although 13C decoupling of the 1H NMR was attempted, we were not able to completely remove all couplings, especially with the added contributions from 2H couplings. To address this problem directly and effectively eliminate the 13C satellites, a bilevel adiabatic decoupling 1H NMR pulse sequence was used.23,24 Although this diminished the signal-to-noise capacity of our experiments, we were able to identify spurious peaks and eliminate them by comparing each metabolite’s 1H NMR data with its corresponding 1H–13C HSQC spectrum. Using this method, we determined that the level of deuterium incorporation was relatively even across all positions in each compound with about 90% deuterium incorporation in 1–5 (Figures S15, S22, S30, S38, S46).
Methicillin-Resistant Staphylococcus aureus Activity of Compound 6.
Compounds 1–6 were evaluated for anticancer activity against MIA PaCa-2 pancreatic cells;25 antimicrobial activities against methicillin-resistant Staphylococcus aureus (MRSA), Enterococcus faecium, Acinetobacter baumannii, and Klebsiella pneumonia; as well as antifungal activity against Candida albicans and Aspergillus fumigatus.26 Only compound 6 displayed anti-MRSA activity at 25 μg/mL (Figure S58). Unfortunately, the MIC value of 6 was not determined due to its limited yield and propensity for decomposition. The anti-MRSA activity of nonisotopically enriched pigmentosin A has not been reported; however, the anti-MRSA activity of 6 is consistent with published bioactivity reports for its analogue, lichenicolin A (MIC of 16 μg/mL).27
CONCLUSIONS
Deuterium enrichment of the fungal growth medium appears to have a range of effects on an organism’s secondary metabolic production. In this study, it was notable that we observed a compound (pigmentosin A, 6) that represented a biosynthetic product that was previously undetected by LC-ESIMS (selected-ion trace) from the fungus, although this isolate has been cultured under many different conditions by our group. In addition, deuterium enrichment unleashed the production of numerous indole alkaloid analogues (1, 3–5) previously not detected from the fungus.12 These results suggest that deuterium enrichment might offer a simple and helpful method for further expanding a fungus’s chemical diversity potential.
As a corollary to these studies, it is noteworthy that deuterium-labeled compounds have shown significant promise in the clinic due to their altered C–D bond strength properties, which changes their susceptibility to in vivo metabolic transformations. For example, Auspex Pharmaceuticals launched SD-809, a deuterated analogue of the drug tetrabenazine, in a phase III clinical trial to treat Huntington’s disease.28,29 In another case, Concert Pharmaceuticals has developed CTP-499, a deuterated analogue of 1-(5-hydroxyhexyl)-3,7-dimethylxanthine, and initiated its phase II clinical trials for treating diabetic kidney disease.28,30 Likewise, Avanir Pharmaceuticals is conducting phase II clinical studies of AVP-786, deuterated dextromethorphan, for depressive disorders.31 The growing appeal of developing pharmaceutical candidates with strategically incorporated deuterium atoms attests to the power that this approach has for the control of a drug’s susceptibility to in vivo metabolic processes. The current experiments provide a direction for exploring additional deuterium-labeling strategies that could be applied to situations requiring improvements in the metabolic stability of drugs and related clinical candidates.
EXPERIMENTAL SECTION
General Experimental Procedures.
UV data were recorded on a Hewlett-Packard 8452A diode array spectrophotometer. Optical rotation measurements were made on an AUTOPOL III automatic polarimeter. IR spectra were recorded on a Shimadzu IRAffinity-1 FT-IR spectrometer. The LC-ESIMS analyses were performed on a Shimadzu UFLC system with a quadrupole mass spectrometer using a Phenomenex Kinetex C18 column (3.0 mm × 75 mm, 2.6 μm) and MeCN−H2O (with 0.1% HCOOH) gradient solvent system. HRESIMS spectra were measured using an Agilent 6538 Ultra High Definition (UHD) Accurate-Mass Q-TOF system. NMR spectra were obtained on Varian spectrometers (500 MHz for 1H and 125 MHz for 13C at the University of Oklahoma; 600 MHz for 1H and 150 MHz for 13C at the University of Florida) and Bruker Avance III spectrometer equipped with a nitrogen-cooled cryoprobe (600 MHz for 1H and 150 MHz for 13C at Bruker Biospin Corp.) using DMSO-d6 and CDCl3 (Aldrich) as solvents. Column chromatography was conducted using Sephadex LH-20. HPLC was carried out using a Waters System equipped with a 1525 binary HPLC pump coupled to a 2998 PDA detector with a Phenomenex C18 column (21.2 × 250 mm or 10 × 250 mm, 5 μm particle size). Glucose-13C6,d7, sodium acetate-d3, and D2O were purchased from Cambridge Isotope Laboratories, Inc. All other materials were purchased from Sigma-Aldrich.
Fungal Isolates, Deuterium Acclimation, and Culture Conditions.
The Aspergillus sp. isolate was obtained from a soil sample collected from Waikiki Beach (Honolulu, HI, USA) as previously described.12 The other fungal isolates, Fusarium solani, Alternaria longissima, Emericellopsis terricola, two Tolypocladium spp., Aspergillus terreus, and Bionectria ochroleuca, were acquired from our cryopreserved fungal collection. The Aspergillus sp. isolate was grown initially on malt extract agar (MEA): 10 g/L malt extract, 1 g/L yeast extract, 15 g/L agar, 0.05 g/L chloramphenicol, and 1 L of distilled deionized water. A portion of the fungus was transferred to an MEA plate made with 25% D2O. Over the course of 3-week increments, the Aspergillus sp. isolate was subcultured onto plates containing MEA with 50%, 75%, and 100% D2O. Similarly, the other fungal isolates were sequentially acclimated on an MEA plate containing 33%, 66%, and 100% D2O. Fungi exhibiting active hyphal expansion on 100% D2O MEA plates were considered “acclimated” strains.
For the screening experiment, ~125 mm3 pieces of mycelium and agar were taken from fungal cultures grown under control (MEA with no deuterium enrichment) and deuterium acclimation (MEA prepared with D2O) conditions and placed into separate 1 L Erlenmeyer flasks containing 25 mL of non-deuterium-enriched (2.0 g of NaNO3, 1.0 g of NaH2PO4·2H2O, 0.5 g of MgSO4·7H2O, 0.5 g of KCl, 0.2 g of CaCO3, 0.01 g of FeSO4·7H2O, 0.005 g of ZnSO4, 4.0 g of glucose, and 0.25 g of sodium acetate per 1 L of H2O) or deuterium-enriched (2.0 g of NaNO3, 1.0 g of NaD2PO4·2D2O, 0.5 g of MgSO4·7D2O, 0.5 g of KCl, 0.2 g of CaCO3, 0.01 g of FeSO4·7D2O, 0.005 g of ZnSO4, 4.0 g of d-glucose (U−13C6, 1,2,3,4,5,6,6-d7), and 0.25 g of sodium acetate-d3 per 1 L of D2O) modified Czapek broth. The deuterium-enriched inorganic salts solution was made by taking nonenriched, preweighed portions of each salt and dissolving it in D2O. The excess D2O was then removed under vacuum at approximately 45 °C. Two or more replicates of each fungus/condition were incubated for 4 weeks at room temperature, at which point the cultures were subjected to partitioning with 1:1 (vol/vol) EtOAc (×3). The organic layers were recovered, the solvent was removed under vacuum, and the residue was analyzed by LC-ESIMS.
For the scale-up production of deuterated Aspergillus sp. metabolites, 45 Erlenmeyer flasks (1 L vol) containing 25 mL of the deuterium-enriched modified Czapek broth were prepared and sterilized by autoclaving. Each flask was inoculated with a 125 mm3 piece of mycelium taken from a culture that had been maintained on MEA agar made with 100% D2O. The flasks were covered with aluminum foil and incubated at room temperature for 8 weeks under a lighting schedule of 12 h light/12 h dark.
Natural Product Purification.
Secondary metabolites were obtained by partitioning the pooled fungal culture broth against EtOAc (3 ×, 1:1 vol/vol). The organic layers were retained and the solvent was removed under vacuum to provide 0.87 g of residue. The solids were applied to a column of Sephadex LH-20 and eluted with MeOH−DCM (1:1) to yield 22 fractions. The fractions were recombined into five fractions based on their LC-ESIMS profiles. The fractions were tested for anti-MRSA activity, and the two active samples were combined to give 0.19 g of material. Further purification was carried out by C18 HPLC (250 mm × 20.2 mm, 5 μm) with a MeOH−H2O gradient (from 30:70 to 100:0 over 30 min), followed by isocratic C18 HPLC (250 mm × 10 mm, 5 μm) with MeCN−H2O (22:78, 54:46, 48:52, or 50:50) to yield 1 (2.5 mg), 2 (2.0 mg), 3 (1.0 mg), 4 (1.5 mg), 5 (1.2 g), and 6 (0.5 mg).
Bilevel Adiabatic Decoupling (BilevelDec) 1H NMR and Calculation of Deuterium Content in Aspergillus sp. Metabolites.
The major parameters considered in the design of the experiment were decoupling bandwidth and decoupler position. Bearing in mind stephacidin A and its analogues represented the most complicated structures in our study, we used compound 2 as a model substrate to test the various experimental parameters. We adjusted the decoupling bandwidth from 150 to 220 ppm and the decoupler position from 75 to 110 ppm. For the experiment, compounds were analyzed by BilevelDec 1H NMR (CDCl3 and/or DMSO-d6) with a relaxation delay of 1 s, observe pulse at 3.3 μs, decoupling bandwidth at 220 ppm, decoupler position at 110 ppm, and the maximum J to be decoupled at 200 Hz. Due to the relatively small amount of protium in the samples, it was difficult to completely remove artifacts, some of which might cause incorrect recognition of real decoupled 1H signals. Therefore, we confirmed each 1H NMR datum by comparison with its corresponding 1H−13C HSQC NMR cross-peak. In both regular and BilevelDec 1H NMR spectra, the amide/hydroxy protons were assigned an integration value of 1. The integration values for the other nonexchangeable protons were determined (Hn), and the percent deuterium was calculated as (1 − Hn) × 100%.
Antibacterial Activity.
The antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) was determined as described.32 Chloramphenicol was used as a positive control. The MRSA strain (ATCC 700787) was maintained on tryptic soy blood agar plates (30 g/L Bacto Tryptic Soy Broth (TSB), 15 g/L Amresco high-purity agar, 5% defibrinated sheep’s blood). An overnight stock culture of S. aureus was diluted in brain-heart infusion (BHI) broth (37 g/L Bacto BHI), 100 μL of which was placed in each well of a 96-well plate. Aliquots (1 μL) of each compound stock solution (in DMSO) were added to the wells of a microtiter plate. An OD600 value was obtained for each well immediately after it was prepared, and the plate was placed into a 37 °C incubator for 16–18 h. The resulting OD600 values for all test and control wells were recorded again, and the data analyzed.
Brevianamide F (U-13C16, 2,5,6,7,8,10,10,11,14,14,15,15,16,16,17-d15, 1):
white, amorphous solid; UV (MeOH) λmax (log ε) 222 (4.36), 280 (3.71) nm; ; IR (film) νmax 3260, 1620, 1420 cm−1; 1H and 13C NMR (see Table S1).
Stephacidin A (U-13C26, 4,5,10,10,14,14,15,15,16,16,20,20,21,−23,23,23,24,24,24,25,26,28, 28,28,29,29,29-d27, 2):
white, amorphous solid; UV (MeOH−CHCl3) λmax (log ε) 208 (4.39), 242 (4.31), 308 (3.83), 328 (3.65, sh) nm; ; IR (film) νmax 3338, 1700, 1666, 1622, 1396 cm−1; 1H and 13C NMR (see Table S1).
Notoamide D (U-13C26, 4,5,10,10,11,14,14,15,15,16,16,17,21,22,−22,23,23,23,24,24,24,25,26,28,28,28,29,29,29-d29, 3):
white, amorphous solid; UV (MeOH) λmax (log ε) 208 (4.28), 236 (4.28), 286 (3.88), 330 (3.74) nm; ; IR (film) νmax 3356, 1710, 1670, 1622, 1362 cm−1; 1H and 13C NMR (see Table S1).
Notoamide L (U-13C25, 4,5,10,10,14,14,15,15,16,16,20,20,21,−23,23,24,24,24,25,26,28,28, 28,29,29,29-d26, 4):
white, amorphous solid; UV (MeOH) λmax (log ε) 206 (4.20), 236 (4.15), 270 (4.13), 368 (3.78) nm; ; IR (film) νmax 3435, 3315, 1700, 1650, 1575, 1380, 1220 cm−1; 1H and 13C NMR (see Table S1).
Notoamide C (U-13C26, 4,5,10,10,11,14,14,15,15,16,16,17,21,22,−22,23,23,23,24,24,24,25, 26,28,28,28,29,29,29-d29, 5):
white, amorphous solid; UV (MeOH) λmax (log ε) 208 (4.27), 248 (4.43), 266 (4.12, sh), 304 (3.70, sh) nm; ; IR (film) νmax 3200, 1700, 1650, 1420, 1290 cm−1; 1H and 13C NMR (see Table S1).
Pigmentosin A (U-13C30, 7,7′,8,8,8′,8′,10,10′,14,14′,15,15,−15,15′,15′,15′,16,16,16,16′,16′,16′-d22, 6):
yellow solid; 1H and 13C NMR (see Table S1).
Supplementary Material
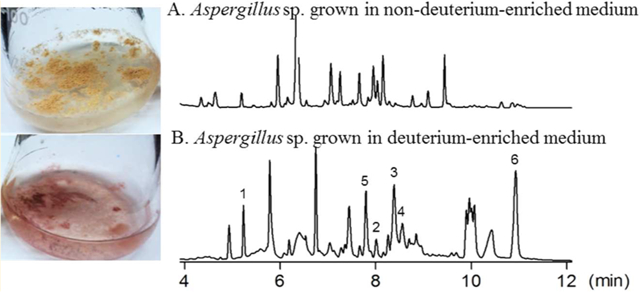
Figure 2.

Secondary metabolome profiles (PDA detection at 190–400 nm) of the Aspergillus sp. grown in non-deuterium-enriched (A) and deuterium-enriched (B) media showing the substantial differences in metabolite composition.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institutes of Health (R01AI085161). The 13C-optimized NMR probe at the University of Florida was supported by the NIH (R01EB009772) and the National High Magnetic Field Laboratory (NSF cooperative agreement DMR-1157490 and the State of Florida). The LC-ESIMS instrument used for this project was provided in part by a Challenge Grant from the Office of the Vice President for Research, University of Oklahoma, Norman Campus, and an award through the Shimadzu Equipment Grant Program.
Footnotes
Supporting Information
Secondary metabolome profiles for 16 fungal strains grown in deuterium-enriched and nonenriched media, 1D, 2D NMR spectra and HRESIMS data for compounds 1–6, as well as primary anti-MRSA activity of isolated compounds. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.5b00337.
The authors declare no competing financial interest.
REFERENCES
- (1).Jones RAY Physical and Mechanistic Organic Chemistry; Cambridge University Press: New York, 1979. [Google Scholar]
- (2).Yakushin FS Russ. Chem. Rev 1962, 31, 123. [Google Scholar]
- (3).Boyer C; Vichot L; Fromm M; Losset Y; Tatin-Froux F; Guetat P; Badot PM Environ. Exp. Bot 2009, 67, 34. [Google Scholar]
- (4).Lécuyer C; Gillet P; Robert F Chem. Geol 1998, 145, 249. [Google Scholar]
- (5).Robert F In Meteorites and the Early Solar System II; Lauretta DS, Mcsween HY, Eds.; University of Arizona Press: Tuscon, 2006. [Google Scholar]
- (6).Hofer KG; Hughes WL Radiat. Res 1971, 47, 94. [PubMed] [Google Scholar]
- (7).Geselowitz DA; McManaway ME; Hofer KG; Neumann RD Radiat. Res 1995, 142, 321. [PubMed] [Google Scholar]
- (8).Thomson JF Biological Effects of Deuterium; Pergamon Press, Macmillan: New York, 1963. [Google Scholar]
- (9).Adams WH; Adams DG J. Pharmacol. Exp. Ther 1988, 244, 633. [PubMed] [Google Scholar]
- (10).Barber J WO9707216A1, 1997.
- (11).Paliy O; Bloor D; Brockwell D; Gilbert P; Barber JJ Appl. Microbiol 2003, 94, 580. [DOI] [PubMed] [Google Scholar]
- (12).Wang X; You J; King JB; Powell DR; Cichewicz RH J. Nat. Prod 2012, 75, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cai S; Du L; Gerea AL; King JB; You J; Cichewicz RH Org. Lett 2013, 15, 4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang B; You J; King JB; Cai S; Park E; Powell DR; Cichewicz RH J. Nat. Prod 2014, 77, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Du L; Robles AJ; King JB; Powell DR; Miller AN; Mooberry SL; Cichewicz RH Angew. Chem., Int. Ed 2014, 53, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cai S; King JB; Du L; Powell DR; Cichewicz RH J. Nat. Prod 2014, 77, 2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Birch AJ; Russell RA Tetrahedron 1972, 28, 2999. [Google Scholar]
- (18).Grundmann A; Li SM Microbiology 2005, 151, 2199. [DOI] [PubMed] [Google Scholar]
- (19).Qian-Cutrone J; Huang S; Shu Y-Z; Vyas D; Fairchild C; Menendez A; Krampitz K; Dalterio R; Klohr SE; Gao QJ Am. Chem. Soc 2002, 124, 14556. [DOI] [PubMed] [Google Scholar]
- (20).Kato H; Yoshida T; Tokue T; Nojiri Y; Hirota H; Ohta T; Williams RM; Tsukamoto S Angew. Chem., Int. Ed 2007, 46, 2254. [DOI] [PubMed] [Google Scholar]
- (21).Tsukamoto S; Kawabata T; Kato H; Greshock TJ; Hirota H; Ohta T; Williams RM Org. Lett 2009, 11, 1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Elix JA; Hardlaw JH Aust. J. Chem 2004, 57, 681. [Google Scholar]
- (23).Kupce E; Freeman R; Wider G; Wuthrich KJ Magn. Reson. A 1996, 122, 81. [Google Scholar]
- (24).Agilent Technologies. Accessed Feb 17, 2014; available from http://www.chem.agilent.com/Library/applications/5990-9896EN.pdf.
- (25).Du L; Risinger AL; King JB; Powell DR; Cichewicz RH J. Nat. Prod 2014, 77, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Henrikson JC; Ellis TK; King JB; Cichewicz RH J. Nat. Prod 2011, 74, 1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).He H; Bigelis R; Yang HY; Chang L-P; Singh MP J. Antibiot 2005, 58, 731. [DOI] [PubMed] [Google Scholar]
- (28).Katsnelson A Nat. Med 2013, 19, 656. [DOI] [PubMed] [Google Scholar]
- (29).Auspex Pharmaceuticals. Accessed Aug 28, 2014 2014; available from http://www.auspexpharma.com/pipeline/.
- (30).Concert Pharmaceuticals. Accessed Sept 10, 2014; available from http://www.concertpharma.com/CTP499WindhoverTop10Project.htm.
- (31).Avanir Pharmaceuticals. Accessed Sept 10, 2014; available from http://ir.avanir.com/phoenix.zhtml?c=61699&p=irolnewsArticle&ID=1960980.
- (32).Cai S; King JB; Du L; Powell DR; Cichewicz RH J. Nat. Prod 2014, 77, 2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.