

Abstract
Objective:
Obesity is associated with myocardial fibrosis and impaired diastolic relaxation, abnormalities that are especially prevalent in women. Normal coronary vascular endothelial function is integral in mediating diastolic relaxation, and recent work suggests increased activation of the endothelial cell (EC) mineralocorticoid receptor (ECMR) is associated with impaired diastolic relaxation. As the endothelial Na+ channel (EnNaC) is a downstream target of the ECMR, we sought to determine whether EC-specific deletion of the critical alpha subunit, αEnNaC, would prevent diet induced-impairment of diastolic relaxation in female mice.
Methods and materials:
Female αEnNaC KO mice and littermate controls were fed a Western diet (WD) high in fat (46%), fructose corn syrup (17.5%) and sucrose (17.5%) for 12–16 weeks. Measurements were conducted for in vivo cardiac function, in vitro cardiomyocyte stiffness and EnNaC activity in primary cultured ECs. Additional biochemical studies examined indicators of oxidative stress, including aspects of antioxidant Nrf2 signaling, in cardiac tissue.
Results:
Deletion of αEnNaC in female mice fed a WD significantly attenuated WD mediated impairment in diastolic relaxation. Improved cardiac relaxation was accompanied by decreased EnNaC-mediated Na+ currents in ECs and reduced myocardial oxidative stress. Further, deletion of αEnNaC prevented WD-mediated increases in isolated cardiomyocyte stiffness.
Conclusion:
Collectively, these findings support the notion that WD feeding in female mice promotes activation of EnNaC in the vasculature leading to increased cardiomyocyte stiffness and diastolic dysfunction.
Keywords: Obesity, Myocardial stiffness, Oxidative stress, Endothelial sodium channel
1. Introduction
The clinical appearance of heart failure is increasing dramatically, in part, due to advancing age of the population and the prevalence of obesity [1]. Of particular importance is the syndrome of heart failure with preserved ejection fraction (HFpEF). In this clinical entity, diastolic dysfunction is an early manifestation and is characterized by the inability of the heart to fill normally with blood because of apparent stiffening of the left ventricular wall [1–5]. Of further note, women, particularly those exhibiting obesity and type II diabetes, are especially predisposed to the development of HFpEF in conjunction with abnormally increased cardiovascular stiffness [6,7].
Previous investigations indicated female mice fed a Western diet (WD) high in saturated fat and refined carbohydrates exhibited accelerated onset of diastolic dysfunction compared with male mice [8]. Notably, female mice also displayed elevated plasma aldosterone levels compared to males [8]. These observations support a distinct sex-related difference in the development of diastolic dysfunction in response to diet-induced obesity [8]. It is generally acknowledged that increased mineralocorticoid receptor (MR) activation plays a key role in the development of cardiovascular disease (CVD), including HFpEF [9–14]. Indeed, large randomized controlled trials have shown that treatment with MR antagonists, such as spironolactone, decreases mortality and morbidity in heart failure patients [6,14–18].
Emerging data support a role for endothelial cell specific MR (ECMR) activation contributing to vascular and cardiac tissue fibrosis ultimately leading to impairments in diastolic relaxation [17–19]. Recent findings also support the paradigm that enhanced ECMR activation associated with diet-induced obesity plays a key role in activation of pro-fibrotic and pro-inflammatory signaling leading to impaired diastolic relaxation in females [19]. It is further appreciated that coronary microvascular endothelial dysfunction promotes myocardial dysfunction and remodeling in the development of diastolic dysfunction [16–19]. However, the exact mechanisms that drive coronary microvascular dysfunction to precipitate diastolic dysfunction are unclear. In this regard, it is known that the endothelium epithelial sodium (Na+) channel (EnNaC) is a signaling target of the ECMR, and that ECMR activation increases expression and function of EnNaC in the heart and vasculature [20–23]. Given the inexorable relationship between obesity and diastolic dysfunction, we sought to determine if cell-specific deletion of the requisite α subunit of EnNaC would mitigate the development of ventricular stiffening and impaired diastolic relaxation associated with diet-induced obesity in females.
2. Methods
2.1. Animals
The EC-specific α subunit EnNaC KO model was generated using fluxed αEnNaC mice (EnNaCf/f) driven by Tie 2 Cre recombinase [23]. Specificity was confirmed by selective downregulation of expression of EnNaC in ECs while levels in macrophages were unaffected [23]. Female αEnNaC−/− and littermate control (αEnNaC+/+) mice (12 weeks of age) were fed a standard mouse chow (CD) or a WD for a further 12 weeks. Mice were allowed ad libitum access to water and food. The WD diet consisted of high fat (46%) and high carbohydrate (41.8%) with sucrose (17.5%) and high-fructose corn syrup (17.5%) (Test Diet modified 58Y1), as previously published [24]. Mice were randomized into 4 groups; group 1: CD (wild-type), group 2: CD-KO, group 3:WD and group 4: WD-KO. All mice were housed under standard temperature (22 °C) and humidity conditions with a 12-h light and dark cycle. Following the period of dietary intervention, conscious mice underwent body composition analysis to determine whole body fat mass and lean mass utilizing an EchoMRI-500 (Echo Medical Systems, Houston, TX, USA) as previously described [23]. Mice were then weighed and euthanized under isoflurane anesthesia. Visceral fat was harvested and weighed to characterize visceral adiposity. A separate group of αEnNaC−/− and αEnNaC+/+ female mice were fed the WD for 16 weeks, after which cardiomyocytes were isolated for determining intrinsic cellular stiffness using atomic force microscopy (AFM). This duration of feeding was chosen to corroborate and extend our earlier findings of increased cardiomyocyte stiffness following 16 weeks of feeding of a WD [19].
2.2. Ultrasound assessment of cardiac function
Doppler ultrasound studies were performed in isoflurane-anesthetized mice utilizing a Vevo 2100 (Visualsonics) instrument in conjunction with a MS550 (22–55 MHz) ultrasound probe [12,19]. Two-dimensional echocardiograms were performed in the apical four chamber view. A region of interest/small sample volume was positioned within the mitral inflow stream just proximal to the mitral leaflets to acquire early (E) and late (A) diastolic blood flow velocities in pulse wave (PW) Doppler mode. From the PW spectra, we determined isovolumic relaxation time (IVRT), isovolumic contraction time (IVCT) and ejection time (ET), parameters needed to calculate the myocardial performance index (MPI), also known as the Tei index. MPI was calculated as the sum of isovolumic contraction and relaxation times divided by ejection time. Next, Tissue Doppler Imaging (TDI) was performed in the apical four chamber view by placing a sample volume at the septal annulus to acquire early (e′) and late (a′) septal annular velocities. B-and M-mode images of the left ventricle (LV) and septum in the short axis view were acquired at the level of the papillary muscles. Left ventricular anterior and posterior wall thicknesses at end systole (LVAWTs and LVPWTs) and diastole (LVAWTd and LVPWTd), luminal diameters (LVIDs and LVIDd) and ejection fraction (EF) were determined offline in M-mode. A modified parasternal long axis view of the left ventricle and aortic root was used to determine stroke volume (SV) and cardiac output (CO). A sample volume was placed in the aortic outflow tract to obtain blood velocity spectra in Doppler PW mode. Offline calculation of the velocity time integral of PW traces and measurement of the maximum diameter of the ascending aorta acquired in B-mode were used to calculate SV and CO.
2.3. EC isolation and patch clamp
As a surrogate for coronary ECs pulmonary ECs were isolated by a two-step magnetic bead protocol using a commercially available kit adapted for use in mice (Miltenyi Biotec Inc., Auburn, CA, USA) [19,22]. Lungs were initially digested with collagenase IV for 90 min. In the first step, contamination with immune cells was eliminated by passage across anti-CD45 antibody bound microbeads. The eluent was then subjected to a second purification step using anti-CD31 antibody-conjugated microbeads [19]. ECs were cultured for 5–8 days at 37 °C, 5% CO2. The purity of isolated ECs was confirmed by CD31 staining. Whole cell Na+ currents were recorded by patch clamp using an EPC-10 amplifier (Heka, Hamden, CT, USA) and Patchmaster software and analyzed using Igor Pro software [22]. Na+ current was recorded from 7 to 9 independent cells per animal/cell isolation. Currents were recorded at membrane potentials between −80 and +80 mV at 40 mV intervals. EnNaC currents were confirmed by inhibition with the Na+ channel inhibitor, amiloride (1 μM).
2.4. Histochemistry and staining
A portion of the LV myocardium was fixed in 3% paraformaldehyde, dehydrated in ethanol and paraffin embedded. 5 μm sections were stained with picrosirus red to assess interstitial fibrosis as previously described [12,19].
2.5. Cardiomyocyte isolation and atomic force microscopy (AFM)
Cardiomyocytes were isolated and AFM performed as previously described [19]. Briefly, hearts were removed from anesthetized mice and the aorta was cannulated for retrograde coronary perfusion of the myocardium. A nominally calcium-free physiological saline solution (PSS) was perfused through the heart for 10 min followed by perfusion of a MEM-based collagenase solution for ~10 min. Solutions were oxygenated with 95% O2/5% CO2 at 37 °C. After coronary perfusion, the left ventricle and septum were isolated, and the tissue was minced, agitated, and filtered at 22–25 °C. Cardiomyocytes were applied to laminin-coated dishes and allowed to adhere for 40 min. Cardiomyocytes were then washed (×3) in nominally calcium-free PSS at 22–25 °C. An AFM nanoindentation protocol [19] was used to estimate the elastic modulus (stiffness) of individual cardiomyocytes.
2.6. Immunoblots
Frozen sections of LV myocardium were thawed and homogenized in lysis buffer containing protease and phosphatase inhibitors. Lysates were then sonicated and centrifuged at 13,000 g for 10 min [8,12]. Protein content of the lysates was determined using the BCA (Pierce) protein assay. 20–30 μg of protein lysate was then electrophoretically separated on a SDS-polyacrylamide gradient gel (4–15%) and transferred to nitrocellulose membranes. After blocking in 5% BSA or 5% nonfat milk in PBS for 1 h, the membranes were incubated with primary antibodies overnight. Primary antibodies used were Nuclear factor erythroid-2-related factor 2 (Nrf2,1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), BTB and CNC homolog 1 (BACH1, 1:250, Santa Cruz Biotechnology, Santa Cruz, CA, USA), HO-1, (1:500, StressGen Biotechnologies, San Diego, CA, USA), NAD(P)H quinone dehydrogenase 1 (NQO1, 1:3000, GeneTex, Irvine, CA, USA), thioredoxin 1 (Trx1, 1:2000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), glutathione reductase 1 (GSR1, 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), 3-nitrotyosine, (3-NT, 1;1000, Millipore, Danvers, MA, USA) or β-actin (1:12000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, horseradish per-oxidase conjugated secondary antibodies were incubated with membranes followed by developing of blots by chemoluminescence [12,19]. Protein bands were quantified by densitometry and normalized to β-actin.
2.7. Real-time qt PCR
Left ventricular cardiac tissue was homogenized in Qiazol (Qiagen, MD, USA) and total RNA was isolated using a Qiagen microRNeasy kit (Qiagen MD, USA). Purity and concentration were assessed using a Nanodrop spectrophotometer [12,19]. First-strand cDNA was synthesized from total RNA using the Improm-II reverse transcription kit (Promega, Madison, WI, USA) and quantitative real-time PCR was performed using a real-Time PCR Detection System (Biorad, Hercules, CA, USA). Primer sequences used were: GAPDH, Forward: 5′-GGAGAAACCTGCCAAGATGA-3′, Reverse: 5′-TCCTCAGTGTACCAGA-3′. Alpha smooth muscle actin (αSMA) Forward 5-′TGTGCTGGACTGTGGAGATG-3′ Reverse: 5′-GAAGGAATAGCCACCTGAG-3′ (IDT, Coralville, IA, USA). The specificity of the primers was analyzed by performing a melting curve. PCR reactions were performed using iTaq UniverSYBR Green SMX (Biorad, Hercules, CA, USA). PCR cycling conditions were 5 min at 95 °C for initial denaturation, 40 cycles of 30 s at 95 °C, 30 s at 58 °C and 30 s at 72 °C. Each real-time PCR was carried out using three individual samples in triplicate, and the threshold cycle values were averaged. Calculations of relative normalized gene expression were performed using Bio-Rad CFX manager software. Results were normalized against the housekeeping gene GAPDH.
2.8. Statistical analysis
Results are reported as the mean ± SEM. Differences in outcomes were determined using a one- or two-way ANOVA and unpaired t-tests, and were considered significant when p < .05. All statistical analyses were performed using Sigma Plot software (Systat Software, version 12). Sample sizes for power analyses were based on our previously published data [8,12,19,21,23].
3. Results
3.1. Effect of αEnNaC subunit deletion on WD-induced weight gain, adiposity, hyperglycemia and blood pressure
We previously reported increases in visceral fat mass and body weight in mice consuming a WD for 16 weeks [12,19,20]. In the current study, after 12 weeks of feeding, we observed increased visceral adiposity in mice fed the WD compared to those receiving a CD (Table 1); however, the WD did not result in a significant increase in overall body weight. αEnNaC deletion did not affect either body weight (Table 1) or visceral adiposity (Table 1) in the WD fed mice. Fasting plasma glucose concentration was not increased in WD fed compared to CD fed mice and αEnNaC deletion had no impact on blood glucose levels (Table.1). No changes in systolic or diastolic BP were observed in the WD or WD-KO groups (Table 1) in line with previous observations that αEnNaC deletion does not alter BP [23].
Table 1.
Effects of EnNaC α deletion on physiological and biochemical parameters.
| Parameter | CD | CDKO | WD | WDKO |
|---|---|---|---|---|
| Body weigh, G | 20.13 ± 0.55 (6) | 20.39 ± 0.89 (6) | 21.62 ± 0.70 (10) | 22.24 ± 0.0.58 (9) |
| Heart weight to body weight, mg/G | 5.38 ± 0.09 (7) | 5.52 ± 0.26 (6) | 5.00 ± 0.11 (10) * | 5.20 ± 0.12 (6) |
| Visceral fat, mg | 338.8 ± 39.0 (6) | 374.3 ± 35.2(6) | 790.9 ± 65.9 * (10) | 707.4 ± 102.0 (6) |
| Fasting blood glucose | 149.0 ± 3.7 (5) | 150.5 ± 15.05 (6) | 169.1 ± 15.3 (8) | 165.0 ± 19.7.1 (5) |
| Systolic BP, mmHg | 97.62 ± 2.3 (4) | 95.68 ± 3.0 (4) | 104.4 ± 4.5 (6) | 103.0 ± 2.3 (5) |
| Diastolic BP, mmHg | 55.4 ± 2.5 (4) | 56.8 ± 0.10 (4) | 65.8 ± 5.3 (6) | 61.4 ± 3.1 (5) |
Summary of values are mean ± SE. Numbers in parentheses are sample sizes. Control diet (CD), CD α EnNaC KO (CDKO), Western diet (WD), WD α EnNaC KO (WDKO)
p < .05 CD versus WD;
p < .05 WD versus WDKO.
3.2. αEnNaC deletion prevents WD-induced cardiac diastolic stiffening
Compared to the CD fed group, WD-fed mice exhibited abnormalities in multiple diastolic parameters (Table 2) including decreases in the tissue Doppler e′/a′ ratio as well as increases in LV filling pressure (E/e′) and diastolic stiffness (E/e′/LVIDd) (Fig. 1A–C). αEnNaC subunit deletion resulted in statistically significant improvement in all of these parameters (Fig. 1 and Table 2). Notably, there were no changes in indices of systolic function including ejection fraction and fractional shortening (Table 2).
Table 2.
Parameters of cardiac function by echocardiography.
| Parameter | CD (5, 6) | CDKO (3, 4) | WD (10) | WDKO (9) |
|---|---|---|---|---|
| Heart rate bpm | 468 ± 27 | 431 ± 32 | 506 ± 16 | 448 ± 26 |
| EF (%) | 69 ± 3 | 71 ± 5 | 70 ± 3 | 69 ± 3 |
| FS (%) | 39 ± 3 | 40 ± 4 | 39 ± 2 | 39 ± 2 |
| LVAWd, mm | 0.82 ± 0.07 | 0.81 ± 0.04 | 0.89 ± 0.04 | 0.83 ± 0.04 |
| LVPWd, mm | 0.71 ± 0.04 | 0.73 ± 0.10 | 0.78 ± 0.03 | 0.72 ± 0.04 |
| LVIDd, mm | 3.85 ± 0.15 | 3.82 ± 0.22 | 3.53 ± 0.09 | 3.55 ± 0.10 |
| LVIDs, mm | 2.34 ± 0.18 | 2.29 ± 0.30 | 2.13 ± 0.12 | 2.20 ± 0.13 |
| LV Mass, mg | 84 ± 10 | 82 ± 7 | 78 ± 3 | 75 ± 6 |
| RWT | 0.40 ± 0.03 | 0.41 ± 0.05 | 0.48 ± 0.03 | 0.44 ± 0.02 |
| CO, ml/min | 31 ± 2 | 27 ± 3 | 28 ± 2 | 25 ± 3 |
| SV, μl | 65 ± 5 | 63 ± 4 | 56 ± 4 | 55 ± 6 |
| E, mm/s | 693 ± 41 | 585 ± 23 | 579 ± 24 * | 623 ± 39 |
| A, mm/s | 446 ± 34 | 426 ± 27‡ | 476 ± 25 | 418 ± 28 |
| E/A | 1.54 ± 0.09 | 1.38 ± 0.06 | 1.25 ± 0.07 * | 1.38 ± 0.04 |
| IVRT, ms | 14.4 ± 0.7 | 16.5 ± 1.7 | 15.0 ± 0.6 | 16.0 ± 1.4 |
| IVCT, ms | 6.7 ± 0.6 | 8.5 ± 1.4 | 8.5 ± 0.9 | 9.1 ± 1.1 |
| ET, ms | 45.6 ± 2.6 | 46.2 ± 2.0 | 39.4 ± 1.6 * | 47.2 ± 2.0† |
| ST, ms | 66.7 ± 3.3 | 71.1 ± 5.0 | 63.0 ± 2.9 | 72.4 ± 4.3 |
| MPI | 0.46 ± 0.02 | 0.53 ± 0.04 | 0.60 ± 0.02 * | 0.52 ± 0.03 |
| e′, mm/s | 29.9 ± 2.1 | 27.7 ± 2.8 | 17.9 ± 1.3 * | 23.9 ± 2.3† |
| a′, mm/s | 20.0 ± 1.6 | 21.2 ± 2.1 | 21.1 ± 1.2 | 21.3 ± 2.1 |
| e′/a′ | 1.43 ± 0.05 | 1.31 ± 0.06 | 0.87 ± 0.07 * | 1.08 ± 0.08† |
| E/e′ | 23.5 ± 1.4 | 21.3 ± 1.0 | 33.7 ± 2.0 * | 27.3 ± 2.2† |
| Diastolic stiffness (E/e′)/LVIDd | 6.13 ± 0.36 | 5.52 ± 0.50 | 9.59 ± 0.66 * | 7.67 ± 0.56† |
Summary of values are mean ± SE. Numbers in parentheses are sample sizes. Control diet (CD), CD EnNaC KO (CDKO), Western diet (WD), WD EnNaC KO (WDKO)
p < .05 CD versus WD;
p < .05 WD versus WDKO;
p < .05 CD versus CDKO.
EF, ejection fraction, FS, fractional shortening, LVAWTd, LV anterior wall thickness-diastole; LVPWTd, LV posterior wall thickness-diastole, LVIDd, LV inner dimension-diastole; LVIDs, LV inner dimension-systole; RWT, relative wall thickness; CO cardiac output; SV, stroke volume; E, velocity of early mitral inflow; A, velocity of late mitral inflow; IVRT, isovolumic relaxation time; IVCT, isovolumic contraction time; ET, ejection time; ST, systolic time; e′, peak diastolic velocity of septal annulus; a′, late diastolic velocity of septal annulus s′, peak systolic velocity of septal annulus; E/e′ index of LV filling pressure; (E/e′)/LVIDd, diastolic stiffness.
Fig. 1.
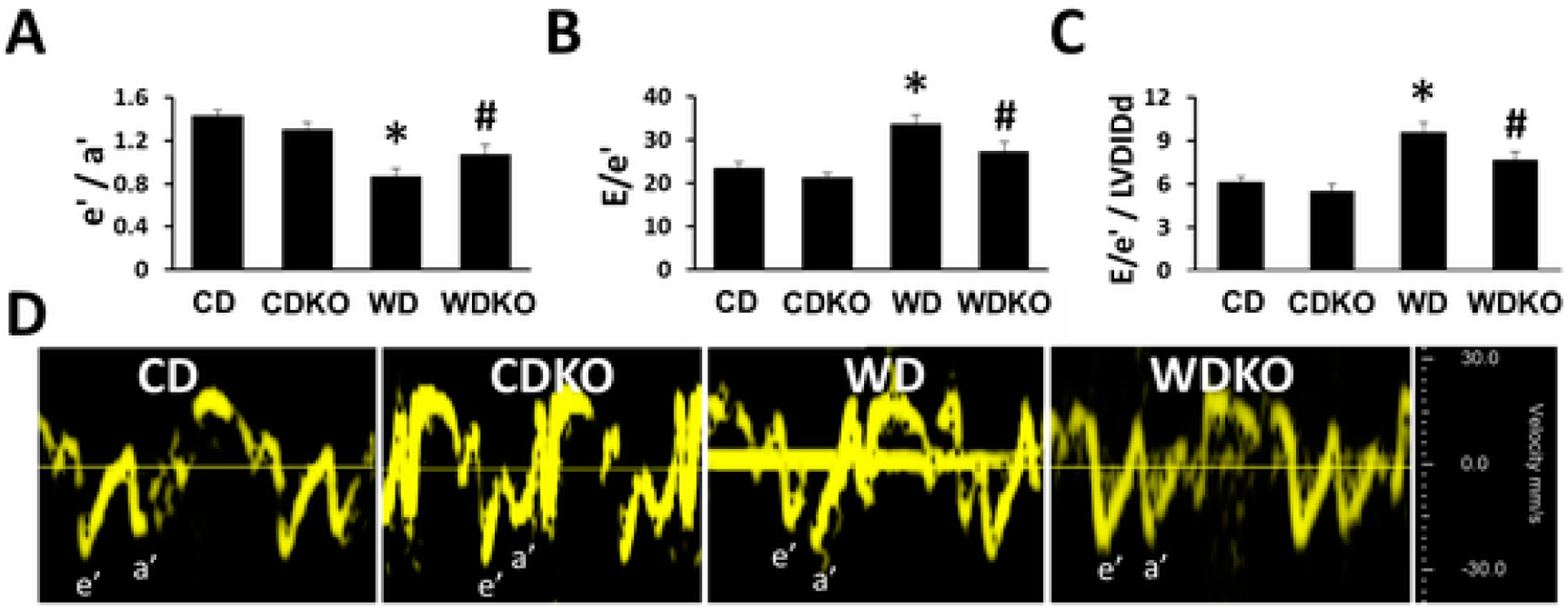
α EnNaC deletion prevents Western diet impairments in diastolic relaxation. A) Tissue Doppler derived e′/a′ ratio and B) increases in E/e′ ratio (LV filling pressure) and C) diastolic stiffness (E/e′/LVIDd). D shows representative tissue Doppler spectra. CD = control diet (n = 5–6); CDKO=CD knock out (n = 3–4) WD = Western diet (n = 10); and WDKO = WD eNaC knock out (n = 9). Data are expressed as means ± SEM. *p < .05 vs CD; #p < .05 vs WD.
3.3. αEnNaC deletion prevents WD-induced oxidant stress
3-Nitrotyrosine (3-NT) staining, a marker of oxidant stress, was significantly increased in cardiac tissue of mice consuming the WD (Fig. 2A and B). αEnNaC subunit deletion significantly reduced 3-NT accumulation in the cardiac tissue (Fig. 2A and B).
Fig. 2.
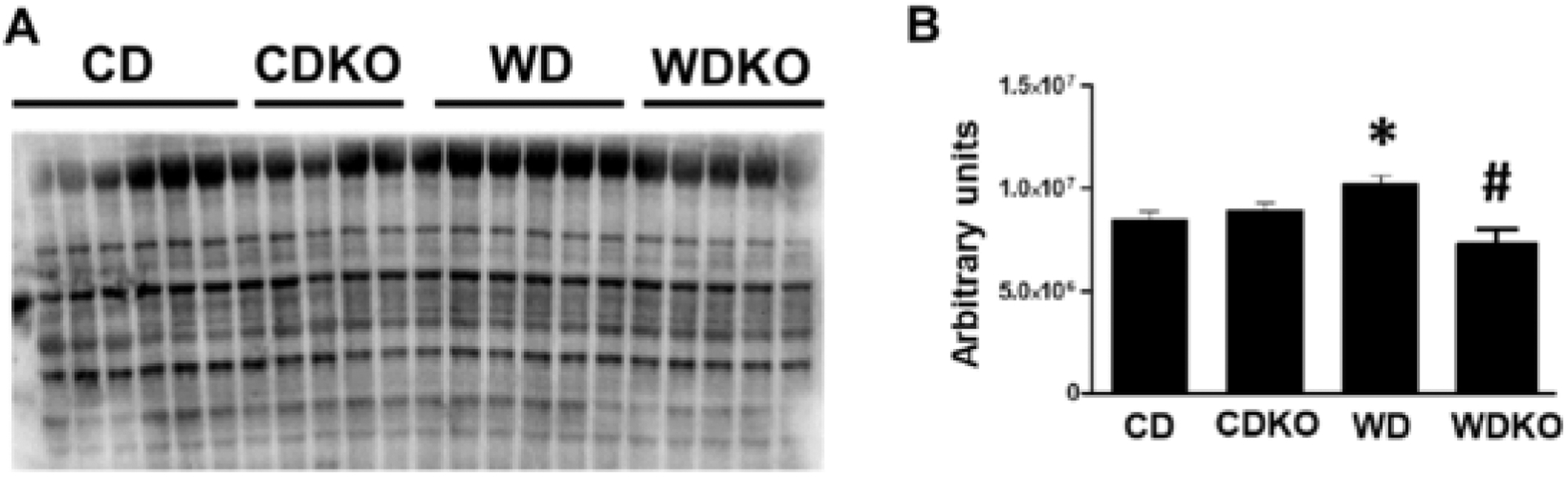
α EnNaC deletion prevents WD induced oxidant stress. Quantification of 3-nitrotyrosine by Western immunoblot. WD induced accumulation of 3–NT was prevented in WD plus KO group. CD = control diet (n = 7); CDKO = CD knock out (n = 5) WD = Western diet (n = 5); and WDKO = WD EnNaC knock out (n = 5). Data are expressed as means ± SEM. *p < .5 vs CD; #p < .05 vs WD.
3.4. Effect of αEnNaC subunit deletion on indices of WD-induced cardiac fibrosis and hypertrophy
The effect of αEnNaC deletion on myocardial fibrosis was evaluated by picrosirus red (PSR) staining of LV tissue. We have previously shown an increase in myocardial fibrosis after 16 weeks of WD feeding compared to a CD [12,19]. In the current study, WD feeding of wild-type mice for the shorter period of 12 weeks, resulted in a comparatively moderate increase in cardiac fibrosis as shown by intensity of PSR staining (WD vs CD, P = .05, Student t-test; Fig. 3A). αEnNaC−/− mice fed the WD showed similar staining levels as seen for WD (Fig. 3A). While increased heart weight was evident in mice fed the WD for 12 weeks (WD vs CD, p = .01, Student t-test; Fig. 3B), along with a significant decrease in heart weight/body weight ratio (WD vs CD, p = .03, Student t-test; Table 1), αEnNaC− deletion had no significant impact on the WD induced change in heart weight (Fig. 3B). Expression of mRNA for αSMA, similarly showed a trend towards being increased by diet (CD vs WD, p = .06, Student t-test; Fig. 3C) and decreased in the case of αEnNaC deletion (WD vs WDKO, p = .05, Student t-test; Fig. 3C). Collectively, these data suggest that at this early time-point, WD is associated with only small changes in myocardial fibrosis and hypertrophy, despite the presence of significant alterations in diastolic dysfunction and oxidative stress.
Fig. 3.
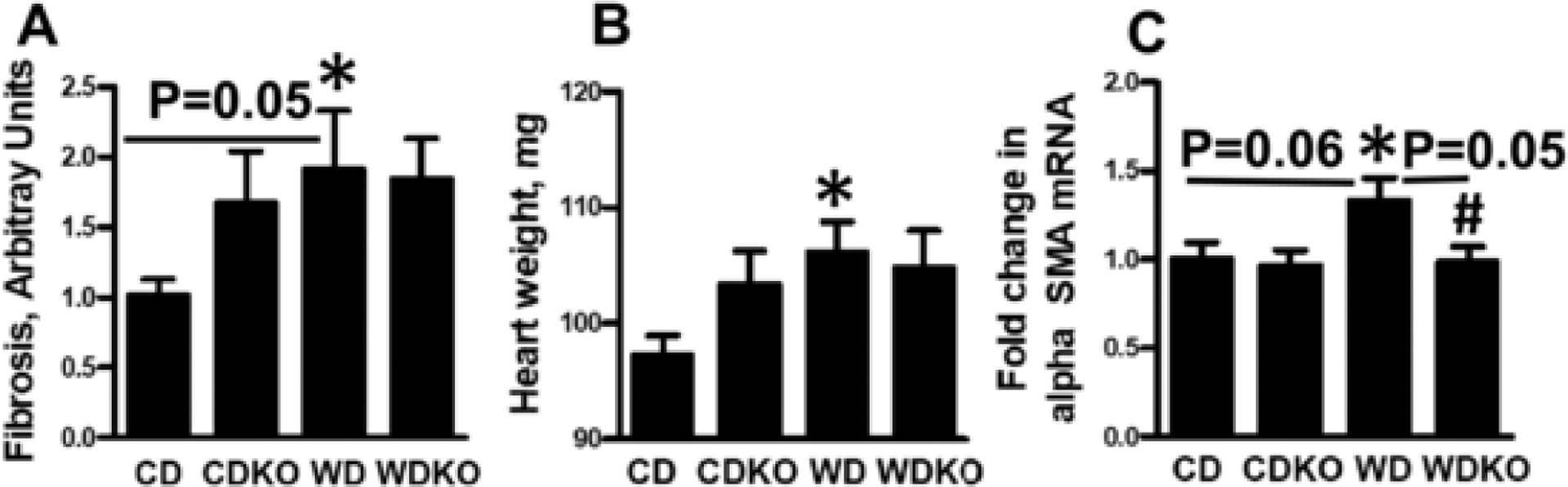
Effect of αEnNaC deletion on WD cardiac fibrosis and hypertrophy. A) Quantification of picrosirus red staining for myocardial fibrosis. B) Heart weight in mg. C) mRNA expression of cardiac hypertrophy marker alpha smooth muscle actin. CD = control diet (n = 7); CDKO = CD knock out (n-5); WD = Western diet (n = 5); and WDKO = WD EnNaC knock out (n = 5). Data are expressed as means ± SEM. *p < .05 vs CD; #p < .05 vs WD.
3.5. Effect of WD feeding and αEnNaC deletion on expression of nuclear factor-E2-related factor (Nrf2) and protein levels of downstream antioxidants HO-1 and thioredoxin (TRX1)
We have previously observed a decrease in the antioxidant HO-1 after 16 weeks of WD feeding [25]. In this regard, Nrf2 is a key regulator of expression of HO-1 and other antioxidant molecules [26–31]. Nrf2 protein levels were increased in mice after WD feeding, and this diet-induced increase was still evident despite αEnNaC deletion (Fig. 4). The levels of BACH1 protein, a regulator of Nrf2 signaling were not altered in any of the groups compared to CD fed mice (Fig. 4). Similarly, the levels of downstream targets of Nrf2, HO-1, NQO1, GSR1 and GSR1 were not significantly changed in αEnNaC−/− mice fed the WD (Fig. 4).
Fig. 4.
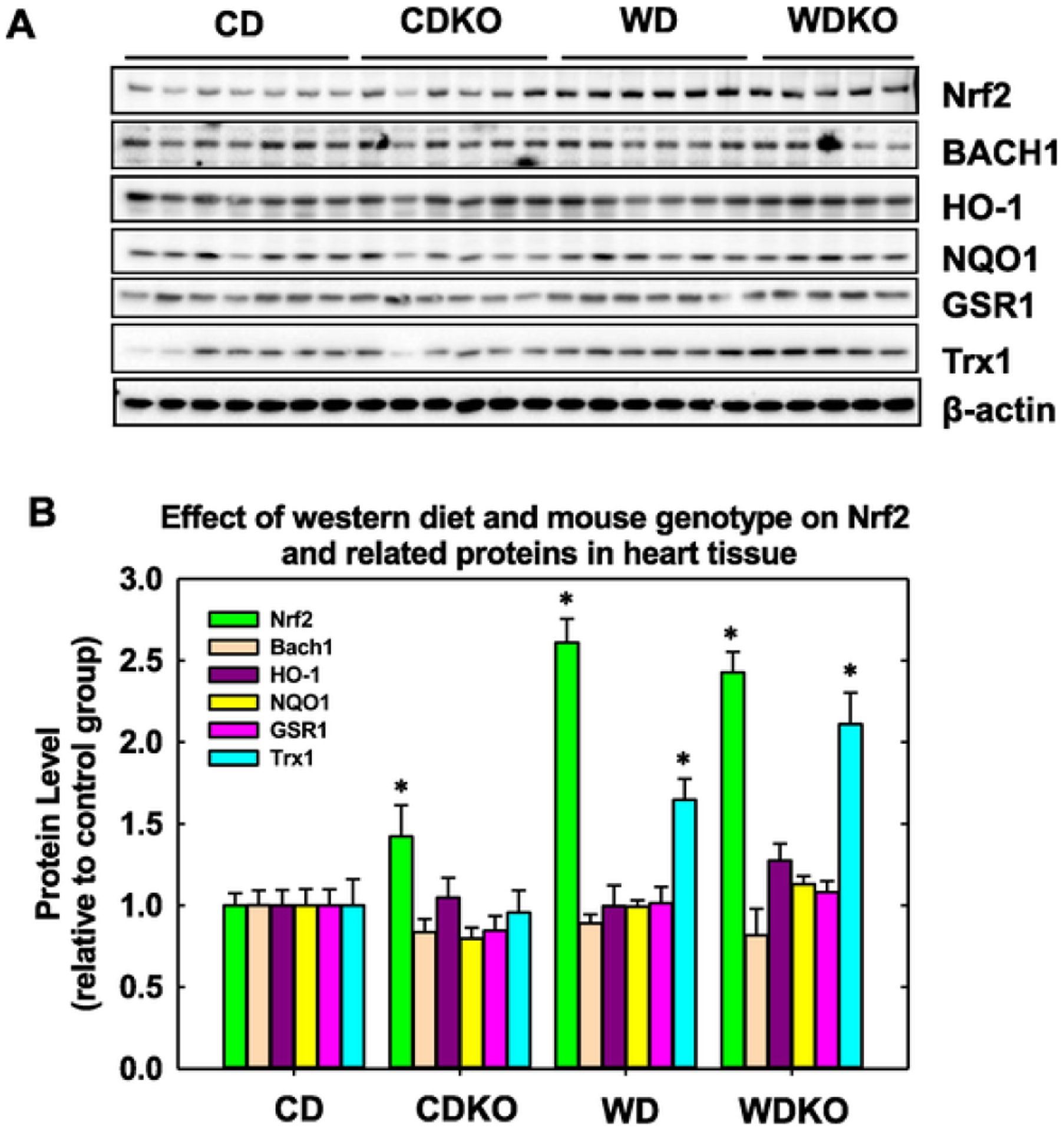
Nrf2 signaling after WD feeding and impact of α EnNaC deletion. A) Representative Western blot for Nrf2 and related molecules. B). Bar diagram showing quantitative changes in protein levels separated by Western blot. β actin was used as a loading control. CD = control diet (n = 7); CDKO = CD knock out (n = 6); WD = Western diet (n = 6); and WDKO = WD EnNaC knock out (n = 5). Data are expressed as means ± SEM. *p < .05 vs CD.
3.6. αEnNaC deletion prevents WD-induced increases of EC inward Na+ currents
Our previous data demonstrated that EC MR activation increases the expression of αEnNaC in ECs in the setting of WD feeding [20], whereas αEnNaC KO mice showed suppressed EC Na+ currents [23]. In the current study, WD feeding for 12 weeks resulted in a significant increase in inward Na+ currents in isolated pulmonary ECs compared to those of mice fed the CD (Fig. 5A–B). In contrast, the WD-induced increases in EC Na+ currents in αEnNaC+/+ mice was significantly attenuated in the αEnNaC−/− mice (Fig. 5A–B). In vessels such as coronary arteries, this attenuation of inward Na+ currents would be anticipated to decrease the development of vascular stiffness and associated myocardial fibrosis and stiffness [19,23].
Fig. 5.
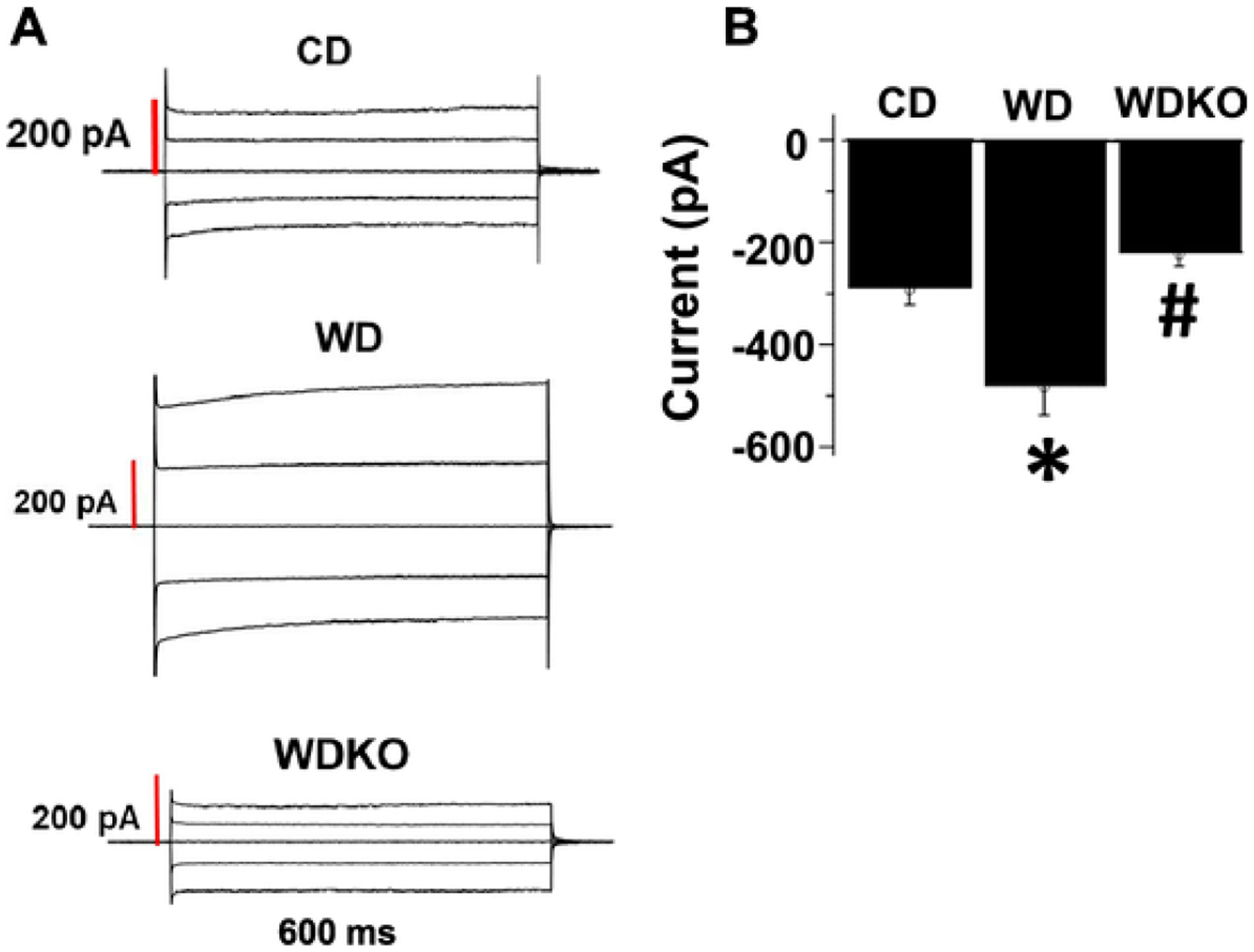
EnNaC α subunit deletion attenuates WD-induced increases of EC inward Na+ currents assessed by patch clamp. A). Representative tracings of whole cell Na+ currents. Currents were recorded at membrane potentials between −80 and +80 mV at 40 mV intervals. B). Group data showing peak inward Na + current (pA) at −80 mV. CD = control diet (n = 3); CDKO = CD knock out (n = 3); WD = Western diet; and WDKO = WD EnNaC knock out (n = 3). For each animal, Na+ currents were recorded from 7 to 9 independent cells and the data averaged to give an n = 1. Data are expressed as means ± SEM. *p < .05 vs CD; #p < .05 vs WD.
3.7. αEnNaC deletion prevents WD-induced increases of intrinsic cardiomyocyte stiffness
We have previously shown in female mice that ECMR deletion prevents WD-induced increases in intrinsic stiffness of cardiomyocytes after 16 weeks of feeding [19]. Therefore, in a cohort of WD fed mice with or without αEnNaC deletion feeding was extended to 16 weeks. In this cohort, cardiomyocyte stiffness was determined using AFM (Fig. 6A). As shown in Fig. 6B, stiffness of cardiomyocytes isolated from WD fed mice was significantly increased compared to CD fed mice. Importantly, deletion of αEnNaC significantly reduced WD-induced cardiomyocyte stiffening (Fig. 6B).
Fig. 6.
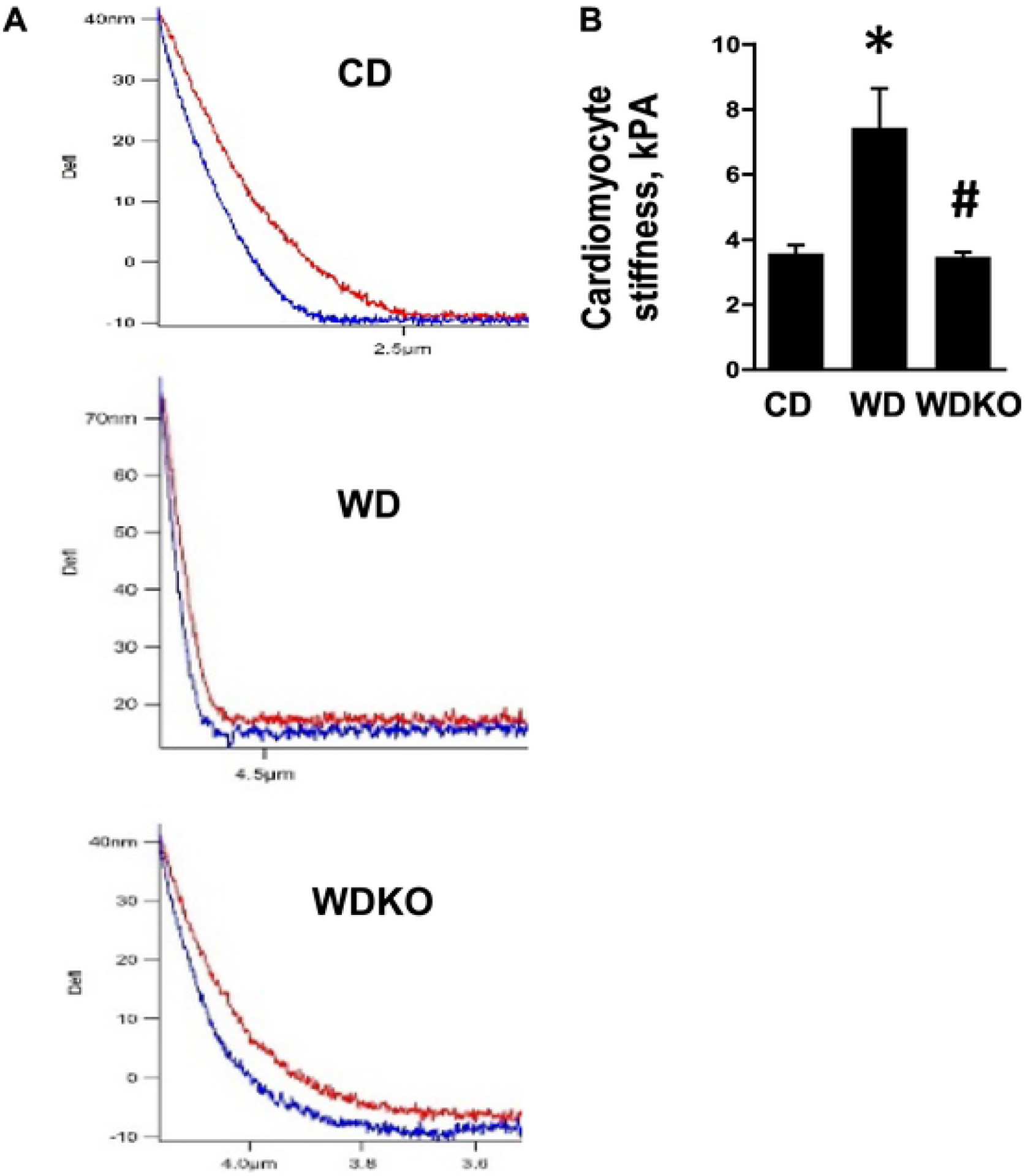
EnNaC α subunit deletion prevents WD induced intrinsic stiffness of isolated cardiomyocytes. A). Force curve by AFM and B) representative histogram to the right. CD = control diet (n = 3); WD = Western diet; (n = 3) and WDKO = WD EnNaC α subunit knock out (n = 3). Data are expressed as means ± SEM. *p < .05 vs CD; #p < .05 vs WD.
4. Discussion
The current investigation demonstrated that deletion of the critical functional subunit of EnNaC prevents cardiomyocyte stiffness and associated impairment of diastolic relaxation in WD-fed female mice. Thus, it is likely that the cardiac target of αEnNaC deletion is the coronary microcirculation of the heart [11–13]. Diastolic dysfunction is one of the early manifestations of HFpEF, especially in obese and diabetic women, and occurs prior to significant impairments in systolic dysfunction [7,8,12]. It was previously observed that diastolic dysfunction occurs earlier in WD fed obese females than males, findings associated with increased aldosterone levels and increased cardiovascular MR expression in females [8,12]. Here, we report that WD feeding for 12 weeks promotes early development of diastolic dysfunction [8,12] in concert with only modest evidence for changes in indices of fibrosis and left ventricular hypertrophy. Increased intrinsic stiffness of single cardiomyocytes in response to WD feeding was demonstrated using atomic force microscopy, and that this abnormality was substantially attenuated in mice with αEnNaC deletion. Finally, our current data indicate that one key mechanism by which EnNaC mediates cardiomyocyte stiffness and impaired diastolic relaxation during WD feeding occurs through increased myocardial oxidative stress [19,21].
Emerging evidence suggests an important role for Nrf2 in the regulation of oxidative stress and cardiovascular protection through induction of downstream antioxidants and cellular protective proteins [25–31]. Altered tissue Nrf2 levels and its downstream antioxidant targets have been associated with obesity and diabetes and may contribute to the increase in oxidative stress under these conditions. Previously we reported a decrease in the Nrf2 target, HO-1, after 16 weeks of WD feeding in female mice [25]. In this investigation, we observed that the levels of Nrf2 protein were increased in mice after 12 weeks of WD feeding, and this increase was not significantly altered with αEnNaC deletion in WD fed mice. Interestingly, the rise in Nrf2 protein in WD fed mice lead to a muted downstream antioxidant molecule response, insufficient to counteract the rise in the production of reactive oxygen species in obese females. The mechanism underlying the attenuated transcriptional antioxidant molecular responses to Nrf2 in WD fed animals is not known, but was not due to increases in the levels of BACH1, an established transcriptional inhibitor of Nrf2 (32). That Nrf2 and its downstream target antioxidant enzymes were not altered in mice with αEnNaC subunit deletion exposed to WD feeding, suggests that the observed improvements in oxidative stress with EnNaC deletion were related to unexplored oxidant/antioxidant mechanisms.
Our previous studies support the notion that EnNaC activation contributes to impairments in NO-dependent aortic and microvascular, i.e. coronary artery relaxation [20,22,23]. The αEnNaC subunit is regarded as the core functional component of the heterotrimeric sodium channel, and increases in αEnNaC subunit expression and membrane abundance in ECs is considered to lead to enhanced inward sodium (Na+) influx, polymerization of G-actin to F-actin (increasing endothelial stiffness) and reduces endothelial cell eNOS activity and NO production [22,23]. Our current data, in concert with the above observations, support the proposition that in diet-induced obesity EnNaC activation in the coronary vasculature leads to cardiomyocyte and ventricular stiffening. The present genetic approach also supports the notion that the development of specific inhibitors of the endothelial sodium channel may be an important strategy to prevent or retard the development of HFpEF, especially in obese females [16–23].
A limitation of the current study relates to the trends for increased expression of NRF2 downstream antioxidant molecules such as HO-1 with EnNaC subunit deletion not reaching statistical significance, presumably because of inadequate cohort sizes. Further, this study did not explore the role of other antioxidant pathways in mediating the cardiac antioxidant effects of EnNaC alpha subunit deletion in this diet induced obesity cardiomyopathy model. Thus, future studies employing larger cohorts and analysis of other antioxidant pathways will be necessary to more completely interrogate the mechanisms by which activation of coronary artery endothelial channels promotes diastolic dysfunction in association with diet-induced obesity.
In conclusion, deletion of αEnNaC prevents the development of cardiomyocyte stiffness and resultant diastolic dysfunction in a translationally relevant model of obesity-associated cardiomyopathy. The cardiovascular protection afforded by deletion of the αEnNaC subunit likely occurs by preventing coronary functional abnormalities and related suppression of myocardial oxidative stress. These results add to the growing literature suggesting that EnNaC activation is an important instigator of obesity cardiomyopathy and a potential important target for prevention and management of HFpEF especially in obese females.
Acknowledgments
We also acknowledge Matthew B. Martin and Michelle D. Lambert for their help in animal experiments and Ernesto Martinez-Martinez for assistance in transfer of α EnNaC−/− and littermate mice. This work was supported with resources and the use of facilities at the Harry S Truman Memorial Veterans Hospital and the Dalton Cardiovascular Center in Columbia, MO.
Funding
This study was funded, in part, by a bridge grant to ARA and JRS from the University of Missouri. JRS received funding from NIH (R01 HL73101-01A and R01 HL107910-01). JRS and GL received funding from the Department of Veterans Affairs Merit System (2I01BX001981-05A1). AWC and CMA received funding from the Department of Veterans Affairs Merit System (BX003391). GL also received funding from NIH (KO 5K08HL132012-02). GJ received funding from American Diabetes Association (Innovative Basic Science Award #1-17-IBS-201). WD received funding from the American Diabetes Association (Innovative Basic Science Award #1-17-IBS-290). MH received funding from NIH (RO1HL085119). FJ received funding from the Fondation de France (2014-00047968) and ANR Investissement Avenir CARMMA (ANR15-RHUS-0003).
Abbreviations:
- CVD
cardiovascular disease
- WD
Western diet
- DIO
Diet-induced obesity
- MR
mineralocorticoid receptor
- NO
nitric oxide
- EC
endothelial cell
- αENaC
epithelial Na+ channel alpha subunit
- αEnNaC
αENaC in ECs
- eNOS
endothelial NO synthase
- BP
blood pressure
- 3-NT
3-Nitrotyrosine
- αSMA
alpha smooth muscle actin
- Nrf2
nuclear factor related erythroid factor 2
- BACH 1
BTC and CTC homolog 1
- GRS1
glutathione reductase 1
- TRX1
thioredoxin
Footnotes
Disclosure statement
No relevant disclosures exist for this work.
References
- [1].Redfield MM Heart failure with preserved ejection fraction. N Engl J Med 2016;375:1868 1867. [DOI] [PubMed] [Google Scholar]
- [2].Borlaug BA The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 2014;11:507–515. [DOI] [PubMed] [Google Scholar]
- [3].Kenchaiah S, Evans JC, Levy D, et al. Obesity and the risk of heart failure. N Engl J Med 2002;347:305–513. [DOI] [PubMed] [Google Scholar]
- [4].Kitzman DW, Shah SJ The HFpEF obesity phenotype: the elephant in the room. J Am Coll Cardiol 2016;68:200–203. [DOI] [PubMed] [Google Scholar]
- [5].Obokata M, Reddy YNV, Pislaru SV, et al. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 2017;136:6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shah SJ, Kitzman DW, Borlaug BA, et al. Phenotype-specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation 2016;134:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Beale AL, Meyer P, Marwick TH, et al. Sex differences in cardiovascular pathophysiology: why women are overrepresented in heart failure with preserved ejection fraction. Circulation 2018;138:198–205. [DOI] [PubMed] [Google Scholar]
- [8].Manrique C, DeMarco VG, Aroor AR, et al. Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a Western diet. Endocrinology 2013;154:3632–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Messaoudi S, Gravez B, Tarjus A, et al. Aldosterone-specific activation of cardiomyocyte mineralocorticoid receptor in vivo. Hypertension 2013;61:361–367. [DOI] [PubMed] [Google Scholar]
- [10].Shen JZ, Morgan J, Tesch GH, et al. Cardiac tissue injury and remodeling is dependent upon MR regulation of activation pathways in cardiac tissue macrophages. Endocrinology 2016;157:3213–3223. [DOI] [PubMed] [Google Scholar]
- [11].Favre J, Gao J, Zhang AD, et al. Coronary endothelial dysfunction after cardiomyocyte-specific mineralocorticoid receptor overexpression. Am J Physiol Heart Circ Physiol 2011;300:H2035–H2043. [DOI] [PubMed] [Google Scholar]
- [12].Bostick B, Habibi J, DeMarco VG, et al. Mineralocorticoid receptor blockade prevents Western diet-induced diastolic dysfunction in female mice. Am J Physiol Heart Circ Physiol 2015;308:H1126–H1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bender SB, DeMarco VG, Padilla J, et al. Mineralocorticoid receptor antagonism treats obesity-associated cardiac diastolic dysfunction. Hypertension 2015;65:1082–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Papagiannis A, Alkagiet S, Tziomalos K The role of mineralocorticoid receptor antagonists in the management of heart failure with preserved ejection fraction. Curr Pharm Des 2018;24:5525–5527. [DOI] [PubMed] [Google Scholar]
- [15].Patel RB, Shah SJ, Fonarow GC, et al. Designing future clinical trials in heart failure with preserved ejection fraction: lessons from TOPCAT. Curr Heart Fail Rep 2017;217–222. [DOI] [PubMed] [Google Scholar]
- [16].Bristow MR, Sharma K, Assmann SF, et al. Data and safety monitoring board evaluation and management of a renal adverse event signal in TOPCAT. Eur J Heart Fail 2017;19:457–465. [DOI] [PubMed] [Google Scholar]
- [17].Paulus WJ, Tschöpe C A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- [18].Taqueti VR, Solomon SD, Shah AM, et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur Heart J 2018;39:840–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jia G, Habibi J, DeMarco VG, et al. Endothelial mineralocorticoid receptor deletion prevents diet-induced cardiac diastolic dysfunction in females. Hypertension 2015;66:1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Martinez-Lemus LA, Aroor AR, Ramirez-Perez FI, et al. Amiloride improves endothelial function and reduces vascular stiffness in female mice fed a Western diet. Front Physiol 2017;8:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jia G, Habibi J, Aroor AR, et al. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metab Clin Exp 2018; 78:69–79. [DOI] [PubMed] [Google Scholar]
- [22].Jia G, Habibi J, Aroor AR, et al. Epithelial sodium channel in aldosterone-induced endothelium stiffness and aortic dysfunction. Hypertension 2018;72:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sowers JR, Habibi J, Aroor AR, et al. Epithelial sodium channels in endothelial cells mediate diet-induced endothelium stiffness and impaired vascular relaxation in obese female mice. Metab Clin Exp 2019;99:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Aroor AR, Habibi J, Nistala R, et al. Diet-induced obesity promotes kidney endothelial stiffening and fibrosis dependent on the endothelial mineralocorticoid receptor. Hypertension 2019;73:849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bostick B, Aroor AR, Habibi J, et al. Daily exercise prevents diastolic dysfunction and oxidative stress in a female mouse model of Western diet induced obesity by maintaining cardiac heme oxygenase-1 levels. Metab Clin Exp 2017;66:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ali F, Zakker M, Karu K, et al. Induction of the cytoprotective enzyme hemeoxygenase-1 by statins is enhanced in vascular endothelium exposed to laminar shear stress and impaired by disturbed flow. J Biol Chem 2009;284:18882–18892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Durante W Targeting heme oxygenase-1 in vascular disease. Curr Drug Targets 2010;11:1504–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Abraham NG, Junge JM, Drummond GS Translational significance of heme oxygenase in obesity and metabolic syndrome. Trends Pharmacol Sci 2016;37:17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Smith RE, Tran K, Smith CC, et al. The role of the Nrf2/ARE antioxidant system in preventing cardiovascular diseases. Diseases 2016; 11: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang X, Park SH, Chang HC, et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J Clin Invest 2017;127:1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cao J, Singh SP, McClung JA, et al. HO-1 signaling prevents obesity-induced cardiomyopathy in obese mice. Am J Physiol Heart Circ Physiol 2017;313:H368–H380. [DOI] [PMC free article] [PubMed] [Google Scholar]