

Supplemental Digital Content is available in the text.
Keywords: actins, genotype, hypertrophic cardiomyopathy, myosin, sarcomere
Background:
Pathogenic variants in MYBPC3, encoding cardiac MyBP-C (myosin binding protein C), are the most common cause of familial hypertrophic cardiomyopathy. A large number of unique MYBPC3 variants and relatively small genotyped hypertrophic cardiomyopathy cohorts have precluded detailed genotype-phenotype correlations.
Methods:
Patients with hypertrophic cardiomyopathy and MYBPC3 variants were identified from the Sarcomeric Human Cardiomyopathy Registry. Variant types and locations were analyzed, morphological severity was assessed, and time-event analysis was performed (composite clinical outcome of sudden death, class III/IV heart failure, left ventricular assist device/transplant, atrial fibrillation). For selected missense variants falling in enriched domains, myofilament localization and degradation rates were measured in vitro.
Results:
Among 4756 genotyped patients with hypertrophic cardiomyopathy in Sarcomeric Human Cardiomyopathy Registry, 1316 patients were identified with adjudicated pathogenic truncating (N=234 unique variants, 1047 patients) or nontruncating (N=22 unique variants, 191 patients) variants in MYBPC3. Truncating variants were evenly dispersed throughout the gene, and hypertrophy severity and outcomes were not associated with variant location (grouped by 5′–3′ quartiles or by founder variant subgroup). Nontruncating pathogenic variants clustered in the C3, C6, and C10 domains (18 of 22, 82%, P<0.001 versus Genome Aggregation Database common variants) and were associated with similar hypertrophy severity and adverse event rates as observed with truncating variants. MyBP-C with variants in the C3, C6, and C10 domains was expressed in rat ventricular myocytes. C10 mutant MyBP-C failed to incorporate into myofilaments and degradation rates were accelerated by ≈90%, while C3 and C6 mutant MyBP-C incorporated normally with degradation rate similar to wild-type.
Conclusions:
Truncating variants account for 91% of MYBPC3 pathogenic variants and cause similar clinical severity and outcomes regardless of location, consistent with locus-independent loss-of-function. Nontruncating MYBPC3 pathogenic variants are regionally clustered, and a subset also cause loss of function through failure of myofilament incorporation and rapid degradation. Cardiac morphology and clinical outcomes are similar in patients with truncating versus nontruncating variants.
Familial hypertrophic cardiomyopathy (HCM) is an autosomal dominant condition, and pathogenic variants in cardiac MyBP-C (myosin binding protein C; encoded by the gene, MYBPC3) are the most common cause.1 MyBP-C is a sarcomeric protein that binds both actin and myosin and regulates cardiac contractility by modulating myofilament sliding velocity.2,3 Because a large number of unique MYBPC3 variants have been associated with HCM, small, single-center cohorts have had limited capacity to systematically analyze genotype-phenotype relationships, particularly given the marked variability in penetrance of MYBPC3-associated HCM.4–7 Resolving these gaps in knowledge will be critical to further personalized risk assessment and management of patients with HCM.
Most MYBPC3 pathogenic variants are frameshift, nonsense, or splice-site variants that result in premature termination codons (PTCs). PTC-containing transcripts are targeted for degradation through nonsense mediated RNA decay, and hence may cause disease through allelic loss of function (resulting in reduced levels of MyBP-C). Consistent with allelic insufficiency, we and others have shown a ≈40% reduction in MyBP-C in heart tissue from patients with HCM,8,9 due to a rate-limiting reduction in MYBPC3 mRNA.10 These studies support the hypothesis that truncating variants in MYBPC3 likely exert a similar primary effect, independent of the specific variant locus. However, comparative analyses across the full genotypic and phenotypic spectrum of truncating variants have not been possible due to the small size of previously available cohorts. Distinct from truncating MYBPC3 variants, nontruncating pathogenic variants (including missense and short in-frame deletions/insertion variants) account for ≈15% of MYBPC3 HCM. The mechanism(s) of MYBPC3 nontruncating pathogenic variants are largely unknown, and it is unclear whether phenotypic expression or clinical outcomes are different in patients carrying missense variants.7,11 A greater understanding of the disease-causing mechanism(s) of nontruncating MYBPC3 pathogenic variants through functional analyses could improve adjudication of variant pathogenicity and expand the pool of clinically actionable gene test results.
Here, we use the largest registry of combined genetics and clinical data for HCM to date, the Sarcomeric Human Cardiomyopathy Registry1 (SHaRe), to generate an adjudicated and comprehensive compendium of MYBPC3 variation, analyze regional variation within MYBPC3, and correlate clinical phenotypes. We find that pathogenic truncating variants are homogeneously distributed throughout the gene, in contrast to nontruncating MYBPC3 pathogenic variants that cluster in specific protein domains. Disease severity is highly variable in MYBPC3 HCM, and we show that this variability is largely independent of variant location or the specific truncating or nontruncating variant based on both disease severity metrics and clinical outcomes. Finally, we experimentally test functional effects of nontruncating pathogenic variants in the identified variant-enriched domains and identify a subset that exhibit allelic loss of function.
Methods
The methods used are described for purposes of replicating the study procedure. Individual patient data will not be made available for purposes of reproducing the results. The study was independently approved by the institutional review board at each center. A detailed methods section is available in the Data Supplement.
Results
Clinical Profile of MYBPC3 Mutation HCM
Among these patients with pathogenic MYBPC3 variants, 1238 (94%) carried single pathogenic variants in MYBPC3 without pathogenic variants in other sarcomere genes and comprised the primary study group. The largest subset of these patients (N=1047, 71%) carried truncating MYBPC3 variants (234 unique variants), while 29% (N=191) had nontruncating variants (22 unique variants).
The demographic and clinical profile of patients with MYBPC3 pathogenic variants is shown in Table 1. The majority (76%) of patients presented in early-mid adulthood (age 18–60 years) with a minority of pediatric (13%) or late adulthood (10%) presentations. The average age of diagnosis was younger among patients with nontruncating pathogenic variants due to a greater percentage with pediatric diagnoses (24% versus 11%, P=0.0001). Maximum left ventricular (LV) wall thickness was greater in the relatively small subset of pediatric patients with nontruncating variants but was similar in other age groups. LV ejection fraction was similarly elevated at a young age in both groups and declined similarly in later age groups. Left atrial diameter progressively increased to a similar extent with increasing age in both truncating and nontruncating groups (with the single exception of the smaller sized group of nontruncating variant patients at age >60; N=17). The distributions of maximum wall thickness, left atrial size, and age of diagnosis among nontruncating and truncating pathogenic variant cases are shown in Figure 1A through 1C. Time to event analysis for composite adverse outcomes revealed no difference between patients with nontruncating or truncating pathogenic variants (Figure 1D), and this result was not different when including probands only (Figure IA in the Data Supplement). There were similarly no differences between the groups for heart failure or ventricular arrhythmia composite outcomes (not shown).
Table 1.
Demographic Characteristics of Patients With Truncating and Nontruncating MYBC3 Pathogenic Variants
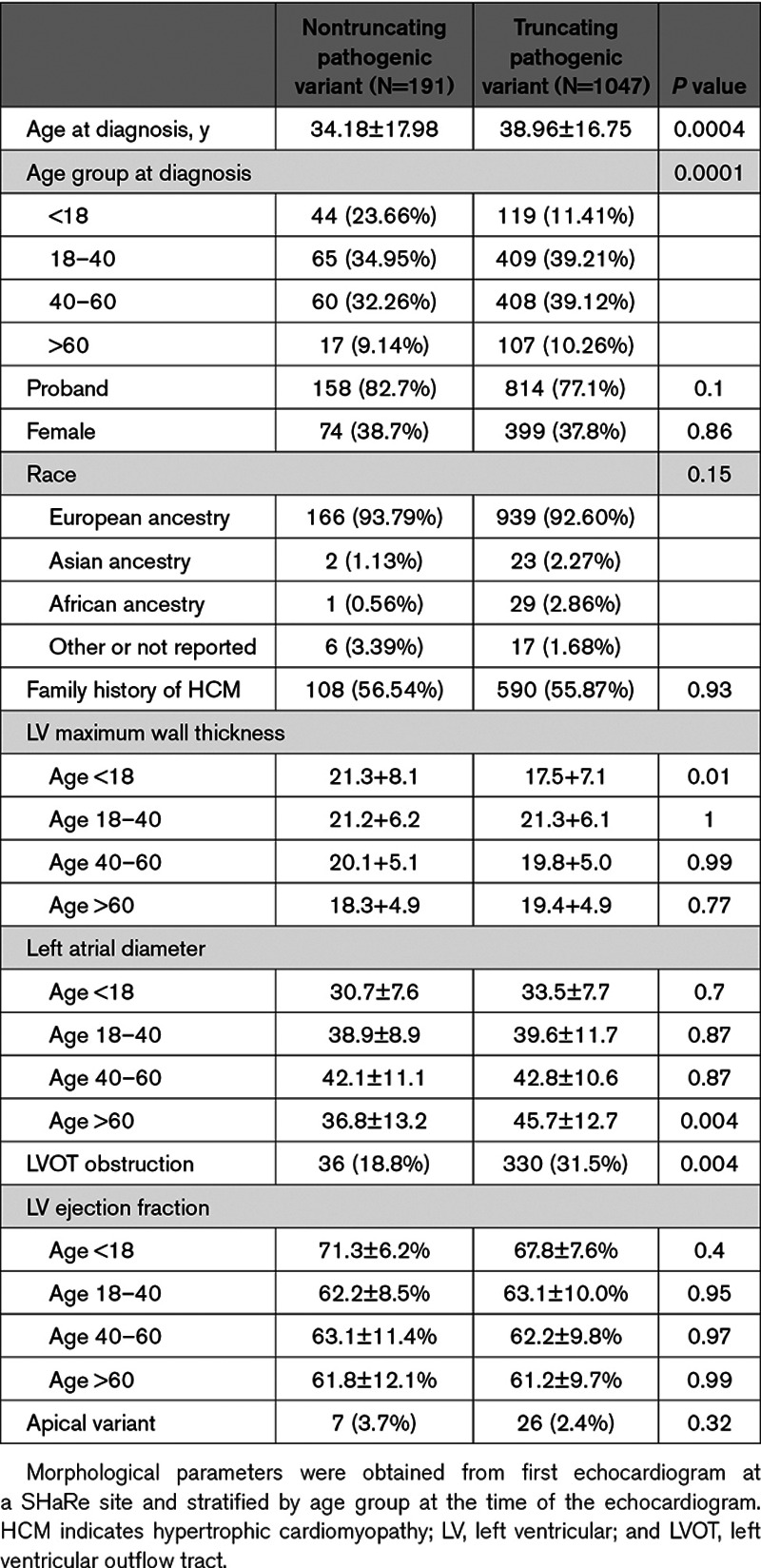
Figure 1.
MYBPC3 nontruncating pathogenic variants cause similar phenotypic severity and adverse event rates as truncating variants. A, Distributions in maximum wall thickness demonstrate broad phenotypic variance and similarity between truncating and nontruncating MYBPC3 pathogenic variant groups. Data is shown in violin plots with median and interquartile range. B, Average age-adjusted left atrial diameter was smaller among nontruncating pathogenic variant carriers. C, Broad variability in disease severity is reflected by range in age of diagnosis in both MYBPC3 groups, with a modestly lower average age of diagnosis among nontruncating pathogenic variant carriers. D, Kaplan-Meier survival analysis shows no difference in the composite adverse event rate from time of birth between truncating and nontruncating pathogenic variant groups. Composite outcome consisted of first occurrence of any of the following: sudden cardiac death, resuscitated cardiac arrest, appropriate implantable cardioverter-defibrillator therapy, cardiac transplantation, left ventricle (LV) assist device implantation, LV ejection fraction <35%, or New York Heart Association class III/IV symptoms, atrial fibrillation (AF), stroke, or death.
Morphological Severity and Adverse Events Are Similar Across Truncating MYBPC3 Pathogenic Variants
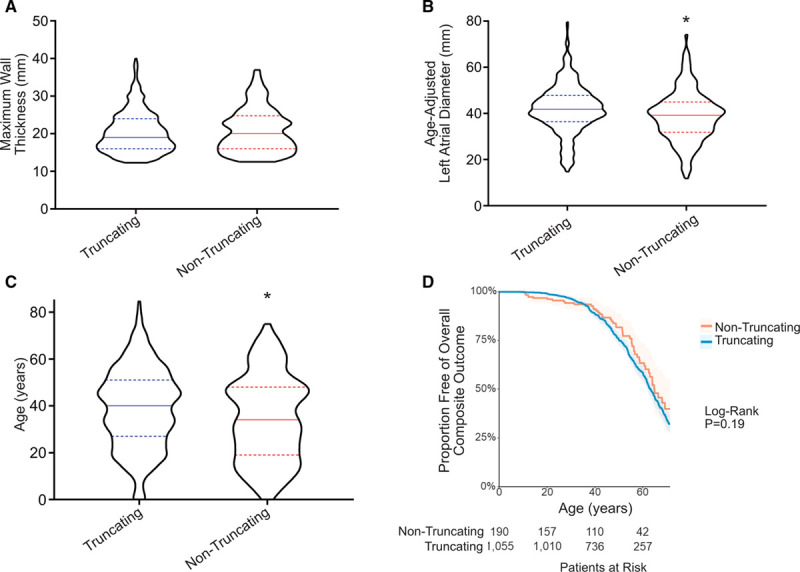
If truncating MYBPC3 variants cause allelic insufficiency as their primary consequence, then the location of the variant within the gene would not be expected to influence the disease severity. To test this, we categorized truncating MYBPC3 pathogenic variants into quartiles by 5′ to 3′ location and compared morphological markers of severity and adverse outcomes. We found no statistically significant difference in maximum wall thickness or age-adjusted left atrial diameter among these groups (Figure 2A and 2B). Composite adverse events were also similar when stratified by variant location quartile (Figure 2C) or by truncating variant type (Figure IB in the Data Supplement).
Figure 2.
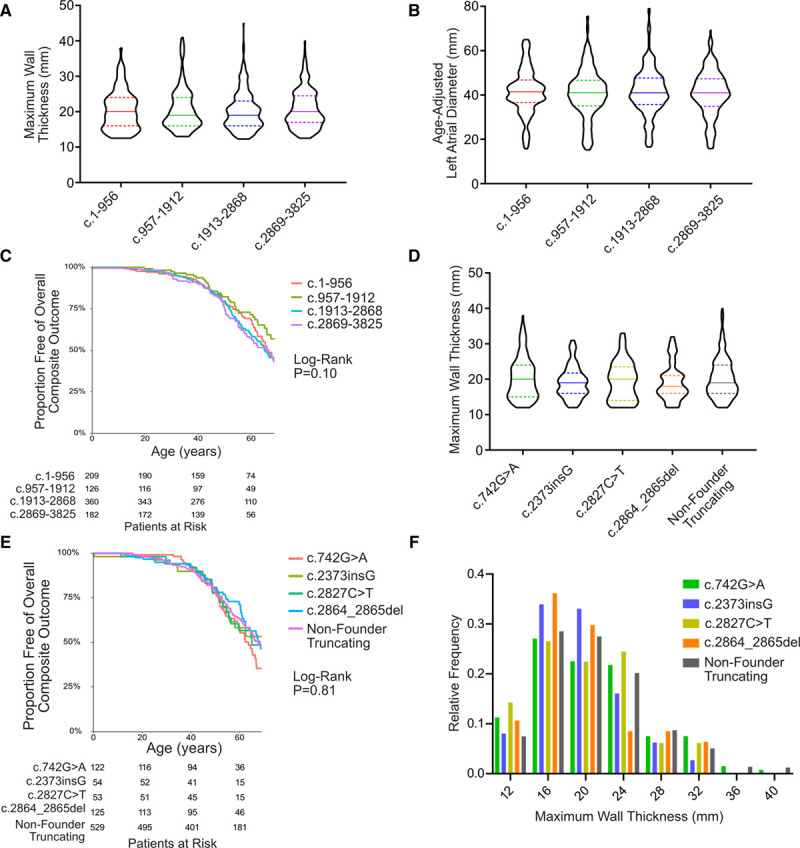
MYBPC3 truncating pathogenic variants cause similar phenotypic severity regardless of variant locus or type. A and B, Truncating MYBPC3 variants were categorized by locus quartiles within the gene to examine whether N-terminal or C-terminal truncations exert different effect sizes. No difference in extent of hypertrophy (A) or left atrial diameter (B) are observed. C and D, Four founder populations within Sarcomeric Human Cardiomyopathy Registry (SHaRe) were compared with determine whether phenotypic severity is different in the setting of these 4 distinct truncating variant types (c.742G>A=exonic splice variant, c.2373insG=frameshift, c.2827C>T=nonsense, c.2864_2865del=frameshift). No difference was observed either in the variance/distribution of hypertrophy (C) or in the magnitude of hypertrophy (D).
Morphological Severity, Adverse Events, and Variability in Phenotype Are Similar Among Founder and Nonfounder Truncating MYBPC3 Pathogenic Variants
Several founder truncating pathogenic variants in MYBPC3 have a high prevalence among patients with HCM. In SHaRe, 4 distinct founder truncating variants exist in large numbers, enabling comparison across subgroups that share the same primary causative sarcomere gene mutation. These founder populations consisted of 142 individuals with the c.742G>A variant (exonic splice variant causing exon skipping and PTC12), 143 with the c.2373insG variant (insertion variant causing frameshift and PTC),13 67 with the c.2827C>T variant (nonsense variant), and 58 with the c.2864_2865del variant (deletion causing frameshift and PTC). LV hypertrophy was similar across each of these 4 founder populations and the remaining nonfounder truncating variant patients (N=638), further supporting that different truncating variants exert a similar effect (Figure 2D). Additionally, adverse events were similar in each founder population compared with patients with nonfounder truncating variants (Figure 2E).
HCM is known to have broad variance in phenotypic severity across individuals. This variance in expressivity has been thought to be due to heterogeneity of effect size of underlying pathogenic variants, the influence of background genetic variation (ie, genetic modifiers), and clinical comorbidities.14–17 Taking advantage of the founder populations, we compared variances across these subgroups each carrying identical pathogenic variants. As shown in the histogram plot of maximum wall thickness in Figure 2F, the 4 founder populations demonstrate similar variance (mean of SDs, 5.96±0.79 mm) compared with the remainder of the truncating variant population (SD, 5.98 mm; P=NS). Taken together, these findings indicate that truncating variants likely exert a similar primary effect, and the marked variance in disease phenotype among truncating variant patients is caused by additional genetic and nongenetic factors, independent of the driving MYBP3 variant.
Variant Classification and Distribution of Truncating and Nontruncating MYBP3 Pathogenic Variants in HCM
MYBPC3 truncating variant types in SHaRe patients consisted of 110 unique insertion/deletion variants, 55 unique nonsense variants, and 69 unique splice variants (Table I in the Data Supplement). Classification of potential splice variants is complicated by the fact that only a portion of splice consensus sites are strictly conserved. The 69 unique splice pathogenic variants were classified through application of the American College of Medical Genetics and Genomics Association for Molecular Pathology criteria, combined with enrichment in SHaRe, prior experimental confirmation, and independent experimental confirmation in select cases (Tables I and II and Figure II in the Data Supplement). These criteria left a total of 26 of 99 potential intronic variants classified at variant of unknown significance (VUS) status. Six exonic splice variants were identified at the last base pair position in their respective exons (donor -1 position), 4 of which have had prior experimental confirmation of splice disruption in human heart tissue.12,18 These splice variants (c.655G>C, c.772G>A, c.772G>C, c.1090G>A, c.1624G>C, c.1790G>A) were consequently classified as truncating—an important distinction since erroneous classification as missense variants would impact clustering analysis of the nontruncating variants. Comparison of our clinical-genetics assignment of variant pathogenicity to the MYBPC3 splice variant prediction mini-gene splice assay developed by Ito et al19 demonstrated a high, though not perfect, level of concordance, with 20 out of 23 variants (87%) in agreement (Tables I and III in the Data Supplement). Nontruncating pathogenic variants were less common than truncating variants, with only 22 unique variants meeting criteria for pathogenicity, present in a total of 191 patients carrying a single sarcomere gene pathogenic variant (15% of MYBPC3 pathogenic variant patients). The potential pathogenicity of 147 nontruncating VUS’s could not be resolved with clinical data from SHaRe.
To determine regional variation in the distribution of MYBPC3 pathogenic variants, we mapped all unique MYBPC3 pathogenic variants in SHaRe by location within the coding sequence, stratified by truncating or nontruncating variant type (Figure 3). Truncating variants were dispersed throughout the coding regions of the gene without evidence of regional clustering. In addition, unique truncating variants were similarly prevalent in the N-terminus (including a variant that disrupts the start codon). In contrast, nontruncating pathogenic variants were primarily localized in the C3, C6, and C10 domains (18 of 22, 82%)—as compared with nontruncating common variants in the Genome Aggregation Database that were distributed throughout the gene (Figure 1, Tables III and IV in the Data Supplement). The C3 domain alone accounted for most individuals with MYBPC3 nontruncating variants in SHaRe (177 of 191, 93%). Among Genome Aggregation Database common variants, a lower percentage (17%, 23 of 135) localized to the C3, C6, or C10 domains (P<0.0001 compared with SHaRe).
Figure 3.
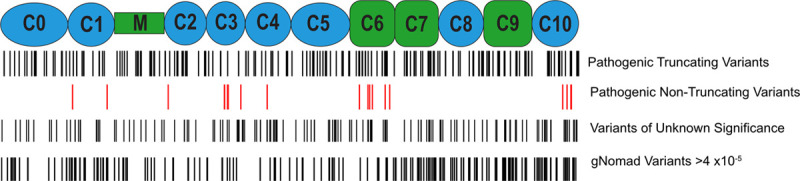
Distribution of MYBPC3 pathogenic variants, variants of unknown significance, and common Genome Aggregation Database (gnomAD) variants relative to MyBP-C (myosin binding protein C) protein domains. Truncating MYBPC3 pathogenic variants are dispersed homogeneously throughout the gene, while nontruncating pathogenic variants exhibit clustering in the C3, C6, and C10 domains (18 of 22, 82%). Nontruncating variants of unknown significance are dispersed throughout the gene, as are gnomAD common variants (ie, allele frequency >4×10−5).
Experimental Confirmation of Domain Specific Effects of MyBP-C Nontruncating Pathogenic Variants on Myofilament Incorporation and Degradation Rate
While strong evidence supports allelic insufficiency is the primary mechanism across the spectrum of truncating MYBPC3 variants, the mechanism(s) of nontruncating MYBPC3 pathogenic variants has not been resolved. We hypothesized that some nontruncating MYBPC3 pathogenic variants may also cause loss of function, but through lack of normal protein localization or structural stability rather than reduced expression. Therefore, we first tested whether exogenously expressed MyBP-C with nontruncating pathogenic variants incorporates normally into the myofilaments. We expressed FLAG-epitope labeled MyBP-C with or without pathogenic nontruncating variants in neonatal rat ventricular myocytes and analyzed localization by immunofluorescence. We found that MyBP-C containing representative C3 or C6 domain nontruncating variants localized normally to the sarcomere A bands while MyBP-C containing C10 domain nontruncating variants was essentially absent from the myofilaments (Figure 4).
Figure 4.
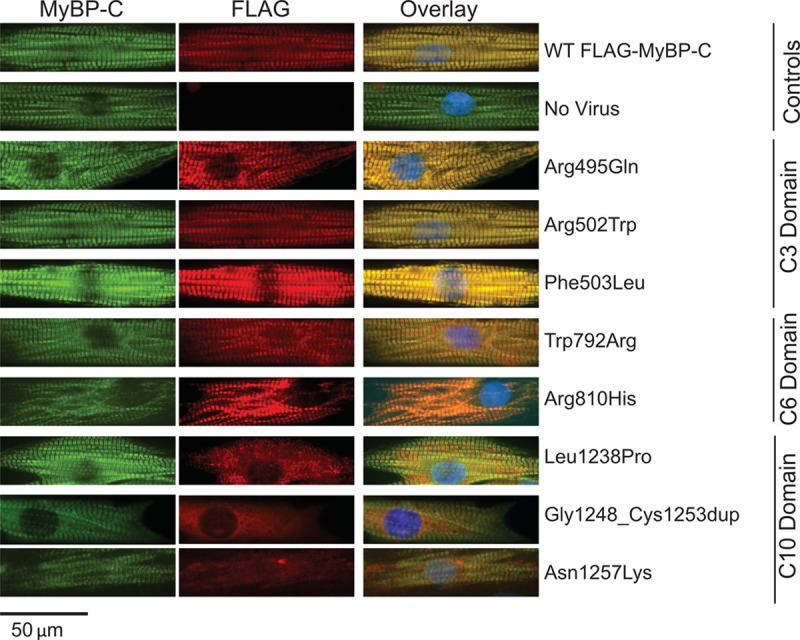
Nontruncating MyBP-C (myosin binding protein C) mutant protein localizes to the myofilament for C3 and C6 domain mutants but does not incorporate into myofilaments for C10 domain mutants. To determine whether mutant MyBP-C proteins integrate normally into the myofilaments, both FLAG-tagged control and mutant constructs were cloned into an adenoviral vector that was then used to transduce neonatal rat ventricular myocytes (NRVMs). Forty-eight hours following transduction, NRVMs were immunofluorescently labeled with an anti-MyBP-C antibody to detect both endogenous and exogenously expressed MyBP-C (left column) and an anti-FLAG antibody to detect only the transduced MyBP-C (middle column). This system achieved stable integration of FLAG-control MyBP-C into myofilaments (top row) with no FLAG signal detected without viral transduction (second row). Nontruncating mutant MyBP-C for C3 and C6 domain pathogenic variants exhibited normal myofilament integration while C10 mutant MyBP-C exhibited poor or no myofilament localization.
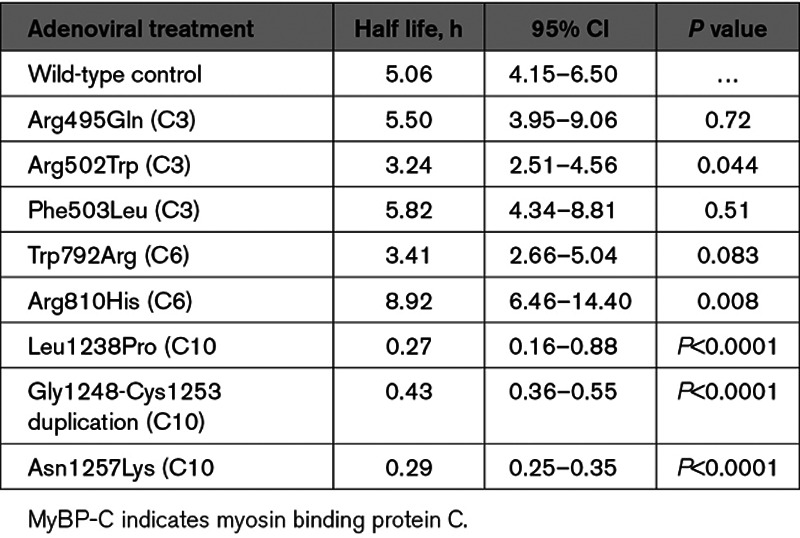
A lack of mutant MyBP-C myofilament incorporation could be either due to perturbation of binding sites required for correct localization or protein instability. To determine if pathogenic variants in the C10 domain result in protein destabilization, we performed cyclohexamide pulse-chase experiments using neonatal rat ventricular myocytes transduced with FLAG-tagged mutant MyBP-C for representative variants. Consistent with MyBP-C destabilization as a consequence of pathogenic variants in the C10 domain, we found a marked 90% reduction in protein half-life (Figure 5 and Table 2). In contrast, most pathogenic variants in the C3 and C6 domains resulted in MyBP-C protein half-lives that were not significantly different from wild-type MyBP-C, though the Arg502Trp variant resulted in a modest 36% shorter protein half-life compared with wild-type (P=0.04). Paradoxically, the Arg810His variant resulted in a 44% prolonged MyBP-C protein half-life compared with wild-type (P=0.008).
Figure 5.
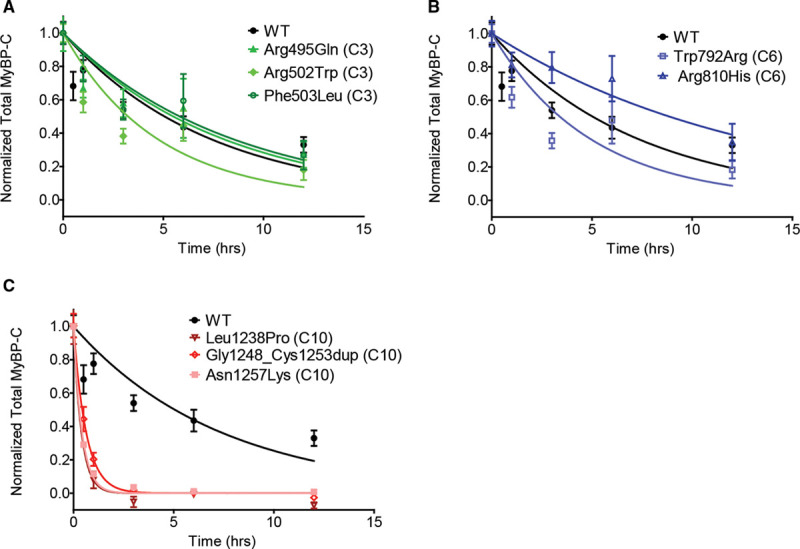
Nontruncating mutant MyBP-C (myosin binding protein C) degradation rates measured by cyclohexamide pulse chase demonstrate rapid degradation for C10 domain nontruncating mutant MyBP-C. To determine whether nontruncating MYBPC3 pathogenic variants alter protein stability, neonatal rat ventricular myocytes were transduced with adenoviral constructs expressing wild-type (WT) control and nontruncating mutant MyBP-C. Cyclohexamide was administered at 0, 30 min, 1 h, 3 h, 6 h, and 12 h to inhibit protein synthesis and MyBP-C was measured (see Methods in the Data Supplement). Data from 2 or more independent experiments performed in quadruplicate were fit to a first order exponential decay curve. The same control data (from FLAG-labeled wild-type expressed MyBP-C) is depicted on each graph (A–C). A and B, C3 and C6 mutant MyBP-C demonstrates similar degradation rates as control. C, C10 mutant MyBP-C demonstrates rapid degradation compared with control. Data is represented as mean±SEM. The calculated half-lives with 95% CIs are shown in Table 2.
Table 2.
Nontruncating Mutant MyBP-C Degradation Rates Measured by Cyclohexamide Pulse Chase
Discussion
Despite genetic variants in MYBPC3 being the most common cause of familial HCM, identifying genotype-phenotype correlations has been elusive, due to the large number of individual pathogenic variants and small numbers of patients previously available to study from single centers. Here, we harness the largest cohort of genotyped patients with HCM to comprehensively describe MYBPC3 genetic variation and associated clinical phenotypes.
A convergent theory of allelic insufficiency from truncating MYBPC3 variants has emerged from human tissue, rodent, and induced pluripotent stem cell model systems.8,10,20–22 Reduction in MyBP-C relative to myosin alters sliding velocities as actin-myosin sliding reaches the C-zones, where MyBP-C is specifically present, resulting in a more rapid contractile deceleration toward peak force development.2,8,10,23 However, clinical-genetics data to confirm this theory have been notably absent. Our findings of a homogeneous distribution of HCM-causing truncating variants throughout MYBPC3, similar phenotypic severity across spatial quartiles in the coding sequence, and similar adverse event rates support the theory that disease results from a biologically similar loss of function mechanism across truncating variants, as opposed to dominant negative consequences from truncated MyBP-C protein (which has not been detectable in human heart or cellular models9,10,12). Furthermore, we found that 4 founder populations, with distinct truncating variants and sizable numbers in SHaRe, exhibit similar disease severity and adverse event rates as compared with nonfounder truncating variant patients. This result extends findings from a single site investigation of the Netherlands founder cohort,24 and counters smaller series that have suggested less pathogenic effects in truncating variant founder cohorts.25,26 A major implication of these results is that patients with truncating MYBPC3 variants would likely derive similar benefit from targeted treatment approaches irrespective of the specific location of the truncating variant.
We further leveraged the truncating variant founder populations in SHaRe to investigate the variability in expressivity in HCM. HCM exhibits vast genetic and phenotypic heterogeneity, which has been a major challenge in determining genotype-phenotype relationships.27 We found that patients with founder variants had a similar distribution of phenotypic features and clinical outcomes as nonfounder patients with HCM with truncating variants. This finding suggests that the variability in disease phenotype among MYBPC3 truncating variant carriers is not dictated solely by the primary pathogenic variant. An important implication of this finding is that additional genetic and nongenetic modifiers likely account for the broad variance in phenotypic severity among patients with MYBPC3 HCM.
We also demonstrate that MYBPC3 nontruncating pathogenic variants, accounting for 15% of MYBPC3 pathogenic variants, generally had a similar phenotypic effect as truncating variants. Minor differences between the groups included a modestly greater proportion of pediatric diagnoses in the nontruncating group and modestly reduced prevalence of LV outflow tract obstruction. However, maximal LV wall thickness across all other age groups and adverse event rates were highly similar.
Because nontruncating variants are robustly adjudicated in SHaRe, we were able to identify strong evidence of domain clustering. We then demonstrated that a subgroup of nontruncating pathogenic variants (those in the C10 domain) renders the resultant mutant protein susceptible to rapid degradation, resulting in a loss of function mechanism similar to truncating variants. In contrast, we show no destabilization in the majority of C3 and C6 domain mutant proteins, which integrate normally in myofilaments. The C3 variant Arg502Trp alters the electrostatic properties of the domain, but how this alteration affects MyBP-C function is not known.28 In engineered heart tissue, overexpression of the C3 mutant Gly531Arg (not present in SHaRe), caused hypercontractility at low calcium levels and was not able to rescue MyBP-C knock-out tissues.29 Further study is required to fully elucidate the impact of C3 and C6 pathogenic variants on contractile function.
In contrast to the clustering evident for pathogenic nontruncating variants, VUS’s in MYBPC3 were relatively common in the SHaRe cohort (N=148, 87% of all unique MYBPC3 nontruncating variants). Accurate prediction of pathogenicity of sarcomere VUS’s is a major challenge for interpretation of genetic testing results and determination of the suitability for cascade testing in family members. Although we confirmed enrichment of nontruncating pathogenic variants in specific MyBP-C domains, as also shown in an independent cohort by Walsh et al,30 the presence of common variants in Genome Aggregation Database in these same domains should preclude a complete reliance on a generalized domain-centric approach to determine variant pathogenicity. Nevertheless, the presence of a variant in the C3, C6, or C10 domains in a patient with HCM increases the probability of pathogenicity and could be used as a supportive criterion with other clinical variables in variant classification. Moreover, identifying VUS’s that cause protein instability could be a useful strategy for functional annotation of variants.
Several limitations to our study should be considered. This was a retrospective, observational study. Although we analyzed by far the largest cohort of patients with HCM with MYBPC3 pathogenic variants to date, the study may be underpowered to detect small differences in phenotype severity or adverse events between groups. In addition, we analyzed pathogenic variant carriers in groups based on variant type and location, but further subdivision to individual pathogenic variants was only feasible for the founder subpopulations. As such, differences in effect size for specific pathogenic variants, particularly in the case of the nontruncating variants, could still exist. Both the SHaRe population and Genome Aggregation Database populations predominantly consist of individuals from European ancestry. Although these attributes lend confidence to the calculation of the odds ratios for HCM-associated versus common population variants reported here, the results are not necessarily representative of genetic variation in other ancestries. Relatedly, the SHaRe population has a greater proportion of patients with HCM with truncating founder variants due to inclusion of certain European sites (the Netherlands, Italy). Lastly, we strategically focused experimental testing of nontruncating pathogenic variants to the impact on protein stability and only examined a subset of representative variants. Future work will be needed to further resolve the functional effects of pathogenic nontruncating MYBPC3 variants that do not destabilize the protein structure and extending these analyses more comprehensively across MYBPC3 nontruncating variants.
In conclusion, we leverage the largest cohort of patients with MYBPC3 pathogenic variants to date to develop a compendium of benign, pathogenic, and uncertain MYBPC3 variants and identify genotype-phenotype correlations. Our results demonstrate that phenotypic severity and clinical outcomes are similar across the range of MYBPC3 pathogenic variant carriers, without obvious associations based on the location of truncating variants, founder, or nonfounder truncating variant carriers, or truncating versus nontruncating variants. These findings highlight the need to identify additional background genetic and nongenetic modifiers that influence the broadly variable HCM disease phenotype. In addition, we show that nontruncating pathogenic variants cluster in particular MyBP-C domains, with those variants in the C10 domain exhibiting protein destabilization leading to loss of function, in contrast to a second subset exhibiting normal myofilament incorporation and stability.
Sources of Funding
Funding for SHaRe has been provided through an unrestricted research grant from Myokardia, Inc, a startup company that is developing therapeutics that target the sarcomere. MyoKardia, Inc, had no role in approving the content of this article. Dr Helms is supported by funding from the National Institutes of Health (K08HL130455). Dr Thompson is supported by the National Institutes of Health (T32 HL007853). Dr Ware is supported by the Wellcome Trust (107469/Z/15/Z) and the Medical Research Council (United Kingdom). Dr Ingles a recipient of a National Health and Medical Research Council (NHMRC) Career Development Fellowship (No. 1162929). Dr Semsarian is the recipient of a NHMRC Practitioner Fellowship (No. 1059156). Dr Olivotto is supported by the Italian Ministry of Health (“Left Ventricular Hypertrophy in Aortic Valve Disease and Hypertrophic Cardiomyopathy: Genetic Basis, Biophysical Correlates and Viral Therapy Models” [RF-2013-02356787] and NET-2011-02347173 [Mechanisms and Treatment of Coronary Microvascular Dysfunction in Patients sith Genetic or Secondary Left Ventricular Hypertrophy]) and by the Tuscany Registry of Sudden Cardiac Death (ToRSADE) project (FAS-Salute 2014, Regione Toscana). Dr Ho is supported by funding from the National Institutes of Health (1P50HL112349 and 1U01HL117006). Dr Day is supported by funding from the National Institutes of Health (R01 11572784), the American Heart Association (grant in aid), the Children’s Cardiomyopathy Foundation, and the Protein Folding Disease Initiative (University of Michigan).
Disclosures
Dr Helms, Dr Ho, Dr Day, Dr Saberi, Dr Olivotto, Dr Colan, Dr Ingles, and E.A. Ashley receive research support from MyoKardia, Inc. The other authors report no conflicts.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- HCM
- hypertrophic cardiomyopathy
- LV
- left ventricle
- MyBP-C
- myosin binding protein C
- PTC
- premature termination codon
- SHaRe
- Sarcomeric Human Cardiomyopathy Registry
This article was sent to Ruth McPherson, MD, PhD, Guest Editor, for review by expert referees, editorial decision, and final disposition.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.002929.
For Sources of Funding and Disclosures, see page 404.
References
- 1.Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation. 2018;138:1387–1398. doi: 10.1161/CIRCULATIONAHA.117.033200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215–1218. doi: 10.1126/science.1223602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Previs MJ, Prosser BL, Mun JY, Previs SB, Gulick J, Lee K, Robbins J, Craig R, Lederer WJ, Warshaw DM. Myosin-binding protein C corrects an intrinsic inhomogeneity in cardiac excitation-contraction coupling. Sci Adv. 2015;1:e1400205 doi: 10.1126/sciadv.1400205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045 [DOI] [PubMed] [Google Scholar]
- 5.Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–8. doi: 10.1038/gim.2014.205 [DOI] [PubMed] [Google Scholar]
- 6.Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 2015;573:188–197. doi: 10.1016/j.gene.2015.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Page SP, Kounas S, Syrris P, Christiansen M, Frank-Hansen R, Andersen PS, Elliott PM, McKenna WJ. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet. 2012;5:156–166. doi: 10.1161/CIRCGENETICS.111.960831 [DOI] [PubMed] [Google Scholar]
- 8.O’Leary TS, Snyder J, Sadayappan S, Day SM, Previs MJ. MYBPC3 truncation mutations enhance actomyosin contractile mechanics in human hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2019;127:165–173. doi: 10.1016/j.yjmcc.2018.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440 [DOI] [PubMed] [Google Scholar]
- 10.Helms AS, Tang VT, O’Leary TS, Friedline S, Wauchope M, Arora A, Wasserman AH, Smith ED, Lee LM, Wen X, et al. Effects of MYBPC3 loss of function mutations preceding hypertrophic cardiomyopathy. JCI Insight. 2019;26:133782 doi: 10.1172/jci.insight.133782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erdmann J, Raible J, Maki-Abadi J, Hummel M, Hammann J, Wollnik B, Frantz E, Fleck E, Hetzer R, Regitz-Zagrosek V. Spectrum of clinical phenotypes and gene variants in cardiac myosin-binding protein C mutation carriers with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2001;38:322–330. doi: 10.1016/s0735-1097(01)01387-0 [DOI] [PubMed] [Google Scholar]
- 12.Helms AS, Davis FM, Coleman D, Bartolone SN, Glazier AA, Pagani F, Yob JM, Sadayappan S, Pedersen E, Lyons R, et al. Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:434–443. doi: 10.1161/CIRCGENETICS.113.000448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alders M, Jongbloed R, Deelen W, van den Wijngaard A, Doevendans P, Ten Cate F, Regitz-Zagrosek V, Vosberg HP, van Langen I, Wilde A, et al. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J. 2003;24:1848–1853. doi: 10.1016/s0195-668x(03)00466-4 [DOI] [PubMed] [Google Scholar]
- 14.Daw EW, Chen SN, Czernuszewicz G, Lombardi R, Lu Y, Ma J, Roberts R, Shete S, Marian AJ. Genome-wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum Mol Genet. 2007;16:2463–2471. doi: 10.1093/hmg/ddm202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claes GR, van Tienen FH, Lindsey P, Krapels IP, Helderman-van den Enden AT, Hoos MB, Barrois YE, Janssen JW, Paulussen AD, Sels JW, et al. Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur Heart J. 2016;37:1815–1822. doi: 10.1093/eurheartj/ehv522 [DOI] [PubMed] [Google Scholar]
- 16.Helms AS, Day SM. Hypertrophic cardiomyopathy: single gene disease or complex trait? Eur Heart J. 2016;37:1823–1825. doi: 10.1093/eurheartj/ehv562 [DOI] [PubMed] [Google Scholar]
- 17.Wooten EC, Hebl VB, Wolf MJ, Greytak SR, Orr NM, Draper I, Calvino JE, Kapur NK, Maron MS, Kullo IJ, et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2013;6:10–18. doi: 10.1161/CIRCGENETICS.112.965277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singer ES, Ingles J, Semsarian C, Bagnall RD. Key value of RNA analysis of MYBPC3 splice-site variants in hypertrophic cardiomyopathy. Circ Genom Precis Med. 2019;12:e002368 doi: 10.1161/CIRCGEN.118.002368 [DOI] [PubMed] [Google Scholar]
- 19.Ito K, Patel PN, Gorham JM, McDonough B, DePalma SR, Adler EE, Lam L, MacRae CA, Mohiuddin SM, Fatkin D, et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc Natl Acad Sci U S A. 2017;114:7689–7694. doi: 10.1073/pnas.1707741114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toepfer CN, Wakimoto H, Garfinkel AC, McDonough B, Liao D, Jiang J, Tai AC, Gorham JM, Lunde IG, Lun M, et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci Transl Med. 2019;11:eaat1199 doi: 10.1126/scitranslmed.aat1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giles J, Patel JR, Miller A, Iverson E, Fitzsimons D, Moss RL. Recovery of left ventricular function following in vivo reexpression of cardiac myosin binding protein C. J Gen Physiol. 2019;151:77–89. doi: 10.1085/jgp.201812238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stöhr A, Friedrich FW, Flenner F, Geertz B, Eder A, Schaaf S, Hirt MN, Uebeler J, Schlossarek S, Carrier L, et al. Contractile abnormalities and altered drug response in engineered heart tissue from Mybpc3-targeted knock-in mice. J Mol Cell Cardiol. 2013;63:189–198. doi: 10.1016/j.yjmcc.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 23.de Lange WJ, Grimes AC, Hegge LF, Ralphe JC. Ablation of cardiac myosin-binding protein-C accelerates contractile kinetics in engineered cardiac tissue. J Gen Physiol. 2013;141:73–84. doi: 10.1085/jgp.201210837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Velzen HG, Schinkel AFL, Oldenburg RA, van Slegtenhorst MA, Frohn-Mulder IME, van der Velden J, Michels M. Clinical characteristics and long-term outcome of hypertrophic cardiomyopathy in individuals with a MYBPC3 (myosin-binding protein C) founder mutation. Circ Cardiovasc Genet. 2017;10:e001660 doi: 10.1161/CIRCGENETICS.116.001660 [DOI] [PubMed] [Google Scholar]
- 25.Teirlinck CH, Senni F, Malti RE, Majoor-Krakauer D, Fellmann F, Millat G, André-Fouët X, Pernot F, Stumpf M, Boutarin J, et al. A human MYBPC3 mutation appearing about 10 centuries ago results in a hypertrophic cardiomyopathy with delayed onset, moderate evolution but with a risk of sudden death. BMC Med Genet. 2012;13:105 doi: 10.1186/1471-2350-13-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jääskeläinen P, Miettinen R, Kärkkäinen P, Toivonen L, Laakso M, Kuusisto J. Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann Med. 2004;36:23–32. doi: 10.1080/07853890310017161 [DOI] [PubMed] [Google Scholar]
- 27.Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105:397–408. doi: 10.1093/cvr/cvv025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang XL, De S, McIntosh LP, Paetzel M. Structural characterization of the C3 domain of cardiac myosin binding protein C and its hypertrophic cardiomyopathy-related R502W mutant. Biochemistry. 2014;53:5332–5342. doi: 10.1021/bi500784g [DOI] [PubMed] [Google Scholar]
- 29.Wijnker PJ, Friedrich FW, Dutsch A, Reischmann S, Eder A, Mannhardt I, Mearini G, Eschenhagen T, van der Velden J, Carrier L. Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue. J Mol Cell Cardiol. 2016;97:82–92. doi: 10.1016/j.yjmcc.2016.03.003 [DOI] [PubMed] [Google Scholar]
- 30.Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, Li N, Felkin L, Ingold N, Govind R, et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. 2019;11:5 doi: 10.1186/s13073-019-0616-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.