

Summary
The gut epithelium is populated by intraepithelial lymphocytes (IELs), a heterogeneous T cell population with cytotoxic and regulatory properties, which can be acquired at the epithelial layer. However, the role of T cell receptor (TCR) in this process remains unclear. Single-cell transcriptomic analyses revealed distinct clonal expansions between cell states, with CD4+CD8αα+ IELs being one of the least diverse populations. Conditional deletion of TCR on differentiating CD4+ T cells or of MHCII on intestinal epithelial cells prevented CD4+CD8αα+ IEL differentiation. However, TCR ablation on differentiated CD4+CD8αα+ IELs, or long-term cognate antigen withdraw, did not affect their maintenance. TCR re-engagement of antigen-specific CD4+CD8αα+ IELs by Listeria monocytogenes did not alter their state but correlated with reduced bacterial invasion. Thus, local antigen recognition is an essential signal for differentiation of CD4+ T cells at the epithelium yet differentiated IELs are able to preserve an effector program in the absence of TCR signaling.
eTOC Blurb
Immune cell adaptation to the harsh intestinal environment is key for healthy gut maintenance. Bilate et al. showed that a subpopulation of intraepithelial lymphocytes is clonally expanded and depends on antigen receptor signaling for their differentiation. Once differentiated, these cells can thrive without antigen, highlighting the ability of these cells to maintain function under different contexts.
Graphical Abstract
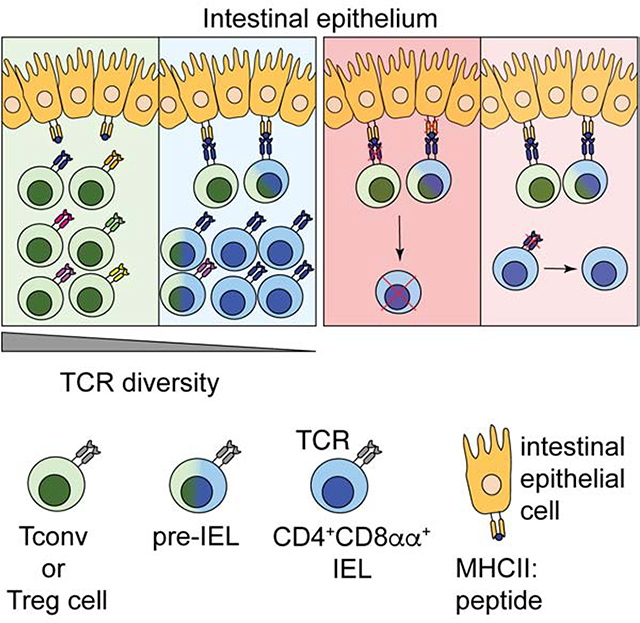
Introduction
T cell receptor (TCR) diversity of circulating T lymphocytes is ontogenically determined by thymic selection while T cell localization and repertoire are largely dependent on exposure to, and recognition of, tissue-specific antigens (Hogquist and Jameson, 2014). In addition to these features, tissue imprinting influences T cell subset differentiation and function (Faria et al., 2017). In the intestine, T cells located in the lamina propria (LP) and the intraepithelial (IE) compartment display gut-specific characteristics, such as expression of gut-homing integrins, but differ markedly in TCR repertoire, migration patterns, and function (McDonald et al., 2018).
Intestinal intraepithelial lymphocytes (IELs) comprise a heterogeneous population of T cells, yet a substantial proportion of IELs, or “fully differentiated IELs”, share common features such as tissue residency, cytotoxic potential, activated phenotype, and expression of CD8αα homodimers (Cheroutre et al., 2011; McDonald et al., 2018). Previous studies propose that CD8αα homodimers, in contrast to conventional CD8αβ heterodimers, work as TCR co-repressors by binding to thymus leukemia (TL) antigen expressed on epithelial cells, resulting in decreased antigen sensitivity of the TCR (Cheroutre and Lambolez, 2008). Similar to regulatory T cells (Treg cells) and invariant natural killer cells (iNKT), CD8αα+TCRαβ+ (or natural IELs, nIEL) are agonist-selected in the thymus (Leishman et al., 2002; Moran et al., 2011; Yamagata et al., 2004), suggesting an important role for TCR specificity in IEL development. Additionally, transgenic mice carrying an αβTCR from naturally-occurring CD8αα+TCRαβ+ T cells preferentially differentiate towards this lineage, further indicating that TCR signaling strength itself may drive IEL fate (Mayans et al., 2014; McDonald et al., 2014).
In addition to the developmentally-imprinted TCR features of intestinal T cells, luminal stimulation and other gut-enriched factors, such as microbial metabolites and epithelial cell factors, further influence peripheral T cell differentiation and plasticity (Atarashi et al., 2013; Lathrop et al., 2011; Sujino et al., 2016; Yang et al., 2014). Conventional CD4+ T cells and Treg cells can differentiate into CD4+CD8αα+ IELs (also referred as differentiated CD4IELs or double-positive, DPIELs) in a microbiota-dependent manner upon migration to the gut epithelium (Bilate et al., 2016; Sujino et al., 2016). However, the role of TCR signaling in IEL differentiation, location, and function has not been established. This is of particular interest given the relatively low abundance of MHC class II-expressing cells in the gut epithelium and other barrier surfaces (Faria et al., 2017).
We sought to address how TCR properties and signaling modulate the location and plasticity of CD4+ T cells in the intestine. We combined TCR repertoire analysis with single-cell transcriptomics using a fate-mapping strategy that allowed us to track Treg cells and conventional CD4+ T cells (Tconv) as they migrate to the intestinal epithelium and differentiate into CD8αα-expressing IELs. Reduced TCR diversity was associated with terminal differentiation of CD4+ T cells into CD4+CD8αα+ IELs via a pre-IEL state. Ablation of surface TCR complexes on Treg cells impaired CD4+CD8αα+ IEL differentiation, suggesting that TCR expression is required for terminal T cell differentiation at the intestinal epithelium. However, TCR ablation or absence of cognate TCR ligand in fully differentiated CD4+CD8αα+ IELs had little, if any, impact on their accumulation or the maintenance of the IEL program under homeostatic conditions or during enteric infection. Our findings indicate that TCR expression and local MHC class II on intestinal epithelial cells (IECs) are required for T cell plasticity at the intestinal epithelium, but not for the maintenance of the IEL program.
Results
TCR diversity follows the transcriptional trajectories of intraepithelial CD4+ T cells
CD4+ T cells acquire an IEL program at the gut epithelium while losing hallmarks of peripheral CD4+ T cells or Treg cells, including the expression of the transcription factors ThPOK and Foxp3, respectively (Sujino et al., 2016). To correlate intestinal epithelium-induced CD4+ T cell plasticity with specific TCR usage in this process, we performed 5’ single-cell RNA sequencing (scRNAseq) coupled to TCRseq analysis using the Chromium Single Cell V(D)J platform (10X Genomics). We analyzed tamoxifen-treated Foxp3eGFP-creERT2 Rosa26|s|-td-Tomato (iFoxp3Tomato) mice by sorting all Treg cell-derived Tomato+ (library 1) or Tconv cell-derived Tomato− (library 2) CD4+CD8β− T cells, therefore examining the whole spectrum of heterogeneity of CD4+ T cells within the epithelium. We confirmed the presence of CD4+CD8αα+ IELs and Treg cells in sorted cells by expression of CD8α and Foxp3, respectively (Figure S1A). This strategy allowed analysis of intra-mouse TCR sharing between Tconv cells, Treg cells, transitioning “pre-IELs” (London et al., 2020), and CD4+CD8αα+ IELs differentiated from them. From the two libraries, we obtained a total of 1,294 scRNAseq profiles (898 for Tomato+ library 1 and 396 for Tomato− library 2) with paired αβTCR sequences for 952 cells (651 for Tomato+ library 1 and 301 for Tomato− library 2) (Figure S1B).
We identified 8 clusters ordered by cell number (0–7) visualized by Uniform Manifold Approximation and Projection (UMAP), and distributed among Tomato+ and Tomato− libraries (Figure 1A, Figure S1C, Table S1), which was overall comparable to what we identified in our parallel study describing the molecular mechanisms of the differentiation towards CD4+CD8αα+ IELs (London et al., 2020). Cluster (7) was characterized by cycling cells and segregated by a strong proliferation gene signature. Due to the low number of cells in this cluster (20 cells), we opted to exclude them from our downstream analysis. Clusters 3, 5, and 6 were composed primarily of Tomato+ cells that express genes ascribed to Treg cells (Figure 1A, Figure S1C, D, Table S1). Treg cell-like (cluster 1) was ascribed to cells expressing a Treg cell profile, yet to a lower extent than the Treg cells of cluster 3, while also expressing some IEL genes (Cd7 and Gzmb) (Table S1). We also identified three non-Treg cell clusters, one of them composed mostly of Tomato+ cells (cluster 0), indicating Treg cell origin, while the other two (clusters 2 and 4) contained a mix of Tomato+ and Tomato− cells (Figure 1A, Figure S1C, Table S1).
Figure 1. Clonal distribution of intraepithelial CD4+ T cells follows single-cell trajectories.

(A-F) Tomato+ (library 1) and Tomato− (library 2) CD4+ T cells from the intestinal epithelium (IE) of a iFoxp3Tomato mouse were sorted for scRNAseq. Cells clustered into 8 (C_0–7) populations, including sub-clusters (3a, 5a) and visualized by UMAP. Cluster names correspond to colors as indicated. (A) Slingshot pseudotime trajectory of Tomato+ (top) and Tomato− (bottom) cells on the UMAP. CD4+CD8αα+-specific slingshot lineages (black lines) depicted in differentiation order. White numbers indicate clusters. (B) Cells ordered along diffusion component 1, separated by clusters of Tomato+ (TP, top) and Tomato− (TN, bottom) cells. (C) RNA velocity analysis vectors (arrows) of Tomato+ (top) and Tomato− (bottom) cells on the UMAP. (D) Diversity 50 (D50) estimate based on paired αβTCRs. (E) Circos plot of paired αβTCR CDR3 sequences; clones ordered clockwise in decreasing size. Links denote clonal sharing between populations of adjacent clusters; clonal expansions of less than 10 cells in purple and at least 10 cells in green among Tomato+ (left) and Tomato− (right) cells. (F) Top expanded clones per Tomato+ (TP, red and blue) and Tomato− (TN, orange and green) cells. Dashed line marks top limit of CD4+CD8αα+ cluster 2. N=1 mouse, 1,294 sequenced cells. See also Figure S1, Table S1, S2.
The homogeneous cluster 2 contained CD8α-expressing cells with a “full IEL program” (McDonald et al., 2018), which included the expression of Itgae (CD103) (Figure 1A, Figure S1C–F, Table S1). The acquisition of IEL markers was inversely correlated with expression of genes downstream of TCR signaling and co-stimulation, such as Tnfrsf4 (encoding Ox40) (Figure S1E, F). Pseudotime analysis combined with RNA velocity extrapolation allowed inference of a peripheral CD4+ T cell differentiation hierarchy within the epithelium (Figure 1A–C, Figure S1G). We observed potential trajectories that lead to the differentiation of CD4+CD8αα+ IELs through a common intermediate stage from either Treg cells (Tomato+) or from Tconv cells (Tomato−) (Figure 1A). Clusters 0 and 4 directly preceded the CD4+CD8αα+ IEL cluster 2, and we refer to them as “pre-IEL1” (cluster 0) and “pre-IEL2” (cluster 4). Trajectory analysis showed that the pre-IEL2 cluster precedes the pre-IEL1 cluster suggesting a less differentiated state (Figure 1A). By applying diffusion component pseudotime ordering of cells, we observed a similar degree of differentiation among clusters containing cells differentiated from both Treg and Tconv cells (Figure 1B). Relative to pre-IELs, the ratio of spliced to unspliced RNA of IEL signature genes was higher in CD4+CD8αα+ IELs, suggesting the latter were in a final stage of differentiation where nascent mRNA molecules are no longer being produced (Figure S1G). Together, these findings suggest that CD4+CD8αα+ IELs represent a final stage of differentiation and that peripheral CD4+ T cells acquire a similar IEL program regardless of their origin.
TCRαβ repertoire information data obtained from each cell from our scRNAseq dataset, revealed variable amounts of clonal expansion and sharing across the identified clusters (Figure 1D–F, Figure S1H–J, Table S2). We used the diversity 50 score (D50) to estimate diversity (scores varied from least diverse 0 to most diverse 0.5) and Morisita-Horn index (MHI) to quantify clonal overlap (100% clonal overlap = 1). Pre-IEL1 (cluster 0) and CD4+CD8αα+ IEL (cluster 2) contained the least diverse cells, with a similar amount of clonal expansion, or dominance (Figure 1D). Pre-IEL2 (cluster 4) displayed intermediate clonal dominance between resting Treg cells (recent epithelial immigrants, REI; cluster 5) and Treg-like cells (cluster 1), further supporting the relatively less differentiated state of cluster 4 as suggested by the pseudotime and velocity analysis. Our analysis also revealed TCR clonal overlap between populations (Figure 1E, F, Figure S1I, J): pre-IEL1 and Treg-like cells had the highest clonal overlap (MHI = 0.41) among Tomato+ cells, while pre-IEL1 and pre-IEL2 displayed the highest clonal overlap (MHI = 0.34) among Tomato− cells. Pre-IEL1 and CD4+CD8αα+ IELs also showed clonal overlap among Tomato+ (MHI=0.29) and Tomato− (MHI=0.17), corroborating the less differentiated state for cells in pre-IEL2 (Figure 1A, E, F, Figure S1 J, Table S2). Tomato+ cells differentiated into pre-IELs, and then into CD4+CD8αα+ IELs (referred to as exTreg-IELs). All of the top expanded clones among the CD4+CD8αα+ IELs were also found in at least one more cluster, with distributions that followed the pseudotime trajectories (Figure 1A, E, F), suggesting expanded CD4+ T cells undergo differentiation at the epithelium. For example, the top expanded Treg cell-derived Tomato+ clones (clones 1TP and 7TP) were present in Treg cells, Treg-like cells, pre-IEL1 and CD4+CD8αα+ IEL clusters, while some other expanded clones (clones 3TP, 4TP, 6TP, 9TP,10TP, 12TP) were not found among CD4+CD8αα+ IELs but were shared between Treg cells, Treg-like cells and pre-IEL1 clusters. Of note, the top expanded Tconv cell-derived Tomato− clones were found within pre-IEL2 and CD4+CD8αα+ IEL clusters (Figure 1E, F, Table S2). It is possible that a fraction of precursors did not receive sufficient signals to convert into CD4+CD8αα+ IELs, or that the time required for differentiation was longer than the timeframe of our analysis. The combination of trajectory analysis, clonal distribution and overlap, with dominance scores, suggests that reduced TCR diversity and increased clonal expansion correlate with differentiation towards CD4+CD8αα+ IELs. Overall, our findings reveal TCR sharing among rather heterogeneous intraepithelial CD4+ T cell populations, with reduced diversity as cells differentiate into CD4+CD8αα+ IELs.
TCR repertoire dictates CD4+CD8αα+ IELs differentiation from epithelial T cell precursors
We next asked the extent to which the TCR can shape the fate of intraepithelial CD4+ T cells. We surveyed the TCRαβ repertoire of CD4+ T cells in the small intestine epithelium of 6 polyclonal mice using several inducible Foxp3 fate-mapping and Foxp3-reporter approaches, single-cell sorting the main subpopulations of CD4+ T cells found in the epithelium based on the expression of CD103, Foxp3 and CD8α: Tconv cells (CD103−CD8α−), Treg cells (Foxp3+), pre-IELs (CD103+CD8α−) and CD4+CD8αα+ IELs (CD103+CD8α+).
First, we used Foxp3RFPNur77GFP double reporter mice to determine clonal expansion and the extent of sharing between Tconv cells, Treg cells, pre-IELs and CD4+CD8αα+ IELs (Figure S2A). This approach does not allow for distinguishing of pre-IELs and CD4+CD8αα+ IELs originated from Tconv or Treg cells. While Tconv and Treg cells were very diverse in both mice analyzed, pre-IELs and CD4+CD8αα+ IELs were clonally expanded, similar to the results shown above. Expanded clones of pre-IELs and CD4+CD8αα+ IELs were shared among all sequenced populations (Figure 2 A, B, Figure S2B–D). Next, we used tamoxifen-treated iFoxp3Tomato mice to determine the extent of clonal overlap between CD4+CD8αα+ IELs differentiated from Treg or Tconv cells (exTreg-IELs Tomato+ vs CD4+CD8αα+ Tomato−) (Figure S2E). The CD8α−Tomato+ population in iFoxp3Tomato mice included not only Treg cells, but also Treg cell-derived pre-IELs, which likely contributed to the clonal expansion and reduced diversity of this population (Figure 2C, Figure S2F). The top expanded clones were shared between CD4+CD8αα+ IELs and exTreg-IELs, suggesting that they differentiated from the same precursors (Figure 2C–E, Figure S2F).
Figure 2. CD4+CD8αα+ IELs are clonally expanded with decreased TCR diversity.

(A-F) TCRβ and TCRα of single intestinal epithelium (IE) CD4+ T cells were sequenced via the MiSeq platform. (A, B) CD4+ conventional (Tconv; RFP−CD103−CD8α−), regulatory T cells (Treg; RFP+), pre-IEL (RFP−CD103+CD8α−) and CD4+CD8αα+ (RFP−CD103+CD8α+) from Foxp3RFPNur77GFP mice were analyzed. (A) αβTCR clonal diversity from 1 of 2 sequenced mice. Each slice represents a distinct αβTCR CDR3. Shared clones within each mouse in green, unique clones in white, and grey-scale represents expanded clones. Enclosed numbers indicate number of clones (numerator) and total number of cells (denominator). (B) Circos plot of paired αβTCR CDR3 sequences, with clones ordered clockwise in decreasing size. Links indicate clonal sharing between populations from 1 of 2 sequenced mice; clonal expansions of less than 10 cells in purple, and at least 10 cells in green. (C-E) CD4+ conventional (Tconv; Tomato− CD103−CD8α−), regulatory T cells mixed with Treg cells-derived pre-IELs (Treg and pre-IEL; Tomato+CD8α−), Tconv-derived pre-IELs (pre-IEL; Tomato−CD103+CD8α−), Treg cells-derived IELs (exTreg-IEL; Tomato+CD103+ CD8α+) and Tconv derived IELs (CD4+CD8αα+; Tomato− CD103+CD8α+) from Foxp3eGFP-cre-ERT2 x Rosa26lsl-tdTomato (iFoxp3Tom) mice were analyzed. (C) αβTCR clonal diversity from 1 of 2 sequenced mice. Label scheme as in (A). (D) Circos plot as in (B) of 1 of 2 sequenced mice. (E) Normalized Morisita-Horn index of paired αβTCR per population from 1 of 2 sequenced mice. (F) Diversity 50 (D50) estimate, pooled from all sequenced mice: Tconv (CD103−CD8α−), Treg cell (Foxp3RFP+ or Foxp3GFP+), pre-IEL (CD103+CD8α−), and CD4+CD8αα+ (CD103+CD8α+). (G-J) TCRα of Vα2+ Vβ6+ CD4+ T cells from three fixed-Vβ6 x Foxp3GFP mice were sequenced via the MiSeq platform as follows: Tconv (CD103−CD8α−), pre-IEL (CD103+CD8α−) and CD4+CD8αα+ (CD103+CD8α−). (G) D50 per population and mouse. (H, I) Circos plots as in (B) and (D) show clonal distribution and overlap of TCRα CDR3 sequences between populations within each mouse (private clones) (H) and sharing between mice (public clones) (I). Grey links denote clonal overlap. (J) Normalized Morisita-Horn index of TCRα per population and mouse. N=2 (A, B; C-E; F), N=3 (G-J). Significant p values as indicated [one-way ANOVA and Bonferroni (F, G)]. See also Figure S2 and Table S3.
Finally, we crossed iFoxp3Tomato with Foxp3GFP mice (iFoxp3Tomato Foxp3GFP) to specifically analyze the TCRαβ repertoire of distinct subsets of IELs, including Treg cells and exTreg-IELs derived from them. The following populations were analyzed after continuous tamoxifen administration: Treg cells (CD8α−, GFP+) and CD4+CD8αα+ IELs originated from either Tconv cells (CD8α+, Tomato−) or from Treg cells (CD8α+, Tomato+; exTreg-IEL) (Figure S2G). Treg cells isolated from the epithelium had a diverse repertoire, whereas CD4+CD8αα+ IELs and exTreg-IELs showed large clonal expansion and reduced TCR diversity (Figure S2H). Moreover, several expanded clones were shared between exTreg-IELs (Tomato+) and CD4+CD8αα+ IELs (Tomato−) (Figure S2H). In summary, our various strategies revealed that while Tconv and Treg cells consist of diverse and largely unique clones, pre-IELs and CD4+CD8αα+ IELs are more clonally restricted and exhibit TCR clonal overlap to a greater extent than their less differentiated precursors, regardless of origin (Figure 2E, F, Figure S2B, D, Table S3). These results indicate that a single Tconv cell precursor can sequentially differentiate into a Treg cell before becoming a CD4+CD8αα+ IEL through a pre-IEL stage.
To further address whether TCR identity controls the differentiation of migrating CD4+ T cells into pre-IELs and CD4+CD8αα+ IELs, we took advantage of the transnuclear (TN) monoclonal strain harboring a commensal-specific Vα2Vβ6 TCR rearrangement that predisposes cells to become CD4+CD8αα+ IELs in the epithelium, in a microbiota-dependent manner (Bilate et al., 2016; Bousbaine et al., 2020). We crossed these mice to WT animals to obtain mice carrying only the Vβ6 chain (referred as fixed-Vβ6 mice) from their offspring, as a means to interrogate TCRα diversity in a diverse setting yet with an imposed repertoire bias dictated by the fixed TCRβ chain. The frequency of Vα2+ cells among Vβ6+ CD4+ T cells was similar in blood, mesenteric lymph nodes (mLN) and IE among fixed-Vβ6 mice (Figure S2I). Single-cell TCRseq analysis of sorted epithelial Vα2+Vβ6+ T cells showed that the same Tconv cell clones could be found in all three mice sequenced, yet the Tconv cells were diverse, as seen in polyclonal mice (Figure 2G–J). Likewise, pre-IELs and CD4+CD8αα+ IELs were clonally expanded (Figure 2G–I, Figure S2J). Clones were extensively shared not only between the sub-populations, but also among the three mice analyzed (Figure 2H–J, Figure S2J). All three mice contained IELs sharing the same TCRα rearrangement used by the TN monoclonal strain, and another CDR3 sequence that differed in only one amino acid from the TN TCR (Figure S2J, Table S3). Thus, by decreasing recombination potential, we found public TCRs among the different IEL states across multiple mice despite the variation in proportion of cells in each state among mice (Figure 2I, J, Figure S2J, K). Taken together, our results indicate that selected clones of Tconv and Treg cells migrate to the epithelium and expand as pre-IELs before they can fully differentiate into CD4+CD8αα+ IELs.
TCR signaling is required for CD4+CD8αα+ IEL development
Our TCR repertoire analyses suggest the existence of a “TCR-bottleneck” between Tconv or Treg cells and pre-IELs, which precedes complete IEL expansion and differentiation in the epithelium. To confirm that downstream TCR signaling is associated with peripheral IEL differentiation, we analyzed Nur77 expression along differentiating peripheral IELs using Foxp3RFPNur77GFP double reporter mice (Figure S3A–C). CD4+CD8αα+ IELs (CD4+CD103+CD8α+) and pre-IELs (CD4+CD103+CD8α−) expressed lower amounts of Nur77 when compared to recently-immigrated CD4+CD103− (Tconv) cells or to Treg cells found in the epithelium (Figure 3A, B). Thus, Nur77 expression is inversely associated with acquisition of CD103 and CD8αα by CD4+ T cells in the epithelium, suggestive of a role for TCR signaling in IEL differentiation from peripheral CD4+ T cells.
Figure 3. TCR signaling is required for CD4+CD8αα+ IEL differentiation.

(A, B) Nur77 as measured by GFP fluorescence (A) and frequency (B) among Foxp3+ regulatory T cells (Treg, RFP+; blue), conventional CD4+ T cells (Tconv, RFP−CD8α−; grey), pre-IELs (RFP−CD103+CD8α−; black) and CD4+CD8αα+ IELs (RFP− CD8α+TL-Tetramer+; red) in the intestinal epithelium (IE) of Nur77GFPFoxp3RFP mice. (C-E) Flow cytometry analysis of CD4+ T cells in the IE of Ox40WT(Trac) (Trac+/+ OX40cre+/− or Tracf/f Ox40cre−/−) or OX40Δ(Trac) (Tracf/f Ox40cre+/−) mice. (C) Dot plots of surface CD8α and TCRβ of CD4+ T cells. Data are expressed as mean +/− SEM of individual mice (n=5 of two independent experiments) (D, E) Frequency of CD4+CD8αα+IELs (CD8α+TL-Tetramer+) among total CD4+ T cells (D) and within TCRβ-sufficient cells from Ox40WT(Trac) (white bar) or Ox40 ΔTrac) (grey bar) mice, or TCRβ-deficient cells from Ox40 Δ (Trac) (red bar) mice (E). Data are expressed as mean +/− SEM of individual mice (n=5–8 of two independent experiments). Significant p values as indicated [student’s t test (D) or one-way ANOVA and Bonferroni (B, E)]. See also Figure S3.
To directly assess the requirement of the TCR for CD4+CD8αα+ IEL differentiation, we employed multiple cre–mediated TCR ablation strategies targeting different stages or subsets of CD4+ T cells. First, we specifically addressed whether TCR signaling was required for Treg cell plasticity in the gut epithelium by crossing Tracf/f mice to iFoxp3Tomato mice (iFoxp3TomΔ(Trac)), which allowed for the tracking of ex-Treg cells as they lost surface TCR expression upon tamoxifen administration (Figure S3D–F). We further compared TCR-sufficient and TCR-deficient CD4+ T cells in each iFoxp3TomΔ(Trac) mouse based on surface TCRβ expression. Whereas CD4+CD8αα+ IEL differentiation from TCR-expressing (TCRβ S+; Tomato− or Tomato+) cells was similar between iFoxp3Tom and iFoxp3TomΔ(Trac) mice, it was significantly impaired among TCR-deficient (TCRβ S− Tomato+) cells of iFoxp3TomΔ(Trac) mice (Figure S3F–H).
Our scRNAseq data showed that Tnfrsf4 (Ox40) expression changed in a developmentally controlled manner, peaking in Treg cells and then rapidly decaying in pre-IELs (as shown in Figure S1F). Thus, we used the Ox40cre driver (Reis et al., 2013) to delete the TCR from pre-IEL or CD4+CD8αα+ IEL precursors. We confirmed TCR deletion in activated CD4+ T cells and Treg cells in the mLN of Tracf/f x Ox40cre (Ox40Δ(Trac)) mice by flow cytometry (Figure S3I–M). In the epithelium, while the total frequency of CD4+ T cells remained the same in Ox40WT(Trac) and Ox40Δ(Trac) mice (Figure S3N), TCR ablation significantly decreased the CD4+CD8αα+ population (Figure 3C, D Figure S3O). Moreover, CD4+CD8αα+ IELs were only observed among TCR-sufficient cells (Figure 3C, E). These data indicate that the differentiation of both Treg and Tconv cells to CD4+CD8αα+ IELs requires TCR expression.
MHCII expression on epithelial cells modulates CD4+CD8αα+ IEL differentiation
Recent studies have suggested a role for intestinal epithelial cell-mediated antigen presentation via MHCII in the regulation of intestinal CD4+ T cell function (Biton et al., 2018; Koyama et al., 2019). We asked whether local MHCII expression by IECs was required for CD4+CD8αα+ IEL differentiation or maintenance. We targeted MHC class II expression exclusively on IECs by crossing Vil1creERT2 mice to H2-Ab1f/f mice (iVil1Δ(MHCII)) (Figure S4A–C). Tamoxifen treatment of iVil1Δ(MHCII) mice starting at 5–7 weeks of age (prior to the appearance of CD4+CD8αα+ IELs in the epithelium) did not affect the frequency of total CD4+ T cells in the epithelium 5–6 weeks later; however, it led to a significant reduction in the frequency of CD4+CD8αα+ as well as pre-IELs, which was accompanied by an increase in Treg cell frequency (Figure 4A). Tamoxifen treatment of 10.5–12 or 16-week-old mice, which typically carry a sizable population of CD4+CD8αα+ IELs, also significantly impacted CD4+CD8αα+ IEL frequencies without affecting the pre-IEL population (Figure 4B, C), suggesting that MHCII expression on IECs is required for continuous differentiation and/or maintenance of CD4+CD8αα+ IELs in adult mice.
Figure 4. MHCII expression by epithelial cells is required for CD4+CD8αα+ IEL differentiation.

(A-C) Flow cytometry analysis of the intestinal epithelium (IE) of iVil1WT(MHCII) (H2-Ab1+/+ Vil1creERT2+/− or H2-Ab1f/f Vil1creERT2−/−) and iVil1 Δ(MHCII) (H2-Ab1f/f Vil1creERT2+/+) mice after tamoxifen administration to mice of different ages as indicated. Dot plots of surface CD8α and TL-Tetramer expression among CD4+ T cells (left). Frequencies of CD4+CD8αα+ IELs (CD4+CD8α+TL-Tetramer+) or Foxp3+ regulatory cells (Treg) among CD4+ T cells (middle). Frequencies of pre-IELs (CD4+CD103+) among CD4+SP (CD4+CD8α−Foxp3−) cells and frequencies of total CD4+ cells among TCRαβ+ cells (right). Data are expressed as mean +/− SEM of individual mice (n=7–15, 3–6 independent experiments per timepoint). Significant p values as indicated [student’s t test (A-C)]. See also Figure S4.
To dissociate the role of MHCII on IECs in the development versus maintenance of CD4+CD8αα+ IELs, we assessed whether continuous generation of CD4+CD8αα+ IELs takes place through the adult life of specific pathogen free (SPF) mice using our fate mapping model (iFoxp3Tomato mice). We administered tamoxifen at different ages either as a single dose or continuous labeling from a young age, when very little to no CD4+CD8αα+ IELs are present, or at an older age (12 weeks old) they can be readily found (Figure S4D). We could still detect Tomato+ CD4+CD8αα+ IELs even when tamoxifen was administered to 12-week old mice, confirming the continuous generation of CD4+CD8αα+ IELs from Treg cells (Figure S4E). Therefore, we conclude that deletion of MHCII on epithelial cells impairs the new generation of CD4+CD8αα+ IELs, rather than the maintenance of these cells.
TCR signaling is largely dispensable for IEL program maintenance
The transition to an IEL program includes upregulation of NK- and cytolytic- molecules as well as CD8αα (Cheroutre et al., 2011; McDonald et al., 2018), with concomitant downregulation of TCR signaling. To evaluate the role of TCR signaling in establishing the IEL program, and in the maintenance of differentiated IELs, we crossed Tracf/f mice to those expressing cre under the enhancer I of the Cd8a gene (E8IΔ(Trac)). E8I is required for CD8α expression on mature T cells and CD8αα+ IELs, but not required for CD8αβ or CD8αα expression in developing thymocytes (Ellmeier et al., 1997). In E8IΔ(Trac) mice, TCR deletion was accompanied by a decrease in CD8α−expressing TCRαβ+ cells in the mLN and IE, including CD4−CD8β+CD8α+ T cells and CD4−CD8β−CD8αα+ TCRαβ+ natural IELs (nIEL) (Figure S5A–F). In the case of peripheral CD4+ T cells, which only express CD8αα in the final stages of their differentiation into IELs, this model allowed us to selectively address the role of the TCR in the maintenance of differentiated CD4+CD8αα+ IELs. We did not observe any significant changes in the accumulation of CD4+CD8αα+ IELs in E8IΔ(Trac) mice when compared to WT littermates (E8IWT(Trac)), even in older animals (Figure 5A, B), suggesting that upon terminal differentiation, these cells do not rely on the TCR for their maintenance in the epithelium. Whereas proliferation, measured by Edu incorporation, was similar among Tconv cells from E8IWT(Trac) and E8IΔ(Trac) mice, remaining TCR-expressing CD4+CD8αα+ IELs in E8IΔ(Trac) mice proliferated more than those of E8IWT(Trac) mice, but only in older animals (Figure 5C, D). Therefore, similar frequencies of CD4+CD8αα+ IELs cannot be exclusively attributed to differential proliferative abilities of TCR-sufficient and TCR-deficient cells. Similar proliferation rates were observed in other CD8αα-expressing IEL subsets, including CD8αβ+CD8αα+ TCRαβ+ (also referred as CD8IELs) and CD8αα+ nIELs (Figure S5G, H). TCR-deficient cells did not undergo apoptosis at a higher rate than TCR-sufficient cells, as annexin V staining showed similar frequencies of apoptotic cells regardless of surface TCRβ expression in all IEL populations (Figure 5E, F, Figure S5 I, J).
Figure 5. TCR signaling is not essential for CD4+CD8αα+ IEL maintenance.

(A-H) Flow cytometry analysis of intestinal epithelium (IE) of 8–22 week-old E8IWT(Trac) (Trac+/+ E8Icre+ or Trac+/+ E8Icre− or Tracf/f E8Icre−) or E8I Δ(Trac) (Tracf/f E8Icre+) mice, grouped by age. (A) Dot plots of surface CD8α and TL-Tetramer expression among CD4+ T cells in E8IWT(Trac) (left) or E8IΔ(Trac) (right) mice. (B) Frequency of CD4+CD8αα+ (CD8α+TL-Tetramer+) among CD4+ T cells in E8IWT(Trac) and E8IΔ(Trac) mice. (C, D) Proliferation frequency (measured by EdU incorporation) of CD4+CD8α−Foxp3− cells (C) among cells with or without surface TCRβ expression (D). (E, F) Annexin V expression among TCRβ sufficient and deficient CD4+CD8αα+ IELs. (E) Dot plots of Annexin V and surface TCRβ among CD4+CD8αα+ IELs in E8IWT(Trac) (left) or E8IΔ(Trac) (right) mice. (F) Frequencies of Annexin V+ cells among TCR sufficient and deficient CD4+CD8αα+IELs. (G, H) Frequencies of IFNγ (G) and granzyme B (Gzmb) (H) production upon PMA and ionomycin ex-vivo stimulation among CD4+CD8αα+s. (I-K) Bulk RNA-sequencing of TCRβ+ CD4+CD8αα+ IELs from E8IWT(Trac) and TCRβ− CD4+CD8αα+ IELs from E8IΔ(Trac) mice (n=3 per group). Volcano plot of differentially expressed genes between indicated populations (p<0.05, in color) (I), selected differentially-enriched gene ontology (GO) pathways between groups (J), and gene set enrichment analysis (GSEA) of CD4+CD8αα+ IEL program, defined by the top differentially-expressed genes in the CD4+CD8αα+ cluster in our single cell RNA-Sequencing from Figure 1 (K). (L-N) Flow cytometry analysis of bone marrow chimeras in sub-lethally irradiated Rag1−/− hosts reconstituted with 1:1 ratio of WT CD45.1+ and E8IΔ(Trac) CD45.2+ cells. Dot plots (L) and frequency (M) of WT CD45.1+ (red) and E8IΔ(Trac) CD45.2+ (blue) cells among non-αβT cells (γδT cells and non-T cells), natural IELs (nIEL, CD4−CD8αα+CD8β−TL-Tetramer+), CD8αβ+CD8αα+ cells (CD4−CD8αα+CD8β+TL-Tetramer+), and CD4+CD8αα+ IELs in the epithelium. (N) Histograms of surface TCRβ expression in WT CD45.1+ (red line) and E8IΔ(Trac) CD45.2+ (blue line) cells of indicated populations. Frequency data are expressed as mean +/− SEM (B-F). Significant p values as indicated [student’s t test (B, C, G, H) or one-way ANOVA and Bonferroni (D, F)]. n=10–16, 6 experiments in total (B), n=4–14, 2 experiments (C, D), n=5–8, 2 experiments (F) n=4–6, 2 experiments (G, H) and n=12, 3 independent experiments (M). See also Figure S5 and Tables S4–S6.
Common IEL functional readouts such as IFNγ production and granzyme B expression showed that hallmarks of the CD4+CD8αα+ IEL phenotype are maintained in the absence of the TCR (Figure 5G, H). Analysis of CD8αβ+CD8αα+ and nIELs also revealed intact IFNγ and granzyme B production, despite TCR loss (Figure S5K–N). Taken together, our results suggest that CD4+CD8αα+ IEL accumulation and at least some of the characteristic features of IELs are maintained in the absence of TCR signaling.
To address the extent to which the IEL program can be affected or maintained in the absence of TCR signaling, we performed bulk RNAseq on TCR-expressing CD4+CD8αα+ IELs from E8IWT(Trac) mice and on TCR-deficient CD4+CD8αα+ IELs from E8IΔ(Trac) mice (Figure S5O). While genes related to TCR activation or signaling, cell cycle or proliferation, anti-apoptosis and protein transport showed reduced expression in cells lacking their TCR, most IEL-related genes were not impacted in E8IΔ(Trac) mice, suggesting the IEL program is maintained in the absence of TCR expression (Figure 5I–K, Table S4). Gene ontology (GO) enrichment analysis showed that only a few pathways, such as protein phosphorylation, were upregulated in TCR-deficient cells (Figure 5J). IEL genes were not changed among CD8αβ+CD8αα+ IELs or nIELs in E8IΔ(Trac) mice, but genes related to TCR signaling were decreased in TCR-deficient cells (Figure S5P, Q, Table S5,6). Of note, Lag3 (Lymphocyte activation gene-3), a TCR inhibitory co-receptor, was downmodulated in all 3 IEL subsets lacking TCR expression (Figure 5I, Table S4–6).
To further evaluate whether the TCR confers any competitive advantage to IELs, we reconstituted sub-lethally irradiated Rag1−/− mice with a 1:1 mix of congenically-marked bone marrow from WT CD45.1+ and E8IΔ(Trac) CD45.2+ mice (Figure 5L). Whereas the ratio of WT to E8IΔ(Trac) remained approximately 1:1 in non-αβT cells, this ratio was significantly skewed towards WT among CD8αβ+CD8αα+ IELs (Figure 5L–N), indicating that TCR-deficient CD8αβ+CD8αα+ IELs were outcompeted by TCR-expressing cells, as expected since their peripheral precursors are affected by this deletion strategy. In contrast, within both nIELs, present since early age (McDonald et al., 2018), and CD4+CD8αα+ IELs, the WT: E8IΔ(Trac) ratio was much less skewed, and surface-TCRβ− cells were readily detected among these populations (Figure 5L–N). Thus, CD4+CD8αα+ IELs are less sensitive than CD8αβ+CD8αα+ IELs to TCR loss. These data indicate that TCR signaling is mostly dispensable for CD4+CD8αα+ IEL maintenance.
Functional features of CD4+CD8αα+ IELs are preserved in the absence of cognate TCR interactions
To specifically address the requirement for cognate TCR ligands in pre-IEL and CD4+CD8αα+ IEL maintenance, we utilized the ovalbumin (OVA)-specific OTII TCR transgenic model. In this system, a fraction of OTII T cells can readily down-modulate ThPOK and differentiate into CD4+CD8αα+ IELs after one week of oral OVA exposure (Reis et al., 2013; Sujino et al., 2016). We asked how long pre-IELs and CD4+CD8αα+ IELs could persist in the absence of cognate TCR interactions. Rag1−/− hosts received cells from OTII-Zbtb7bGFP Rag1−/− donors and fed a 1% OVA-containing diet for 10 days. Animals were then switched to a regular diet and pre-IEL and CD4+CD8αα+ IEL frequencies and their cytokine potential was analyzed at different time points up to 10 weeks post OVA removal (Figure 6A). The relative abundance of transferred OTII cells found in mLN and IE decreased over time (Figure S6A, B). In contrast, the frequency of CD4+CD8αα+ IELs increased until 20 days after OVA withdrawal, then reached a plateau (Figure 6B). In this setting, we cannot discern if pre-IELs acquire the CD4+CD8αα+ IEL phenotype in the absence of TCR engagement. Moreover, functional IEL parameters such as production of IFNγ, expression of granzyme B, NK receptor 2B4 (CD244) and down-modulation of ThPOK, were preserved in the absence of the cognate antigen, albeit to lower extents in later timepoints (Figure 6C and Figure S6C, D). We conclude that pre-IELs and CD4+CD8αα+ IELs induced by dietary antigen are long-lived and do not require cognate TCR interaction to survive or to maintain their functional phenotype.
Figure 6. Cognate ligand interaction is dispensable for the maintenance of CD4+CD8αα+ IELs under steady state or after Listeria monocytogenes infection.

(A-C) Total splenocytes and lymph node cells from OTII Zbtb7bGFP Rag1−/− mice were transferred to Rag1−/− animals prior to ovalbumin (OVA) diet treatment and analyzed at different time points after OVA withdrawal as indicated. (A) Experimental layout. (B) Dot plots of surface CD8α and TL-Tetramer expression among total OTII CD4+ T cells (left) and of CD8α and CD103 expression among CD8β− CD4+ T cells (middle) 36 days after OVA removal. Graphs show frequencies of CD4+CD8αα+IELs among total OTII (Vα2+) CD4+ T cells (middle) and of pre-IELs (CD8α− CD103+) among CD4 single positive (CD4+SP) cells (OTII CD4+ CD8α−) (right) at indicated times after OVA removal in white and after continuous OVA diet for 30 or 82 days in grey bars. (C) Dot plots of surface CD8α and CD103 expression among total OTII CD4+ T cells (left), granzyme B (Gzmb) and Zbtb7b-GFP among CD4+CD8αα+ (CD103+CD8α+, middle) and IFNγ and IL-17A among CD4+CD8αα+ (right) 36 days after OVA removal and ex-vivo stimulation with PMA and ionomycin. Graphs represent frequencies of IFNγ and Gzmb production among CD4+CD8αα+ (CD8α+ CD103+) as in (B). (D-J) Rag1−/− animals were treated as in (A) and 20 days after OVA removal, animals were infected with Listeria monocytogenes expressing full length SFB protein 3340 or OVA (Lm-SFB or Lm-OVA, respectively) or were left uninfected. All mice were treated with streptomycin 24h prior to infection and analyzed 9 days after infection. (D) Experimental layout. (E) Frequencies of CD4+CD8αα+ among total OTII CD4+ T cells. (F) Frequencies of pre-IELs among CD4+SP cells. (G, H) Frequencies of IFNγ (G) and Gzmb (H) among ex-vivo PMA and ionomycin stimulated CD4+CD8αα+IELs. (I, J) Colony forming units (CFU) per liver (I) or spleen (J) 9 days post infection. Data are expressed as mean +/− SEM of individual mice (n=3–5, 2 experiments (A-C) and n= 3–9, 2 experiments (D-J)). Significant p values as indicated [student’s t test (I, J) or one-way ANOVA and Bonferroni (B, C, E-H)]. See also Figure S6.
We used this TCR ligand withdrawal model to address how CD4+CD8αα+ IELs with and without cognate TCR re-engagement respond in a context of enteric infection. Rag1−/− mice received OTII cells and were fed on OVA-containing diet (as above) before oral infection with Listeria monocytogenes (Lm) expressing OVA (Lm-OVA), or an irrelevant protein from segment filamentous bacteria (Lm-SFB) (Yang et al., 2014) (Figure 6D). We infected mice with Lm-OVA or Lm-SFB 20 days after OVA withdrawal, as CD4+CD8αα+ IELs reached a plateau at this timepoint (Figure 6B) and analyzed the outcome at day 9 post-infection (Figure 6D). Infection with Lm-OVA or Lm-SFB did not affect pre-IEL or CD4+CD8αα+ IEL frequency (Figure 6E, F) or the IEL program of OVA-specific CD4+CD8αα+ IELs, as measured by IFNγ, granzyme B, 2B4 (CD244) and ThPOK expression (Figure 6G, H, Figure S6E, F). We found equal proportions of transferred cells in the epithelium and mLN of mice infected with Lm-OVA or Lm-SFB (Figure S6G, H). However, bacterial burden in liver and spleen at day 9 post-infection was lower in mice infected with Lm-OVA than Lm-SFB despite similar bacterial recovery in feces at day 3 post-infection (Figure 6I, J, Figure S6I). Taken together, our data establish the crucial role for TCR specificity, and engagement, in CD4+CD8αα+ IEL differentiation. However, after the full IEL program is established in the epithelium, TCR expression or cognate antigen recognition by CD4+CD8αα+ IELs appear mostly dispensable for their maintenance and functional properties, which may include protection of the epithelial lining against intracellular pathogens.
Discussion
Our study revealed a previously unappreciated degree of TCR sharing between largely distinct CD4+ T cell subsets in the periphery, distinct amounts of clonal expansion and TCR diversity along differentiating IELs, and a specific requirement for TCR signaling during the early stages of the IEL differentiation process. Specific TCR usage has been reported in both natural- and peripherally-induced Treg cells located in distinct tissues, including the intestine (Cebula et al., 2013; Lathrop et al., 2011). Early analyses of TCR diversity in nIEL subsets reveal a restriction in TCR repertoire, referred to as “oligoclonal repertoire” (Guy-Grand et al., 1991; Regnault et al., 1994; Rocha et al., 1991). More recently, work using a TCRmini mouse model harboring restricted TCRα and TCRβ repertoires showed TCR overlap between CD4+CD8αα+ IELs and Treg cells (Wojciech et al., 2018). In addition to the agonist selection of thymic IEL precursors, studies that generated transgenic mouse strains carrying existing IEL αβTCRs suggest that TCR specificity may be sufficient to drive IEL fate (Mayans et al., 2014; McDonald et al., 2014).
It remained unclear, however, how specific TCRs correlate with IEL differentiation. Our analyses revealed that intraepithelial clones displayed intra-clonal plasticity: highly expanded clones were found among pre-IELs and the homogenous cluster of CD4+CD8αα+ IELs. A possible explanation for this finding is that the less expanded clones lacked additional signals, such as TCR ligands or environmental components, required for full differentiation into CD4+CD8αα+ IELs (Cervantes-Barragan et al., 2017; Cortez et al., 2014; Mucida et al., 2013; Reis et al., 2014). The clonal analyses presented here, including the TCR sharing of CD4+CD8αα+ IELs derived from Treg or from Tconv cells, corroborate previous studies suggesting a lineage relationship between Treg cells and IELs (Bilate et al., 2016; Sujino et al., 2016). Additionally, their clonal distribution suggests that IEL differentiation may favor particular TCR specificities, also evidenced by the TCRα repertoire of fixed-Vβ6 mice, perhaps in a process analogous to peripheral Treg cell differentiation, where TCR recognition in a context–dependent manner leads to Foxp3 expression (Mucida et al., 2005).
Despite being “chronically activated” as defined by the expression of activation markers such as CD69 and CD44 (Mucida et al., 2013), CD4+CD8αα+ IELs do not show signs of strong TCR activation. This is consistent with an increased threshold for TCR activation for mature IELs agonistically selected in the thymus (Cheroutre and Lambolez, 2008; Guehler et al., 1998; Meresse et al., 2004; Yamagata et al., 2004). Conversely, high amounts of Nur77 expression were reported in agonist-selected T cells such as iNKT cells during thymic selection, which subsequently decays upon migration of these cells to the spleen or liver (Moran et al., 2011). In the periphery, recent studies have indicated that among natural TCRγδ IELs, features in their TCR allow them to use spatially distinct regions to discriminate non-clonal (agonist-selecting elements) from ligands affecting each clone specifically; features that allow tissue-specific migration (including Vγ7+ in the small intestine epithelium), yet potentially a recognition of a diverse set of ligands by clonally-restricted TCRγδ (Melandri et al., 2018). It remains to be determined whether these findings in TCRγδ cells, which allow them to share both innate and adaptive functions, relate to peripheral IEL differentiation.
While the majority of CD4+CD8αα+ IELs express low amounts of Nur77, a small fraction of them maintained Nur77 expression, suggesting that TCR re-engagement can be modulated within the epithelium. It is possible that, similar to thymic agonist selection of CD8αα+ IELs, the strength of TCR stimulation within the epithelium could also influence the “TCR bottleneck” described here during peripheral IEL differentiation. Indeed, availability of antigens presented by IECs could function in the late stage of IEL differentiation, possibility supported by the decreased CD4+CD8αα+ IEL population upon MHCII targeting on IECs. The modulation of MHCII expression by IECs has been linked to the capacity of microbes to attach to the epithelium, and to IFNγ production in both human and murine models (Panja et al., 1998; Umesaki et al., 1995). MHCII expression by IECs has been recently associated with a variety of physiological, such as regulation of the stem cell niche, and pathological functions, such as CD4+ T cell-mediated inflammation during graft-versus-host disease, in part through interacting with gut resident T cells that provide cytokines (Biton et al., 2018; Koyama et al., 2019; Ladinsky et al., 2019). Our data suggest a model in which antigen presentation by IECs is instrumental for the differentiation of CD4+CD8αα+ IELs, which may further enhance MHCII expression by IECs via IFNγ production. Whether this is exclusively dependent on antigen presentation by IECs needs further demonstration.
However, our results show that TCR ablation on CD4+CD8αα+ IELs, or long-term withdrawal of their cognate antigen, does not impair their persistence in the epithelium nor the production of IFNγ and granzyme B, or the maintenance of an IEL program. While this is unexpected in terms of T cell biology, it is consistent with the decreased antigen sensitivity and increased threshold for TCR activation in cells expressing CD8αα homodimers. This is in contrast to the requirement of TCR expression on mature Treg cells for their suppressive function and maintenance of an effector Treg cell program (Levine et al., 2014). The extent to which CD4+CD8αα+ IELs depend on TCR signaling for specific functions and whether it is needed in discrete modules during different stages of differentiation from Treg cells to CD4+CD8αα+ IELs remains to determined. While our earlier studies have revealed several local anti-inflammatory functions of CD4+CD8αα+ IELs, our results here suggest a possible unappreciated role for these cells in resistance mechanisms against intracellular pathogens in a TCR-specific context. We have previously demonstrated that CD4+CD8αα+ IELs could quickly gain cytotoxic potential under certain conditions, like IL-15 exposure upon TCR-engagement (Mucida et al., 2013). However, the specific mechanisms utilized by CD4+CD8αα+ IELs in the enhancement of barrier function against Lm, and whether additional preexisting Lm-OVA specific CD4+ T cells, such as memory or tissue-resident memory (TRM) cells (Beura et al., 2019; Romagnoli et al., 2017), played a synergistic role, are yet to be determined.
Given the potential abundance of antigens to which the intestinal epithelium is exposed, the restricted TCR diversity of CD4+CD8αα+ IELs is counterintuitive. It is possible that pre-IEL and CD4+CD8αα+ IEL clones recognize a particular set of peptide-MHCII complexes. The origins (microbial or dietary), and the range of TCR specificities and affinities for these antigens recognized by CD4+CD8αα+ IELs still remains to be elucidated. This will help address the impact of MHC class II-restricted immune responses at the intestinal epithelium.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Angelina M. Bilate (abilate@rockefeller.edu) or Daniel Mucida (mucida@rockefeller.edu).
Materials Availability
All animal strains used in this study are available from The Jackson Laboratory or were provided by the indicated investigators. This study did not generate any new unique reagents.
Data Availability
All the raw sequencing data and processed files produced for this study is accessible through the accession number PRJNA661330.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Animal care and experimentation were consistent with NIH guidelines and were approved by the Institutional Animal Care and Use Committee at the Rockefeller University. Rag1−/− (002216), OTII (004194), Zbtb7beGFP (027663), Rosa26lsl-tdTomato (007914), Foxp3eGFP-CreERT2 (016961), CD45.1(B6.SJL Ptprca, 002014), Nr4a1EGFP/cre (Nur77GFP, 016617), Foxp3IRES-mRFP (008374), Ox40IRES-cre (012839) mice were purchased from Jackson Laboratories and housed in our facility. Tracf/f mice were kindly provided by A. Rudensky (MSKCC). Vil1creERT2 mice were generated by (el Marjou et al., 2004) and kindly provided by D. Artis (Cornell). Foxp3IRES-GFP mice were provided by V. Kuchroo (Harvard) and H2-Ab1f/f mice were provided by M. Nussenzweig (Jax 013181). E8Icre were kindly provided by I. Taniuchi (Jax 008766). Transnuclear mice were provided by H. Ploegh and generated by (Bilate et al., 2016). Several of these lines were interbred in our facilities to obtain the final strains described elsewhere in the text. Genotyping was performed according to the protocols established for the respective strains by Jackson Laboratories or by donor investigators. Mice were maintained at the Rockefeller University animal facility under specific pathogen-free (SPF) conditions. Both male and female littermates were used, age 8–22 weeks old.
Oral infection with Listeria monocytogenes
L. monocytogenes 10403S-inlA strain expressing full length OVA (Lm-OVA) or SFB 3340 protein (Lm-SFB) were grown overnight in brain heart infusion media. Lm-OVA was provided by L. Lefrançois and LM-SFB provided by D. Littman. Mice were infected with 1010 colony forming units (CFU) 24h after oral treatment with 20mg of Streptomycin (Sigma-Aldrich) diluted in water. At day 9 post-infection, intraepithelial lymphocytes were harvested and analyzed by flow cytometry.
METHOD DETAILS
Isolation of intestinal T cells
Intraepithelial and lamina propria lymphocytes were isolated as previously described (Bilate et al., 2016; Reis et al., 2013). Briefly, small intestines were harvested and washed in PBS and 1mM dithiothreitol (DTT) followed by 30 mM EDTA. Intraepithelial cells were recovered from the supernatant of DTT and EDTA washes and mononuclear cells were isolated by gradient centrifugation using Percoll. Lymphocytes from lamina propria were obtained after collagenase digestion of the tissue. Single-cell suspensions were then stained with fluorescently-labeled antibodies for 25min at 4°C prior to downstream flow cytometry (analysis or sorting) as specified in figure legends.
Staining strategy
For analysis of all ΔTrac strains (and their controls), the following gating strategy was utilized to examine CD4+ T cells: single live lymphocytes (based on size and live/dead fixable dye Aqua stain), CD45+, TCRγδ−, intracellular TCRβ+, CD8β−/low, CD4+. For analysis of mixed bone marrow chimeras using congenic strains the following gating strategy was used: intracellular TCRβ+/− CD4+/−, CD8β−/low, TL-Tetramer+/− CD8α+/−, CD45.1+ CD45.2− or CD45.2+ CD45.1−, surface TCRβ+/−. For single-cell sorting of cells subjected to scTCRseq the following gating strategy was used: single live lymphocytes, CD45+, TCRγδ−, TCRβ+, CD8β−/low, CD4+, CD103+/−, Tomato+/− CD8α+/−, and Foxp3-GFP+/− or Foxp3-RFP+/− as indicated in figure legends. For sorting of total Vα2Vβ6 T cells from fixed-Vβ6 mice the following strategy was used: single live lymphocytes CD45+, TCRβ+, CD4+ CD8β−/low, Vα2+ Vβ6+, CD103+/−, CD8α+/−. Based on the index-sorting information and fluorescence intensity, single cells were assigned as Tconv cells (CD103−CD8α−), pre-IEL (CD103+CD8α−) or CD4+CD8αα+ IEL (CD103+CD8α+). For sorting of cells subjected to bulk RNAseq we used single live lymphocytes CD45+, TCRγδ−, TCRβ+/−, CD8β+/−, CD4+/−, TL-tetramer+/− CD8α+/− as indicated elsewhere in the text. For sorting of cells subjected to 10X Genomics, we gated on single live lymphocytes CD45+ TCRγδ−, TCRβ+, CD8β−/low, CD4+, Tomato+/−. IELs and Treg cells were confirmed by post-sort staining for surface CD8α and intranuclear Foxp3, respectively.
Intranuclear and intracellular staining and flow cytometry
Intranuclear staining for Foxp3 was conducted using Foxp3 / Transcription Factor Staining Buffer Set according to manufacturer’s instructions (eBioscience, USA). For analysis of cytokine secretion, total mononuclear cells isolated from epithelium were plated in 48-well plates and incubated at 37°C with 100ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma) and 200ng/mL ionomycin (Sigma) for 4 hours. Golgi stop solution containing Monensin (2μM, BD Biosciences) was added 1.5h after PMA and ionomycin. Intracellular staining for IFNγ and granzyme B was conducted in Perm/Wash buffer after fixation and permeabilization in Fix/Perm buffer (BD Biosciences, USA) according to kit instructions. Flow cytometry data was acquired on an LSR-II flow cytometer (Becton Dickinson, USA) and analyzed using FlowJo software package (Tri-Star, USA).
Edu treatment and detection
1mg EdU was injected intravenously at 5mg/mL in PBS 16 and 4 hours prior to analysis. Detection was performed using the Click-iT™ Plus EdU Flow Cytometery Assay kit (Thermo Fisher Scientific, C10632), according to manufacturer’s instructions.
Annexin V staining
Annexin V staining on single-cell suspensions was performed according to manufacturer’s instructions (eBioscience) and analyzed by flow cytometry.
Cell transfer
Single-cell suspensions from spleens and mesenteric lymph nodes of OTII Zbtb7bGFP Rag1−/− donor animals were depleted of red blood cells, filtered through a 70μm filter, and resuspended in phosphate buffered saline (PBS). 5 × 105 cells were were intravenously injected into Rag1−/− recipient animals in a final volume of 100μL.
OVA feeding
Animals were fed with Modified AIN-76A Rodent diet with 1% ovalbumin (w/w) (Cat D12051002, Research Diets Inc) diet ad libitum for 10 days or as indicated in figure legends.
Enumerating bacterial burden
Liver and spleens were homogenized in PBS containing 1% saponin (Calbiochem) through a 70mm cell strainer. Homogenates were incubated for 1h at 4°C. Fecal pellets were homogenized in 250ml of PBS. Dilutions were plated on Oxford Listeria selective agar plates (Merck KGaA) and incubated for 2 days at 37°C to quantify CFUs.
Tamoxifen treatment
Tamoxifen (Sigma) was dissolved in corn oil (Sigma) and 10% ethanol and shaking at 37°C for 30min-1h. Five doses of Tamoxifen (1mg/dose) was administered to Vil1creERT2 animals intraperitonially at 10mg/mL during one week, with 2 extra 1mg boosts 3 days apart 2 weeks before analysis, when analysis was more than 4 weeks after initial dosing, as indicated in figure legends. Four doses of Tamoxifen (5mg/dose) were administered to Foxp3eGFP-creERT2 animals intragastrically 3 times 2 days apart in the first week, and then 2 times (3 days apart) every other week up to 2 weeks before analysis as indicated in figure legends. Unless otherwise noted, time between first tamoxifen administration and read-out was 10 weeks.
Generation of mixed bone marrow chimeras
Bone marrow cells were harvested from WT CD45.1 or Tracf/f E8Icre CD45.2 donors and depleted of T cell precursors using CD90.2 beads (Miltenyi) according to manufacturer’s instructions. An equal mix of 5×106 total cells from WT CD45.1 and Tracf/f E8Icre CD45.2 donors was intravenously injected into sub-lethally irradiated (6 Gy) Rag1−/− hosts. Mice were analyzed 12–16 weeks after reconstitution.
Single-cell TCR sequencing
Single cells were index-sorted using a FACS Aria into 96-well plates containing 5μL of lysis buffer (TCL buffer, Qiagen 1031576) supplemented with 1% β-mercaptoethanol) and frozen in −80°C prior to RT-PCR. RNA and RT-PCRs for TCRα and TCRβ were prepared as previously described (Dash et al., 2011). PCR products for TCRα and TCRβ were either subjected to Sanger sequencing or multiplexed with barcodes and submitted to MiSeq sequencing (Han et al., 2014) using True Seq Nano kit (Illumina). For Miseq data, fastq files were de-multiplexed and paired-end reads were assembled at their overlapping region using PANDASEQ (Masella et al., 2012) and FASTAX toolkit. Demultiplexed and collapsed reads were assigned to wells according to barcodes. Fasta files from both Sanger and Miseq sequences were aligned and analyzed on IMGT (imgt.org/HighV-QUEST) (Brochet et al., 2008). Cells with identical TCRβ CDR3 nucleotide sequences were considered as the same clones. Clonality was confirmed by sequencing TCRα of the expanded clones as assessed by TCRβ sequencing for mice shown in Figure S2F and H. Clonality was assigned based on paired TCRαβ for mice shown in Figure 2 and S2C. Only in frame junction sequences of TCRβ were included in the analysis. For fixed-Vβ6 mice shown in Figure 2 and S2, each TCRα CDR3 was considered a clone.
Bulk RNAseq library preparation
Sorted cells (300–800 cells) were lysed in a guanidine thiocyanate buffer (TCL buffer, Qiagen) supplemented with 1% β-mercaptoethanol. RNA was isolated by solid-phase reversible immobilization bead cleanup using RNAClean XP beads (Agentcourt, A63987), reversibly transcribed, and amplified as described (Trombetta et al., 2014). Uniquely barcoded libraries were prepared using Nextera XT kit (Illumina) following manufacturer’s instructions. Sequencing was performed on an Illumina NextSeq500 for a total yield of 400M reads.
Bulk RNAseq analysis
Raw fastq files were processed by using the mouse transcriptome (gencode M23) with the kallisto (v0.46) software (Bray et al., 2016). Analysis of transcript quantification was performed at the gene level by using the sleuth (v0.30) package for R (Pimentel et al., 2017). Shortly, we modeled batch effect and our experimental design using the sleuth_fit function and detected differentially expressed genes between all groups by the likelihood ratio test (lrt). To spot statistically significantly expressed genes between group pairs, we used the wald-test function. Genes with false discovery rate less than 0.05 and 1 log2 fold-change were used in downstream analysis. The batch effect was removed from the expression matrix by using the removebatcheffect function available in the limma package (Ritchie et al., 2015). Gene set enrichment analysis (GSEA) was performed by using signature gene sets in gmt format and a pre-ranked gene list by log2 fold-change between two groups as input for the fgsea. Gene ontology (GO) analysis was executed by comparing all detected genes as our background and the lists of differentially expressed genes against the biological processes gene sets by using topGO(Alexa and Rahnenfuhrer, 2019; Carlson M, 2019).
Single cell RNAseq library preparation
Tomato+ and Tomato− CD4+ cells were sorted from epithelium, counted for viability and immediately subjected to library preparation. The scRNA-seq and scTCR-seq libraries were prepared using the 10x Single Cell Immune Profiling Solution Kit, according to the manufacturer’s instructions at the Genomics core of Rockefeller University. The scRNA libraries were sequenced on an Illumina NextSeq500 to a minimum sequencing depth of 50,000 reads per cell using read lengths of 26 bp read 1, 8 bp i7 index, 98 bp read 2. The single-cell TCR libraries were sequenced on an Illumina NextSeq500 to a minimum sequencing depth of 5,000 reads per cell using read lengths of 150 bp read 1, 8 bp i7 index, 150 bp read 2.
Data processing of single cell RNAseq and single cell TCRseq libraries
Raw fastq files derived from our RNA-seq libraries were processed with cellranger count (v3.1.0) using the 10x Genomics prebuilt mouse reference (v3.0.0 mm10). Our libraries were processed independently and merged into a single experiment for analysis using Seurat (v3.1.1) (Stuart et al., 2019). Quality control was performed by removing cells with high (> 5%) mitochondrial UMI content. Cells having more than 4000 or less than 200 genes were excluded from our analysis. TCR contigs and annotation were performed with the Cellranger vdj workflow from 10x Genomics and the prebuild mouse reference (v3.1.0 mm10). Paired TCR clones were defined by the V, (D), J and CDR3 nucleotide composition for alpha and beta chains. TCR clone sharing was assigned to cells expressing identical V, (D), J, and CDR3 nucleotide and amino acid sequences.
Single cell RNAseq normalization and statistical analysis
The raw UMI counts were normalized by applying a regression model with negative binomial error distribution, available through the SCTransform function in the Seurat (v3.1.1) package (Hafemeister and Satija, 2019). The top 3000 variable genes were first used for dimensional reduction by PCA using the scaled data. The first 30 principal components were further used for visualization using the Manifold Approximation and Projection (UMAP) and cell clustering (Hafemeister and Satija, 2019; Stuart et al., 2019). Gene markers were detected by comparing each cluster against all cells through the Wilcoxon rank sum test. Genes with log2 fold-changes of 0.4 and adjusted p values of 0.05 or less were considered for downstream analysis. To determine the cell cycle phase, we scored each cell based on gene sets for the S and G2M phases using the CellCycleScoring function within the Seurat package (Tirosh et al., 2016). Cells that did not score in at least one set, were marked as in the G1 phase.
Trajectory analyses
We inferred cell differentiation using the pseudotime slingshot and diffusion map algorithm adapted for sc-RNAseq analysis and implemented through the package destiny (Angerer et al., 2016). Normalized expression values were used as input for the Diffusion Map function. Cells were ordered based on the first diffusion component. To visualize lineage differentiation within our UMAP projection and find differentially expressed genes correlating with the pseudotime, we used the slingshot package (Street et al., 2018). We also performed RNA velocity extrapolation to determine cell differentiation trajectories by measuring the ratio of new (nascent/unspliced) and old (spliced) RNA molecules in each cell. We used the software Velocyto to quantify unspliced and spliced RNA molecules from our 10x dataset (La Manno et al., 2018). The resultant loom matrices were imported in the R environment and added to the Seurat object. Lastly, we performed the RNA velocity analysis by using the SeuratWrappers package.
Generation of circos plots
TCR sharing (clonal overlap) was visualized using Circos to create circular plots aesthetics (Krzywinski et al., 2009). Each ideogram denotes a cell population as indicated in figures, with bands representing clonal distribution sorted clockwise from more expanded to less expanded. Circos plots for the 10x dataset were created by only displaying the links connecting adjacent ideograms to avoid overplotting. Circos plots for the Miseq data were created displaying all possible shared links.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Statistical analysis was carried out using GraphPad Prism v.8. Flow cytometry analysis was carried out using FlowJo software. Data in graphs show mean +/− SEM and p values <0.05 were considered significant. Repertoire diversity was analyzed by the Chao1 index and by Diversity 50 (D50). Diversity 50 (D50) was calculated on Excel as the fraction of dominant clones that account for the cumulative 50% of the total paired CDR3s identified in each UMAP cluster. CDR3 similarity (TCR sharing/clonal overlap) was calculated using the Morisita-horn overlap index by using the divo package (Rempala and Seweryn, 2013). GraphPadPrism v.8 was used for graphs and Adobe Illustrator 2019 used to assemble and edit figures.
Supplementary Material
Table S1. Top genes per single cell RNA-Seq cluster. Related to Figure 1.
Top significantly differentially expressed genes per cluster as determined by the wilcoxon rank sum test. Columns indicate cluster name, gene, average log fold change (avg_logFC), principle components 1 and 2 (pct.1and pct.2, respectively), and adjusted p value (p_val_adj).
Table S2. Clones per single cell RNA-Seq cluster. Related to Figure 1.
Number of distinct clones (clones with a unique number) per single cell RNA-Seq cluster (C0-C7), separated by Tomato−(left) and Tomato+(right) shown in figure 1. Clones shared between clusters are indicated in the column “sharing clusters” depicting in how many clusters a given clone appears. “TCRa_junction” and “TCRb_junction” columns show the CDR3 amino acid sequence for TCRα and TCRβ for each clone, respectively.
Table S3. List of amino acid sequences of TCRαβ junctions. Related to Figure 2.
Table shows all CDR3 amino acid sequences included in figures 2 and S2 for polyclonal mice and for fixed-Vβ6 mice sorted by mouse genotype and number. Clone size column indicates the number of times a given clone appears in a given population per mouse.
Highlights.
Intraepithelial CD4+CD8αα+ IEL differentiation is accompanied by clonal restriction
TCR and local antigen presentation are required for CD4+CD8αα+ IEL differentiation
T cell receptor (TCR) engagement is dispensable for CD4+CD8αα+ IEL maintenance
Acknowledgements
We are grateful to A. Rogoz and S. Gonzalez for exceptional animal care, mouse colony management and genotyping, Yasmeen Khan, K. Gordon, K. Chhosphel, C. Zhao, the Genomics Core and additional Rockefeller University employees for continuous assistance. We thank NIH tetramer facility for providing the TL tetramer. We thank D. Littman for providing Lm-SFB. We thank R. Parsa and A. Lockhart for providing advice on Listeria infection experiments. We are grateful to J. Lafaille and B. Reis for suggestions, and all the members of the Mucida lab for fruitful discussions.
Funding
This work was supported by NIH grants R01DK093674, R01DK113375 and R21A144827. DM is also supported by the Mathers Foundation and Pershing Square Foundation, the Black Family Metastasis Center and the FASI/FARE Consortium.
Footnotes
Declaration of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexa A, and Rahnenfuhrer J (2019). topGO: Enrichment Analysis for Gene Ontology. R package version 2.24.0. [Google Scholar]
- Angerer P, Haghverdi L, Buttner M, Theis FJ, Marr C, and Buettner F (2016). destiny: diffusion maps for large-scale single cell data in R. Bioinformatics 32, 1241–1243. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Beura LK, Fares-Frederickson NJ, Steinert EM, Scott MC, Thompson EA, Fraser KA, Schenkel JM, Vezys V, and Masopust D (2019). CD4(+) resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J Exp Med 216, 1214–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilate AM, Bousbaine D, Mesin L, Agudelo M, Leube J, Kratzert A, Dougan SK, Victora GD, and Ploegh HL (2016). Tissue-specific emergence of regulatory and intraepithelial T cells from a clonal T cell precursor. Sci Immunol 1, eaaf7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, Ashenberg O, Su CW, Smillie C, Shekhar K, et al. (2018). T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 175, 1307–1320 e1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousbaine D, Bhagchandani P, London M, Mimee M, Olesen S, Poyet M, Cheloha RW, Sidney J, Ling J, Gupta A, et al. (2020). Antigen-specific induction of CD4+CD8αα+ intraepithelial T lymphocytes by Bacteroidetes species. bioRxiv, 2020.2008.2005.236513. [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brochet X, Lefranc MP, and Giudicelli V (2008). IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36, W503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M (2019). GO.db: A set of annotation maps describing the entire Gene Ontology. R package version 3.8.2. [Google Scholar]
- Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, Bry L, Kraj P, Kisielow P, and Ignatowicz L (2013). Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 497, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J, Cortez VS, Caparon MG, Donia MS, Gilfillan S, et al. (2017). Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 357, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheroutre H, and Lambolez F (2008). Doubting the TCR coreceptor function of CD8alphaalpha. Immunity 28, 149–159. [DOI] [PubMed] [Google Scholar]
- Cheroutre H, Lambolez F, and Mucida D (2011). The light and dark sides of intestinal intraepithelial lymphocytes. Nature reviews. Immunology 11, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, Cervantes-Barragan L, Song C, Gilfillan S, McDonald KG, Tussiwand R, Edelson BT, Murakami Y, Murphy KM, Newberry RD, et al. (2014). CRTAM controls residency of gut CD4+CD8+ T cells in the steady state and maintenance of gut CD4+ Th17 during parasitic infection. J Exp Med 211, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P, McClaren JL, Oguin TH 3rd, Rothwell W, Todd B, Morris MY, Becksfort J, Reynolds C, Brown SA, Doherty PC, and Thomas PG (2011). Paired analysis of TCRalpha and TCRbeta chains at the single-cell level in mice. J Clin Invest 121, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, and Robine S (2004). Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193. [DOI] [PubMed] [Google Scholar]
- Ellmeier W, Sunshine MJ, Losos K, Hatam F, and Littman DR (1997). An enhancer that directs lineage-specific expression of CD8 in positively selected thymocytes and mature T cells. Immunity 7, 537–547. [DOI] [PubMed] [Google Scholar]
- Faria AMC, Reis BS, and Mucida D (2017). Tissue adaptation: Implications for gut immunity and tolerance. J Exp Med 214, 1211–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guehler SR, Finch RJ, Bluestone JA, and Barrett TA (1998). Increased threshold for TCR-mediated signaling controls self reactivity of intraepithelial lymphocytes. Journal of immunology 160, 5341–5346. [PubMed] [Google Scholar]
- Guy-Grand D, Cerf-Bensussan N, Malissen B, Malassis-Seris M, Briottet C, and Vassalli P (1991). Two gut intraepithelial CD8+ lymphocyte populations with different T cell receptors: a role for the gut epithelium in T cell differentiation. J Exp Med 173, 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. bioRxiv, 576827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han A, Glanville J, Hansmann L, and Davis MM (2014). Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nature biotechnology 32, 684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, and Jameson SC (2014). The self-obsession of T cells: how TCR signaling thresholds affect fate ‘decisions’ and effector function. Nature immunology 15, 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hulsdunker J, Varelias A, Vetizou M, Kuns RD, Robb RJ, Zhang P, et al. (2019). MHC Class II Antigen Presentation by the Intestinal Epithelium Initiates Graft-versus-Host Disease and Is Influenced by the Microbiota. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Araujo LP, Zhang X, Veltri J, Galan-Diez M, Soualhi S, Lee C, Irie K, Pinker EY, Narushima S, et al. (2019). Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, and Hsieh CS (2011). Peripheral education of the immune system by colonic commensal microbiota. Nature 478, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leishman AJ, Gapin L, Capone M, Palmer E, MacDonald HR, Kronenberg M, and Cheroutre H (2002). Precursors of functional MHC class I- or class II-restricted CD8alphaalpha(+) T cells are positively selected in the thymus by agonist self-peptides. Immunity 16, 355–364. [DOI] [PubMed] [Google Scholar]
- Levine AG, Arvey A, Jin W, and Rudensky AY (2014). Continuous requirement for the TCR in regulatory T cell function. Nature immunology 15, 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London M, Bilate AM, Castro TBR, and Mucida D (2020). Stepwise chromatin and transcriptional acquisition of an intraepithelial lymphocyte program. bioRxiv, 2020.2006.2004.134650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masella AP, Bartram AK, Truszkowski JM, Brown DG, and Neufeld JD (2012). PANDAseq: PAired-eND Assembler for Illumina sequences. Bmc Bioinformatics 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayans S, Stepniak D, Palida SF, Larange A, Dreux J, Arlian BM, Shinnakasu R, Kronenberg M, Cheroutre H, and Lambolez F (2014). alphabetaT cell receptors expressed by CD4(−)CD8alphabeta(−) intraepithelial T cells drive their fate into a unique lineage with unusual MHC reactivities. Immunity 41, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BD, Bunker JJ, Ishizuka IE, Jabri B, and Bendelac A (2014). Elevated T cell receptor signaling identifies a thymic precursor to the TCRalphabeta(+)CD4(−)CD8beta(−) intraepithelial lymphocyte lineage. Immunity 41, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BD, Jabri B, and Bendelac A (2018). Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nature reviews. Immunology 18, 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melandri D, Zlatareva I, Chaleil RAG, Dart RJ, Chancellor A, Nussbaumer O, Polyakova O, Roberts NA, Wesch D, Kabelitz D, et al. (2018). The gammadeltaTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nature immunology 19, 1352–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, Raulet DH, Lanier LL, Groh V, Spies T, et al. (2004). Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21, 357–366. [DOI] [PubMed] [Google Scholar]
- Moran AE, Holzapfel Kl Fau - Xing Y, Xing Y Fau - Cunningham NR, Cunningham Nr Fau - Maltzman JS, Maltzman Js Fau - Punt J, Punt J Fau - Hogquist KA, and Hogquist KA (2011). T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med 208, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, Reis BS, Huang Y, Lambolez F, Docherty M, et al. (2013). Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nature immunology 14, 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, and Curotto de Lafaille MA (2005). Oral tolerance in the absence of naturally occurring Tregs. The Journal of clinical investigation 115, 1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panja A, Goldberg S, Eckmann L, Krishen P, and Mayer L (1998). The regulation and functional consequence of proinflammatory cytokine binding on human intestinal epithelial cells. Journal of immunology 161, 3675–3684. [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nature methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Regnault A, Cumano A, Vassalli P, Guy-Grand D, and Kourilsky P (1994). Oligoclonal repertoire of the CD8 alpha alpha and the CD8 alpha beta TCR-alpha/beta murine intestinal intraepithelial T lymphocytes: evidence for the random emergence of T cells. J Exp Med 180, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis BS, Hoytema van Konijnenburg DP, Grivennikov SI, and Mucida D (2014). Transcription Factor T-bet Regulates Intraepithelial Lymphocyte Functional Maturation. Immunity 41, 244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, and Mucida D (2013). Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nature immunology 14, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempala GA, and Seweryn M (2013). Methods for diversity and overlap analysis in T-cell receptor populations. J Math Biol 67, 1339–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha B, Vassalli P, and Guy-Grand D (1991). The V beta repertoire of mouse gut homodimeric alpha CD8+ intraepithelial T cell receptor alpha/beta + lymphocytes reveals a major extrathymic pathway of T cell differentiation. J Exp Med 173, 483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli PA, Fu HH, Qiu Z, Khairallah C, Pham QM, Puddington L, Khanna KM, Lefrancois L, and Sheridan BS (2017). Differentiation of distinct long-lived memory CD4 T cells in intestinal tissues after oral Listeria monocytogenes infection. Mucosal immunology 10, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. Bmc Genomics 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao YH, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sujino T, London M, Hoytema van Konijnenburg DP, Rendon T, Buch T, Silva HM, Lafaille JJ, Reis BS, and Mucida D (2016). Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science 352, 1581–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, and Regev A (2014). Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr Protoc Mol Biol 107, 4 22 21–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesaki Y, Okada Y, Matsumoto S, Imaoka A, and Setoyama H (1995). Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol Immunol 39, 555–562. [DOI] [PubMed] [Google Scholar]
- Wojciech L, Szurek E, Kuczma M, Cebula A, Elhefnawy WR, Pietrzak M, Rempala G, and Ignatowicz L (2018). Non-canonicaly recruited TCRalphabetaCD8alphaalpha IELs recognize microbial antigens. Scientific reports 8, 10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata T, Mathis D, and Benoist C (2004). Self-reactivity in thymic double-positive cells commits cells to a CD8 alpha alpha lineage with characteristics of innate immune cells. Nature immunology 5, 597–605. [DOI] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. (2014). Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510, 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Top genes per single cell RNA-Seq cluster. Related to Figure 1.
Top significantly differentially expressed genes per cluster as determined by the wilcoxon rank sum test. Columns indicate cluster name, gene, average log fold change (avg_logFC), principle components 1 and 2 (pct.1and pct.2, respectively), and adjusted p value (p_val_adj).
Table S2. Clones per single cell RNA-Seq cluster. Related to Figure 1.
Number of distinct clones (clones with a unique number) per single cell RNA-Seq cluster (C0-C7), separated by Tomato−(left) and Tomato+(right) shown in figure 1. Clones shared between clusters are indicated in the column “sharing clusters” depicting in how many clusters a given clone appears. “TCRa_junction” and “TCRb_junction” columns show the CDR3 amino acid sequence for TCRα and TCRβ for each clone, respectively.
Table S3. List of amino acid sequences of TCRαβ junctions. Related to Figure 2.
Table shows all CDR3 amino acid sequences included in figures 2 and S2 for polyclonal mice and for fixed-Vβ6 mice sorted by mouse genotype and number. Clone size column indicates the number of times a given clone appears in a given population per mouse.
Data Availability Statement
All the raw sequencing data and processed files produced for this study is accessible through the accession number PRJNA661330.