

Abstract
Nanoparticle (NP) based drug delivery systems have broadened our horizon of translational research for decades. However, conventional bulk mixing methods have impeded successful clinical translations of nanomedicines due to the limited ability of controlled, scalable production of nanomedicines with high uniformity. Herein, we present an on-chip preparation of self-assembled, drug-encapsulated polymeric NPs for their improved uniformity and homogeneity, leading to enhanced anti-cancer effect in vitro and in vivo compared to bulk synthesis. The NPs were formulated through rapid convective mixing of two aqueous solutions of a hydrophilic polymer and an anti-cancer drug, doxorubicin (DOX), in the swirling microvortex reactor (SMR). Compared to conventional bulk-mixed NPs (BMPs), the microvortex-synthesized NPs (MVPs) exhibited narrower size distributions and better size tunability with varied Reynolds numbers. The improved uniformity and homogeneity of the produced MVPs enhanced cellular uptake and anti-cancer effect with pH-responsive drug release in vitro. Furthermore, therapeutic application of MVPs in tumor-bearing mice models presented much higher accumulation at the tumor region, resulting in an improved tumor regression and decreased side effects at off-targeted organs. Our findings suggest that uniformly designed NPs with more homogeneous properties can induce a significant enhancement of an anti-cancer effect in vivo. We believe that our validation of the correlation between an enhanced therapeutic effect and improved uniformity of NPs can provide the potential of a high-speed on-chip synthesis as a scalable manufacturing platform for reliable clinical translations of nanomedicines.
Keywords: microvortex, nanoparticles, self-assembly, polymers, anti-cancer drug
Graphical Abstract
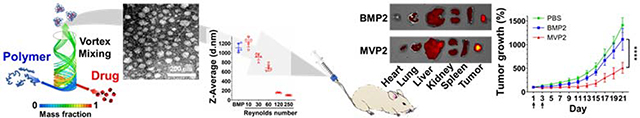
Facile preparation of uniform and homogenous drug-encapsulated polymeric nanoparticles (NPs) is achieved with on-chip self-assembly. The microvortex-synthesized NPs (MVPs) demonstrate superior properties in cellular uptake and anti-cancer effect compared to bulk mixed NPs (BMPs). In particular, the MVPs exhibit high accumulation at tumor region and enhanced tumor regression in tumor-bearing mouse model, presenting potential of reproducible preparation of nanomedicines for reliable clinical translation.
1. Introduction
Nanotechnology has enabled biomedical applications of engineered nanomedicines and demonstrated the potential of drug candidates for clinical translation. Despite several successful translations of the findings from bench to bedside, controlled reproduction of designed nanomedicines with consistent therapeutic properties remains a challenge.[1] One suitable approach to mass production of nanomedicines is to leverage simple precursor mixing to induce the self-assembly mechanism for NP formulation. Typical bulk mixing (BM) method is to use a plastic tube or glass vial in a lab-scale or a few-liter reactor in a pilot-scale for easy synthesis and application in laboratories, but this approach usually leads to a polydisperse or wide size distribution and batch-to-batch variation of the synthesized NPs.[1a, 2] This less controllability of variables and synthesis conditions in space and time remains to be addressed for advanced nanomedicine manufacturing in the future.
Microfluidic technologies have provided various approaches to precision control of precursor mixing flows and rates for continuous synthesis of a variety of NPs, showing the great potential to produce highly uniform NPs at a higher production rate.[1b, 2a, 3] Recently, researchers have reported reproducible preparation of monodisperse NPs by on-chip synthesis compared to conventional bulk mixing NPs (BMPs), and further presented their enhanced therapeutic effect at the cellular level.[4] Several studies demonstrated an improved precursor mixing and resulting reproducibility of the produced NPs[5] but most remain limited to the in vitro validation of the higher cellular uptake and anti-cancer effect than bulk synthesis of the same drug-loaded NPs or nanomedicines due to the inherent complexity of in vivo system.[6] Evaluation of therapeutic NPs using animal models is required to translate nanomedicines, since a therapeutic effect of a nanomedicine is a combined outcome of not only a single factor of cellular uptake but also the absorption, distribution, metabolism, and elimination (ADME) of administrated NPs.[7] Therefore, it is important to show whether the high size uniformity of NPs controlled by microfluidic synthesis hold the effectiveness in animal model studies, which becomes critical in manufacturing and therapeutic applications for clinical translation.
Herein, we demonstrate that NPs synthesized using the swirling microvortex reactor (SMR) exhibit higher size uniformity and property homogeneity, resulting in both improved cellular uptake in vitro and drug delivery efficacy in vivo for the treatment of cancer.[8] This NP is prepared by simple self-assembly of polymeric phenylboronic acid (pPBA) and doxorubicin (DOX) through formation of phenylboronic ester in a high-speed on-chip synthesis mechanism.[9] We optimize formation of NPs with SMR at various DOX/PBA ratios and Reynolds numbers (Re) and compare the cellular uptake and cytotoxicity of the microvortex-synthesized NPs (MVPs) and bulk mixed NPs (BMPs). We use tumor-bearing nude mice to monitor bio-distributions of the MVPs and BMPs after intravenous injection and demonstrate an enhanced therapeutic effect of the MVPs. We evaluate the anti-cancer effect and possible adverse effect of the NPs in the mouse model by monitoring tumor growth and histological assay of the major organs. Lastly, we discuss the potential of self-assembled therapeutic NPs synthesized by on-chip-based high-speed mixing for the scalable mass production and reliable clinical translation.
2. Results and discussion
2.1. Self-assembly of pPBA and DOX for nanoparticle formation
Formulation of self-assembled NPs composed of pPBA and DOX was achieved by an interaction between the PBA moiety of pPBA and diol of DOX. To synthesize polymeric PBA (pPBA), the PBA moiety was conjugated on the hydrophilic polymer backbone, pMVEMA, at the ratio of approximately 31.7% PBA, which was calculated by 1H NMR (Figure S1a, S1b). The synthesized pPBA was simply mixed with DOX to form a phenylboronic ester bond, resulting in BMPs (Scheme S1b). Optimal molar ratios of PBA to DOX were determined by size analyses of BMPs, which was also applied for MVPs (Figure S2a). In the DLS size analysis, the NPs showed aggregation at PBA/DOX ratios of 0.25, 0.5 and 1, while ratios of 2, 4 and 8 presented relatively stable NP formation. To use relatively lower polymer amounts, we selected the two ratios of 2 and 4 in the present study to compare the in vitro and in vivo therapeutic effects of BMPs, which ratios were also used for MVPs below.
We used our SMR device that shows a high mixing efficiency of 92% to synthesize MVPs with the two ratios of 2 and 4 (Figure 1a).[8] The MVPs showed much narrower size distributions and smaller sizes than BMPs (Figure 1b and S2b), and the higher size uniformity was confirmed in MVPs than in BMPs with the spherical shape of the NPs (Figure 1c). BMPs were considerably heterogeneous in size, but MVPs were highly homogeneous (Figure 1d). In addition, we verified the interaction between PBA of pPBA and 1,3-cis-diol of DOX as a driving force of self-assembly by measuring the fluorescence of PBA that is quenched when the PBA interacts with diol moiety to form a phenylboronic ester bond.[10] The fluorescence of all the NPs decreased compared to free pPBA, indicating that the interaction between DOX and PBA was a main factor to form the NPs (Figure S3b).
Figure 1.

Nanoparticle formation and comparison between BMPs and MVPs at PBA/DOX ratios of 2 and 4. (a) Scheme of nanoparticle formation through swirling microvortex flow of pPBA and DOX in aqueous solutions. (b) Size distributions of NPs were analyzed by DLS and (c) morphologies of the NPs were observed by TEM with negative staining to compare the obtained nanoparticles depending on ratios and preparation method. (d) Representative size distribution of nanoparticles measured from TEM images (n = 50). (e) Z-average sizes and (f) fluorescence quenching of PBA derived by interaction of pPBA and DOX depending on Reynolds number of SMR chip.
2.2. Controlled Self-assembly of MVPs
To elicit the controllability of SMR, we prepared the MVPs at Reynolds numbers (Re) by adjusting the flow rate. It is widely known that the size of NPs is strongly affected by the growth time during the process of molecular assembly, which is highly related to the mixing efficiency of fluids depending on the flow rate and Re.[1a, 11] Computational fluid dynamics (CFD) results for mixing efficiency of SMR presented an increase in mixing efficiency as Re increased, implying that the smaller size of NPs resulted from the shorter growth time at a higher mixing speed (Figure S3d). As expected, the size of the NPs gradually decreased and the higher fluorescence quenching was observed as Re increased from 10 to 250 (Figure 1e and S3c). Interestingly, BMPs showed similar size and fluorescence quenching to MVP2 at Re of 10 where the BM method conventionally used in the bulk synthesis has similar mixing efficiency to SMR chip-based synthesis. On the other hand, the higher Re caused a smaller NP size as well as a lower fluorescence of PBA due to more assemblies of PBA and DOX, implying that pPBA and DOX were well mixed with the rapid convective flows. Taken together, we confirmed the controllability of SMR for NP formulation that the Re value is one of the crucial factors to govern the size and interaction efficiency between PBA and DOX, whereas the macroscale mixing in the BM method cannot implement the corresponding Re to better mix precursors and result in NPs with better controlled quality.
2.3. pH-responsive DOX release
A major driving force of self-assembly was formation of phenylboronic ester bond between diol of DOX and PBA of pPBA, the strength of which is reversibly correlated to the pH.[8, 12] The phenylboronic ester is stable at physiological pH 7.5 but it readily dissociates at an acidic condition, i.e. pH 5.0. Our NPs has the acid-labile property of phenylboronic ester bond with which drugs can be released favorably in tumor-targeted drug delivery, as well as intracellular drug release, thanks to the acidic conditions of tumor microenvironment and intracellular endosomes.[13] We monitored the corresponding DOX release out of the NPs at pH 5.0 and pH 7.5 for 48 h (Figure 2a). The NPs incubated at pH 5.0 showed a rapid release of DOX, whereas a negligible DOX release was observed at pH 7.5. We also confirmed that there is no significant difference in the drug release profile when applying MVPs and BMPs, showing that the destruction of a particular structure is primary due to the dissociation of phenylboronic ester bond between DOX and pPBA, in spite of intact spherical shapes with different size distribution of both NPs at pH 7.5 (Figure 2b).
Figure 2.

Drug release profiles of NPs with regard to pH at PBA/DOX ratios of 2 and 4. (a) pH-dependent DOX release profiles of each NP incubated at pH 5.0 and pH 7.5. Amount of loaded DOX was regarded as 100%. (b) Morphology of each NP incubated at pH 5.0 and pH 7.5 for 2 h were observed by TEM with negative staining. Scale bar = 200 nm.
2.4. In vitro cell viability and cellular uptake
The therapeutic effect of NPs at PBA/DOX ratios of 2 and 4 was evaluated at the cellular level. The NPs were readily dissociated at acidic endosomal pH, leading to the DOX release and anti-cancer effect. Accordingly, all the NPs showed measurable anti-cancer effects, while free pPBA itself did not induce cytotoxicity (Figure S4a). We hypothesized that more uniform structures of MVPs than those of BMPs affect cellular uptake of the NPs to exhibit an efficient anti-cancer effect.[14] IC50 values of the MVPs were 1.23–1.97 times lower than that of the BMPs (Figure 3a), meaning that MVPs have greater anti-cancer effects than those of BMPs. In addition, the MVPs showed higher cellular uptake of both DOX and pPBA labelled with fluorescence dye (pPBA-F) than do the BMPs regardless of the polymer-drug ratio. Furthermore, the relative mean fluorescence intensities (MFIs) of DOX and pPBA-F were significantly higher at the groups treated with MVPs, indicating increased cellular uptakes of the MVPs than BMPs (Figure 3b, c and S5a, b). We note that the better IC50 values of the MVPs were attributed to precisely controlled uniform properties of the NPs synthesized by SMR. Taken together, NPs fabricated by SMR are not only uniform and homogeneous in the property but also exhibit an enhanced therapeutic effect on drug release.
Figure 3.

Assessment of in vitro anti-cancer effect of NPs with the delivery efficiency into cancer cell lines. (a) IC50 values (nM) of the NPs against U87-MG, D556, and Daoy cell lines evaluated by MTT assays after 48 h incubation. (b) Confocal fluorescence images of D556 cell showing cellular uptake of NPs for 4 h. Blue, red, and green images represent nucleus, DOX, and pPBA labelled with fluorescence dye, respectively. Scale bar is 50 μm. (c) Relative DOX and pPBA mean fluorescence intensities of (b). Each MFI was normalized by maximum intensity.
2.5. In vivo bio-distribution of NPs
Bio-distribution of NPs strongly depends on their size, surface charge, and morphology.[15] With our interesting results in vitro, we expected different bio-distribution of the MVPs and BMPs. As studies have reported that nanoparticles tend to accumulate in a tumor tissue through the abnormal and loose blood vessels called the enhanced permeability and retention (EPR) effect,[16] we observed an increased accumulation of the NPs and pH-responsive release of DOX in the acidic tumor site at 24 h and 48 h (Figure 4). Importantly, the MVPs exhibited significantly higher accumulation of DOX at the tumor (i.e., enhanced tumor-targeting effect in vivo) than BMPs at both 24 h and 48 h (Figure 4b), while the BMPs caused relatively inefficient accumulation in the tumor site due to the heterogeneous size distribution. This demonstration further underscores the importance of NP size uniformity by the MV synthesis.
Figure 4.

Monitoring bio-distributions of NPs in tumor-bearing nude mice. (a) Representative images of DOX accumulation in major organs at 24 h and 48 h after i.v. injection of each NP (2.5 mg DOX kg−1). Fluorescence intensity was expressed as radiant efficiency (photons sec−1 cm−2 Sr−1). (b) Quantitative analysis of DOX content evaluated by an average radiant efficiency of each organ at 24 h and 48 h (n = 3). Data represents the mean ± SD (*p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
2.6. In vivo anti-cancer effect of NPs
With the exciting demonstration of an enhanced tumor-targeting effect by MVPs, we further evaluated the therapeutic effect of the NPs to verify whether an enhanced tumor-targeting effect of the MVPs induces the anti-cancer effect (Figure 5). MVP2 exhibited not only a much higher anti-cancer effect but also a more prolonged tumor regression profile than BMP2 (Figure 5a). Likewise, MVP4 showed a better anti-cancer effect than BMP4, although the degree of tumor regression was comparable between MVP4 and BMP4. The greater effect on tumor regression by MVP2 than by MVP4, compared to each BMP with the same polymer-drug ratio, is primary because the relatively smaller size of MVP4 leads to greater chance to be eliminated from the body by hepatobiliary clearance.[15a, 17] Since the therapeutic effect solely depended on delivery efficiency of DOX to cancer cells in this experiment, we confirmed that the resulting anti-cancer effect of NPs was well-correlated with accumulation and retention of DOX at the tumor site, as demonstrated in the bio-distribution results (Figure 4). Tumor weight and shape observed on day 21 were well correlated with the tumor growth (Figure 5b, c). There were no serious changes in the body weight, implying negligible systemic toxicity of the NPs themselves (Figure 5d).
Figure 5.

Evaluation of in vivo anti-cancer effect of NPs at PBA/DOX ratios of 2 and 4 using tumor-bearing nude mice. (a) Tumor growth after i.v. injection of each NP (arrows, twice injection of 2.5 mg DOX kg−1 at day 1 and day 3, n = 6). (b) Weights and (c) images of tumors dissected from each group at day 21. (d) Body weight changes of tumor-bearing mice. (e) Histological images of heart, liver, and tumor with H&E staining. Vacuolization (white arrows) and necrosis (red arrows) were depicted. Scale bar = 100 μm. Data represents the mean ± SD (*p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
In addition, induced histological alterations of organs by the NPs were evaluated with histological analysis of haematoxylin and eosin (H&E) staining to consider nonspecific delivery of NPs because DOX can induce damage in the heart and liver.[18] In the heart, vacuolization, a representative damage by DOX, was observed from the BMP groups. Moreover, BMPs induced several vacuolization and necrosis in the liver as well. On the other hand, MVPs induced no noticeable damage in both organs, but severe damage in tumors, as inferred from the condensed and eosinophilic cytoplasm as well as dense purple chromatin fragments (Figure 5e). Consequently, MVPs exhibited greatly enhanced anti-cancer effect derived from their high tumor targeting in vivo rather than BMPs.
3. Conclusion
We presented highly controlled therapeutic NPs created by polymer-drug self-assembly through high-speed on-chip synthesis. The MV approach produced narrower size distributions of NPs with an excellent reproducibility and controllability where the size of the MVPs can be determined by an increased flow rate of SMR. As a result, MVPs exhibited higher cellular uptakes and more enhanced anti-cancer effects in vitro and significantly higher tumor accumulation and regression in vivo than those of BMPs. Furthermore, SMR production rate up to 3.0–3.5 kg MVPs h−1 facilitated animal studies with no chip-to-chip variation, while the highest production rate of other previous studies was 3.15 kg day−1 through paralleled microfluidic devices.16 In summary, we proved that MVPs, which is prepared by highly controlled synthesis, exhibited not only uniform properties but an enhanced therapeutic effect in tumor-bearing mice, attributed to a higher accumulation of monodisperse NPs at tumor region. Our findings can highlight the importance of highly controlled on-chip synthesis of NPs for successful clinical translation. As well, there is a possibility to extend our microfluidics-based synthetic approach to other self-assembled nanomaterials fabricated by simple mixing and nanoparticles formed by chemical bonds occurring in physiological conditions. Therefore, we expect that our strategy can suggest the solution for the limited large-scale production of the NPs for clinical trials to enable the reproducible synthesis of the nanoparticles at a suitable scale for the rapid clinical development.
4. Experimental Section
Materials:
Doxorubicin (DOX) was purchased from MedChem Express (Monmouth Junction, NJ, USA). Poly(methyl vinyl ether-alt-maleic acid) (pMVEMA, 80 kDa) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). 3-aminophenylboronic acid monohydrate (PBA-amine) was obtained from TCI Co. (Tokyo, Japan). Flamma®675 amine was supplied by Bioacts (Incheon, Korea). Matrigel® Matrix was purchased from Corning (NY, USA). Polydimethylsiloxane (PDMS, Sylgard 184 silicone elastomer kit) for fabrication of the SMR was purchased from Dow Corning Corporation (Auburn, MI, USA). All commercial reagents were used without additional purification.
Instrumentation:
TEM images were obtained using an HT7700 TEM (Hitachi, Tokyo, Japan) and JEM-2210 (JEOL, Tokyo, Japan) coupled with the DigitalMicrograph camera and software suite from Gatan (Pleasanton, CA). 1H-NMR spectra were obtained using a Bruker Advance 300 MHz NMR spectrophotometer (Bruker, MA, USA). Hydrodynamic size of nanomaterials was measured using Zetasizer Nano ZS from Malvern Instruments (Worcestershire, United Kingdom). All UV/vis absorption spectra and fluorescence spectra were measured using a Cytation 5 plate reader from BioTek (Winooski, VT, USA). The CLSM images were observed using a LSM 700 confocal microscope from ZEISS (Oberkochen, Germany).
Synthesis of polymeric PBA:
polymeric PBA (pPBA) was synthesized by spontaneous conjugation of PBA to pMVEMA as previously reported (Figure S1a).[9, 19] Briefly, pMVEMA and PBA-amine were dissolved in DMSO and stirred at room temperature overnight. After the conjugation of PBA into the polymer, an unreacted PBA-amine was removed by dialysis (Spectra/Por®, MWCO = 10 kDa) against deionized water for 48 h while replacing the external medium several times. The synthesized pPBA was obtained by lyophilization and a conjugation ratio of PBA to polymer was analyzed using 1H NMR (Figure S1b). 1H NMR (300 MHz, D2O, δ): 7.9–7.2 (m, 4xH, Ph), 3.9–3.6 (m, 1H, -CH-), 3.6–3.1 (m, 3H, -OCH3), 3.1–2.4 (m, 2H, CHCOO), 2.4–1.4 (m, 2H, -CH2-). For the fluorescence labelled polymer (pPBA-F), Flamma®675 amine was added with PBA-amine and prepared by the same procedure as aforementioned.
Fabrication of SMR chip:
The SMR uses two inlets and a single outlet, with a height of 50 mm, outlet width of 2 mm, inlet width of 1mm, and channels with the dimensions 0.2 mm x 0.2mm, (mixing efficiency was 0.92, volumetric average in a SMR).[8] To fabricate the SMR with the PDMS, elastomer and curing agent with a 10:1 mass ratio were poured on a patterned silicon wafer and cured at 80°C overnight. The molded SMR chip was torn off and bonded to glass slides using plasma cleaner (PDC-32G, Harrick Plasma, Ithaca, NY) for 60 seconds.
Numerical simulations:
Computational fluid dynamics (CFD) was applied to calculate outlet mass fraction of mixture between two miscible liquids in swirling microvortex reactor (SMR) with commercial software, Fluent (Ansys, Inc. Concord, MA). Water information at room temperature was used for two inlets boundary condition in order to simply complex liquid conditions, and no-slip boundary condition was imposed at walls. Simulations were implemented with four difference Reynolds number cases to explore the relation between mixing efficiency of NPs and mass flow rate. For simulation analysis, the post-processing program, Tecplot 360 (Tecplot, Inc. Bellevuo, WA) was used to calculate mixing efficiency at each SMR sections.
Preparation of the doxorubicin and pPBA nanoparticle (DOX/pPBA NP):
DOX/pPBA NP was prepared by MV or BM method. The pPBA and DOX solution were prepared at various molar ratios of DOX to PBA with fixed PBA concentration (2.5 mM) in PBS. For MV method, the solutions were mixed rapidly at various Re in the SMR chip, where the flow ratio between two solutions was 1:1 at Re of 250. Syringe pumps (Harvard Apparatus, MA, USA) were used to pump the solutions into the SMR, and then the mixed solution was collected. For BM method, the solutions were simply mixed in 1.5 mL tube with the volume ratio of 1:1. The production rate was calculated at Re 250; MVP2: 3.5 kg NP h−1, 1 kg DOX h−1, MVP4: 3.0 kg NP h−1, 0.5 kg DOX h−1.
After forming the NP, the solution was appropriately diluted depending on its final purpose. The size and the morphology of the NP were measured by DLS and TEM, respectively. For TEM sampling, a solution of the NPs was dropped onto a copper grid and negatively stained with a filtered 2% PTA (pH 7.5) solution for 30 seconds. To observe the pH-responsive deformation of NP, the NPs were incubated in PBS with pH 5.0 and pH 7.5 for 2 h. An interaction between DOX and PBA was confirmed by a fluorescence quenching of PBA with the fixed final pPBA concentration at 2.5 mM [PBA].
Drug release:
According to the pH-responsive behavior of PBA-DOX interaction, DOX release profile was monitored at PBS with different pH. Briefly, the prepared NP was diluted in PBS with pH 7.5 and 5.0 at the final concentration of 200 μM [DOX]. The released DOX was separated by ultrafiltration (3 kDa MWCO) at the desired time point, and fresh PBS with corresponding pH was added into the solution containing the NPs. Released DOX was quantified by measuring fluorescence (ex 495 nm/em 595 nm) of the filtrate and calculated as [(released drug)/(loaded drug)×100].
Cell culture:
U-87 MG (human glioblastoma) cell line was supplied from Korean Cell Line Bank (Seoul, Korea). Daoy and D556 (human meduloblastoma) cell lines were supplied from ATCC (Manassas, VA, USA). For whole cell culture experiments, the cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Gaithersburg, MD, USA) containing 9% fetal bovine serum (Gibco, Gaithersburg, MD, USA) and 100 U mL−1 penicillin/streptomycin (Gibco, Gaithersburg, MD, USA) and incubated in humidified incubators maintaining 37 °C and 5% CO2.
Cell viability test:
Cell viability was measured by MTT assay to evaluate the cytotoxicity of DOX, pPBA, MVP2/4, and BMP2/4. Cells were seeded on a 96-well plate at a density of 9 × 103 cells per well for Daoy and 1.0 × 104 cells per well for U-87 MG and D556 and incubated for overnight. After cell adhesion, the cells were washed with DPBS and treated with each sample in 200 μL of serum-containing medium. After 48 h, the media was removed and washed with cold DPBS. Media containing the MTT agent (0.5 mg mL−1) were added to each well. After further incubation for 3 h, the media was carefully removed, and 200 μL of DMSO was added to dissolve formazan salts. 100 μL of the formazan solution was transferred to new 96-well plates and an absorbance at 595 nm was measured by microplate spectrofluorometer (Cytation 5 plate reader from BioTek, Winooski, VT, USA). The viability of non-treated cells was regarded as 100%.
In vitro cellular uptake monitoring by CLSM:
Daoy and D556 cell lines were seeded on glass coverslip placed in 6-well culture plates at an initial density of 5 × 104 cells per well. After 12 h, cells were washed by DPBS and treated with 1 μM [DOX] equivalence MVP or BMP in serum-free media. In order to normalize the fluorescence intensity of each NP, an appropriate ratio of pPBA-F and pPBA was utilized for the fabrication of BMP/MVP2 and BMP/MVP4. After 4 h of incubation, cells were washed with DPBS three times and fixed with 10 % neutral buffered formalin (NBF) at 4 °C for overnight. Cells on the coverslips were mounted using Vectashield anti-fade mounting medium with DAPI (Vector Labs, CA, USA). Intracellular localization of DOX and pPBA-F were detected by the CLSM at excitation / emission wavelength of 488 / 600 nm and 639 / 700 nm, respectively. The images were analyzed by ZEN 3.0 (blue edition) software. Mean fluorescence intensities (MFIs) were calculated by dividing the sum of fluorescence intensity of the cells with the number of the cells (n = 30).
In vivo bio-distribution imaging:
All the animal experiments were approved by the Postech Biotech Center Ethics Committee (POSTECH-2019–0021). Female BALB/c nude mice were supplied by Orient Bio (Gyeonggi, Korea). The mice weighing 17 ± 1 g were inoculated subcutaneously (SC) with 7 × 106 U-87MG cells in 100 μL of 50% Matrigel® (50 μL cell suspension + 50 μL Matrigel®). When the size of tumor reached 115 mm3, the mice were intravenously (i.v.) injected with 100 μL of each NPs containing 2.5 mg kg−1 DOX equivalent. After 24 h and 48 h, the fluorescence images of excised tumors and organs were acquired using the IVIS spectrum small-animal in vivo imaging system (Califer Lifescience, Hopkinton, MA, USA). Average radiant efficiencies of region of interest (ROI) were measured by living image software (PerkinElmer, USA).
In vivo anti-cancer experiments:
All animal experiments were approved by the Postech Biotech Center Ethics Committee (POSTECH-2019–0021). Female BALB/c nude mice were provided by Orient Bio (Gyeonggi, Korea). The female BALB/c nude mice weighing 17 ± 1 g were inoculated SC into the right flank with 7 × 106 U-87 MG cells in 100 μL of 50% Matrigel®. When the average tumor volume reached 150 mm2, the mice were randomly divided into five groups of six mice per group. PBS, BMP2, MVP2, BMP4 and MVP4 solutions (2.5 mg kg−1 DOX equivalence) were i.v. injected in tumor-bearing nude mice on day 1 and day 3, and tumor size and weight change of mice were monitored for 21 days. To monitor the anti-cancer effect, two dimensions of tumor growth were measured every other day using electronic calipers. Volume of tumors was calculated using the formula of an elongated spheroid: [tumor volume = 0.5 × a × b2], where a is the longest dimension and b is the shortest dimension. The volume of tumors was measured until the average of the longest and the shortest dimension of tumor from PBS group (average mice weight of 20 g) was over 14 mm (21 days after sample injection), and the animals were sacrificed. To determine statistical differences, all results were analyzed using GraphPad Prism 7 software by performing one-way or two-way ANOVA.
Immunohistological analysis:
To analyze tumor histology, representative mice from each group sacrificed on day 21 were selected and the tumors were excised. Tumors were fixed by 10% NBF for 1 day, embedded in paraffin, and sectioned with Finesse ME microtome. Each tumor section was stained with hematoxylin and eosin to observe the histological changes depending on the sample. Histological images were obtained using high-resolution camera (Adimec Q-12A-180, Netherlands) with 20× magnification.
Supplementary Material
Acknowledgement
This research was supported by the National Research Foundation of Korea (NRF) grant (NRF-2017R1E1A1A01074088), the Creative Materials Discovery Program (NRF-2018M3D1A1058813) funded by the Korean government (Ministry of Science and ICT), the National Institutes of Health Director’s New Innovator Award 1DP2HL142050 (Y.K.) and the National Science Foundation under CAREER CMMI 1653006 (Y.K.). This work was in part performed at the Institute for Electronics and Nanotechnology at the Georgia Institute of Technology, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542174).
Footnotes
Conflicts of interest
The authors declare the following competing financial interests: In compliance with the institutional guidelines of the Georgia Institute of Technology, Y.K. discloses his financial interest in Mepsgen and Mepsgenlab, two biotechnology companies developing microengineered physiological systems and biomimetic nanoparticles for medical applications. Mepsgen and Mepsgenlab did not support the aforementioned research, and currently these companies have no rights to any technology or intellectual property developed as part of this research.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Sungjin Jung, School of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea; George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Junseok Lee, Department of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Junha Lim, Department of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Jeeyeon Suh, Department of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Taeyoung Kim, George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Jungho Ahn, George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Won Jong Kim, School of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea; Department of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
YongTae Kim, George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA; Wallace H. Coulter Department of Biomedical Engineering, Parker H. Petit Institute for Bioengineering and Bioscience, Institute for Electronics and Nanotechnology, Georgia Institute of Technology, Atlanta, GA 30332, USA..
References
- [1].a) Zhang L, Chen Q, Ma Y, Sun J, ACS Applied Bio Materials 2019, 3, 107; [DOI] [PubMed] [Google Scholar]; b) Ahn J, Ko J, Lee S, Yu J, Kim Y, Jeon NL, Adv Drug Deliv Rev 2018, 128, 29; [DOI] [PubMed] [Google Scholar]; c) Tsui JH, Lee W, Pun SH, Kim J, Kim DH, Adv Drug Deliv Rev 2013, 65, 1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Chiesa E, Dorati R, Pisani S, Conti B, Bergamini G, Modena T, Genta I, Pharmaceutics 2018, 10; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hong L, Sesen M, Hawley A, Neild A, Spicer PT, Boyd BJ, Soft Matter 2019, 15, 9565. [DOI] [PubMed] [Google Scholar]
- [3].a) Li W, Zhang L, Ge X, Xu B, Zhang W, Qu L, Choi CH, Xu J, Zhang A, Lee H, Weitz DA, Chem Soc Rev 2018, 47, 5646; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Damiati S, Kompella UB, Damiati SA, Kodzius R, Genes (Basel) 2018, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lattuada M, Hatton TA, Nano Today 2011, 6, 286. [Google Scholar]
- [4].a) Lim JM, Swami A, Gilson LM, Chopra S, Choi S, Wu J, Langer R, Karnik R, Farokhzad OC, ACS Nano 2014, 8, 6056; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu D, Cito S, Zhang Y, Wang CF, Sikanen TM, Santos HA, Adv Mater 2015, 27, 2298; [DOI] [PubMed] [Google Scholar]; c) Rhee M, Valencia PM, Rodriguez MI, Langer R, Farokhzad OC, Karnik R, Adv Mater 2011, 23, H79; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu D, Bernuz CR, Fan J, Li W, Correia A, Hirvonen J, Santos HA, Advanced Functional Materials 2017, 27; [Google Scholar]; e) Majedi FS, Hasani-Sadrabadi MM, VanDersarl JJ, Mokarram N, Hojjati-Emami S, Dashtimoghadam E, Bonakdar S, Shokrgozar MA, Bertsch A, Renaud P, Advanced Functional Materials 2014, 24, 432; [Google Scholar]; f) Cao Y, Silverman L, Lu C, Hof R, Wulff JE, Moffitt MG, Mol Pharm 2019, 16, 96. [DOI] [PubMed] [Google Scholar]
- [5].a) Sun J, Zhang L, Wang J, Feng Q, Liu D, Yin Q, Xu D, Wei Y, Ding B, Shi X, Jiang X, Adv Mater 2015, 27, 1402; [DOI] [PubMed] [Google Scholar]; b) Hu C, Bai Y, Hou M, Wang Y, Wang L, Cao X, Chan CW, Sun H, Li W, Ge J, Ren K, Sci Adv 2020, 6, eaax5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Yong KT, Law WC, Hu R, Ye L, Liu L, Swihart MT, Prasad PN, Chem Soc Rev 2013, 42, 1236; [DOI] [PubMed] [Google Scholar]; b) Verjans ET, Doijen J, Luyten W, Landuyt B, Schoofs L, J Cell Physiol 2018, 233, 2993; [DOI] [PubMed] [Google Scholar]; c) Yoon M, Efremenko A, Blaauboer BJ, Clewell HJ, Toxicol In Vitro 2014, 28, 164. [DOI] [PubMed] [Google Scholar]
- [7].a) Su C, Liu Y, Li R, Wu W, Fawcett JP, Gu J, Adv Drug Deliv Rev 2019, 143, 97; [DOI] [PubMed] [Google Scholar]; b) Yuan D, He H, Wu Y, Fan J, Cao Y, J Pharm Sci 2019, 108, 58; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hagens WI, Oomen AG, de Jong WH, Cassee FR, Sips AJ, Regul Toxicol Pharmacol 2007, 49, 217. [DOI] [PubMed] [Google Scholar]
- [8].Toth MJ, Kim T, Kim Y, Lab Chip 2017, 17, 2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee J, Kim J, Lee YM, Park D, Im S, Song EH, Park H, Kim WJ, Acta Pharmacol Sin 2017, 38, 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kim J, Lee YM, Kim H, Park D, Kim J, Kim WJ, Biomaterials 2016, 75, 102. [DOI] [PubMed] [Google Scholar]
- [11].a) Kimura N, Maeki M, Sato Y, Note Y, Ishida A, Tani H, Harashima H, Tokeshi M, ACS Omega 2018, 3, 5044; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jahn A, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M, Langmuir 2007, 23, 6289; [DOI] [PubMed] [Google Scholar]; c) Thiermann R, Mueller W, Montesinos-Castellanos A, Metzke D, Löb P, Hessel V, Maskos M, Polymer 2012, 53, 2205. [Google Scholar]
- [12].Yan J, Springsteen G, Deeter S, Wang B, Tetrahedron 2004, 60, 11205. [Google Scholar]
- [13].a) Maxfield FR, Yamashiro DJ, Adv Exp Med Biol 1987, 225, 189; [DOI] [PubMed] [Google Scholar]; b) Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ, Cancer Res 2013, 73, 1524; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) van Sluis R, Bhujwalla ZM, Raghunand N, Ballesteros P, Alvarez J, Cerdn S, Galons J-P, Gillies RJ, Magnetic Resonance in Medicine 1999, 41, 743. [DOI] [PubMed] [Google Scholar]
- [14].a) Albanese A, Chan WC, ACS Nano 2011, 5, 5478; [DOI] [PubMed] [Google Scholar]; b) Wang Z, Tiruppathi C, Minshall RD, Malik AB, ACS Nano 2009, 3, 4110; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, Brown D, Alkilany AM, Farokhzad OC, Mahmoudi M, Chem Soc Rev 2017, 46, 4218; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rabanel JM, Adibnia V, Tehrani SF, Sanche S, Hildgen P, Banquy X, Ramassamy C, Nanoscale 2019, 11, 383. [DOI] [PubMed] [Google Scholar]
- [15].a) Zamboni WC, Torchilin V, Patri AK, Hrkach J, Stern S, Lee R, Nel A, Panaro NJ, Grodzinski P, Clin Cancer Res 2012, 18, 3229; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blanco E, Shen H, Ferrari M, Nat Biotechnol 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Matsumura Y, Maeda H, Cancer Research 1986, 46, 6387; [PubMed] [Google Scholar]; b) Maeda H, Bharate GY, Daruwalla J, Eur J Pharm Biopharm 2009, 71, 409; [DOI] [PubMed] [Google Scholar]; c) Maeda H, Sawa T, Konno T, Journal of Controlled Release 2001, 74, 47. [DOI] [PubMed] [Google Scholar]
- [17].a) Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW, J Control Release 2016, 240, 332; [DOI] [PubMed] [Google Scholar]; b) Li SD, Huang L, Mol Pharm 2008, 5, 496; [DOI] [PubMed] [Google Scholar]; c) Danaei M, Dehghankhold M, Ataei S, Hasanzadeh Davarani F, Javanmard R, Dokhani A, Khorasani S, Mozafari MR, Pharmaceutics 2018, 10; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Haute DV, Berlin JM, Ther Deliv 2017, 8, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Song S, Chu L, Liang H, Chen J, Liang J, Huang Z, Zhang B, Chen X, Front Pharmacol 2019, 10, 1030; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ky B, Putt M, Sawaya H, French B, Januzzi JL Jr., Sebag IA, Plana JC, Cohen V, Banchs J, Carver JR, Wiegers SE, Martin RP, Picard MH, Gerszten RE, Halpern EF, Passeri J, Kuter I, Scherrer-Crosbie M, J Am Coll Cardiol 2014, 63, 809; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shivakumar P, Rani MU, Reddy AG, Anjaneyulu Y, Toxicol Int 2012, 19, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kim J, Lee J, Lee YM, Pramanick S, Im S, Kim WJ, J Control Release 2017, 259, 203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.