

Abstract
Natural products have historically been a rich source of diverse chemical matter with numerous biological activities, and have played an important role in drug discovery in many areas including infectous disease. Synthetic and medicinal chemistry have been, and continue to be, important tools to realize the potential of natural products as therapeutics and as chemical probes. The formation of biofilms by bacteria in an infection setting plays a significant factor in the recalcitrance of many bacterial infections, conferring increased tolerance to many antibiotics and to the host immune reposnse, and as yet there no approved theraputics for combatting biofilm-based bacterial infections. Small molecules that interfere with the ability of bacteria to form and maintain biofilms can overcome antibiotic tolerance conferred by the biofilm phenotype, and have the potential to form combination therapies with conventional antibiotics. Many natural products with anti-biofilm activity have been identified from plants, micobes, and marine life, including: elligic acid glycosides, hamamelitannin, carolacton, skyllamycins, promysalin, phenazines, bromoageliferin, flustramine C, meridianin D, and brominated furanones. Total synthesis and medicinal chemistry programs have facilitated structure confirmation, identification of critical structural motifs, better understanding of mechanistic pathways, and the development of more potent, more accessible, or more pharmacologically favorable derivatives of anti-biofilm natural products.
Graphical Abstract
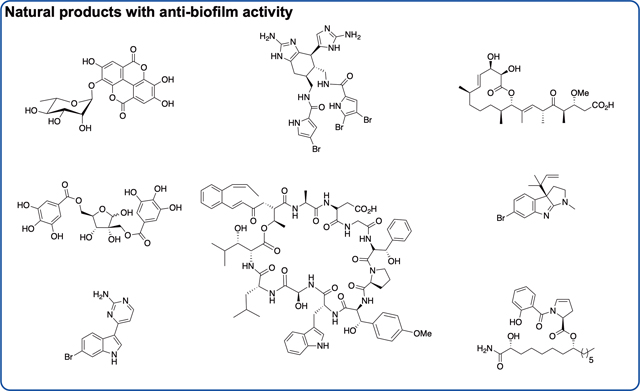
Introduction
Natural products are molecules produced by a biological source, and include secondary metabolites, molecules that are not directly involved in growth, development or reproduction, and are not essential for survival under normal conditions. Instead, they typically provide competitive advantage, or mediate interaction between and/or defense against other organisms. The biosynthesis of natural products is often unique to a specific organism and the environment within which it resides. Although the number of building blocks are limited, the number of novel secondary metabolites is almost infinite.1
Natural products have long been a source of therapeutic entities for a myriad of disease areas including: oncology, neurological disease, cardiovascular and metabolic disease, immunological and inflammatory diseases, and infectious disease.2 Natural products have played a particularly predominant role in the identification of antibacterial compounds, from the discovery of penicillin, produced by fungi of the genus Penicillium, in 1928,3 to the approval of fidaxomicin, a first in class macrocyclic antibiotic produced by the actinomycete Dactylosporangium aurantiacum, in 20114. Nature continues to be an important source of antibacterial agents; of the 126 small molecule antibacterial agents approved between January 1981 and September 2019, eleven were natural products, and 78 were natural product derivatives.5
Much of the reason for the utility of natural products stems from the fact that they typically occupy chemical space not usually found in synthetic-based screening libraries. They are typically chemically more complex than synthetic “drug-like”’ molecules, possessing a greater proportion of sp3 -hybridized bridgehead atoms and chiral centers, a lower nitrogen but higher oxygen content on average, a greater number of aliphatic versus aromatic rings, and accordingly, a greater degree of three-dimensionality.6
Chemical synthesis has played an important role both in the development of natural products for biomedical applications, and in their use as chemical tools to answer biological questions and identify new targets. Total synthesis of natural products in some cases enables the acquisition of sufficient quantities of material that cannot be accessed from the source organism. In other cases, total synthesis has been utilized to confirm the structure of natural products.7, 8 Synthetic chemistry has been used to identify structural motifs within the natural product that are required for activity, and medicinal chemistry campaigns have utilized this information to develop analogues with improved activity, reduced toxicity, more desirable pharmacological properties, or that are more readily accessible.
Many natural products have been investigated both for the ability to disrupt bacterial biofilms, or to sensitize biofilm cells to conventional antibiotics. These compounds continue to serve as inspiration for the development of novel antibiofilm agents with improved activity and pharmacological profiles. The development of synthetic strategies to access active natural products and their analogues is important if the potential of these scaffolds, both as chemical probes and therapeutic entities, is to be harnessed. In this review, we discuss the role of chemical synthesis in the identification and development of natural products for combatting bacterial biofilms. We give a brief background explaining biofilms and their role in infection and antibiotic resistance. We describe natural products isolated from plants, microbes, and marine organisms that have demonstrated anti-biofilm activity, and for select examples, describe how chemistry has played a role in their study, development and application. A summary of activities of the featured natural products is shown in Table 1. This review is not meant to be a comprehensive description of all natural product and natural product-derived compounds with anti-biofilm activity, several other reviews have covered this area,9–12 but is intended to be an analysis of the impact of chemical synthesis in both developing therapeutic strategies to overcome biofilm-based bacterial infections, and in answering fundamental biological questions about the biofilm phenomenon.
Table 1.
Summary of natural product activity
| Natural product or natural product class | Source | Antibiofilm activity | Mechanism | Any other biological activity | ||
|---|---|---|---|---|---|---|
| Bacterial species | Activity | Analogue activity | ||||
| Ellagic acid glycosides 15, 16, 20 | Plant (Rubus ulmifolius Schott. | S. aureus | 15 MBIC50 64 μg/mL | - | Inhibits cellular adhesion | - |
| Hamaelitannin 21 | Plant (Hamamelis virginiana) |
S. aureus | EC50 145.5 μM (pretreatment) 165.1 μM (treatment of preformed biofilms) with vancomycin |
33 EC50 0.389 μM (pretreatment) 7.976 μM (treatment of preformed biofilms) with vancomycin |
Inhibits TRAP phosphorylation | Inhibits TNF-mediated endothelial cell death Cytotoxic activity colon cancer cells |
| Carolacton 53 | Microbe (Sorangium cellulosum ) | S. mutans | Decreases biofilm cell viability 3078% % at 62 nM - 2 μM | 77 decreases biofilm cell viability 75–93% at 62 nM - 2 μM | Affects VicKRX and ComCDE via PknB | Inhibits human cancer cell lines via MTHFD1 and MTHFD2 |
| Skyllamycins B 78 and C 79 | Microbe (Streptomyces sp.) | P. aeruginosa | 78 EC50 30 μM 79 60 μM | - | Unknown | - |
| Promysalin 99 | Microbe (Pseudomonas putida) | P. aeruginosa | IC50 1 μM (PAO1) 125 nM (PA14) Disperses PAO1 and PA14 biofilms at 6.25 μM | 108 IC50 7.7 μM (PAO1) 19 nM (PA14) | Binds Sdh | Inhibits pyoverdine production in P. putida KT2440 |
| Phenazines | Microbe (various bacteria) | Gram-positive bacteria | - | 114 MBEC 9.38, 2.35, 0.20 μM (MRSA, MRSE, VRE) | Potentially through binding iron (II) | - |
| Bromoageliferin 128 | Marine (Agelas conifera) | Many | IC50 10.6 μM (MRSA) 28.3 μM (A. baumannii) | 144 IC50 0.89 μM (MRSA) 28.3 μM 151 IC50 26 μM (A. baumannii) | Binding response regulators | Reduces voltage-dependent calcium entry in PC12 cells |
| Flustramine C 160 | Marine (Flustra foliacea) | Many | 30% A. baumannii biofilm inhibition at 100 μM | 179 IC50 16.3 μM (E. coli) 13.4 μM (S. aureus) | Potentially through indole signaling pathways | - |
| Meridianin D 180 | Marine (Aplidium meridianum) | S. aureus M. smegmatis | IC50 87.4 μM (MRSA) IC50 21.5 μM EC50 51.0 μM (M. smegmatis) | 188 IC50 9.62 μM (MRSA) 189 IC50 7.5 μM EC50 27.9 μM (M. smegmatis) | Unknown | Inhibits multiple kinases |
| Brominated Furanones | Marine (Delisea pulchra) | S. enterica | - | 190 IC50 10 μM | QS Inhibition | Cytotoxic |
Bacterial biofilms
Biofilms are sessile, surface associated, heterogeneous structures formed by multicellular communities of bacteria (and other microorganisms.13 Bacteria form biofilms in many environments in response to a number of external and internal stimuli. The ability to form biofilms is a universal attribute of bacteria,14 and biofilms play a role in a number of infections including: infection of indwelling medical devices, wound infections, bacterial endocarditis, otitis media, dental carries, and lung infections of cystic fibrosis patients.9 Bacteria within a biofilm are better able to evade the host immune response, and can display upwards of 1000-fold decreased sensitivity to antibiotics compared to planktonic bacteria,15 which contributes to the recalcitrance of biofilm based infections towards traditional approaches to eradication. The antibiotic tolerance exhibited by bacteria residing within a biofilm can render antibiotics ineffective even against bacterial strains that do not harbour genetic resistance determinants. Tolerance differs from antibiotic resistance in that it involves a reduction in the rate of antibiotic-induced killing of a whole bacterial population in comparison to cultures of the strain from which the tolerant population was derived.16 It is important to note that cells displaying biofilm mediated antibiotic tolerance will exhibit susceptibility to the antibiotic upon dissociation from the biofilm.17
There are several factors responsible for the decreased sensitivity of biofilm cells to antibiotic therapy. One of the most significant factors is the presence of the biofilm matrix. This self-produced matrix, which is predominantly comprised of extracellular polymeric substances (EPS) including carbohydrates, proteins, lipids and extracellular DNA, forms the three-dimensional architecture of the biofilm, and performs several functions. These include: providing mechanical stability, mediating adhesion, enabling transportation of nutrients and waste within the biofilm, and providing protection against desiccation, ultraviolet radiation, some predators, metallic cations, host immune defences, and biocides. The matrix also limits penetration of certain antibiotic classes into the biofilm 18–20 Another contributing factor to antibiotic tolerance in a biofilm setting is the fact that nutrient and oxygen levels within a biofilm vary both spatial and temporally, leading to varied rates of microbial growth. Given that most conventional antibiotics target biosynthetic processes that only occur in actively growing bacteria and display limited efficiency in killing non-multiplying bacteria, cells within the biofilm exhibiting reduced metabolic activity show greater tolerance to such antibiotics.18, 21–23 Bacterial cells within a biofilm also exhibit altered gene expression compared to planktonic cells, including genes that mediate antibiotic resistance such as efflux pumps.24 Increased mutation rates and the presence of hypermutable strains as a result of oxidative stress in the biofilm environment,25, 26 and increased horizontal gene transfer as a result of high population density27, 28 additionally contribute to increased rates of resistance development and spread. Finally, the presence of persister cells, cells that are genetically homogeneous to the rest of the population yet phenotypically heterogeneous and exhibit antibiotic tolerance through a mechanism generally thought to be a result of adopting a state of dormancy in which they become metabolically inactive, is also thought to contribute to the lack of efficacy of many antibiotics toward biofilm-based infections.29–31 All the above mentioned tolerance mechanisms make the eradication of biofilm-based infections a considerably challenging endeavour.32
Current clinical approaches for the treatment of biofilm-based bacterial infections are severely limited and dependent upon causative agent and infection site. Select antibiotics have demonstrated some activity against bacterial biofilms, for example polymyxin antibiotics such as colistin, which does not depend upon cells being metabolically active to effect bacterial killing. However, colistin typically has to be administered as part of a combination regimen due to rapid resistance acquisition by metabolically active cells within the biofilm.15 The RNA polymerase inhibitor rifampin has also been reported to have some activity against biofilm cells, namely staphylococcal biofilms,33 but again is not recommended as monotherapy due to rapid selection of resistant mutants.34 Other current treatment approaches include: removal of colonized foreign bodies, surgical debridement and aggressive antibiotic therapy,35, 36 drainage of abscesses, or long-term high dose combination antibiotic regimens. These approaches often still result only in biofilm reduction rather than eradication, and continued chronic suppressive treatment is required.35, 37 Another issue with the use of conventional antibiotics is the fact that several studies have shown that the presence of sub-inhibitory levels of several commonly prescribed antibiotics can induce in vitro biofilm formation. Such antibiotic levels occur not only at the Beginning and end of a dosing regimen, but importantly also in areas of the biofilm that antibiotics cannot readily access as a result of limited penetration.38
Alternative approaches to combatting biofilms involve the identification and development of compounds that act specifically on biofilm cells, typically in one (or more) of three ways: 1) inhibition of biofilm formation (to be classified as an anti-biofilm compound this should ideally occur with no or minimal toxicity toward planktonic bacteria), 2) dispersion of pre-formed biofilms, and 3) killing of biofilm cells (either selectively or in addition to planktonic toxicity). Such compounds have the potential to act in an adjuvant manner and be paired with conventional antibiotics, as has been successfully achieved with the pairing of ß-lactamase inhibitors with ß-lactam antibiotics such as Augmentin, and would be a valuable therapeutic option to overcome biofilm-based infections. Many small molecules with anti-biofilm activity have been identified, either through rational design to interfere with one of the many processes essential for the formation and maintenance of biofilms, or as in the case of many of the natural products detailed in this review, through phenotypic screening.9
Many of the anti-biofilm small molecules for which the mechanism of action is known, including some natural products, act by interfering with one or more of the bacterial signaling and communication systems that regulate biofilm formation and maintenance.39 Some of these are analogues of the native signaling molecules, while others are based upon distinct scaffolds. Probably the most well characterized bacterial communication systems are the quorum sensing (QS) systems utilized by bacterial communities to coordinate gene expression, and consequently phenotypes including biofilm formation, in response to changes in population density.40 Different QS systems are employed by different bacterial species, such as acyl homoserine lactone (AHL) based systems found in Gram-negative bacteria,41 autoinducing peptides (AIP) based systems found in Gram-positive bacteria,42, 43 and the universal autoinducer-2 (AI-2) based system employed by both Gram-negative and Gram-positive bacteria.44, 45 The different QS systems act through the same basic premise, which is centered upon release and detection of small diffusible signaling molecules known as autoinducers. QS systems comprise two primary proteins: a synthase that produces the autoinducer in response to population changes and environmental signals, and a receptor protein that enacts a response (change in gene expression) upon binding of the autoinducer above a threshold level.46 Both the synthase and receptor, as well as many auxiliary proteins involved in QS systems, represent potential targets for modulating biofilms. Two-component system (TCS) signaling also plays a role in biofilm formation and regulation (in addition to many other phenotypes) across many bacterial species by allowing the bacterium to sense and respond to environmental changes. TCS consist of a sensor histidine kinase that responds to an extracellular stimulus and, through a series of phosphorylation events, activates the cognate DNA-binding response regulator, leading to a change in gene expression.47 Both the two primary proteins of TCS, and many auxiliary proteins can be targets for controlling biofilm formation. Other signaling pathways that are involved in biofilm regulation and have been targeted as an anti-biofilm strategy include: the diguanylate cyclase (DGC) and phosphodiesterase (PDE) pathway that utilizes the second messenger bis-(3’5’)-cyclic di-guanylic acid (c-di-GMP), 48, indole signaling pathways,49–51 and long chain fatty acid signaling pathways.52
Plant natural products as antibiofilm agents
Plants have long been a source of natural products with a myriad of biological activities. Most plants produce hundreds to thousands of unique phytoalexins and other secondary metabolites belonging to various chemical families including: flavonoids, phenols, terpenoids, furanoacetylenes, steroid glycoalkaloids, stilbenes, sulfur-containing compounds and indoles.53 Plant natural products serve purposes such as self-defense, and interaction with other organisms in the environment.54 The biological utility of phytochemicals has been recognized and exploited for centuries in the practice of traditional medicine, where plant extracts have been used to treat numerous ailments, and medicinal plant usage remains widespread today.55
Despite the spectrum of biological activities that plant natural products exhibit, there are no approved plant-derived antibiotics - most plant-derived compounds have only weak antibiotic activity that is several orders of magnitudes weaker than that of microbially produced antibiotics.56 Yet, particularly in their natural setting, plants are generally able to successfully mount a defense against infecting organisms. This is believed to be a result of the fact that many plant natural products possess anti-virulence properties, comprising the main category of anti-infective activity identified from this source.54 Several plant natural products that exhibit antibiofilm activity have been reported, as described below.
Flavonoids are a large, structurally diverse group of polyphenolic natural products that, along with semi-synthetic and synthetic derivatives, have been extensively studied for their antibacterial activity.57 Baicalin (5,6-dihydroxy-7-O-glucuronide flavone) 1 (Figure 1), isolated from the roots of Scutellaria baicalensis, has a long history of use in Chinese medicine and has been reported to increase the susceptibility of Burkholderia cenocepacia biofilms to tobramycin.58, 59 This activity was initially thought to be a result of QS inhibition,58 but was later revealed to be due to modulation of the oxidative stress response. It was shown that baicalin affects multiple pathways, including oxidative phosphorylation, ultimately leading to increased reactive oxygen species (ROS) production in the presence of tobramycin, which accounts for increased tobramycin-mediated killing of biofilm cells.59 Naringenin 2, and quercetin 3, are found in many plants and have been shown to act as QS antagonists in Vibrio harveyi, and to inhibit biofilm formation by V. harveyi and Escherichia coli.60 Phloretin 4, a dihydrochalcone intermediate of the biosynthetic pathway of flavonoids found in apple peel and strawberries,61 reduces biofilm formation by enterohemorrhagic E. coli (EHEC) without affecting either planktonic growth or biofilms of commensal E. coli, and is thought to act via the repression of AI-2 importer genes.62 Proanthocyanidins, represented by procyanidin A2 5, a class of flavonoids that are produced by cranberry plants, have been reported to inhibit biofilm formation by Pseudomonas aeruginosa and to potentiate gentamicin activity in a Galleria mellonella model of P. aeruginosa infection.63
Figure 1.

Natural products isolated from plants that have demonstrated anti-biofilm activity.
The stilbenoid resveratrol 6, (found in the skin of grapes and berries) inhibits biofilm formation by Vibrio cholerae,64 while this and the related oxyresveratrol 7 also inhibit biofilm formation by uropathogenic E. coli (UPEC) at low concentrations (10–50 μg/mL) by reducing fimbriae production and swarming motility.65 The triterpene ursolic acid 8 (found in many plants, in particularly large quantities apple peel), inhibits E. coli biofilm formation and upregulates genes involved in biofilm related processes such as chemotaxis and motility.66 The diterpene 4-epi-pimaric acid 9, which is produced by Aralia cachemirica L. (Araliaceae) inhibits biofilm formation by Streptococcus mutans, an oral pathogen implicated in dental carries and endocarditis.67
Garlic extracts have been reported to increase the susceptibility of P. aeruginosa biofilms to tobramycin in vitro, and to clear P. aeruginosa lung infections in vivo when used prophylactically.68 The active compounds are thought to be ajoenes 10 and 11, which exert their effects through modulation of quorum sensing.69, 70 Another sulfur containing phytochemical that has been reported to display anti-biofilm activity is the isothiocyanate iberin 12, produced by horseradish, which inhibits biofilm formation by P. aeruginosa, potentially by inhibiting the Gac/Rsm quorum sensing systems.71
Ellagic acid glycosides
Rubus ulmifolius Schott. (Rosaceae) is a plant that produces edible berries, and has also traditional medicine applications for the treatment of skin and soft tissue infections (SSTI).72 The anti-staphylococcal activity of R. ulmifolius was validated by Quave et al. as part of a study to evaluate extracts of south Italian plants for antibacterial and anti-biofilm activity against methicillin-resistant Staphylococcus aureus (MRSA). The ethanolic extract of the roots of this plant were reported to inhibit biofilm formation with an IC50 of 8 μg/mL, significantly lower than its minimum inhibitory concentration (MIC) of 512 μg/mL.73 Subsequent bioassay-guided fractionation of the ethanolic root extract identified fraction 220D-F2, which inhibited biofilm formation across a panel of genotypically-diverse clinical S. aureus isolates and also enhanced the susceptibility of biofilms to the functionally distinct antibiotics daptomycin, clindamycin and oxacillin. This fraction was also reported to reduce pre-formed biofilms of Streptococcus pneumoniae as well as display antibiotic activity against planktonic cultures of this bacterium.74 Fraction 220D-F2 was subject to LC-UV/MS/MS analysis to identify the active chemical architecture responsible for anti-biofilm activity, revealing the presence of ellagic acid 13 (Scheme 1) along with several ellagic acid derivatives, in addition to sapogenin-related compounds.75
Scheme 1.

Synthesis of ellagic acid glycosides 15, 16, and 20.
While mass spectrometry was able to confirm the presence of generic xylopyranoside or xylofuranoside ellagic acid as well as mannopyranoside ellagic acid, this technique is unable to determine the identity of the sugar, anomeric configurations, nor the site of glycosylation. To address this ambiguity, the Weinert and Quave labs synthesized 15 and 16, and examined their anti-biofilm activity. Ellagic acid itself is extremely insoluble and somewhat unreactive, thus per-O-acetyl α-glycosyl iodide donors, generated from reaction of per-O-acetyl sugars with trimethylsilyl iodide (TMSI), were used for the preparation of ellagic acid glycosides. The regioselectivity of glycosylation was ensured by first protecting ellagic acid 13 as its perO-t-butyldimethylsilyl ether 14 (Scheme 1), followed by selective tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF)mediated deprotection at the 3-position to afford the 3monophenolate in situ (the selectivity is thought to be mediated by inductive effects of the proximal endocyclic lactone oxygen). Glycosylation of this intermediate with α-L-rhamnopyranosyl iodide or α-D-xylopyranosyl iodide afforded protected α-rhamnopyranoside ellagic acid and β-xylopyranoside ellagic acid. For the formation of the xyloside derivative, it was necessary to carry out the reaction in the presence of tetrabutylammonium iodide (TBAI) to promote in situ anomerization of the sugar to the more reactive β-iodide which, following iodide displacement by the adjacent acetoxy group, ensured generation of solely the β-xyloside upon reaction of the acetoxonium intermediate with protected ellagic acid. De-Osilylation of the glycosides with K2CO3 in wet DMF followed by deacetylation with K2CO3 in MeOH/H2O, followed by treatment with cation-exchange resin afforded ellagic acid 3-O-rhamnoside 15 and ellagic acid 3-O-xyloside 16 which were used as LCMS standards to confirm occurrence of both in the 220D-F2 extract.72
Ellagic acid rhamnoside 15 inhibited biofilm formation with a 50% minimum biofilm inhibitory concentration (MBIC50) of 64 μg/mL, compared to 128 μg/mL for ellagic acid and 25 μg/mL for 220D-F2 against the strain used in this study, and exhibited lower growth inhibition than 220D-F2. Ellagic acid xyloside 16 exhibited similar biofilm inhibition activity to 15 but much more potent growth inhibition. The authors posit that the high anti-biofilm activity and limited growth inhibition of ellagic acid rhamnoside suggest that it is a key component of 220D-F2 in the context of antibiofilm activity. Confocal laser scanning microscopy (CLSM) revealed similar biofilm architecture and levels of attachment upon treatment with 15 to that observed following treatment with 220D-F2.72
The ellagic acid glycoside scaffold has been further explored by the Townsend lab via a synthetic approach that centers around de novo ellagic acid formation utilizing a copper mediated Ullmann coupling and phenolic O-glycosylation. This approach can provide access to novel ellagic acid derivatives and potentially allow for incorporation of aryl moieties bearing unnatural substitutions. The synthesis was initiated by per-O-benzylation of methyl gallate 17 followed by electrophilic aromatic bromination with NBS. Ullmann coupling of the resultant aryl bromide in the presence of excess copper at elevated temperature delivered biaryl 18. Debenzylation and subsequent bis-lactonization provided ellagic acid, which was per-O-silylated with triisopropylsilyl chloride to give 19. As in the Weinert synthesis, the ellagic acid glycosyl acceptor was generated via selective de-O-silylation at the 3-position in situ followed by reaction of the quaternary ammonium salt of 19 under Gervay-Hague conditions with the desired glycosyl iodide and TBAI at elevated temperature. Finally, a one-pot, two-step global deprotection afforded both the previously reported ellagic acid xyloside and rhamnoside in addition to the arabinose derivative 20.76
The synthetic ellagic acid glycosides displayed modest antibiofilm activity against the Gram-positive pathogen Streptococcus agalactaie (Group B Streptococcus, GBS). Arabinoside 20 was the most active of the three, and was shown by scanning electron microscopy (SEM) to act via early-stage inhibition of cellular adhesion.76
Hamamelitannin
Hamamelitannin (2’,5-di-O-galloyl-D-hamamelose) 21 (Scheme 2 is a component of the bark of Hamamelis virginiana (witch hazel) that has been reported as an inhibitor of TNF-mediated endothelial cell death and DNA fragmentation,77 and to display specific cytotoxic activity against colon cancer cells78. Hamamelitannin was identified as an analogue of the S. aureus quorum sensing inhibitor RNAIII-inhibiting peptide (RIP) through a virtual screen of commercially available small molecules.79 RIP mediates biofilm formation by inhibiting phosphorylation of TRAP (target of RNA-III activating protein) and subsequent expression of the agr operon.80 When combined with vancomycin, hamamelitannin has been reported to effect a significant increase in killing of biofilm cells in vitro in several biofilm model systems.
Scheme 2.

Hamamelitannin 21 and synthesis of first generation hamamelitannin analogues.
Hamamelitannin has also been reported to increase the susceptibility of S. aureus biofilm cells to antibiotics form other classes including: several ß-lactams, daptomycin, linezolid, tobramycin, and fusidic acid without affecting their activity toward planktonic cells81. In vivo, the combination of hamamelitannin and vancomycin significantly enhanced survival compared to either compound alone in both Caenorhabditis elegans and G. mellonella infection models,58 as well as in a mouse mammary gland infection model. Additionally, hamamelitannin has been shown to prevent device-associated infections in a rat graft model of both S. aureus and S. epidermidis infections.79 The TraP receptor has been reported to be involved in the mechanism by which hamamelitannin sensitizes S. aureus biofilms, by affecting cell wall synthesis and decreasing its thickness, and by mediating extracellular DNA release.81
Hamamelitannin has been utilized as a scaffold for the development of novel vancomycin potentiators against staphylococcal biofilms. Given the absence of any structural information regarding the inhibitor–target interaction, much of the work has involved iterative bioactivity guided analogue design, exploiting synthetic and medicinal chemistry to not only identify compounds with greater activity, but also potentially providing information about target interactions.
Initially aiming to address the liabilities within the hamamelitannin scaffold with regards to therapeutic potential, namely the high polarity due to the large number of hydroxyl group, which might compromise bioavailability, the propensity of aromatic hydroxyl groups to undergo oxidation and glucuronidation, and the potential instability of the ester linkages, Vermote et al. synthesized a series of hamamelitannin analogues (Scheme 2).82 In this study, three modifications to the hamamelitannin scaffold were explored: isosteric replacement of the ester groups, systematic modification or elimination of the aromatic hydroxyl groups, and removal of the anomeric hydroxyl group to replace the hamamelose core with a tetrahydrofuran ring. Library synthesis was initiated from branched azidolactol 22 and proceeded through a three-step protection-benzylation-deprotection sequence to afford 23 (along with its anomer). Intermediate 23 was converted to the bis-azido compound 24 via methanesulfonyl activation of the 2’ hydroxyl, followed by azide displacement. Staudinger reduction, EDC-mediated acylation of diamine 25, and finally deprotection provided seven amido analogues with various hydroxyl patterns on the aromatic rings. Azidolactol 22 was also utilized to generate 51 total analogues, which in addition to replacement of the ester linkers of hamamelitannin with amide linkers, the anomeric hydroxyl was also removed (two analogues with identical acyl groups at the 2’ and 5-positions, and 49 analogues with a benzamide at the 2’-position and varying acyl groups at the 5-position). Reduction of 22 to triol 27 and subsequent tosylation was followed by cyclization, which proceeds to give solely the cis-isomer 28. Azide displacement of the tosylate followed by reduction of the resulting bis-azido compound 29 and subsequent acylation of the diamine 30 allowed for the generation of two analogues that contained identical acyl groups at the 2’ and 5positions. In a divergent approach, phthalimide displacement of tosylate 28 formed 31, in which the 2’ and 5-nitrogen moieties could be orthogonally revealed. Hydrazine cleavage of the phthalimide was followed by benzoylation to afford 32, which underwent azide reduction and acylation to provide a library of 49 analogues.
The effect of the hamamelitannin analogues upon the susceptibility of S. aureus biofilms to vancomycin both under pretreatment and combination treatment regimens was determined, revealing that ester replacement with an amide moiety is well tolerated, as is removal of the anomeric hydroxyl, and finally that removal of phenolic groups is also well tolerated. The 5-orthochlorobenzamide derivative 33 was the most active compound to emerge from the in vitro assay, displaying EC50 values (defined as the concentration needed to double the effect of antibiotic) of 0.389 μM and 7.976 μM for pretreatment and treatment of preformed biofilms respectively. By comparison, methyl isostere of the ortho-chloro phenyl moiety of 33 exhibited EC50 values of 13.67 μM and 82.62 μM, and the parent hamamelitannin exhibited EC50 values of 145.5 μM and 165.1 μM for pretreatment and treatment of preformed biofilms respectively. Compound 33 was subsequently shown to be stable in human plasma and in the presence of both rat and human S9 microsomal fractions, and did not display cytotoxicity in MRC-5 lung fibroblast cells up to a concentration of 128 μM. Furthermore, in both a C. elegans and murine infection model, the combination of compound 33 with vancomycin or cephalexin respectively effected a significantly greater reduction in CFUs compared to antibiotic alone, and the effect was greater than that observed for hamamelitannin.82
To gain further insight into the structure activity relationship (SAR) of the hamamelitannin scaffold, a library of 21 derivatives was next prepared, consisting of analogues in which the 5-amido group was replaced with alternative nitrogen-based linkers, and analogues with various substituents at the ortho position of the 5-phenyl ring. Replacement of the 5-amide linker with a sulfonamide group was found to be tolerated, as seen by the comparable activity observed between compounds 34 and 35 (Figure 2), but did not improve upon the activity of 33. 5-Benzamide ortho substitution with other halides resulted in compounds with increased activity over compound 34, but still less potent than compound 33. Finally, the 5-N-methylated analogue of 34, compound 36, exhibited increased activity compared to compound 34, which the authors attribute to an out-of-plane distortion of the benzamide moiety leading to better interaction with the binding site.83 In a subsequent report that centered on delineating the SAR at the 2ʹ-position of lead compound 33, as well as the construction of hybrid molecules in which the most active moieties at the 2’ and 5-positions were combined, led to the identification of phthalimide 37, which exhibited greater potency than 33, returning EC50s of 0.26 μM and 1.27 μM for pretreatment and combination treatment respectively.84
Figure 2.

Second generation hamamelitannin analogues.
In a follow-up study with the aim of identifying compounds with improved synthetic accessibility and activity the same group investigated structural alternatives for the central tetrahydrofuran ring of 33.85 They screened a subset of the publicly available ZINC database using the 2-chloro-N-(2-methoxyethyl)benzamide moiety of 33 as a reference fragment, and identified five potential central scaffolds. Two of those retained the tetrahydrofuran ring but either transposed the benzamidomethyl group to the 1-position (38), or deleted the hydroxyl group at the 3-position (39), the other three scaffolds replaced the tetrahydrofuran ring with either a substituted 1,4-dioxane (40), a 1,3-substituted morpholine (41) or a 2,4-substituted pyrrolidine (42). Deletion of the 3-hydroxyl group, or replacement of the tetrahydrofuran with a morpholine or pyrrolidine ring, resulted in a loss of activity (the latter was also reported in a recent study that examined the activity of N-substituted pyrrolidine analogues)86. Only compounds 38 and 40 displayed the ability to potentiate vancomycin against S. aureus biofilms. Compound 38, in which the benzamidomethyl group was moved to the 1-position delivered improved activity compared to compound 33 for the combination treatment of preformed biofilms with vancomycin, and comparable activity when administered as a pretreatment. Compound 40, which contains a dioxane core, displayed comparable activity under both conditions.85
Microbial natural products as antibiofilm agents
Many natural products produced by microorganisms have found application in a variety of therapeutic areas, including as immunosuppressive, anticancer, and anti-inflammatory agents. Furthermore, most antibiotics are, or are derived from, microbial secondary metabolites that are produced by bacteria and fungi as a means of competing for resources with other species in densely populated environments.87 Microbial natural products have likely been highly successful as a source of antibiotic scaffolds because they are derived from millions of years of evolution, and natural selection has ensured that they not only possess high potency and selectivity, but also the necessary physicochemical properties to be able to penetrate the bacterial cell envelope, something synthetic compounds often lack.88 It has been suggested that natural-product antibiotics, at sub inhibitory concentrations, may affect biofilm formation by neighboring microbes89, and several non-antibiotic microbial natural products have proved effective as scaffolds for the identification of anti-biofilm compounds, as described below.
Walkmycin C 43 (Figure 3), produced by Streptomyces sp. strain MK632–100F11, causes the formation of abnormal S. mutans biofilms and a reduction in biofilm mass. Walkmycin C was identified from a screen for inhibitors of the TCS histidine kinase WalK in Bacillus subtilis, and has also been shown to inhibit autophosphorylation of WalK in S. aureus by targeting the conserved catalytic domain.90 The anti-biofilm activity of this compound against S. mutans is a result of inhibition of autophosphorylation of the TCS histidine kinases VicK and CiaH, which play a role in sucrose-dependent biofilm formation.90, 91 Another Streptomyces secondary metabolite that has demonstrated anti-biofilm activity is streptorubin B 44, a prodiginine produced by Streptomyces sp. strain MC11024. Streptorubin B was identified from a screen of actinomycete culture extracts and inhibits biofilm formation by S. aureus at concentrations as low as at 1μg/mL.92 An enantioselective total synthesis of streptorubin B has been completed by the Thomson lab, which enabled confirmation of the absolute and relative stereochemistry of this natural product.93
Figure 3.
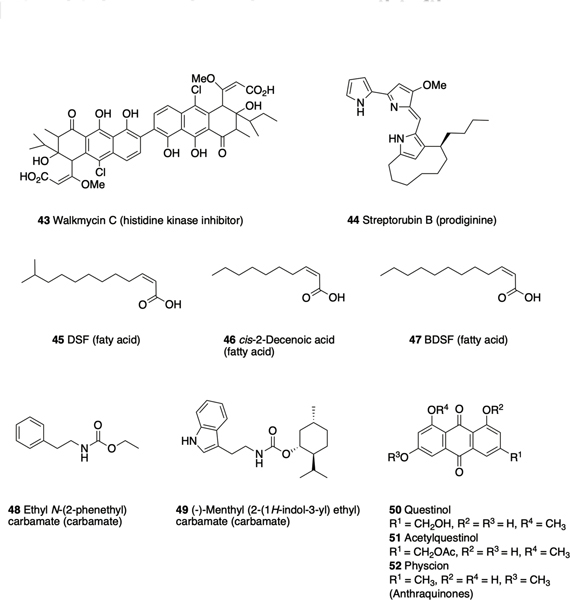
Microbial natural products that have demonstrated anti-biofilm activity.
The plant pathogen Xanthomonas campestris produces cis-11methyl-2-dodecenoic acid 45, known as diffusible signal factor (DSF)94, which has been reported to inhibit biofilm formation by Bacillus cerus.95 Other DSF-family signaling molecules that have demonstrated anti-biofilm activity include cis-2-decenoic acid 46, produced by P. aeruginosa, which disperses both its own biofilms and those of multiple other species including the human pathogens E. coli, K. pneumoniae, and S. aureus.96 The Burkholderia diffusible signal factor (BDSF), cis-2-dodecenoic acid 47, produced by B. cenocepacia, inhibits and disperse biofilms of Francisella novicida.97
The simple ethyl N-(2-phenethyl) carbamate 48, is produced by the marine bacterium SCRC3P79 (Cytophaga sp.), and was initially reported to exhibit anti-biofilm activity against another marine bacterium, Rhodospirillum salexigens.98 This activity was subsequently shown to translate to several medically relevant pathogens including S. aureus, and analogue synthesis based upon this scaffold led to the identification of several compounds with increased activity, the most active of which being the menthyl derivative 49.99 Several metabolites isolated from the endophytic fungus Eurotium chevalieri KUFA 0006 have reported anti-biofilm activity, including the anthraquinones questinol 50 and acetylquestinol 51, which inhibit biofilm formation by E. coli, and physcion 52, which inhibits biofilm formation by S. aureus and Enterococcus faecalis.100
Carolacton
Carolacton 53 (Scheme 3) is a macrolactone secondary metabolite produced by the myxobacterium Sorangium cellulosum and has been reported to selectively kill biofilm cells of S. mutans, exhibiting only minor effects on both planktonic cell growth of S. mutans101 and on other oral bacteria102. Carolacton has also been reported to exhibit growth inhibition of S. pneumoniae, and its target in this bacterium was determined to be the folate-dependent C1 metabolism enzyme FolD. Interestingly, carolacton has also been shown to inhibit the human FolD orthologs, MTHFD1 and MTHFD2 and inhibit the growth of several human cancer cell lines.103
Scheme 3.

Total synthesis of carolacton 53.
In S. mutans, carolacton affects several regulatory systems including the VicKRX and ComCDE two-component signal (TCS) transduction systems. VicKRX is required for the response to oxidative stress,104 and also plays an essential role in biofilm formation and maintenance of cell wall and membrane integrity. ComCDE plays an essential role in several pathogenic behaviors including: biofilm formation, virulence, survival under acidic conditions, and genetic competence.105 These effects are thought to be mediated through the serine/threonine protein kinase PknB, and it is hypothesized that carolacton effects membrane defects, which causes leakage of cytoplasmic content and subsequent cell death, during the pH decrease that occurs during biofilm growth of this bacterium.105
The structure of carolacton was elucidated in 2010 by the Kirschning and Müller labs, during which chemical derivatization was utilized to aid in the assignment of configuration for all the stereogenic centers. Carolacton 53, was treated with pbromobenzoyl chloride, leading to macrolactonization between the C1 carboxy group and the C18 hydroxy group to form the bislactone 54. Compound 54 was treated with both enantiomers of methoxy(trifluoromethyl)phenylacetyl chloride (MTPA-Cl), affording the diastereomeric esters (the (R)-diastereomer 55 is depicted in Scheme 3). Mosher analysis was applied and the absolute configuration at C17 was determined from the calculated ∆δSR values to be (R), and relative configurations for the remaining stereogenic centers of 54 could then be established using a combination of extended NMR analyses and molecular modeling and confirmed by X-ray analysis once a crystal was obtained.106
The first total synthesis of carolacton was published in 2012 by Schmidt and Kirschning and utilized several metal-mediated key transformations, including: Ley and Duthaler–Hafner aldol reactions and asymmetric Nozaki–Hiyama–Kishi and asymmetric Negishi–Fu CC coupling reactions.107 Seeking a synthetic route that more readily allowed for exploration of the carolacton SAR, Hallside et al. reported a concise synthesis in which key elements include ring-closing metathesis followed by a selective olefin hydrogenation, and macrocycle formation by esterification (Scheme 3). The known gulonolactone-derived diol 56 was subject to oxidative cleavage to form aldehyde 57, and a subsequent Z-selective Wittig olefination yielded alkene 58. Benzoate removal followed by Swern oxidation afforded lactone 59, which was treated with allylmagnesium bromide and CuBr·SMe2 to effect an SN2ʹ opening of the lactone and form 60 as a single diastereomer. The second subunit of the natural product was assembled by first carrying out enolization of the Evans β-ketoimide 61 under conditions favoring formation of the (E)isomer, followed by reaction with aldehyde 62 to form 63 in an inseparable mixture with a dr of 5:1.16. However, the subsequent three-step sequence of methylation, reduction, and oxidation− Wittig olefination allowed for isolation of 64 as a single diastereomer. Silylation to form 65, followed by conversion of the ester to an aldehyde and application of Leighton’s (R,R)-trans EZ-Crotyl Mix afforded 66 in a 5:1 dr. The two subunits 60 and 66 were then coupled via a Steglich esterification to give 67, which underwent selective terminal alkene metathesis upon treatment with the second-generation Grubbs catalyst, forming 68. Selective olefin reduction and concomitant PMB group removal was followed by TBS removal to provide diol 69. Finally, simultaneous oxidation of the primary and secondary alcohols under Swern conditions, followed by a Lindgren–Pinnick oxidation yielded keto-acid 70, and removal of the acetonide afforded carolacton 53. This synthetic route allowed for the biological investigation of intermediates 69 and 70, and while 70 was inactive, diol 69 was shown to significantly affected both the integrity and morphology of the biofilm matrix (albeit at higher concentrations than the natural product).102
Both the Kirschning and Wuest labs have developed synthetic routes to analogues of carolacton to delineate the SAR of this scaffold, and to readily access simplified active analogues that could be used as chemical probes to further understanding of the mechanism of action of carolacton. In 2015 the Kirschning lab reported the synthesis of several new carolacton analogues, revealing important SAR data. Inversion of configuration at C9 abolished activity, while methyl ester 71 retained activity but the analogous C17 carbonyl derivative 72 did not, implying that (R)configured carbinol moiety at C17 is crucial. Additionally, the bicyclic bislactone derivative 73106 also retained activity, and it was subsequently shown that the activity of 71 and 73 was a result of enzymatic hydrolysis by S. mutans to the natural product; these derivatives essentially act in a prodrug-like manner. 108
In 2017, the Wuest lab reported several carolacton analogues, accessed through a diverted total synthesis (DTS) approach, that displayed a range of biological activities. Compound 74 inhibits biofilm formation, while compound 75 acts in a similar manner to carolacton, killing cells during biofilm growth, while compound 76 inhibits biofilm maturation.109 More recently, the same group reported that compound 77, a further simplified carolacton analogue, inhibits biofilm formation and decreases biofilm cell viability to a greater degree than carolacton. This simplified analogue was employed in a proof of concept forward chemical genetic screen of S. mutans mutants to identify the CcpA signaling pathway as likely harboring the target of 77.110
Skyllamycins
Skyllamycins B 78 and C 79 were identified as non-antibiotic inhibitors of P. aeruginosa biofilms in a high-throughput screen of 312 natural-product prefractions that utilized the redox-sensitive dye XTT to determine bacterial cellular metabolic activity in addition to with biofilm mass. The skyllamycin family of natural products are cyclic depsipeptides produced by Streptomyces sp. Skyllamycins B and C exhibited M respectively, and when co-dosed with azithromycin, were shown to eliminate biofilms and depress cellular metabolic activity.111
The skyllamycins are highly modified and possess a number of unusual amino acids, including an extremely rare α-OH-glycine residue.112 Giltrap et al. have reported a synthesis of the skyllamycins, which involved the initial preparation of a series of structurally simplified analogues and subsequent adaption of the route to afford skyllamycins B and C. The synthesis required synthesis of the (2S,3S)-β-OH-Phe monomer that began with Grignard addition of PhMgBr to (R)-Garner’s aldehyde 80, giving a mixture of diastereomers (Scheme 4). Swern oxidation to 81, followed by diastereoselective DIBAL-H reduction yielded the desired antidiastereomer 82, which was treated with p-toluenesulfonic acid to remove the oxazolidine moiety and afford diol 83. Selective oxidation of the primary alcohol was achieved using a TEMPO-mediated oxidation with NaOCl as the stoichiometric oxidant to form 84. Subsequent exchange of amine protecting group delivered the protected (2S,3S)-β-OH-Phe 85. The protected (2S,3S)-β-OH-O-Me-Tyr residue was also prepared from (R)-Garner’s aldehyde 80, and involved addition of the organo-lithium species generated from para-bromoanisole to give alcohol 86 as a mixture of diastereomers. Oxazolidine removal, oxidation, and swapping protecting group as above afforded 87. Final protection of the hydroxyl using a novel pseudoproline protecting group strategy provided the fully protected β-OH-Tyr monomer 88, which could be separated from its diastereomer by silica column chromatography. The final non-proteogenic amino acid, (2R,3S)-β-OH-Leu, was accessed in a similar manner, starting with the reaction between (S)-Garner’s aldehyde 89 and iPrMgBr to yield alcohol 90 with exclusive (3S)-stereochemistry. Removal of the oxazolidine moiety to give diol 91 was followed by selective oxidation of the primary alcohol. Finally, swapping of the amine protecting group, and protection as before afforded the fully protected β-OH-Leu monomer 92.
Scheme 4.

Total synthesis of skyllamycins B 78 and C 79. Top: synthesis of non-proteogenic amino acids. Bottom: Construction of natural products.
Construction of the natural products was achieved by first assembling the linear peptide precursors 93 and 94 via Fmoc-solid phase peptide synthesis utilizing both conventional and microwave assisted HATU-mediated peptide coupling conditions. An on-resin esterification yielded the branched peptides, to which the two remaining residues were coupled. Cleavage from the resin and deprotection afforded the unprotected linear peptides 95 and 96. The linear peptides then underwent oxidative cleavage to aldehydes 97 and 98 by treatment with NaIO4 under pH-controlled conditions and quenching with ethylene glycol. Finally, cyclization and concomitant formation of the α-OH-Gly residue afforded skyllamycins B 78 and C 79.112
The synthetic skyllamycins, along with the simplified analogues synthesized during method development, were examined for antibiofilm activity against P. aeruginosa. It was revealed that removal of the hydroxyl group from the hydroxyglycine motif, removal of the βOH moieties from the three β-hydroxy amino acids, or elimination of the methyl group from the OMe-tyrosine had a significant impact upon activity, indicating that the skyllamycin scaffold is amenable to manipulation, which could allow for the exploration of simplified analogues.112
Promysalin
Promysalin 99 was isolated from the saprotrophic soil bacterium Pseudomonas putida RW10S1, and reported to exhibit anti-bacterial activity against P. aeruginosa and other pseudomonads.113 Promysalin was later reported to disperse P. aeruginosa biofilms and to inhibit pyoverdine production in P. putida KT244.114, 115 It was subsequently determined that the biological target of promysalin in P. aeruginosa is succinate dehydrogenase (Sdh), which plays a key role in both the tricarboxylic acid cycle (TCA) and respiration.116 The compound also binds iron115. The postulated structure given in the initial disclosure lacked stereochemical assignment, and while bioinformatic analysis of the gene cluster indicated that the absolute configuration of the C16 stereocenter should be assigned as (S), the C2 and C8 stereocenters remained unassigned. Steele et. al developed a synthetic route to access the four possible diastereomers generated by these two centers, and were able to confirm the stereochemistry of promysalin as (2R,8R,16S).114
Synthesis of the promysalin diastereomers commenced with construction of the myristic acid fragment (Scheme 5). The known compound (−)-100, formed from 5-hexenoic acid and the phenylalanine-derived Evans oxazolidinone, underwent diastereoselective oxidation using the Davis oxaziridine, and was then silyl protected to provide (−)-101. Subsequent crossmetathesis with homoallylic alcohol (+)-102 or (−)-102 in the presence of Grubbs catalyst C711, followed by hydrogenation then ammonolysis, afforded diastereomers (+)-103a and (+)-103b, respectively. In the same manner, myristic acid diastereomers (−)-103c and (−)-103d were synthesized from the enantiomeric starting material (+)-100.
Scheme 5.

Synthesis of promysalin 99 and structures of analogues 108-110.
The proline–salicylate fragment was accessed from SEM protected methyl salicylate 104, which was subjected to ester hydrolysis, HATU mediated coupling with trans-4-L-hydroxyproline methyl ester, and Dess–Martin oxidation to provide (+)-105. Regioselective conversion to the enamine followed by reduction using a modified Stille reaction and then ester hydrolysis provided the proline−salicylate coupling fragment (−)-106. Compound (−)-106 was then subject to EDC-mediated esterification with each of the four myristic acid diastereomers which, followed by deprotection under mild conditions that employed 1 M TBAF in THF with DMPU as a cosolvent, afforded the four promysalin diastereomers (−)-107a-d.114 1H and 13C NMR spectral data for compound 107a best aligned with that of the isolated material, and this diastereomer also exhibited the greatest anti-biofilm activity, exhibiting IC50 values of 1 μM and 125 nM against PAO1 and PA14 respectively, and dispersing biofilms of both strains at 6.25 and 12.5 μM , leading the authors to conclude that the structure of the natural product is that of (−)-107a.
In a follow-up study aiming to elucidate the chemical moieties responsible for the species-specific activity of promysalin, the Wuest lab synthesized a series of promysalin analogues using another diverted total synthesis (DTS) approach. They determined that removal of the proline ring double bond, replacement of the proline with a piperidine ring, or substitutions on the proline ring other than fluorination (compound 108 IC50s 7.7 and 0.019 μM against PAO1 and PA14 respectively), led to a reduction or complete loss of activity. Alterations to the salicylate moiety, including varying the position, removal, replacement, or esterification of the phenol were not well tolerated. Side chain modifications however, did produce active analogues including alkene 109, and de-hydroxy analogue 110, which demonstrated equipotent activity to the natural product. Compound 110 is more synthetically accessible than promysalin, requiring two less steps, one less protecting group and avoiding the necessity of a chiral auxiliary.115
Halogenated Phenazines
Phenazines, a class of secondary metabolites identified in both Gram positive and Gram-negative bacteria,117 have demonstrated antibiofilm activity in addition to antibiotic, antifungal, and anticancer activities.118, 119 Murine model studies have demonstrated that production of pyocyanin 111 (Figure 5) by P. aeruginosa regulates the release of the EPS component extracellular DNA.120 Studies have shown that, in cystic fibrosis (CF), young patients initially get infected by S. aureus and later P. aeruginosa co-infects CF patients and competes with S. aureus through phenazine mediated generation of reactive oxygen species (ROS).121 Phenazines also participate in iron acquisition, energy-generation, redox homeostasis and even act as signaling molecules, prolonging the originators lifespan in comparison to non-phenazine-producing bacteria.119 The discovery that bacteria utilize the redox-active nature of phenazines as a means of competing with other microbes within their environment led their investigation as potential antibacterial agents.
Figure 5.

Halogenated phenazines and quinolones 101-120.
The Huigens lab synthesized and screened a panel of halogenated phenazines and their ester analogues as potential antimicrobial agents.122 Compared to pyocyanin (MIC = 50 μM), halogenated phenazines 112 (MIC = 6.25 μM) and 113 (MIC = 50 μM) demonstrated comparable or improved antibacterial activity against S. aureus and S. epidermidis. To investigate the hypothesis that P. aeruginosa in chronic CF infection could be eradicating S. aureus biofilms, the potential of halogenated phenazines as antibiofilm agents was examined. Diverse 1-methoxyphenazine scaffolds were accessed through either a Wohl-Aue reaction or condensation of quinone with symmetric phenylenediamines, yielding halogenated phenazines with diversity at the 2-, 4-, 7-, and 8- positions.122–126 Using a Calgary Biofilm Device (CBD) assay, multiple biofilm eradicators against Gram-positive bacteria were identified, with compound 114 being the most potent biofilm-eradicator against Gram -positive bacteria (MBEC = 9.38 μM, 2.35 μM, and 0.20 μM against MRSA, methicillin-resistant S. epidermidis (MRSE), and vancomycin resistant Enterococcus faecium (VRE) respectively). It was revealed that a bromine at the 2-position is essential for both planktonic and biofilm-eradicating activities, with compound 115 exhibiting four- to 32-fold higher MBECs than 114.124 Halogens at the 7- and 8- position were also found to enhance biofilm-eradication activity against Gram-positive bacteria.
In a subsequent report, that broadened the synthetic scope, a two-step Buchwald–Hartwig cross-coupling and reductive-cyclization was utilized to construct multiple 1methoxyphenazines.127 Additionally, using the potent biofilm eradicator 116 (MBECs 4.69 μM, 37.5 μM, and 0.59 μM against MRSA, MRSE, and VRE respectively) as a starting point, several prodrugs were synthesized with the aim of enhancing water-solubility (e.g. carbonate analogue 117) or selectivity against bacteria (e.g. alkyloxycarbonyloxymethyl analogue 118). While both 117 and 118 retained biofilm eradication activity, carbonate 117 showed enhanced planktonic activity against MRSA, compared to parent 116 which was thought to be a result of enhanced membrane permeability. Hemolysis assay, metal-chelation studies, and RNA transcription analysis, indicate that HPs could eradicate biofilms via binding iron (II) and thereby inducing iron starvation.123, 128
Following these findings, similar metal-chelating scaffolds including halogenated quinolines and nitroxolines were investigated, revealing that the 2-position of halogenated quinolines plays an important role in tuning activity against Gram-positive bacteria.129 After generating a series of halogenated quinolines using microwave assisted Friedländer synthesis,130 the 2-position was rapidly diversified via alkylation and reductive-amination chemistry.131, 132 While several halogenated quinolines demonstrated planktonic toxicity, compound 119 exhibited potent biofilm-eradication activity against S. aureus, S. epidermidis, and E. faecium (MBECs 7.8 μM, 5.9 μM and 1.0 μM against MRSA, MRSE and VRE respectively) without significant HeLa-cell-cytotoxicity or human red blood cell (RBC) lysis. Attempts to improve water-solubility of this scaffold, through the introduction of polar groups at the 2-position utilizing Williamson ether synthesis with compound 120, resulted in a reduction in biofilm eradication activity (MBECs 200 μM, 75 μM and 2.35 μM against MRSA, MRSE and VRE respectively).133
Marine natural products as antibiofilm agents
The marine environment encompasses tremendous biodiversity and marine organisms have long been a rich source of bioactive compounds.134 It is thought that the unique conditions found in marine environments are responsible for the production of molecules bearing significant diversity in terms of both structural and functional features, which consequently exhibit a high degree of bioactivity.135 In particular, the sessile nature of marine invertebrates such as sponges and corals has driven the evolution of chemical defense systems to evade predation and prevent biofouling. Marine natural product derived drugs with a broad spectrum of therapeutic applications including: cancer, anti-viral, chronic pain, and hypertriglyceridemia have been approved, and many others are in development.136 The study of marine natural products is hampered by the fact that typically only small quantities can be isolated from the source organism, and synthetic methods to access these natural products are therefore highly valuable if the potential of this rich source of chemical space is to be realized.
Examples of marine natural products that have demonstrated anti-biofilm activity include: the diterpene alkaloid (−)-ageloxime D 121 (Figure 6), which was isolated from the marine sponge Agelas nakamurai, and inhibits biofilm formation by S. epidermidis without affecting bacterial growth.137 Another diterpene, darwinolide, 122, which was isolated from the Antarctic sponge Dendrilla membranosa, inhibits MRSA biofilm formation with an IC50 value of 33.2 μM, four-fold lower than its MIC.138 Total synthesis of darwinolide was recently reported by Siemon et al.139 The tryptamine derivative bufotenine 123, isolated from the Mediterranean gorgonian Paramuricea clavate, has been reported to exhibit anti-adhesion activity against the marine bacterium Pseudoalteromonas spp D41.140 Synoxazolidinones are brominated guanidinium oxazolidinones isolated from the sub-arctic ascidian Synoicum pulmonaria. Synoxazolidinone A 124 exhibits moderate anti-bacterial activity against Gram-positive bacteria,141 while synoxazolidinone C 125 has been reported to inhibit adhesion by several species of marine bacteria.142 Synthetic analogues of synoxazolidinone A reported by the Pierce lab, compounds 126 and 127, inhibit MRSA biofilm formation with low or sub micromolar IC50 values (0.78 and 1.2 μM respectively), while compound 126 disperses pre-formed biofilms with an EC50 of 4.7 μM.143
Figure 6.

Marine natural products that have demonstrated anti-biofilm activity.
Bromoageliferin
Bromoageliferin 128 (Scheme 6) belongs to the pyrrole-imidazole class of alkaloids, which has been exclusively isolated from marine sponges144, and has been reported to reduce voltage-dependent calcium entry in PC12 cells.145 The structurally simple oroidin 129 (Figure 7), is a key intermediate in the biosynthesis of many of the more complex pyrrole-imidazole alkaloids, and was first isolated from Agelas oroides in 1971,146 (it has subsequently been isolated from several other sponges), while bromoageliferin was isolated from Agelas conifera.147 Oroidin and bromoageliferin were reported to inhibit the growth of R. salexigens SCRC 113 by Yamada et al. in 1997.98 Both bromoageliferin and the related pyrrole-imidazole alkaloid sceptrin 130 inhibit biofilm formation by the commonly encountered human pathogens MRSA and Acinetobacter baumannii with IC50s of 10.6 μM and 28.3 μM respectively.148
Scheme 6.

Total synthesis of bromoageliferin 128.
Figure 7.

Bromoageliferin inspired compounds with anti-biofilm activity.
Following the first enantioselctive total synthesis of ageliferin by the Baran lab in 2007, via a vinyl cyclobutane rearrangement of sceptrin,149 the Chen lab developed a biomimetic synthesis of the ageliferins that allowed for flexible introduction of the pyrrole side chains thus allowing synthesis of dibromoageliferin.150 The synthesis was initiated by coupling azidoimidazole 131 with allylic ester 132 to give 133 (Scheme 6), which was then subject to oxidative cyclization via treatment with Mn(OAc)3 in acetic acid at 50–60 °C, or with Mn(picolinate)3 in methanol at 90 °C to form the ageliferin core skeleton 134. Optimal yield of 135 was achieved upon using crude 134, as a mixture of C9’ diastereomers, that upon decarboxylation yielded a single (cis) diastereomer, which was subsequently mesylated to deliver 135. Conversion to the iodide and finally displacement with sodium azide gave 136. The orthogonally masked amine groups of this key intermediate allowed for the selective introduction of the differing pyrrole units of bromoageliferin. The dibromopyrrole was introduced first at the N7 position through Staudinger reduction of the azide and coupling with the dibromopyrrole to form 137. Selective coupling to the less hindered N7 position was achieved with careful control of reaction time. Introduction of the monobromopyrrole unit at the N7’ position was achieved through Boc-deprotection and coupling under the same conditions as before to form 138. Construction of the second 2-AI group was carried out by installation of a guanidine group on 138. This was followed by oxidation of the hydroxyl group with 2iodoxybenzoic acid (IBX) to the aldehyde which underwent intramolecular cyclization to form 139. Following correction of the C9ʹ configuration of 139 through a slow equilibration under acidic conditions (60−72 h, d.r. > 10:1), the C10’ carbonyl group was removed by sequential reductions to afford 140. Removal of the BOM protecting group was achieved through a two-pot process consisting of conversion to the corresponding chloromethyl derivative (BCl3) followed by cleavage with ammonium hydroxide., Finally, hydrolysis of the triphenylphosphine imide group with hydrochloric acid afforded bromoageliferin 128.150
Both bromoageliferin and the simple pyrrole-imidazole alkaloid oroidin 129 have been employed as templates for the design of a number of classes of simplified, more easily accessible analogues, many of which exhibit anti-biofilm activity across a broad spectrum of bacteria. The first indication that simplified bromoageliferin analogues could modulate bacterial biofilms occurred with the report that trans-bromoageliferin analogue (TAGE) 141 and cis-bromoageliferin (CAGE) 142 inhibited biofilm formation by P. aeruginosa (IC50‘s of 100 μM against PAO1).151 Structural modification led to the identification of the di-brominated acylpyrrole containing analogue 143, which inhibits the formation of biofilms by P. aeruginosa more potently than does TAGE, and also inhibits biofilm formation by A. baumannii. Bordetella bronchiseptica and S. aureus.152 These analogues retain both the bicyclic core of the natural product, along with the 2-aminoimidazole (2-AI) subunit. The 2-AI subunit would prove to be particularly important in conferring anti-biofilm activity, and there have been numerous subsequent reports of 2-AI containing compounds that demonstrate anti-biofilm activity. Simplification of the chiral bicyclic structure found in bromoageliferin produced a series of 2-aminobenzimidazole (2-ABI) analogues that exhibit anti-biofilm activity against Gram-positive, Gram-negative, and mycobacteria. Compound 144 inhibits biofilm formation by S. aureus E. faecium and S. epidermidis with low micromolar IC50s and was shown to act via a zinc dependent mechanism31, as was 2-ABI 145, for the inhibiton of biofilm formation by MRSA153. In 2012 it was reported that a homologue of 145, compound 146, inhibits biofilm formation by Mycobaacterium smegmatis.154 Analogue synthesis identified compound 147, which in additon to inhbiting biofilm formation by M. smegmatis155, also inhibits biofilm formation by Salmonella enterica.156 Addtionally, the simple 2-ABI 5,6-dimethylbenzimidazole (DMABI) 148 was reported by the Blackwell lab to potently inhibit biofilm formation by P. aeruginosa, potentially through interference with the P. aeruginosa Las and Rhl QS systems.157
Several classes of oroidin-like compounds have also been synthesized and examined for anti-biofilm activity. Dihydrosventrin (DHS) 149 inhibits biofilm formation by P. aeruginosa, A. baumannii, and B. bronchiseptica158, while the para-bromophenyl derivative 150, exhibits increased anti-biofilm activity against A. baumannii as compared to DHS.159 Compound 151 inhibits and disperse biofilms of both P. aeruginosa and A. baumannii,160 and a biotinylated analogue of 151 was used to identify the target of this class of compounds in A. baumannii as the response regulator BfmR (of the BfmRS TCS),161 which plays an important role in biofilm formation162 in this bacterium.
The related 2-AI 152, inhibits biofilm formation by S. mutans and downregulates the histidine kinase ComD in this bacterium,163, 164 while several aryl 2-AI compounds including 153 bind both BfmR and the Francisella spp. response regulator QseB, and inhibit biofilm formation by both species.165, 166 The Van der Eycken and Steenackers labs have also conducted medicinal chemistry programs on the 2-AI scaffold, and reported a series of aryl 2-AIs including 154 that exhibit anti-biofilm activity against P. aeruginosa and S. typhimurium.167, 168 A series of tri-substituted 2-AI compounds, including 155, synthesized as analogues of Naamine A 156, also displayed antibiofilm properties against the same two bacteria. 169 Inspired by the dimeric nature of naturally occurring 2-AI containing compounds, these same groups investigated the activity of 5-aryl-2AIs dimers linked by an alkyl chain between either the N1-position or the N2-positions. They observed an increase in anti-biofilm activity for N1,N1 -linked dimers compared to the respective monomers against S. typhimurium, E. coli and S. aureus, while certain N2,N2 -linked dimers additionally displayed increased biofilm activity against P. aeruginosa compared to the respective monomers. For example, dimer 157 exhibited IC50 values of 0.3, 10.1, 5.4, and 8.9 μM against S. typhimurium, P. aeruginosa, E. coli, and S. aureus respectively compared to values of 84.4, 15, 75.5, and >800 μM respectively for the monomer.170
Expanding upon the key 2-AI and 2-ABI moieties contained in the compounds described above, libraries of compounds possessing the related 2-aminopyrimidine and 2-aminoquinazoline moieties have also been examined. 2-aminopyrimidine 158 inhibits biofilm formation by MRSA171 and M. smegmatis,154 while 2aminoquinazoline 159 inhibits biofilm formation by M. smegmatis172
Flustramine C
Flustramine alkaloids, a family of indole-derived secondary metabolites isolated from the bryozoan Flustra foliacea,173, 174 were reported by Riedel et al. to possess antimicrobial activity against marine bacterial strains, suggesting a role combating fouling organisms.175 While most flustramines do not exhibit significant activity against terrestrial bacteria, some derivatives were reported to antagonize AHL-regulated QS systems,175 which inspired the investigation of the flustramine scaffold for the identification of antibiofilm agents.
Following total syntheses of flustramine C 160 by the Sakamoto176 and Funk177 groups, Lindel et al. reported a synthetic approach from the related flustramine alkaloid deformylflustrabromine 161 via a Danishefsky inverse prenylation (Scheme 7).178 The synthesis initiated with the conversion of N-methyltryptamine 162 to N-formyl-N-methyltryptamine 163 followed by treatment with t-BuOCl and freshly prepared prenyl-9BBN to effect the inverse prenylation and provide 1,1-dimethylallyl indole 164. Compound 164 was monobrominated at the 6-position via treatment with NBS in AcOH/HCO2H, to form the natural product flustrabromine 165, which was converted to deformylflustrabromine 161 via hydrolysis of the formamide moiety with sodium hydroxide. The key step in the conversion of deformylflustrabromine 161 to flustramine C 160 was the 1,2-rearrangement of the inverse prenyl group, which was successfully achieved via treatment with one equivalent of NBS.178
Scheme 7.

Synthesis of flustramine C 160 and deformylflustrabromine 161.
The Lindel synthetic route was utilized to investigate the antibiofilm activity of flustramine family members, revealing that flustramine C modestly inhibited biofilm formation by A. baumannii (30% at 100 μM).179 The synthesis of analogues of flustramine C in which the inverse prenyl moiety was replaced by a propargyl group, thus allowing for diversification via click chemistry, led to the identification of several compounds that displayed anti-biofilm activity against several species. Compounds 166-168 (Figure 8) inhibited biofilm formation by MRSA with IC50 values of 86 μM, 25 μM, and 5.4 μM respectively, although toxicity toward planktonic bacteria increased as chain length increased. Modest inhibition of biofilm formation by E. coli and A. baumannii (166 and 167 only), without planktonic toxicity was also observed. Aromatic analogues 169-171 demonstrated a similar activity trend to the aliphatic analogues, in which anti-biofilm activity and planktonic toxicity increased with increasing chain length, and compounds 169-171 inhibited biofilm formation by both methicillin sensitive S. aureus (IC50 values of 32.0 μM, 12.5 μM, and 6.6 μM respectively) and MRSA (IC50 values of 13.5 μM, 6.6 μM, and 3.4 μM respectively). Compound 172, a furindoline analogue of 170, was synthesized using similar chemistry, and inhibited biofilm formation by MRSA with an IC50 of 18.7 μM) in a non-toxic manner.179 Subsequently, related series of flustramine C analogues in which a propargyl group was installed at the at 3-position of indole, with or without bromination of the indole, and again derivatized via click chemistry were reported by Minvielle et al.180 Compound 173 inhibited biofilm formation by A. baumannii, S. aureus and MRSA, yet interestingly promoted E. coli biofilm formation, while compound 174 modestly inhibited biofilm formation by S. aureus yet its 2-bromo analogue 175 promoted formation.
Figure 8.

Flustramine inspired compounds with anti-biofilm activity.
Desformylflustrabromine 161 has also been investigated as a scaffold for the identification of compounds with anti-biofilm activity. Desformylflustrabromine itself exhibits moderate inhibition of biofilm formation by E. coli and S. aureus, however it also exhibits toxicity toward planktonic bacteria. A comprehensive SAR study of this scaffold revealed that the C-6 bromine and the C-2 reverse prenyl group are essential for anti-biofilm activity. Alkylation of the indole nitrogen was tolerated, while activity could be increased through modulation of the N-methyl moiety.181, 182 Compounds 176 (IC50 53 μM and 5.9 μM against E. coli and S. aureus respectively) and 177 (IC50 15.6 μM and 7.7 μM against E. coli and S. aureus respectively) were found to be highly active non-toxic inhibitors of biofilm formation. Compound 178, in which the inverse prenyl group was transposed to the indole nitrogen, demonstrated moderate biofilm-inhibition activity (IC50 80 μM and 65 μM against E. coli and S. aureus respectively). The three-carbon homologue of 176, compound 179, exhibited potent biofilm-inhibition activity (IC50 16.3 μM and 13.4 μM against E. coli and S. aureus respectively), demonstrating the tunability of the alkyl-linker. Mechanistic studies with 176 revealed that, analogous to indole itself, the activity of compound 176 is dependent on temperature, TnaA, and the transcriptional regulator SdiA, suggesting that 176 may be modulating biofilm formation via indole-based signaling pathways.181
Meridianin D
The meridianins are secondary metabolites isolated from the marine invertebrate Aplidium meridianum,183 that have demonstrated a number of biological activities,184–186 including inhibition of CDKs, GSK-3, PKA and other protein kinases at low micromolar concentration,187 and antibacterial activity against S. aureus186 and M. tuberculosis.188 Given that meridianin D 180 (Scheme 8) possesses both an indole and 2-aminopyrimidine moiety, it was investigated as a scaffold for the identification of novel anti-biofilm compounds. Huggins et al. utilized the synthetic approach developed by Bredereck189 to access meridianin D and analogues.190 Synthesis of meridianin D commenced with acylation of 6-bromo indole 181 at the 3-position using acetyl chloride and tin chloride to afford 182, followed by tosyl protection of the indolic nitrogen giving 183. The enaminone 184 was prepared by reaction with DMF/dimethylformamide-dimethylacetal (DMF-DMA), and finally cyclization and deprotection of the enaminone in 2-methoxyethanol using potassium carbonate and guanidine hydrochloride provided meridianin D 180, which inhibited biofilm formation by MRSA with an IC50 value of 87.4 μM.190 SAR exploration centered on varying the position and identity of the halogen on the indole ring, alkylation of the indole nitrogen, alkylation of the exocyclic nitrogen of the 2-AP, and replacement of the 2-AP with a 2-AI. Transposition of the bromine moiety from the 6-position in the natural product to the 5-position in compound 185 (Figure 9) led to increased activity, with this compound exhibiting an IC50 value of 17.9 μM for the inhibition of biofilm formation by MRSA. Compound 185 also modestly dispersed pre-formed MRSA biofilms, with an EC50 value of 138.4 μM, which was lowered by 33.4% when dosed with vancomycin at a concentration that does not affect the biofilm as a standalone treatment. Alkylation of the exocyclic amine of 185 with methyl (186) or ethyl (187) led to an increase in dispersal activity albeit with a tandem increase in toxicity and diminished synergy with vancomycin, while benzylation at this position (188) increased inhibition activity (IC50 9.62 μM) but abolished dispersal activity. Replacement of the 2-aminopyrimidine with a 2-AI in both meridianin D and compound 185 resulted in increased planktonic toxicity, and no antibiofilm activity below toxic concentrations. The meridianin D analogue library was also investigated for its effect on mycobacterium biofilms, which revealed that the natural product exhibited moderate dispersal activity (EC50 51.0 μM) but more potent inhibition (IC50 21.5 μM) against M. smegmatis.191 Several analogues displayed increased activity compared to meridianin D, including 2AI containing 189, which inhibited biofilm formation with an IC50 of 7.5 μM, and dispersed pre-formed biofilms with an EC50 of 27.9 μM. As mentioned above, compound 189 did not disperse MRSA biofilms, indicating that synthetic manipulation of the meridianin scaffold can be utilized to tune selectivity.
Scheme 8.

Synthesis of meridianin D 180.
Figure 9.

Meridianin D analogues with anti-biofilm activity.
Brominated furanones
Halogenated furanones are secondary metabolites isolated from the marine macro-alga Delisea pulchra, first reported by The Roche Research Institute of Marine Pharmacology,192 and found to have broad-spectrum antimicrobial as well as biofilm-inhibition activities,193–195 while some brominated furanones have been reported to display cytotoxic effects toward human neuroblastoma SK-N-SH cells196. Halogenated furanones share structural similarities with AHLs and have been shown to inhibit quorum sensing through competitive interaction with LuxR-type receptors.197, 198 The exocyclic vinyl bromide has been reported to be an essential structural feature, mediating activity through nucleophilic attack and subsequent degradation of the target protein.198
Sulfuric acid mediated synthesis of halogenated furanones from bromo-substituted levulinic acid was first reported by Sims et al., 199 and later optimized by Steinberg et al.200 The synthesis involves condensation of ethyl acetoacetates with ethyl 2-bromoalkanoates, subsequent hydrolysis and decarboxylation of the diester in presence of sodium hydroxide, followed by reaction with molecular bromine. Furanone 190 (Figure 10) was reported to inhibit QS in P. aeruginosa and to effect bacterial clearance in a pulmonary mouse model,198, 201, 202 however the therapeutic application of halogenated furanones is hindered by toxicity concerns, with high doses of 190 resulting in mortality in a fish model despite a lack of HeLa cell cytotoxicity.203 Janssens et al. synthesized 190 along with a series of analogues with varied alkyl chain lengths, and reported that 190 inhibits biofilm formation by S. enterica with an IC50 of 10 μM, and exhibits mild planktonic toxicity (MIC 500 μM).204 The ethyl analogue 191 demonstrated moderate biofilm inhibition (IC50 50 μM) without any planktonic toxicity, while the isomer 192 was inactive in both assays.
Figure 10.

Brominated furanones with anti-biofilm activity.
To further investigate structure-activity of this class of compounds, Han et al. synthesized 193 starting from α-methyllaevulinic acid, which was then converted to brominated analogues 194 and 195 using NBS.205 Interestingly, both 193 and its dibromo counterpart 194 displayed biofilm inhibition against E. coli without any planktonic toxicity. However, mono-bromo analogue 195, while able to inhibit biofilm formation, also displayed toxicity. Chang et al. synthesized 5-hydroxyl-3,4-halogenated-5H-furan-2ones via an acid catalyzed oxidative cyclization of furfural, which was used as a building block to access a series of alkyl analogues, ultimately identifying multiple compounds with moderate biofilm inhibition activity against P. aeruginosa.206 Computational investigation of hydrogen bonding interactions with LasR indicated that aryl esters may interact more strongly with the binding site than alkyl esters, and synthesis of several analogues identified compound 196, which demonstrated 63% biofilm inhibition against P. aeruginosa at 1/16 MIC (32 μg/ml, MIC 512 μg/ml). Molecular docking revealed similar interactions between 196 and LasR to those of AHLs, which, coupled with the fact that 196 reduces pyocyanin production, indicates that 196 acts through a LasR depended mechanism. Compound 196, which does not possess an exocyclic vinyl bromide, did not demonstrate any cytotoxicity MTT assay in RAW264.7 cells, which may enable circumvention of toxicity issues associated with the brominated furanone class of anti-biofilm compounds.
Conclusions and future perspectives
The ability of bacteria to form biofilms within an infection setting is a significant factor in the recalcitrance of such infections. Small molecules that interfere with the ability of bacteria to form and maintain biofilms during infection can reverse antibiotic tolerance conferred by the biofilm phenotype, and have the potential to form combination therapies with conventional antibiotics. Many of the compounds identified to date with such activity are natural product small molecules, isolated from various sources including plants, marine organisms, and other microbes. Total synthesis can confirm structure and provide material for screening, while semi-synthesis and medicinal chemistry can identify natural product analogues with improved activity and the physicochemical properties necessary to advance compounds towards therapeutic feasibility. We have described the activity, synthesis and analogue development of several natural products from various sources. The plant derived hamamelitannin synergizes with vancomycin against biofilms of S. aureus, and synthesis has identified an analogue of this natural product displays 20-fold increased activity for the treatment of pre-formed biofilms. The marine sponge derived bromoageliferin and related pyrrole imidazole alkaloids inhibit biofilms of many bacterial species, while a synthetic analogue inhibits MRSA biofilm formation with over 10-fold increased potency, highlighting the utility of synthetic chemistry for optimizing biological activity.
Limited chemical space has yet been explored for the identification of natural product anti-biofilm compounds, and, given the predominance of natural product scaffolds in the antibiotic field, it should be assumed that there are many antibiofilm scaffolds still to be identified. The discovery and development of additional compounds will require efforts from multiple disciplines, and synthetic and medicinal chemistry will be vital if the potential of natural products for the treatment of biofilm-based infections is to be realized.
Figure 4.

Carolacton analogues 71-77.
Acknowledgements
The authors would like to thank the National Institutes of Health (AI136904 and DE022350) for generous support.
Footnotes
Conflicts of interest
Dr. C. Melander is co-founder of Agile Sciences, a biotechnology company seeking to commercialize anti-biofilm compounds.
References
- 1.Dias DA, Urban S and Roessner U, Metabolites, 2012, 2, 303–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler MS, Robertson AA and Cooper MA, Nat Prod Rep, 2014, 31, 1612–1661. [DOI] [PubMed] [Google Scholar]
- 3.Hoff B, Pöggeler S and Kück U, Eukaryot Cell, 2008, 7, 465470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cruz MP, P T, 2012, 37, 278–281. [PMC free article] [PubMed] [Google Scholar]
- 5.Newman DJ and Cragg GM, J Nat Prod, 2020. [Google Scholar]
- 6.Rodrigues T, Reker D, Schneider P and Schneider G, Nat Chem, 2016, 8, 531–541. [DOI] [PubMed] [Google Scholar]
- 7.Nicolaou KC and Snyder SA, Angew Chem Int Ed Engl, 2005, 44, 1012–1044. [DOI] [PubMed] [Google Scholar]
- 8.Maier ME, Nat Prod Rep, 2009, 26, 1105–1124. [DOI] [PubMed] [Google Scholar]
- 9.Worthington RJ, Richards JJ and Melander C, Org Biomol Chem, 2012, 10, 7457–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackledge MS, Worthington RJ and Melander C, Curr Opin Pharmacol, 2013, 13, 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rabin N, Zheng Y, Opoku-Temeng C, Du Y, Bonsu E and Sintim HO, Future Med Chem, 2015, 7, 647–671. [DOI] [PubMed] [Google Scholar]
- 12.Fletcher M, Jennings H,MC and Wuest WM, Journal, 2014, 70, 6373–6383. [Google Scholar]
- 13.Berlanga M and Guerrero R, Microb Cell Fact, 2016, 15, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.López D, Vlamakis H and Kolter R, Cold Spring Harb Perspect Biol, 2010, 2, a000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoiby N, Bjarnsholt T, Givskov M, Molin S and Ciofu O, Int J Antimicrob Agents, 2010, 35, 322–332. [DOI] [PubMed] [Google Scholar]
- 16.Tuomanen E, Durack DT and Tomasz A, Antimicrob Agents Chemother, 1986, 30, 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjarnsholt T, APMIS Suppl, 2013, 1–51. [DOI] [PubMed] [Google Scholar]
- 18.Mah TFC and O’Toole GA, Trends in Microbiology, 2001, 9, 34–39. [DOI] [PubMed] [Google Scholar]
- 19.Flemming HC, Neu TR and Wozniak DJ, J Bacteriol, 2007, 189, 7945–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flemming HC and Wingender J, Nat Rev Microbiol, 2010, 8, 623–633. [DOI] [PubMed] [Google Scholar]
- 21.Spoering AL and Lewis K, J Bacteriol, 2001, 183, 67466751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurdle JG, O’Neill AJ, Chopra I and Lee RE, Nat Rev Microbiol, 2011, 9, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coates AR and Hu Y, Trends in pharmacological sciences, 2008, 29, 143–150. [DOI] [PubMed] [Google Scholar]
- 24.Davey ME and O’Toole G A, Microbiol Mol Biol Rev, 2000, 64, 847–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blazquez J, Clin Infect Dis, 2003, 37, 1201–1209. [DOI] [PubMed] [Google Scholar]
- 26.Conibear TC, Collins SL and Webb JS, PLoS One, 2009, 4, e6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hausner M and Wuertz S, Appl Environ Microbiol, 1999, 65, 3710–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savage VJ, Chopra I and O’Neill AJ, Antimicrob Agents Chemother, 2013, 57, 1968–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poole K, J Antimicrob Chemother, 2012, 67, 2069–2089. [DOI] [PubMed] [Google Scholar]
- 30.Wood TK, Knabel SJ and Kwan BW, Appl Environ Microbiol, 2013, 79, 7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conlon BP, Rowe SE and Lewis K, Adv Exp Med Biol, 2015, 831, 1–9. [DOI] [PubMed] [Google Scholar]
- 32.Lebeaux D, Ghigo JM and Beloin C, Microbiol Mol Biol Rev, 2014, 78, 510–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saginur R, Stdenis M, Ferris W, Aaron SD, Chan F, Lee C and Ramotar K, Antimicrob Agents Chemother, 2006, 50, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Podos SD, Thanassi JA, Leggio M and Pucci MJ, Antimicrob Agents Chemother, 2012, 56, 3812–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu H, Moser C, Wang HZ, Hoiby N and Song ZJ, Int J Oral Sci, 2015, 7, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart PS and Costerton JW, Lancet, 2001, 358, 135138. [DOI] [PubMed] [Google Scholar]
- 37.May JG, Shah P, Sachdeva L, Micale M, Kruper GJ, Sheyn A and Coticchia JM, Int J Pediatr Otorhinolaryngol, 2014, 78, 10–13. [DOI] [PubMed] [Google Scholar]
- 38.Kaplan JB, The International journal of artificial organs, 2011, 34, 737–751. [DOI] [PubMed] [Google Scholar]
- 39.Brackman G and Coenye T, Curr Pharm Des, 2015, 21, 5–11. [DOI] [PubMed] [Google Scholar]
- 40.Miller MB and Bassler BL, Annu Rev Microbiol, 2001, 55, 165–199. [DOI] [PubMed] [Google Scholar]
- 41.Parsek MR and Greenberg EP, Proc Natl Acad Sci U S A, 2000, 97, 8789–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldry M, Kitir B, Frokiaer H, Christensen SB, Taverne N, Meijerink M, Franzyk H, Olsen CA, Wells JM and Ingmer H, Plos One, 2016, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malone CL, Boles BR and Horswill AR, Appl Environ Microb, 2007, 73, 6036–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vendeville A, Winzer K, Heurlier K, Tang CM and Hardie KR, Nat Rev Microbiol, 2005, 3, 383–396. [DOI] [PubMed] [Google Scholar]
- 45.Jani S, Seely AL, Peabody VG, Jayaraman A and Manson MD, Microbiology, 2017. [DOI] [PubMed] [Google Scholar]
- 46.Camilli A and Bassler BL, Science, 2006, 311, 1113–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Worthington RJ, Blackledge MS and Melander C, Future Med Chem, 2013, 5, 1265–1284. [DOI] [PubMed] [Google Scholar]
- 48.Yan H and Chen W, Chemical Society reviews, 2010, 39, 2914–2924. [DOI] [PubMed] [Google Scholar]
- 49.Melander RJ, Minvielle MJ and Melander C, Tetrahedron, 2014, 70, 6363–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee J and Lee JH, Fems Microbiology Reviews, 2010, 34, 426–444. [DOI] [PubMed] [Google Scholar]
- 51.Lee J, Jayaraman A and Wood TK, BMC Microbiol, 2007, 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dirusso CC and Black PN, J Biol Chem, 2004, 279, 4956349566. [DOI] [PubMed] [Google Scholar]
- 53.Jeandet P, Hébrard C, Deville MA, Cordelier S, Dorey S, Aziz A and Crouzet J, Molecules, 2014, 19, 18033–18056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salam AM and Quave CL, Curr Opin Microbiol, 2018, 45, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caesar LK and Cech NB, Nat Prod Rep, 2019, 36, 869888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borges A, Abreu AC, Dias C, Saavedra MJ, Borges F and Simões M, Molecules, 2016, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farhadi F, Khameneh B, Iranshahi M and Iranshahy M, Phytother Res, 2019, 33, 13–40. [DOI] [PubMed] [Google Scholar]
- 58.Brackman G, Cos P, Maes L, Nelis HJ and Coenye T, Antimicrob Agents Chemother, 2011, 55, 2655–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slachmuylders L, Van Acker H, Brackman G, Sass A, Van Nieuwerburgh F and Coenye T, PLoS One, 2018, 13, e0190533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vikram A, Jayaprakasha GK, Jesudhasan PR, Pillai SD and Patil BS, J Appl Microbiol, 2010, 109, 515–527. [DOI] [PubMed] [Google Scholar]
- 61.Pandey RP, Li TF, Kim EH, Yamaguchi T, Park YI, Kim JS and Sohng JK, Appl Environ Microbiol, 2013, 79, 3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JH, Regmi SC, Kim JA, Cho MH, Yun H, Lee CS and Lee J, Infect Immun, 2011, 79, 4819–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ulrey RK, Barksdale SM, Zhou WD and van Hoek ML, Bmc Complem Altern M, 2014, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Augustine N, Goel AK, Sivakumar KC, Ajay Kumar R and Thomas S, Phytomedicine : international journal of phytotherapy and phytopharmacology, 2014, 21, 286–289. [DOI] [PubMed] [Google Scholar]
- 65.Lee JH, Kim YG, Raorane CJ, Ryu SY, Shim JJ and Lee J, Biofouling, 2019, 35, 758–767. [DOI] [PubMed] [Google Scholar]
- 66.Ren DC, Zuo RJ, Barrios AFG, Bedzyk LA, Eldridge GR, Pasmore ME and Wood TK, Applied and Environmental Microbiology, 2005, 71, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ali F, Sangwan PL, Koul S, Pandey A, Bani S, Abdullah ST, Sharma PR, Kitchlu S and Khan IA, European Journal of Clinical Microbiology & Infectious Diseases, 2012, 31, 149–159. [DOI] [PubMed] [Google Scholar]
- 68.Bjarnsholt T, Jensen PO, Rasmussen TB, Christophersen L, Calum H, Hentzer M, Hougen HP, Rygaard J, Moser C, Eberl L, Hoiby N and Givskov K, Microbiol-Sgm, 2005, 151, 3873–3880. [DOI] [PubMed] [Google Scholar]
- 69.Jakobsen TH, van Gennip M, Phipps RK, Shanmugham MS, Christensen LD, Alhede M, Skindersoe ME, Rasmussen TB, Friedrich K, Uthe F, Jensen P, Moser C, Nielsen KF, Eberl L, Larsen TO, Tanner D, Høiby N, Bjarnsholt T and Givskov M, Antimicrob Agents Chemother, 2012, 56, 2314–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakamoto M, Kunimura K, Suzuki JI and Kodera Y, Exp Ther Med, 2020, 19, 1550–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan SY, Liu Y, Chua SL, Vejborg RM, Jakobsen TH, Chew SC, Li Y, Nielsen TE, Tolker-Nielsen T, Yang L and Givskov M, Antimicrob Agents Chemother, 2014, 58, 66486659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fontaine BM, Nelson K, Lyles JT, Jariwala PB, García-Rodriguez JM, Quave CL and Weinert EE, Front Microbiol, 2017, 8, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quave CL, Plano LR, Pantuso T and Bennett BC, J Ethnopharmacol, 2008, 118, 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Talekar SJ, Chochua S, Nelson K, Klugman KP, Quave CL and Vidal JE, PLoS One, 2014, 9, e97314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quave CL, Estevez-Carmona M, Compadre CM, Hobby G, Hendrickson H, Beenken KE and Smeltzer MS, PLoS One, 2012, 7, e28737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chambers SA, Gaddy J and Townsend S, Chemistry, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Habtemariam S, Toxicon, 2002, 40, 83–88. [DOI] [PubMed] [Google Scholar]
- 78.Sánchez-Tena S, Fernández-Cachón ML, Carreras A, Mateos-Martín ML, Costoya N, Moyer MP, Nuñez MJ, Torres JL and Cascante M, J Nat Prod, 2012, 75, 26–33. [DOI] [PubMed] [Google Scholar]
- 79.Kiran MD, Adikesavan NV, Cirioni O, Giacometti A, Silvestri C, Scalise G, Ghiselli R, Saba V, Orlando F, Shoham M and Balaban N, Molecular Pharmacology, 2008, 73, 1578–1586. [DOI] [PubMed] [Google Scholar]
- 80.Horswill AR, Stoodley P, Stewart PS and Parsek MR, Anal Bioanal Chem, 2007, 387, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brackman G, Breyne K, De Rycke R, Vermote A, Van Nieuwerburgh F, Meyer E, Van Calenbergh S and Coenye T, Sci Rep, 2016, 6, 20321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vermote A, Brackman G, Risseeuw MD, Vanhoutte B, Cos P, Van Hecke K, Breyne K, Meyer E, Coenye T and Van Calenbergh S, Angew Chem Int Ed Engl, 2016, 55, 65516555. [DOI] [PubMed] [Google Scholar]
- 83.Vermote A, Brackman G, Risseeuw MDP, Coenye T and Van Calenbergh S, Bioorg Med Chem, 2016, 24, 45634575. [DOI] [PubMed] [Google Scholar]
- 84.Vermote A, Brackman G, Risseeuw MD, Cappoen D, Cos P, Coenye T and Van Calenbergh S, ACS Med Chem Lett, 2017, 8, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vermote A, Brackman G, Risseeuw MDP, Coenye T and Van Calenbergh S, Eur J Med Chem, 2017, 127, 757–770. [DOI] [PubMed] [Google Scholar]
- 86.Bouton J, Van Hecke K, Rasooly R and Van Calenbergh S, Beilstein J Org Chem, 2018, 14, 2822–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stubbendieck RM and Straight PD, J Bacteriol, 2016, 198, 2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wright GD, Nat Prod Rep, 2017, 34, 694–701. [DOI] [PubMed] [Google Scholar]
- 89.Townsley L and Shank EA, Trends Microbiol, 2017, 25, 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eguchi Y, Kubo N, Matsunaga H, Igarashi M and Utsumi R, Antimicrob Agents Chemother, 2011, 55, 14751484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qi F, Merritt J, Lux R and Shi W, Infect Immun, 2004, 72, 4895–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Suzuki N, Ohtaguro N, Yoshida Y, Hirai M, Matsuo H, Yamada Y, Imamura N and Tsuchiya T, Biol Pharm Bull, 2015, 38, 889–892. [DOI] [PubMed] [Google Scholar]
- 93.Hu DX, Clift MD, Lazarski KE and Thomson RJ, J Am Chem Soc, 2011, 133, 1799–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dow JM, Crossman L, Findlay K, He YQ, Feng JX and Tang JL, Proc Natl Acad Sci U S A, 2003, 100, 1099511000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Deng Y, Lim A, Lee J, Chen S, An S, Dong YH and Zhang LH, BMC Microbiol, 2014, 14, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davies DG and Marques CNH, Journal of Bacteriology, 2009, 191, 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dean SN, Chung MC and van Hoek ML, Appl Environ Microb, 2015, 81, 7057–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamada A, Kitamura H, Yamaguchi K, Fukuzawa S, Kamijima C, Yazawa K, Kuramoto M, Wang GYS, Fujitani Y and Uemura D, Bulletin of the Chemical Society of Japan, 1997, 70, 3061–3069. [Google Scholar]
- 99.Rogers SA, Whitehead DC, Mullikin T and Melander C, Org Biomol Chem, 2010, 8, 3857–3859. [DOI] [PubMed] [Google Scholar]
- 100.May Zin WW, Buttachon S, Dethoup T, Pereira JA, Gales L, Inácio Â, Costa PM, Lee M, Sekeroglu N, Silva AMS, Pinto MMM and Kijjoa A, Phytochemistry, 2017, 141, 86–97. [DOI] [PubMed] [Google Scholar]
- 101.Kunze B, Reck M, Dotsch A, Lemme A, Schummer D, Irschik H, Steinmetz H and Wagner-Dobler I, BMC Microbiol, 2010, 10, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hallside MS, Brzozowski RS, Wuest WM and Phillips AJ, Org Lett, 2014, 16, 1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fu C, Sikandar A, Donner J, Zaburannyi N, Herrmann J, Reck M, Wagner-Döbler I, Koehnke J and Müller R, Nat Commun, 2017, 8, 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Banu LD, Conrads G, Rehrauer H, Hussain H, Allan E and van der Ploeg JR, Infect Immun, 2010, 78, 2209–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reck M, Rutz K, Kunze B, Tomasch J, Surapaneni SK, Schulz S and Wagner-Dobler I, J Bacteriol, 2011, 193, 56925706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jansen R, Irschik H, Huch V, Schummer D, Steinmetz H, Bock M, Schmidt T, Kirschning A and Müller R, Journal, 2010, 7, 1284–1289. [Google Scholar]
- 107.Schmidt T and Kirschning A, Angew Chem Int Ed Engl, 2012, 51, 1063–1066. [DOI] [PubMed] [Google Scholar]
- 108.Stumpp N, Premnath P, Schmidt T, Ammermann J, Dräger G, Reck M, Jansen R, Stiesch M, Wagner-Döbler I and Kirschning A, Org Biomol Chem, 2015, 13, 5765–5774. [DOI] [PubMed] [Google Scholar]
- 109.Solinski AE, Koval AB, Brzozowski RS, Morrison KR, Fraboni AJ, Carson CE, Eshraghi AR, Zhou G, Quivey RG, Voelz VA, Buttaro BA and Wuest WM, J Am Chem Soc, 2017, 139, 7188–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Solinski AE, Scharnow AM, Fraboni AJ and Wuest WM, ACS Infect Dis, 2019, 5, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Navarro G, Cheng AT, Peach KC, Bray WM, Bernan VS, Yildiz FH and Linington RG, Antimicrob Agents Chemother, 2014, 58, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giltrap AM, Haeckl FPJ, Kurita KL, Linington RG and Payne RJ, J Org Chem, 2018, 83, 7250–7270. [DOI] [PubMed] [Google Scholar]
- 113.Li W, Estrada-de los Santos P, Matthijs S, Xie GL, Busson R, Cornelis P, Rozenski J and De Mot R, Chem Biol, 2011, 18, 1320–1330. [DOI] [PubMed] [Google Scholar]
- 114.Steele AD, Knouse KW, Keohane CE and Wuest WM, J Am Chem Soc, 2015, 137, 7314–7317. [DOI] [PubMed] [Google Scholar]
- 115.Steele AD, Keohane CE, Knouse KW, Rossiter SE, Williams SJ and Wuest WM, J Am Chem Soc, 2016, 138, 5833–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Keohane CE, Steele AD, Fetzer C, Khowsathit J, Van Tyne D, Moynié L, Gilmore MS, Karanicolas J, Sieber SA and Wuest WM, J Am Chem Soc, 2018, 140, 1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Laursen JB and Nielsen J, Chem Rev, 2004, 104, 16631686. [DOI] [PubMed] [Google Scholar]
- 118.Spicer JA, Gamage SA, Rewcastle GW, Finlay GJ, Bridewell DJ, Baguley BC and Denny WA, J Med Chem, 2000, 43, 1350–1358. [DOI] [PubMed] [Google Scholar]
- 119.Guttenberger N, Blankenfeldt W and Breinbauer R, Bioorg Med Chem, 2017, 25, 6149–6166. [DOI] [PubMed] [Google Scholar]
- 120.Das T and Manefield M, PLoS One, 2012, 7, e46718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Machan ZA, Pitt TL, White W, Watson D, Taylor GW, Cole PJ and Wilson R, J Med Microbiol, 1991, 34, 213217. [DOI] [PubMed] [Google Scholar]
- 122.Borrero NV, Bai F, Perez C, Duong BQ, Rocca JR, Jin S and Huigens RW, Org Biomol Chem, 2014, 12, 881886. [DOI] [PubMed] [Google Scholar]
- 123.Garrison AT, Bai F, Abouelhassan Y, Paciaroni NG, Jin SG and Huigens RW, Rsc Advances, 2015, 5, 1120–1124. [Google Scholar]
- 124.Garrison AT, Abouelhassan Y, Kallifidas D, Bai F, Ukhanova M, Mai V, Jin S, Luesch H and Huigens RW 3rd, Angew Chem Int Ed Engl, 2015. [DOI] [PubMed] [Google Scholar]
- 125.Garrison AT, Abouelhassan Y, Norwood VM, Kallifidas D, Bai F, Nguyen MT, Rolfe M, Burch GM, Jin S, Luesch H and Huigens RW, J Med Chem, 2016, 59, 38083825. [DOI] [PubMed] [Google Scholar]
- 126.Yang H, Abouelhassan Y, Burch GM, Kallifidas D, Huang G, Yousaf H, Jin S, Luesch H and Huigens RW, Sci Rep, 2017, 7, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Garrison AT, Abouelhassan Y, Kallifidas D, Tan H, Kim YS, Jin S, Luesch H and Huigens RW, J Med Chem, 2018, 61, 3962–3983. [DOI] [PubMed] [Google Scholar]
- 128.Abouelhassan Y, Zhang Y, Jin S and Huigens RW, Angew Chem Int Ed Engl, 2018, 57, 15523–15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Abouelhassan Y, Garrison AT, Burch GM, Wong W, Norwood VM and Huigens RW, Bioorg Med Chem Lett, 2014, 24, 5076–5080. [DOI] [PubMed] [Google Scholar]
- 130.Garrison AT, Abouelhassan Y, Yang H, Yousaf HH, Nguyen TJ and Huigens Iii RW, Medchemcomm, 2017, 8, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Basak A, Abouelhassan Y and Huigens RW, Org Biomol Chem, 2015, 13, 10290–10294. [DOI] [PubMed] [Google Scholar]
- 132.Basak A, Abouelhassan Y, Norwood VM, Bai F, Nguyen MT, Jin S and Huigens RW, Chemistry, 2016, 22, 9181–9189. [DOI] [PubMed] [Google Scholar]
- 133.Basak A, Abouelhassan Y, Kim YS, Norwood VM, Jin S and Huigens RW, Eur J Med Chem, 2018, 155, 705–713. [DOI] [PubMed] [Google Scholar]
- 134.Kiuru P, D’Auria MV, Muller CD, Tammela P, Vuorela H and Yli-Kauhaluoma J, Planta Med, 2014, 80, 1234–1246. [DOI] [PubMed] [Google Scholar]
- 135.Jiménez C, ACS Med Chem Lett, 2018, 9, 959–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liang X, Luo D and Luesch H, Pharmacol Res, 2019, 147, 104373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hertiani T, Edrada-Ebel R, Ortlepp S, van Soest RW, de Voogd NJ, Wray V, Hentschel U, Kozytska S, Muller WE and Proksch P, Bioorg Med Chem, 2010, 18, 12971311. [DOI] [PubMed] [Google Scholar]
- 138.von Salm JL, Witowski CG, Fleeman RM, McClintock JB, Amsler CD, Shaw LN and Baker BJ, Org Lett, 2016, 18, 2596–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Siemon T, Steinhauer S and Christmann M, Angew Chem Int Ed Engl, 2019, 58, 1120–1122. [DOI] [PubMed] [Google Scholar]
- 140.Pénez N, Culioli G, Pérez T, Briand JF, Thomas OP and Blache Y, J Nat Prod, 2011, 74, 2304–2308. [DOI] [PubMed] [Google Scholar]
- 141.Tadesse M, Strøm MB, Svenson J, Jaspars M, Milne BF, Tørfoss V, Andersen JH, Hansen E, Stensvåg K and Haug T, Org Lett, 2010, 12, 4752–4755. [DOI] [PubMed] [Google Scholar]
- 142.Trepos R, Cervin G, Hellio C, Pavia H, Stensen W, Stensvåg K, Svendsen JS, Haug T and Svenson J, J Nat Prod, 2014, 77, 2105–2113. [DOI] [PubMed] [Google Scholar]
- 143.Edwards GA, Shymanska NV and Pierce JG, Chem Commun (Camb), 2017, 53, 7353–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hoffmann H and Lindel T, Synthesis-Stuttgart, 2003, 17531783. [Google Scholar]
- 145.Bickmeyer U, Toxicon, 2005, 45, 627–632. [DOI] [PubMed] [Google Scholar]
- 146.Forenza S, Minale L and Riccio R, J Chem Soc Chem Comm, 1971, 1129–&. [Google Scholar]
- 147.Keifer PA, Schwartz RE, Koker MES, Hughes RG, Rittschof D and Rinehart KL, Journal of Organic Chemistry, 1991, 56, 2965–2975. [Google Scholar]
- 148.Melander RJ, Liu HB, Stephens MD, Bewley CA and Melander C, Bioorg Med Chem Lett, 2016, 26, 5863–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.O’Malley DP, Li K, Maue M, Zografos AL and Baran PS, J Am Chem Soc, 2007, 129, 4762–4775. [DOI] [PubMed] [Google Scholar]
- 150.Wang X, Tan X, Lu J, Cormier KW, Ma Z and Chen C, J Am Chem Soc, 2012, 134, 18834–18842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huigens RW, Richards JJ, Parise G, Ballard TE, Zeng W, Deora R and Melander C, Journal of the American Chemical Society, 2007, 129, 6966–6967. [DOI] [PubMed] [Google Scholar]
- 152.Huigens RW, Ma L, Gambino C, Basso A, Moeller PDR, Cavanagh J, Wozniak DJ and Melander C, Molecular Biosystems, 2008, 4, 614–621. [DOI] [PubMed] [Google Scholar]
- 153.Lindsey EA, Brackett CM, Mullikin T, Alcaraz C and Melander C, Medchemcomm, 2012, 3, 1462–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ackart DF, Lindsey EA, Podell BK, Melander RJ, Basaraba RJ and Melander C, Pathog Dis, 2014, 70, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nguyen TV, Peszko MT, Melander RJ and Melander C, Medchemcomm, 2019, 10, 456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huggins WM, Vu Nguyen T, Hahn NA, Baker JT, Kuo LG, Kaur D, Melander RJ, Gunn JS and Melander C, Medchemcomm, 2018, 9, 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Frei R, Breitbach AS and Blackwell HE, Angew Chem Int Ed Engl, 2012, 51, 5226–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Richards JJ, Huigens RW, Ballard TE, Basso A, Cavanagh J and Melander C, Chemical communications, 2008, 1698–1700. [DOI] [PubMed] [Google Scholar]
- 159.Richards JJ, Reed CS and Melander C, Bioorganic & Medicinal Chemistry Letters, 2008, 18, 4325–4327. [DOI] [PubMed] [Google Scholar]
- 160.Ballard TE, Richards JJ, Wolfe AL and Melander C, Chemistry-a European Journal, 2008, 14, 10745–10761. [DOI] [PubMed] [Google Scholar]
- 161.Thompson RJ, Bobay BG, Stowe SD, Olson AL, Peng L, Su Z, Actis LA, Melander C and Cavanagh J, Biochemistry, 2012, 51, 9776–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tomaras AP, Flagler MJ, Dorsey CW, Gaddy JA and Actis LA, Microbiol-Sgm, 2008, 154, 3398–3409. [DOI] [PubMed] [Google Scholar]
- 163.Liu C, Worthington RJ, Melander C and Wu H, Antimicrob Agents Chemother, 55, 2679–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wright CJ, Wu H, Melander RJ, Melander C and Lamont J, Mol Oral Microbiol, 2014, 29, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Milton ME, Minrovic BM, Harris DL, Kang B, Jung D, Lewis CP, Thompson RJ, Melander RJ, Zeng D, Melander C and Cavanagh J, Front Mol Biosci, 2018, 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Milton ME, Allen CL, Feldmann EA, Bobay BG, Jung DK, Stephens MD, Melander RJ, Theisen KE, Zeng D, Thompson RJ, Melander C and Cavanagh J, Mol Microbiol, 2017, 106, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Steenackers HP, Ermolat’ev DS, Savaliya B, De Weerdt A, De Coster D, Shah A, Van der Eycken EV, De Vos DE, Vanderleyden J and De Keersmaecker SC, J Med Chem, 2011, 54, 472–484. [DOI] [PubMed] [Google Scholar]
- 168.Robijns SC, De Pauw B, Loosen B, Marchand A, Chaltin P, De Keersmaecker SC, Vanderleyden J and Steenackers HP, FEMS immunology and medical microbiology, 2012, 65, 390–394. [DOI] [PubMed] [Google Scholar]
- 169.Ermolat’ev DS, Bariwal JB, Steenackers HP, De Keersmaecker SC and Van der Eycken EV, Angew Chem Int Ed Engl, 2010, 49, 9465–9468. [DOI] [PubMed] [Google Scholar]
- 170.Trang TTT, Dieltjens L, Hooyberghs G, Waldrant K, Ermolat’ev DS, Van der Eycken EV and Steenackers HPL, Bioorg Med Chem, 2018, 26, 1470–1480. [DOI] [PubMed] [Google Scholar]
- 171.Lindsey EA, Worthington RJ, Alcaraz C and Melander C, Org Biomol Chem, 2012, 10, 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cox KE and Melander C, Medchemcomm, 2019, 10, 1177–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Carle JS and Christophersen C, Journal of the American Chemical Society, 1979, 101, 4012–4013. [Google Scholar]
- 174.Carle JS and Christophersen C, Journal of Organic Chemistry, 1981, 46, 3440–3443. [Google Scholar]
- 175.Peters L, Konig GM, Wright AD, Pukall R, Stackebrandt E, Eberl L and Riedel K, Applied and Environmental Microbiology, 2003, 69, 3469–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kawasaki TT, Romi, Sakaguchi K-e, Sekiguchi H and Sakamoto M, Journal, 1996, 37, 7525–7528. [Google Scholar]
- 177.Fuchs JR and Funk RL, Org Lett, 2005, 7, 677–680. [DOI] [PubMed] [Google Scholar]
- 178.Lindel T, Brauchle L, Golz G and Bohrer P, Organic Letters, 2007, 9, 283–286. [DOI] [PubMed] [Google Scholar]
- 179.Bunders C, Cavanagh J and Melander C, Org Biomol Chem, 2011, 9, 5476–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Minvielle MJ, Bunders CA and Melander C, Medchemcomm, 2013, 4, 916–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bunders CA, Minvielle MJ, Worthington RJ, Ortiz M, Cavanagh J and Melander C, J Am Chem Soc, 2011, 133, 20160–20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Minvielle MJ, Eguren K and Melander C, Chemistry, 2013, 19, 17595–17602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Franco LH, Joffé E. B. d. K, Puricelli L, Tantian M, Seldes AM and Palermo JA, J. Nat. Prod, 1998, 61, 11301132. [DOI] [PubMed] [Google Scholar]
- 184.Giraud F, Alves G, Debiton E, Nauton L, Thery V, Durieu E, Ferandin Y, Lozach O, Meijer L, Anizon F, Pereira E and Moreau P, Journal of medicinal chemistry, 2011, 54, 4474–4489. [DOI] [PubMed] [Google Scholar]
- 185.Radwan MA and El-Sherbiny M, Bioorganic & medicinal chemistry, 2007, 15, 1206–1211. [DOI] [PubMed] [Google Scholar]
- 186.Bharate SB, Yadav RR, Khan SI, Tekwani BL, Jacob MR, Khan IA and Vishwakarma RA, MedChemComm, 2013, 4, 1042. [Google Scholar]
- 187.Bharate SB, Yadav RR, Battula S and Vishwakarma RA, Mini Rev Med Chem, 2012, 12, 618–631. [DOI] [PubMed] [Google Scholar]
- 188.Yadav RR, Khan SI, Singh S, Khan IA, Vishwakarma RA and Bharate SB, European journal of medicinal chemistry, 2015, 98, 160–169. [DOI] [PubMed] [Google Scholar]
- 189.Bredereck H, Effenberger F, Botsch H and Rehn H, Journal, 1964, 98, 1081–1086. [Google Scholar]
- 190.Huggins WM, Barker WT, Baker JT, Hahn NA, Melander RJ and Melander C, ACS Med Chem Lett, 2018, 9, 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Brackett SM, Cox KE, Barlock SL, Huggins WM, Ackart DF, Bassaraba RJ, Melander RJ and Melander C, Journal, 2020, 11, 92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.J. L. Reichelt and M. A. Borowitzka, 1984, 116, 158–168.
- 193.De Nys R, Steinberg PD, Willemsen P, Dworjanyn SA, Gabelish CL and King RJ, Biofouling, 1995, 8, 259–271. [Google Scholar]
- 194.Ren D, Sims JJ and Wood TK, Environ Microbiol, 2001, 3, 731–736. [DOI] [PubMed] [Google Scholar]
- 195.Ren D, Sims JJ and Wood TK, Lett Appl Microbiol, 2002, 34, 293–299. [DOI] [PubMed] [Google Scholar]
- 196.Yang S, Abdel-Razek OA, Cheng F, Bandyopadhyay D, Shetye GS, Wang G and Luk YY, Bioorg Med Chem, 2014, 22, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Manefield M, de Nys R, Naresh K, Roger R, Givskov M, Peter S and Kjelleberg S, Microbiology, 1999, 145, 283291 %@ 1350–0872. [DOI] [PubMed] [Google Scholar]
- 198.Manefield M, Rasmussen TB, Henzter M, Andersen JB, Steinberg P, Kjelleberg S and Givskov M, MicrobiologySgm, 2002, 148, 1119–1127. [DOI] [PubMed] [Google Scholar]
- 199.C. M. Beechan and J. J. Sims, 1979, 20, 1649–1652.
- 200.A. J. Manny, S. Kjelleberg, N. Kumar, R. de Nys, R. W. Read and P. Steinberg, 1997, 53, 15813–15826.
- 201.Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song ZJ, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Hoiby N and Givskov M, Embo Journal, 2003, 22, 3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wu H, Song Z, Hentzer M, Andersen JB, Molin S, Givskov M and Hoiby N, Journal of Antimicrobial Chemotherapy, 2004, 53, 1054–1061. [DOI] [PubMed] [Google Scholar]
- 203.M. Rasch, C. Buch, B. Austin, W. J. Slierendrecht, K. S. Ekmann, J. L. Larsen, C. Johansen, K. Riedel, L. Eberl, M. Givskov and L. Gram, 2004, 27, 350–359. [DOI] [PubMed]
- 204.Janssens JCA, Steenackers H, Robijns S, Gellens E, Levin J, Zhao H, Hermans K, De Coster D, Verhoeven TL, Marchal K, Vanderleyden J, De Vos DE and De Keersmaecker SCJ, Applied and Environmental Microbiology, 2008, 74, 6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Han Y, Hou S, Simon KA, Ren D and Luk YY, Bioorg Med Chem Lett, 2008, 18, 1006–1010. [DOI] [PubMed] [Google Scholar]
- 206.Y. Chang, P.-C. Wang, H.-M. Ma, S.-Y. Chen, Y.-H. Fu, Y.-Y. Liu, X. Wang, G.-C. Yu, T. Huang, D. E. Hibbs, H.-B. Zhou, W.M. Chen, J. Lin, C. Wang, J.-X. Zheng and P.-H. Sun, 2019, 140, 105058. [DOI] [PubMed]