

Abstract
Oxidized cholesterols have emerged as important signaling molecules of immune function, but little is known about the role of these oxysterols during mycobacterial infections. We found that expression of the oxysterol-receptor GPR183 was reduced in blood from patients with tuberculosis (TB) and type 2 diabetes (T2D) compared to TB patients without T2D and was associated with TB disease severity on chest x-ray. GPR183 activation by 7α,25-dihydroxycholesterol (7α,25-OHC) reduced growth of Mycobacterium tuberculosis (Mtb) and Mycobacterium bovis BCG in primary human monocytes, an effect abrogated by the GPR183 antagonist GSK682753. Growth inhibition was associated with reduced IFN-β and IL-10 expression and enhanced autophagy. Mice lacking GPR183 had significantly increased lung Mtb burden and dysregulated IFNs during early infection. Together, our data demonstrate that GPR183 is an important regulator of intracellular mycobacterial growth and interferons during mycobacterial infection.
Keywords: Mycobacterium tuberculosis; diabetes; oxysterols; 7α,25-dihydroxycholesterol; GPR183; EBI2; host-direct therapies; autophagy
Introduction
Patients with tuberculosis and type 2 diabetes (TB–T2D) co-morbidity have increased bacterial burden and more severe disease, characterized by higher sputum smear grading scores and greater lung involvement on chest x-ray compared to TB patients without T2D (1, 2). TB–T2D patients are also more likely to fail TB therapy and to relapse (3). The reason for the increased disease severity has largely been attributed to hyperglycemia-mediated immune dysfunction, but hyperglycemia alone does not fully explain these observations (3, 4). We recently showed that independent of hyperglycemia, cholesterol concentrations in T2D patients vary greatly across different ethnicities (5). However, how cholesterol and its metabolites contribute to Mycobacterium tuberculosis (Mtb) infection outcomes remains to be elucidated.
To gain novel insights into the underlying immunological mechanisms of increased susceptibility of T2D patients to TB and to identify novel targets for host-directed therapy (HDT), we performed whole blood transcriptomic screens on TB patients with and without T2D and identified differential regulation of the transcript for oxidized cholesterol-sensing G protein-coupled receptor (GPCR), GPR183. Also known as Epstein Barr virus-induced gene 2 (EBI2), GPR183 is primarily expressed on cells of the innate and adaptive immune system (6–8). Several oxysterols can bind to GPR183 with 7α,25-hydroxycholesterol (7α,25-OHC) being the most potent endogenous agonist (6, 9, 10). GPR183 has been studied mainly in the context of viral infections (11), immune cells (6, 7, 9, 12–18), and astrocytes (19, 20); it facilitates the chemotactic distribution of lymphocytes, dendritic cells and macrophages to secondary lymphoid organs (12, 15, 16, 21, 22). Little is known about the biological role of GPR183 in the context of bacterial infections, including TB. We show here that GPR183 is a key regulator of intracellular bacterial growth and type-I IFN production during mycobacterial infection and reduced GPR183 expression is associated with increased TB disease severity.
Methods
Study Participants
TB patients and their close contacts were recruited at TB clinics outside Cape Town (South Africa). TB diagnosis was made based on positive GeneXpert MTB/RIF (Cepheid; California, USA) and/or positive MGIT culture (BD BACTED MGIT 960 system, BD, New Jersey, USA) and abnormal chest x-ray. Chest x-rays were scored, based on Ralphs score (23), by two clinicians independently. Participants with LTBI were close contacts of TB patients who tested positive on QuantiFERON-TB Gold in tube assay (Qiagen, Hilden, Germany). All study participants were screened for T2D based on HbA1c ≥6.5% and random plasma glucose ≥200 mg/dl or a previous history of T2D. Further details are available in the Supplementary Materials .
RNA Extractions and Nanostring Analysis
Total RNA was extracted from cell pellets collected in QuantiFERON-TB gold assay tubes without antigen using the Ribopure Ambion RNA isolation kit (Life Technologies, California, USA) and eluted RNA treated with DNase for 30 min. Samples with a concentration of ≥20 ng/µl and a 260/280 and 260/230 ratio of ≥1.7 were analyzed at NanoString Technologies in Seattle, Washington, USA. Differential expression of 594 genes, including 15 housekeeping genes, was performed using the nCounter GX Human Immunology kit V2. NanoString RCC data files were imported into the nSolver 3 software (nSolver Analysis software, v3.0), and gene expression was normalized to housekeeping genes.
Cell Culture
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donor blood by Ficoll-Paque (GE Healthcare, Illinois, USA) gradient centrifugation and monocytes (MNs) isolated using the Pan Monocyte Isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany), with >95% purity assessed by flow cytometry. MNs were plated onto Poly-D-lysine coated tissue culture plates (1.3 × 105 cells/well) and rested overnight at 37°C/5%CO2 in RPMI-1640 medium supplemented with 10% heat-inactivated human AB serum (Sigma Aldrich, Missouri, USA), 2 mM L-glutamine and 1 mM sodium pyruvate before infection. THP-1 cells (ATCC #TIB-202) were differentiated with 25 ng/ml PMA for 48 h and rested for 24 h prior to infection.
In Vitro Mtb (H37Rv)/M. bovis (BCG) Infection
Mtb H37Rv or M. bovis BCG single cell suspensions were added at a multiplicity of infection (MOI) of 1 or 10 with/without 100 nM 7α,25-dihydroxycholesterol (Sigma Aldrich) and with/without 10 µM GSK682753 (Focus Bioscience, Queensland, Australia), followed by 2 h incubation at 37°C/5%CO2 to allow for phagocytosis. Non-phagocytosed bacilli were removed by washing each well twice in warm RPMI-1640 containing 25 mM HEPES (Thermo Fisher Scientific). Infected cells were incubated (37°C/5%CO2) in medium with/without GPR183 agonist and/or antagonist and CFUs determined after 48 h.
To quantify bacterial growth over time, CFUs at 48 h were normalized to uptake at 2 h. Percentages of mycobacterial growth were determined relative to untreated cells. For RNA extraction, MNs were lysed by adding 500 µl of TRIzol reagent. Further details are provided in the supplementary information.
Western Blotting
THP-1 cells were infected with BCG with/without 100 nM 7α,25-OHC and with/without 10 µM GSK682753 and lysed at 6 or 24 h post infection (p.i.) in ice-cold RIPA buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0; Thermo Fisher Scientific), supplemented with complete Protease Inhibitor Cocktail (Sigma Aldrich) (120 µl RIPA/1 × 106 Cells). Protein concentrations were determined using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) as per manufacturer’s protocol. 10 µg of protein per sample was loaded on NovexTM 10–20% Tris-Glycine protein gels (Thermo Fisher Scientific) and transferred onto iBlot2 Transfer Stacks PVDF membrane (Thermo Fisher Scientific). Membranes were blocked with Odyssey Blocking buffer (Milennium Science, Victoria, Australia) for 2 h, probed with rabbit anti-human LC3B (1:1,000, Sigma L7543) and rabbit anti-human GAPDH (1:2,500, Abcam 9485) overnight, followed by detection with goat anti-rabbit IgG DyLight 800 (1:20,000; Thermo Fisher Scientific). Bands were visualized using the Odyssey CLx system (LI-COR Biosciences, Nebraska, USA) and analyzed with Image Studio Lite V5.2 (LI-COR Biosciences).
Immunofluorescence
Differentiated THP-1 cells were seeded onto a poly-D-lysine coated, 96-well glass-bottom black tissue culture plate (4.5 × 104 cells/well) and kept in RPMI-1640 medium minus phenol red (Thermo Fisher Scientific) supplemented with 10% heat-inactivated FBS at 37°C/5% CO2. Cells were infected with BCG at a MOI of 10, with/without 100 nM 7α,25-OHC, with/without 10 µM GSK682753 for 2 h, washed and incubated for a further 4 h with agonists and antagonists. Cells were then fixed with 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.05% saponin (Sigma Aldrich) for 20 min and blocked with 1% BSA, 0.05% saponin (Sigma Aldrich) for 1 h. Cells were immunolabeled with rabbit anti-human LC3B (ThermoFisher L10382; 1:1,000), 0.05% saponin at room temperature for 1 h followed by Alexa FluorTM 647 goat anti-rabbit IgG (ThermoFisher A21245; 1:1,000), 0.05% saponin at room temperature for 1 h followed by nuclear staining with Hoechst 33342 (Thermo Fisher Scientific 62249; 1:2,000) for 15 min. Cells were washed, and confocal microscopy was performed using the Olympus FV3000, 60× magnification. Images obtained were analyzed with the ImageJ software (24).
Murine GPR183 KO vs WT Model
Equal numbers of male and female C57BL/6 WT and Gpr183tm1Lex (age 18–20 weeks, 10 mice per group/timepoint) were aerosol infected with 300 CFU Mtb H37Rv using an inhalation exposure system (Glascol). At 2- and 5-weeks post infection, lungs and blood were collected for RNA and CFU determination. Formalin-fixed lung lobes were sectioned and examined microscopically and scored by a veterinary pathologist. Further details are available in the supplementary information.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism v.7.0.3 (GraphPad Software). T-test and Wilcoxon’s test were used to analyze Nanostring data. Mann–Whitney U test and t-test were used to analyze in vitro infection, qPCR, and ELISA data. Data are presented as means ± SEM. Statistically significant differences between two groups are indicated in the figures as follows ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Ethics Statement
The human studies were approved by the Institutional Review Board of Stellenbosch University (N13/05/064 and N13/05/064A) and all study participants signed pre-approved informed consent documents prior to enrolment into the studies. All animal studies were approved by the Animal Ethics Committee of the University of Queensland (MRI-UQ/596/18) and conducted in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes.
Results
Blood GPR183 mRNA Expression Is Reduced in Patients With TB–T2D Compared to TB Patients Without T2D
Blood was obtained from the study participants with latent TB infection (LTBI, n = 11), latent TB infection with T2D (LTBI + T2D, n = 14), active pulmonary TB disease (TB, n = 9), and active pulmonary TB disease with T2D (TB + T2D, n = 7). Total RNA was extracted and NanoString analyses performed. Among genes differentially expressed between TB and TB + T2D we identified a single GPCR, GPR183. We focused on GPR183 as GPCRs are bona fide drug targets due to their importance in human pathophysiology and their pharmacological tractability.
GPR183 expression was significantly down-regulated at diagnosis (p = 0.03, t-test) in blood from TB + T2D patients compared to TB patients without T2D ( Figure 1A ). The reduced GPR183 expression was not driven by diabetes per se, as there were no differences in GPR183 expression between LTBI and LTBI + T2D ( Figure 1A ). After 6 months, at the end of successful TB treatment, we saw GPR183 expression significantly increased (p = 0.0156) in TB + T2D patients to a level comparable to the TB patients without T2D ( Figure 1B ). Therefore, we speculated that blood GPR183 expression is associated with extent of TB disease, which is frequently more severe in T2D patients. We indeed determined an inverse correlation between GPR183 mRNA expression in the blood and TB disease severity on chest x-ray ( Figure 1C ).
Figure 1.

GPR183 mRNA expression in patients with active and latent TB infection with or without T2D. Total RNA was isolated from whole blood incubated overnight in QuantiFERON-TB Gold. GPR183 mRNA expression was determined and normalized to reference genes using the NanoString technology. GPR183 expression in whole blood of (A) TB (n = 9) and TB + T2D (n = 7) patients, LTBI (n = 11) and LTBI + T2D (n = 14) patients, Wilcoxon test. (B) TB (n = 9) and TB + T2D (n = 7) patients at baseline and 6 month’s treatment, t-test. (C) Correlation between GPR183 expression and chest x-ray score, TB + T2D patients (n = 7) filled squares, TB patients (n =8) open circles. Data are presented as means ± SEM; not significance (ns) P > 0.05; *P ≤ 0.05; ****P ≤ 0.0001.
In order to identify which cell type is associated with decreased expression of GPR183 in the blood, we performed flow cytometry analysis for GPR183 expression on PBMCs from TB patients with and without T2D. We investigated GPR183 expression on CD4+ and CD8+ T-cells, B cells, dendritic cells, NK cells and monocytes. We found that the only cell type with a significant reduction in GPR183 positivity in TB + T2D vs. TB, both in terms of frequency and median fluorescent intensity, was the non-classical monocyte population ( Supplementary Figures 1A, B ). The frequencies of GPR183 + non-classical monocytes from LTBI and LTBI + T2D were not significantly different ( Supplementary Figure 1C ). We therefore next investigated whether GPR183 plays a role in the innate immune response during Mtb infection.
Oxysterol-Induced Activation of GPR183 Reduces Intracellular Mycobacterial Growth
We investigated whether in vitro activation of GPR183 with its endogenous agonist impacts the immune response to mycobacteria in primary human MNs. MNs from 15 healthy donors were infected with BCG (n = 7) or Mtb H37RV (n = 8) ( Figure 2 ) at a MOI of one in the presence or absence of the GPR183 agonist 7α,25-OHC and/or the antagonist GSK682753. Activation of GPR183 by 7α,25-OHC significantly increased the uptake of BCG and Mtb H37RV ( Figure 2A ) at 2 h p.i. This increase in phagocytosis was abolished by the simultaneous addition of the GPR183 antagonist GSK682753, confirming that increased mycobacterial uptake was the result of GPR183 activation. Interestingly, we observed ~50% reduction in the growth of BCG and Mtb H37RV ( Figure 2B ) by 48h p.i. in 7α,25-OHC treated cells, and again, this effect was abrogated by GSK682753. The addition of 7α,25-OHC and/or GSK682753 had no detrimental effect on the viability of human THP-1 cells ( Supplementary Figure 2A ). There was also no effect of 7α,25-OHC and GSK682753 on BCG growth in liquid culture ( Supplementary Figure 2B ), thus confirming that the significant mycobacterial growth inhibition in MN cultures was attributable to the immune modulatory activity of 7α,25-OHC via GPR183. Independently, we observed that H37Rv down-regulates GPR183 in primary MNs ( Supplementary Figure 3 ).
Figure 2.
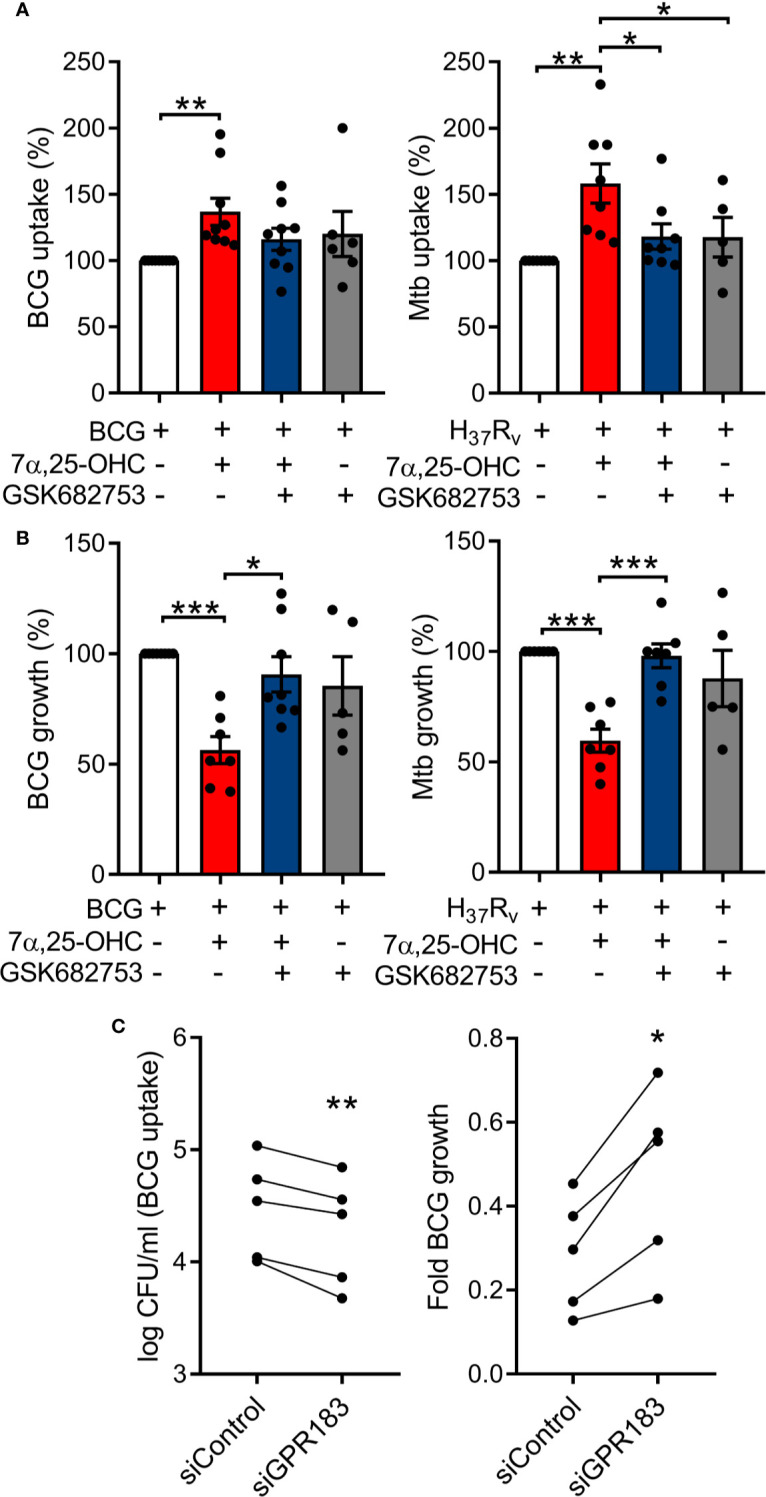
Oxysterol-induced activation of GPR183 in primary MNs significantly inhibits intracellular mycobacterial growth, while GPR183 knockdown increases intracellular mycobacterial growth. Primary MNs from eight donors (A) and seven donors (B) were infected with BCG or Mtb H37Rv (MOI 1), ± 7α,25-OHC (100 nM), ± GSK682753 (10 µM). Uptake of (A) BCG and Mtb H37Rv was determined at 2h p.i. Growth of (B) BCG and Mtb H37Rv was determined at 48h post-infection. Percent of mycobacterial growth was calculated as the fold change of CFU at 48h compared to CFU at 2h, normalized to non-treated cells. PMA-differentiated THP-1 cells were transfected with 20 nM of either negative control siRNA or GPR183 siRNA for 48h before infection with BCG (MOI 1). (C) Mycobacterial uptake was determined at 2h and intracellular mycobacterial growth was determined at 48h p.i. (normalized to uptake). Data are presented as means ± SEM; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; paired t-test.
To confirm the role of GPR183 in phagocytosis and growth inhibition, we next performed GPR183 siRNA knockdown experiments. Differentiated THP-1 cells were transfected with 20 nM of GPR183-targeting siRNA (siGPR183) or negative control siRNA (siControl). We observed ~80% reduction of GPR183 mRNA level and ~50% reduction of protein expression in cells transfected with siGPR183 when compared to siControl-transfected cells ( Supplementary Figures 4A, B ) at 48 h. Forty-eight hours after transfection the cells were infected with BCG at a MOI of one. We observed a marked decrease in BCG uptake in cells transfected with siGPR183 (p = 0.0048) compared to siControl-transfected cells and a significant increase in intracellular mycobacterial growth over time (p = 0.0113, Figure 2C ).
GPR183 Is a Negative Regulator of the Type I Interferon Pathway in Human MNs
In genome wide association studies GPR183 has been implicated as a negative regulator of the IRF7 driven inflammatory network (25). Therefore, we focused subsequent experiments on type-I IFN regulation. To determine whether GPR183, a constitutively active GPCR (26), has a direct effect on IRFs and IFNB1 expression, we performed knockdown experiments in primary MNs. GPR183 knockdown ( Supplementary Figure 4C ) up-regulated IFNB1 (2.7–5.5 fold; P = 0.0115) as well as IRF1, IRF3, IRF5, and IRF7, although the latter did not reach statistical significance ( Figure 3A ).
Figure 3.

GPR183 knockdown increases expression of transcription factors regulating type I interferon responses. (A) Total RNA was isolated from primary MNs following 48 h incubation with 20 nM GPR183 siRNA (or negative control siRNA). Gene expression of IFNB1, IRF1, IRF3, IRF5, IRF7 was measured by qRT-PCR using RPS13 as reference gene. Data are normalized to cells transfected with negative control siRNA. (B) NanoString analyses of RNA isolated from TB and TB + T2D cohort showed similar increase in type I IFN associated genes IRF1, IRF5, IRF7. Data are presented as fold changes ± SEM; *P ≤ 0.05; **P ≤ 0.01; paired t-test.
IRF1, IRF5, and IRF7 transcripts were similarly up-regulated in whole blood from TB + T2D patients compared to TB patients ( Figure 3B ), consistent with the downregulation of GPR183 mRNA expression ( Figure 1 ). IRF3 expression was not significantly different between TB and TB + T2D patients (data not shown).
GPR183 Activation Induces a Cytokine Profile Favoring Mtb Control
Next, we investigated whether the reduced intracellular mycobacterial growth observed in primary MNs treated with 7α,25-OHC was associated with a change in MN secreted cytokines. Gene expression of IFNB1, TNF, and IL-10 was measured 24 h following infection with Mtb H37RV at MOI of one ( Figure 4A ). The concentrations of the corresponding cytokines were measured in cell culture supernatant by ELISA ( Figure 4B ). Mtb infection significantly up-regulated the expression of IFNB1 (P = 0.0068), TNF (P = 0.0001), IL-10 (P < 0.0001) ( Figure 4A ), and IL-1B ( Supplementary Figure 5A ). 7α,25-OHC significantly down-regulated Mtb-induced IFNB1 expression (P = 0.0017), while it did not affect TNF, IL-10 or IL-1B expression. At the protein level, the concentrations of IFN-β and IL-10, but not TNF-α (P < 0.0001 and P = 0.0090, respectively, Figure 4B ) nor IL-1β ( Supplementary Figure 5B ) were significantly lower in the culture supernatant of 7α,25-OHC-treated Mtb-infected primary MNs compared to untreated infected cells.
Figure 4.

Activation of GPR183 leads to cytokine production favoring Mtb control. Primary MN from healthy donors (n = 8) were infected for 2 h with Mtb H37Rv (MOI 10:1), 7a,25-OHC (100 nM), and/or GSK682753 (10 μM). Cells were washed and left with drugs for a further 22 h. Changes in the expression of (A) IFNB1, TNF, and IL10 were measured by qPCR and normalized to untreated infected cells. Concentrations of (B) IFN-b, TNF-a, and IL-10 in the culture supernatant were measured by ELISA. Data are presented as mean fold change ± SEM or min to max for box plots; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001; paired t-test.
The Oxysterol 7α,25-OHC Induces Autophagy
We aimed to identify whether 7α,25-OHC impacts the production of reactive oxygen species (ROS) and the autophagy pathway. ROS production in BCG-infected primary MNs was not affected by 7α,25-OHC ( Supplementary Figure 6 ); however, we observed an increase in accumulation of the autophagosome marker LC3B-II in BCG-infected THP-1 cells treated with 7α,25-OHC (P = 0.0119, Figure 5A ). We next performed the experiments in the absence and presence of the lysosomal inhibitor chloroquine in order to determine autophagic flux. Autophagic flux in BCG-infected cells was significantly increased with 7α,25-OHC treatment (P = 0.0069, Figure 5B ). The simultaneous addition of the GPR183 antagonist GSK682753 with 7α,25-HC, decreased the levels of LC3B-II and autophagic flux; however, this did not reach statistical significance.
Figure 5.

Treatment with 7α,25-OHC induces autophagy. PMA-differentiated THP-1 cells were infected/uninfected and co-incubated with ±7α,25-OHC, ±GSK682753, for 2 h. Extracellular BCG was removed, and cells were incubated for a further 4 h or 22 h in RPMI medium containing drugs. (A) Cells were lysed at 6 h or 24 h (Flux) p.i. (B) The band intensity was then normalized to the reference protein, GAPDH and further normalized to the BCG. Autophagic flux was obtained by subtracting chloroquine positive values with chloroquine negative values. (C) Cells were visualized using the Olympus FV 3000 confocal microscope. At least 30 cells were counted for every condition. Data are presented as ± SEM; ns, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; unpaired t-test.
We next confirmed the induction of autophagy via microscopy. The number of LC3B-II puncta per cell increased in 7α,25-OHC stimulated BCG-infected THP-1 cells compared to the untreated BCG-infected cells (P = 0.0358, Figure 5C ). The 7α,25-OHC effect could be reduced by antagonist GSK682753 (P = 0.0196).
GPR183 KO Mice Have Higher Bacterial Burden During the Early Stage of Infection
To confirm the effect of the GPR183 receptor in vivo, we infected WT and GPR183 KO mice with aerosolized Mtb. At 2 weeks p.i., GPR183 KO mice showed significantly increased mycobacterial burden in the lungs compared to WT mice (P = 0.0084, Figure 6A ), while the bacterial burden was comparable at 5 weeks p.i. ( Supplementary Figure 7 ). GPR183 KO mice also had higher lung pathology scores although this did not reach significance ( Figure 6B ). GPR183 KO mice had significantly increased Ifnb1 expression in the lungs (P = 0.0256; Figure 6C ), along with increased Irf3 (P = 0.0159); however, Irf5 ( Supplementary Figure 8 ) and Irf7 ( Figure 6C ) remained unchanged. Irf7 transcription was increased in the blood from GPR183 KO compared to WT mice (P = 0.0513; Figure 6D ), but Ifnb1, Irf3 and Irf5 expression was not different ( Figure 6D , Supplementary Figure 6 ). At the RNA level Tnf, Ifng, and Il1b were not significantly different between GPR183 KO and WT mice ( Figure 7A ). Unexpectedly, at the protein level, the concentrations of IFN-β (P = 0.0232) and IFN-γ (P = 0.0232) were significantly lower in GPR183 KO mice lung, while TNF-α (P = 0.7394) and IL-1β (P = 0.0753) were not statistically different ( Figure 7B ).
Figure 6.

GPR183KO mice have higher lung CFU, corresponding with increased expression of transcription factors regulating type I interferon responses. Mice were infected with 300 CFU of aerosol Mtb H37Rv. (A) Bacterial lung burden 2 weeks p.i. (B) Total histology lung score. RNA was isolated from Mtb-infected lung and blood samples 2 weeks p.i. (C) Gene expression of Ifnb1, Irf3, and Irf7 in the lungs, (D) Ifnb1, Irf3, and Irf7 in the blood, was measured by qRT-PCR using Hprt1 as reference gene. Data are presented as ± SEM; ns, P > 0.05; *P ≤ 0.05; **P ≤ 0.01.
Figure 7.

Pro-inflammatory cytokine expression at 2 weeks p.i. of Mtb H37Rv-infected mice. Mice were infected with 300 CFU of aerosol Mtb H37Rv. (A) Gene expression of Ifng, Il1b, and Tnf in the lungs. (B) Concentrations of IFN-β, IFN-γ, IL-1β and TNF-α in the culture supernatant were measured by ELISA. Data are presented as ± SEM; ns, P > 0.05; *P ≤ 0.01.
Discussion
Historically oxidized cholesterols, so called oxysterols, were considered by-products that increase polarity of cholesterol to facilitate its elimination. However, they have recently emerged as important lipid mediators that control a range of physiological processes including metabolism, immunity, and steroid hormone synthesis (27).
Our findings define a novel role for GPR183 in regulating the host immune response during Mtb infection (summarized in Figure 8 ). We initially identified GPR183 through a blood transcriptomic screen in TB and TB + T2D patients and found an inverse correlation between GPR183 expression and TB disease severity on chest x-ray. Although we demonstrate that the decrease in blood GPR183 in TB + T2D patients is likely due, in part, to a decreased frequency of non-classical monocytes expressing GPR183, we cannot rule out that reduced GPR183 expression in whole blood is partially attributable to neutrophils and eosinophils as preferential loss of neutrophils and eosinophils occurs upon PBMC isolation. In our study the TB patients with T2D had more severe TB compared to those without T2D; therefore we cannot ascertain whether lower GPR183 expression is linked to TB + T2D comorbidity or TB disease severity.
Figure 8.

Schematic summary of the role of GPR183 in Mtb-infected human monocytes.
We demonstrate that activation of GPR183 by 7α,25-OHC in primary human MNs during Mtb infection results in significantly better control of intracellular Mtb growth. This is in contrast to a recently published study showing increased Mtb growth with 7α,25-OHC when added post-infection in murine RAW264.7 cells (28). The discrepancies between the studies could also be attributed to the different cell types and infection dose, which was 25 times higher in the aforementioned study. Consistent with the findings of Tang et al. (28) in murine cells we show that mycobacterial infection down-regulates GPR183 in human MNs, which may be an immune-evasion strategy specific to mycobacteria since LPS, a constituent of Gram-negative bacteria, upregulates GPR183 (15). Whether the observed increase in phagocytosis in the presence of 7α,25-OHC is a non-specific effect driven by internalization of agonist bound GPR183 and non-specific uptake of bacteria or an increase in pattern recognition receptors remains to be elucidated.
We further demonstrate that GPR183 activation by 7α,25-OHC reduces IFN-β expression and secretion in Mtb-infected primary MNs and targeted GPR183 knockdown significantly upregulating IRFs and IFNB1. Similarly, gene expression of IRF1, IRF5, and IRF7 is up-regulated in TB + T2D patients compared to TB patients and corresponds with down-regulation of GPR183, thereby demonstrating that GPR183 expression is associated with IFN regulatory factors during human TB, and GPR183 is a negative regulator of type I IFNs in Mtb-infected human MNs.
There is mounting evidence that the production of type-I IFNs is detrimental during Mtb infection (29, 30). Up-regulation of type-I IFN blood transcript signatures occurs in TB disease and correlates with disease severity (31). In macrophages, Mtb induces up-regulation of IFNB1 expression as early as 4 h p.i. to limit IL-1β production, a critical mediator in the host defense against Mtb (32). Although 7α,25-OHC significantly reduced IFNB1 mRNA, we did not observe an increase in IL1B mRNA, suggesting that the GPR183-mediated regulation of type-I IFN does not influence IL1B expression. In addition to GPR183 mediated reduction in IFN-β, we observed a decrease in IL-10 in Mtb-infected primary MNs treated with 7α,25-OHC. IL-10 production is induced by type-I IFN signaling (33, 34) and promotes Mtb growth (35) by reducing the bioavailability of TNF-α through the release of soluble TNF receptors and preventing the maturation of Mtb-containing phagosomes (35–38). Collectively, we show that GPR183 is a negative regulator of type-I IFNs in primary MNs, and agonist induced activation of GPR183 reduces Mtb-induced IFN-β production, while leaving expression of cytokines important for Mtb control unchanged.
Further confirming the role of GPR183, GPR183 KO mice infected with Mtb had significantly higher bacterial burden in the lung compared to WT mice 2 weeks p.i. (prior to initiation of the adaptive immune response to Mtb) with this effect disappearing at 5 weeks p.i., when T cell responses against Mtb are fully established. Our results thus strengthen the contention that GPR183 plays an important role in the innate immune control of Mtb irrespective of hyperglycemia. We confirmed the importance GPR183 in regulating type-I interferons during Mtb infection in vivo. GPR183 KO mice infected with Mtb had significantly increased lung Ifnb1 and Irf3 mRNA. Unexpectedly, IFN-β and IFN-γ secretions were both significantly downregulated in the lung. These differences between mRNA and protein levels may be due to kinetic parameters of transcription versus translation or mRNA stability versus protein consumption.
Furthermore, we demonstrate that the GPR183 agonist 7α,25-OHC promotes autophagy in macrophages infected with mycobacteria. Autophagy is a cellular process facilitating the elimination of intracellular pathogens including Mtb (39). Antimicrobial autophagy was shown to be inhibited by Mycobacterium leprae through upregulation of IFN-β and autocrine IFNAR activation which in turn increased expression of the autophagy blocker OASL (2′-5′-oligoadenylate synthetase like) (40). Whether there is a link between the 7α,25-OHC-induced reduction of IFN-β production and the increase in autophagy remains to be investigated in future studies.
Several autophagy promoting re-purposed drugs including metformin are currently being assessed as HDTs for TB (41). We propose that GPR183 is a potential target for TB HDT, warranting the development of specific, metabolically stable small-molecule agonists for this receptor to ultimately improve TB treatment outcomes in TB patients with and without T2D co-morbidity.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The human studies were approved by the Institutional Review Board of Stellenbosch University (N13/05/064 and N13/05/064A), and all study participants signed pre-approved informed consent documents prior to enrolment into the studies. The patients/participants provided their written informed consent to participate in this study. All animal studies were approved by the Animal Ethics Committee of the University of Queensland (MRI-UQ/596/18) and conducted in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes.
Author Contributions
AG, SB, and KR wrote the manuscript. AG, SB, RS, SH, HS, MN, CF, LK, HT, TW, HB-O, AH, CM, LV, and NW carried out the experiments. AG, SB, MN, HS, RS, and SH analyzed the data. TM-P, MR, LS, GW, and KR interpreted the data and developed the theoretical framework. KR conceived the original idea. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID) and the South African Medical Research Council under the US-South African Program for Collaborative Biomedical Research (grant number: R01AI116039) to KR and by the TANDEM Grant of the EUFP7 (European Union’s Seventh Framework Program) under Grant Agreement NO. 305279 to GW for study participant recruitment, by the Novo Nordisk Foundation to MR and TM-P. All other laboratory-based research activities were supported by grants from the Australian Infectious Diseases Research Center, The Australian Respiratory Council and the Mater Foundation to KR. The Translational Research Institute is supported by a grant from the Australian Government.
Conflict of Interest
MMR is a co-founder of Antag Therapeutics and of Synklino.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the clinical research team at Stellenbosch University for assistance with identification and recruitment of study participants as well as coordination of clinical and administrative activities. We thank Matthew Sweet for critical review of the manuscript. Illustrations were created with Biorender.com. This manuscript has been released as a pre-print at bioRxiv (42).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.601534/full#supplementary-material
References
- 1. Magee MJ, Kempker RR, Kipiani M, Gandhi NR, Darchia L, Tukvadze N, et al. Diabetes mellitus is associated with cavities, smear grade, and multidrug-resistant tuberculosis in Georgia. Int J Tuberculosis Lung Dis (2015) 19(6):685–92. 10.5588/ijtld.14.0811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong Z, Shi J, Dorhoi A, Zhang J, Soodeen-Lalloo AK, Tan W, et al. Hemostasis and Lipoprotein Indices Signify Exacerbated Lung Injury in TB With Diabetes Comorbidity. Chest (2018) 153(5):1187–200. 10.1016/j.chest.2017.11.029 [DOI] [PubMed] [Google Scholar]
- 3. Critchley JA, Restrepo B, II, Ronacher K, Kapur A, Bremer AA, Schlesinger LS, et al. Defining a Research Agenda to Address the Converging Epidemics of Tuberculosis and Diabetes: Part 1: Epidemiology and Clinical Management. Chest (2017) 152(1):165–73. 10.1016/j.chest.2017.04.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ronacher K, van Crevel R, Critchley JA, Bremer AA, Schlesinger LS, Kapur A, et al. Defining a Research Agenda to Address the Converging Epidemics of Tuberculosis and Diabetes: Part 2: Underlying Biologic Mechanisms. Chest (2017) 152(1):174–80. 10.1016/j.chest.2017.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Restrepo B, II, Kleynhans L, Salinas AB, Abdelbary B, Tshivhula H, Aguillon-Duran GP, et al. Diabetes screen during tuberculosis contact investigations highlights opportunity for new diabetes diagnosis and reveals metabolic differences between ethnic groups. Tuberculosis (Edinb) (2018) 113:10–8. 10.1016/j.tube.2018.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature (2011) 475(7357):524–7. 10.1038/nature10280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gessier F, Preuss I, Yin H, Rosenkilde MM, Laurent S, Endres R, et al. Identification and characterization of small molecule modulators of the Epstein-Barr virus-induced gene 2 (EBI2) receptor. J Med Chem (2014) 57(8):3358–68. 10.1021/jm4019355 [DOI] [PubMed] [Google Scholar]
- 8. Rosenkilde MM, Benned-Jensen T, Andersen H, Holst PJ, Kledal TN, Luttichau HR, et al. Molecular pharmacological phenotyping of EBI2. An orphan seven-transmembrane receptor with constitutive activity. J Biol Chem (2006) 281(19):13199–208. 10.1074/jbc.M602245200 [DOI] [PubMed] [Google Scholar]
- 9. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct B-cell migration through EBI2. Nature (2011) 475(7357):519–23. 10.1038/nature10226 [DOI] [PubMed] [Google Scholar]
- 10. Benned-Jensen T, Norn C, Laurent S, Madsen CM, Larsen HM, Arfelt KN, et al. Molecular characterization of oxysterol binding to the Epstein-Barr virus-induced gene 2 (GPR183). J Biol Chem (2012) 287(42):35470–83. 10.1074/jbc.M112.387894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daugvilaite V, Arfelt KN, Benned-Jensen T, Sailer AW, Rosenkilde MM. Oxysterol-EBI2 signaling in immune regulation and viral infection. Eur J Immunol (2014) 44(7):1904–12. 10.1002/eji.201444493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature (2009) 460(7259):1122–6. 10.1038/nature08226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity (2009) 31(2):259–69. 10.1016/j.immuni.2009.06.016 [DOI] [PubMed] [Google Scholar]
- 14. Chalmin F, Rochemont V, Lippens C, Clottu A, Sailer AW, Merkler D, et al. Oxysterols regulate encephalitogenic CD4(+) T cell trafficking during central nervous system autoimmunity. J Autoimmun (2015) 56:45–55. 10.1016/j.jaut.2014.10.001 [DOI] [PubMed] [Google Scholar]
- 15. Preuss I, Ludwig MG, Baumgarten B, Bassilana F, Gessier F, Seuwen K, et al. Transcriptional regulation and functional characterization of the oxysterol/EBI2 system in primary human macrophages. Biochem Biophys Res Commun (2014) 446(3):663–8. 10.1016/j.bbrc.2014.01.069 [DOI] [PubMed] [Google Scholar]
- 16. Gatto D, Wood K, Caminschi I, Murphy-Durland D, Schofield P, Christ D, et al. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nat Immunol (2013) 14(5):446–53. 10.1038/ni.2555 [DOI] [PubMed] [Google Scholar]
- 17. Chiang EY, Johnston RJ, Grogan JL. EBI2 is a negative regulator of type I interferons in plasmacytoid and myeloid dendritic cells. PloS One (2013) 8(12):e83457. 10.1371/journal.pone.0083457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emgard J, Kammoun H, Garcia-Cassani B, Chesne J, Parigi SM, Jacob JM, et al. Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity (2018) 48(1):120–32. 10.1016/j.immuni.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rutkowska A, Preuss I, Gessier F, Sailer AW, Dev KK. EBI2 regulates intracellular signaling and migration in human astrocyte. Glia (2015) 63(2):341–51. 10.1002/glia.22757 [DOI] [PubMed] [Google Scholar]
- 20. Rutkowska A, Shimshek DR, Sailer AW, Dev KK. EBI2 regulates pro-inflammatory signalling and cytokine release in astrocytes. Neuropharmacology (2018) 133:121–8. 10.1016/j.neuropharm.2018.01.029 [DOI] [PubMed] [Google Scholar]
- 21. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature (2016) 533(7601):110–4. 10.1038/nature17947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity (2015) 42(4):704–18. 10.1016/j.immuni.2015.03.002 [DOI] [PubMed] [Google Scholar]
- 23. Ralph AP, Ardian M, Wiguna A, Maguire GP, Becker NG, Drogumuller G, et al. A simple, valid, numerical score for grading chest x-ray severity in adult smear-positive pulmonary tuberculosis. Thorax (2010) 65(10):863–9. 10.1136/thx.2010.136242 [DOI] [PubMed] [Google Scholar]
- 24. Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinf (2017) 18(1):529. 10.1186/s12859-017-1934-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benned-Jensen T, Smethurst C, Holst PJ, Page KR, Sauls H, Sivertsen B, et al. Ligand modulation of the Epstein-Barr virus-induced seven-transmembrane receptor EBI2: identification of a potent and efficacious inverse agonist. J Biol Chem (2011) 286(33):29292–302. 10.1074/jbc.M110.196345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benned-Jensen T, Rosenkilde MM. Structural motifs of importance for the constitutive activity of the orphan 7TM receptor EBI2: analysis of receptor activation in the absence of an agonist. Mol Pharmacol (2008) 74(4):1008–21. 10.1124/mol.108.049676 [DOI] [PubMed] [Google Scholar]
- 27. Mutemberezi V, Guillemot-Legris O, Muccioli GG. Oxysterols: From cholesterol metabolites to key mediators. Prog Lipid Res (2016) 64:152–69. 10.1016/j.plipres.2016.09.002 [DOI] [PubMed] [Google Scholar]
- 28. Tang J, Shi YN, Zhan L, Qin C. Downregulation of GPR183 on infection restricts the early infection and intracellular replication of mycobacterium tuberculosis in macrophage. Microbial Pathogenesis (2020) 145:104234. 10.1016/j.micpath.2020.104234 [DOI] [PubMed] [Google Scholar]
- 29. Donovan ML, Schultz TE, Duke TJ, Blumenthal A. Type I Interferons in the Pathogenesis of Tuberculosis: Molecular Drivers and Immunological Consequences. Front Immunol (2017) 8:1633:1633. 10.3389/fimmu.2017.01633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moreira-Teixeira L, Mayer-Barber K, Sher A, O’Garra A. Type I interferons in tuberculosis: Foe and occasionally friend. J Exp Med (2018) 215(5):1273–85. 10.1084/jem.20180325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466(7309):973–7. 10.1038/nature09247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol (2011) 187(5):2540–7. 10.4049/jimmunol.1100926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. 10.1038/nri3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35(6):1023–34. 10.1016/j.immuni.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beamer GL, Flaherty DK, Assogba BD, Stromberg P, Gonzalez-Juarrero M, de Waal Malefyt R, et al. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J Immunol (2008) 181(8):5545–50. 10.4049/jimmunol.181.8.5545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hart PH, Hunt EK, Bonder CS, Watson CJ, Finlay-Jones JJ. Regulation of surface and soluble TNF receptor expression on human monocytes and synovial fluid macrophages by IL-4 and IL-10. J Immunol (1996) 157(8):3672–80. [PubMed] [Google Scholar]
- 37. O’Leary S, O’Sullivan MP, Keane J. IL-10 blocks phagosome maturation in mycobacterium tuberculosis-infected human macrophages. Am J Respir Cell Mol Biol (2011) 45(1):172–80. 10.1165/rcmb.2010-0319OC [DOI] [PubMed] [Google Scholar]
- 38. Armstrong L, Jordan N, Millar A. Interleukin 10 (IL-10) regulation of tumour necrosis factor alpha (TNF-alpha) from human alveolar macrophages and peripheral blood monocytes. Thorax (1996) 51(2):143–9. 10.1136/thx.51.2.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chandra P, Kumar D. Selective autophagy gets more selective: Uncoupling of autophagy flux and xenophagy flux in Mycobacterium tuberculosis-infected macrophages. Autophagy (2016) 12(3):608–9. 10.1080/15548627.2016.1139263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Toledo Pinto TG, Batista-Silva LR, Medeiros RCA, Lara FA, Moraes MO. Type I Interferons, Autophagy and Host Metabolism in Leprosy. Front Immunol (2018) 9:806:806. 10.3389/fimmu.2018.00806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cerni S, Shafer D, To K, Venketaraman V. Investigating the Role of Everolimus in mTOR Inhibition and Autophagy Promotion as a Potential Host-Directed Therapeutic Target in Mycobacterium tuberculosis Infection. J Clin Med (2019) 8(2):232. 10.3390/jcm8020232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bartlett S, Gemiarto AT, Ngo MD, Sajiir H, Hailu S, Sinha R, et al. GPR183 regulates interferons and bacterial growth during Mycobacterium tuberculosis infection: interaction with type 2 diabetes and TB disease severity. bioRxiv (2020). 10.1101/2020.07.15.203398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.