

Abstract
Therapeutic options for patients with treatment-resistant epilepsy represent an important unmet need. Addressing this unmet need was the main factor driving the drug discovery program that led to the synthesis of padsevonil, a first-in-class antiepileptic drug candidate that interacts with two therapeutic targets: synaptic vesicle protein 2 and GABAA receptors. Two PET imaging studies were conducted in healthy volunteers to identify optimal padsevonil target occupancy corresponding to levels associated with effective antiseizure activity in rodent models. Optimal padsevonil occupancy associated with non-clinical efficacy was translatable to humans for both molecular targets: high (>90%), sustained synaptic vesicle protein 2A occupancy and 10–15% transient GABAA receptor occupancy. Rational dose selection enabled clinical evaluation of padsevonil in a Phase IIa proof-of-concept trial (NCT02495844), with a single-dose arm (400 mg bid). Adults with highly treatment-resistant epilepsy, who were experiencing ≥4 focal seizures/week, and had failed to respond to ≥4 antiepileptic drugs, were randomized to receive placebo or padsevonil as add-on to their stable regimen. After a 3-week inpatient double-blind period, all patients received padsevonil during an 8-week outpatient open-label period. The primary endpoint was ≥75% reduction in seizure frequency. Of 55 patients randomized, 50 completed the trial (placebo n = 26; padsevonil n = 24). Their median age was 36 years (range 18–60), and they had been living with epilepsy for an average of 25 years. They were experiencing a median of 10 seizures/week and 75% had failed ≥8 antiepileptic drugs. At the end of the inpatient period, 30.8% of patients on padsevonil and 11.1% on placebo were ≥75% responders (odds ratio 4.14; P = 0.067). Reduction in median weekly seizure frequency was 53.7% and 12.5% with padsevonil and placebo, respectively (unadjusted P = 0.026). At the end of the outpatient period, 31.4% were ≥75% responders and reduction in median seizure frequency was 55.2% (all patients). During the inpatient period, 63.0% of patients on placebo and 85.7% on padsevonil reported treatment-emergent adverse events. Overall, 50 (90.9%) patients who received padsevonil reported treatment-emergent adverse events, most frequently somnolence (45.5%), dizziness (43.6%) and headache (25.5%); only one patient discontinued due to a treatment-emergent adverse event. Padsevonil was associated with a favourable safety profile and displayed clinically meaningful efficacy in patients with treatment-resistant epilepsy. The novel translational approach and the innovative proof-of-concept trial design maximized signal detection in a small patient population in a short duration, expediting antiepileptic drug development for the population with the greatest unmet need in epilepsy.
Keywords: antiepileptic drug, drug-resistant epilepsy, partial-onset seizures, [11C]UCB-J, PET, imaging
Guided by results of two human PET imaging studies, a single 400 mg bid dose of the antiepileptic drug candidate, padsevonil, was evaluated for patients with treatment-resistant epilepsy. At the end of a 3-week double-blind inpatient period, padsevonil displayed a favourable safety profile and conferred clinically meaningful benefit over placebo.
Graphical Abstract
Graphical Abstract.

Introduction
Antiepileptic drugs (AEDs) are the cornerstone of therapy in epilepsy, a spectrum of diseases that affect an estimated 70 million people worldwide (Thijs et al., 2019). While many AEDs have become available over the past decades, seizure-freedom rates obtained with the newer AEDs do not appear to have improved. In two longitudinal, observational cohort studies conducted at the same epilepsy centre 16 years apart, seizure-freedom rate was 64% despite introduction of newer AEDs (Kwan and Brodie 2000; Chen et al., 2018). In the two studies, as in others, the likelihood of achieving seizure freedom diminished sharply with each unsuccessful regimen (Schiller and Najjar 2008). Drug-resistant epilepsy, which affects 30–40% of patients, is associated with significant morbidity and increased risk of mortality (Weaver and Pohlmann Eden 2013; Laxer et al., 2014; Engel 2016; Strzelczyk et al., 2017). Consequently, there is an important unmet need for novel AEDs, developed using new research strategies, that can help improve seizure control for patients with drug-resistant epilepsy (Chen et al., 2018).
While the first AEDs were discovered through serendipity, the principal method for developing newer AEDs has so far been based on empirical screening and testing in animal models; very few have been developed based on hypothesis-driven, target-based rational drug design (Rogawski 2008; Löscher et al., 2013). The latter approach was used to develop padsevonil (PSL), the first in a novel chemical class of drugs (Wood et al., 2020). PSL was designed specifically to interact with two therapeutic targets. Presynaptically, it binds with very high affinity to synaptic vesicle protein 2 (SV2) and postsynaptically, with low-to-moderate affinity to GABAA receptors (GABAARs), where it acts as a partial agonist at the benzodiazepine site (Wood et al., 2020). This specific profile was selected since antiseizure potency correlates with SV2A binding affinity (Kaminski et al., 2008; Kaminski et al., 2009; Gillard et al., 2011), while the low-to-moderate affinity for the benzodiazepine site would be expected to produce therapeutic effects without inducing excessive sedation or tolerance (Rundfeldt and Löscher, 2014). Selective interaction of PSL with its intended targets was demonstrated in in vitro binding assays where PSL displayed a nanomolar affinity for all three SV2 isoforms (SV2A, SV2B and SV2C) and micromolar affinity for the benzodiazepine site (Wood et al., 2020). Results of in vivo target occupancy experiments in mice supported in vitro results (Wood et al., 2020).
Animal models for testing PSL’s antiseizure efficacy were carefully selected to be fit-for-purpose and have high predictive validity for its intended attributes (Denayer et al., 2014; Löscher 2016). PSL showed efficacy not only in standard acute seizure models but also in models representing chronic epilepsy, characterized by suboptimal or no response to existing AEDs (Leclercq et al., 2020). Of note, in the 6 Hz (44 mA) model, at the same target occupancies, PSL provided greater protection than the selective SV2A ligands levetiracetam (LEV) and brivaracetam (BRV), or the benzodiazepine, diazepam, administered alone, and LEV or BRV administered in combination with diazepam (Leclercq et al., 2020). PSL also demonstrated greater efficacy than nine other mechanistically diverse AEDs in the amygdala kindling model (Leclercq et al., 2020), which is predictive of efficacy in human drug-resistant epilepsy (Löscher, 2011, 2017).
Acceptable safety results in first-in-human trials (Otoul et al. 2017) led to PET imaging studies to establish the PSL dose in humans that would achieve target occupancy similar to that observed in non-clinical studies and to enable a Phase IIa proof-of-concept (PoC) trial. PET target occupancy studies can help guide dose selection for drug candidates, reducing the risk of under- or overdosing in clinical trials (Melhem 2013). Testing depends on the availability of PET tracers and has so far not been used in AED development. PET tracers were available to characterize PSL binding at GABAARs and at SV2A, but not at SV2B or SV2C. Target occupancy associated with effective antiseizure activity in non-clinical models, combined with Phase I and human PET data were subsequently used to select the dose expected to produce the best efficacy/tolerability profile in patients. This translational approach, not previously used in the development of AEDs, allowed for rational dose selection, which in turn enabled implementation of an efficient single-dose PoC trial to test efficacy and safety in patients with highly treatment-resistant disease. The current report provides results of the two imaging studies, and the PoC trial, as well as population pharmacokinetic (popPK) modelling and exposure-response analyses.
Materials and methods
PET studies
PSL target occupancy was determined in two Phase I, single-centre, open-label studies conducted in Belgium: one for SV2A (EudraCT Number 2016-001190-32) and the other for GABAARs (EudraCT Number 2012-000231-79). Both studies were approved by an Independent Ethics Committee and conducted in accordance with local Belgian laws, and all applicable regulatory and the International Conference on Harmonization-Good Clinical Practice guidelines. All volunteers provided written informed consent before entering the studies.
The radiotracer [11C]UCB-J was used for the SV2 study; it has been shown to perform as best-in-class in non-human primates (Nabulsi et al., 2016) and humans (Finnema et al., 2016). [11C]-Flumazenil has been used to assess GABAAR occupancy in vivo in humans and non-human primates (Abadie and Baron, 1990) and was used in the current GABAA PET study. Full methodological details of both PET imaging studies are provided in the Supplementary Material.
Proof-of-concept trial
Trial design
This Phase IIa trial was conducted at 10 specialized epilepsy centres across Belgium, Bulgaria, Germany, Hungary, the Netherlands and Spain (NCT02495844). The trial was initiated in August 2015 with patient screening (first patient, first visit), and completed in February 2017 (last patient, last visit). Trial protocol, amendments and patient informed consent were reviewed by a national, regional or Independent Ethics Committee. The trial was conducted in accordance with local laws of countries involved and the Good Clinical Practice guidelines of International Council for Harmonization, based on the principles of the Declaration of Helsinki.
The trial consisted of a 4-week historical and a 2- to 3-week prospective outpatient baseline (Fig. 1). After randomization to PSL or placebo, patients entered a double-blind 3-week inpatient period, consisting of a titration, a maintenance and a transition-to-outpatient week. Patients then entered an open-label outpatient maintenance period of 8 weeks, at the end of which they could opt to enter the open-label extension (OLE) or exit the trial after a 2-week tapering and a 4-week safety follow-up period.
Figure 1.

Proof-of-concept trial design and patient disposition. Patients were randomized 1:1 to receive treatment with padsevonil (PSL) 400 mg bid or placebo. At the end of Week 2, patients in the placebo group transitioned to treatment with PSL 400 mg bid, and at the end of Week 3, all patients transitioned to open-label treatment with PSL 400 mg bid for 8 weeks. *At trial end, patients could either exit the trial and enter a safety follow-up period or continue treatment with PSL in an open-label extension (OLE).
All patients continued using their baseline AED regimen throughout the inpatient period; changes during the outpatient maintenance period were reviewed by the trial physician on a case-by-case basis. Use of benzodiazepines as rescue medication was permitted if ≤3 doses were taken within 7 days.
Patient population
Patients (≥18 years) with a diagnosis of focal epilepsy who had failed to achieve seizure control with ≥4 AED regimens of adequate dose and duration, including their current treatment, were invited to participate in the trial. During the historical and prospective baseline periods, patients had to have experienced ≥4 focal with or without focal-to-bilateral tonic-clonic seizures per week and could not have had any seizure-free period longer than 3 days.
Patients were excluded if they had current or previous liver disease; clinically significant abnormality on ECG; current or previous psychiatric disease or serious psychiatric AEs with a previous exposure to LEV or BRV; history of suicide attempt or suicidal ideation in the past 6 months; history of hypersensitivity reactions, autoimmune disease, unexplained syncope, seizure clusters or status epilepticus; epilepsy surgery <1 year or an epilepsy dietary therapy initiated <3 months before screening; current or previous substance abuse/dependence. Patients were also excluded if they were being treated with tiagabine, felbamate, vigabatrin or enzyme-inducing AEDs. Concomitant use of LEV was permitted, and randomization was stratified by LEV use. Use of benzodiazepines, zolpidem, zaleplon or zopiclone >3 times per week for any indication, as well as non-antiepileptic prescription or non-prescription drugs, and dietary or herbal products that are potent inducers or inhibitors of the cytochrome P450 3A4 pathway for 2 weeks (or 5 half-lives, whichever is longer) before the baseline visit was prohibited. Female patients of childbearing potential were required to use contraception; male patients were required to use barrier contraception during the trial and for 3 months after the final dose.
Randomization and blinding
At trial start, patients, caregivers, investigators and trial personnel were unaware of treatment assignment. Several members of the sponsor company were unblinded to the primary efficacy results for an interim analysis of data following completion of the double-blind inpatient period. Patients were randomized to treatment with PSL 400 mg bid or placebo (1:1) ratio using Interactive Response Technology (IRT) System. Randomization was stratified based on current or previous LEV use to ensure balance across treatment groups.
Dose selection rationale
The known pharmacology of drugs acting at GABAARs and non-clinical evidence correlating SV2A occupancy and antiseizure efficacy, directed the synthesis of PSL to ensure low (∼10%) GABAAR occupancy and quasi full saturation at SV2A, allowing maximum efficacy while avoiding sedation. In the murine amygdala kindling model, PSL ED50 for protection against focal-to-bilateral seizures was 1.2 mg/kg; at this dose, estimated occupancy was >90% at SV2A and approximately 10% at GABAARs (Leclercq et al., 2020; Wood et al., 2020). Results of the PET studies indicated similar target occupancy after therapeutically relevant doses; ie, sustained high‐level SV2A occupancy (>90%) and low, but sufficient GABAAR occupancy (13.4%) at the 400 mg bid dose. The 400 mg bid dose was also the highest dose tested in a first-in-human multiple ascending dose trial (490 mg was tested as a single dose) (Otoul et al., 2017). Therefore, 400 mg bid was deemed to be the optimal, maximal dose that would best translate the murine amygdala kindling data (in vivo SV2A and GABAAR occupancy at ED50) to humans (Fig. 2). Doses higher than 400 mg bid would not likely increase the benefit/tolerability ratio, since SV2A occupancy >95% was not predicted to enhance efficacy while GABAAR occupancy >15% would increase the likelihood of tolerance development and GABA-mediated adverse events.
Figure 2.

Translating SV2A and GABAA receptor target engagement from rodents to humans. In the amygdala kindling model of chronic epilepsy, the ED50, the PSL dose that protected 50% of mice against focal-to-bilateral seizures was 1.2 mg/kg. In vivo occupancy studies in mice showed that at this dose, PSL exhibited >90% SV2A occupancy and ∼10% occupancy at GABAA receptors (GABAARs). The differential proportionality of target engagement was retained in humans as shown in PET studies following administration of PSL 400 mg bid; >90% SV2A and 13% GABAAR occupancy. SV2A occupancy was sustained; in contrast, GABAAR occupancy was transient, and at 2 h post-dose, occupancy for the average of all regions of interest was ≤5% for most volunteers. The 400 mg bid dose was selected for the Phase II proof-of-concept trial, as it was expected to achieve exposure levels similar to those associated with robust efficacy in the amygdala kindling model; high, sustained SV2A, and low, transient GABAAR occupancy.
Trial outcomes
The a priori defined primary efficacy outcome was the ≥75% responder rate (RR), the proportion of patients with a ≥ 75% reduction in focal seizure frequency during the last 2 weeks (PSL group) or the first 2 weeks (placebo group) of the inpatient period compared with baseline. Secondary efficacy variables were median percent reduction in focal seizure frequency from baseline to the 2-week inpatient and outpatient maintenance period and seizure-freedom rate during the 2-week inpatient period, and last 4 weeks of the outpatient maintenance period.
Patients were monitored throughout the trial for treatment-emergent adverse events (TEAEs), coded according to the Medical Dictionary for Regulatory Activities version 19.1. Given disease severity among trial participants, the effect of PSL on cognitive and psychiatric measures was monitored closely. Psychiatric assessment was performed by a staff member trained in the identification of psychiatric symptoms, using the Brief Psychiatric Rating Scale.
Other safety assessments included vital signs measurements, clinical laboratory tests, 12-lead electrocardiograms, 2D Doppler echocardiography, physical and neurological examinations, Mini-Mental State Examination (for changes in memory or cognition), the Clinical Institute Withdrawal Assessment-Benzodiazepines (CIWA–B, for detection of withdrawal symptoms) and the Columbia-Suicide Severity Rating Scale.
Statistical analysis
Results of Phase III trials of adjunctive BRV and lacosamide were used to determine sample size. The desired effect of PSL in the current target population was a doubling of the effect of lacosamide and BRV in similar patients. The 75% RR was estimated to be <5% with placebo in these trials, 12.1% [95% confidence interval (CI) 7.3–19.7%] with BRV and 20.1% (95% CI 12.9–30.1%) with lacosamide (data on file). The target effect was set to have at least 36.5% of patients on PSL achieving 75% RR to provide initial evidence of potential differentiation from other AEDs. Based on a two-group Fisher's exact one-sided test with a ratio active: placebo of 1:1, the required number of patients was determined to be 23 in each arm to detect a significant difference between the two arms with a power of 80%.
The primary outcome (75% RR) was analysed using logistic regression with effects for treatment and the log-transformed baseline seizure frequency as a continuous covariate. The frequency and percentage of 75% responders were reported with the estimated odds ratios and two-sided Wald 95% CI for PSL versus placebo.
Percent reduction in weekly focal seizure frequency from baseline to the end of the inpatient period between the treatment arms was compared using the Wilcoxon–Mann–Whitney test. The Hodges-Lehmann non-parametric estimator was used to estimate the median percent reduction in seizures between groups and its corresponding 95% CI was provided. Percent reduction in focal seizure frequency from baseline to the end of the outpatient maintenance period was assessed using summary statistics. Seizure-freedom rate for the 2-week inpatient period was analysed using logistic regression with effects for treatment and log-transformed baseline seizure frequency as a continuous covariate. Seizure-freedom rate was also evaluated by treatment group for the last 4 weeks of the outpatient period and for the overall PSL treatment period using summary statistics. All analyses were performed using SAS® v9.3 (SAS Institute, Cary, NC, USA).
All patients who were randomized and received ≥1 dose of trial drug formed the safety set (SS), and patients in the SS who had ≥1 post-baseline seizure diary entry formed the full analysis set (FAS). All efficacy analyses were performed using data from FAS.
A post hoc subgroup descriptive analysis was performed to evaluate the potential impact of co-administration of SV2A-targeting AEDs, i.e., LEV or BRV, on PSL safety and efficacy.
Integrated analysis of PK, receptor occupancy and efficacy
A popPK model was developed based on rich PK sampling at the start and end of the inpatient period and sparse sampling during the outpatient period (10 per patient) of the trial. Analyses were conducted via non-linear mixed-effects modelling, performed using NONMEM v7.3.0 (ICON Dev. Solutions) (Lindbom et al., 2005). The model, qualified with available data, was used to simulate the plasma concentration vs time profiles for 50 mg to 400 mg bid, doses taking inter-patient variability into account. Concentration profiles, in particular the steady-state Ctrough, were compared with the estimated typical EC90 values from the SV2A RO modelling.
A preliminary exposure–response analysis was also conducted using PSL plasma concentration and efficacy data collected during the PoC trial.
Data availability
Due to the small volunteer numbers in the PET studies, Individual Patient Data (IPD) cannot be adequately anonymized, and therefore, cannot be shared. Data from the Phase II trial may be requested by qualified researchers 6 months after product approval in the USA and/or Europe, or global development is discontinued. Investigators may request access to anonymized IPD and redacted documents including raw and analysis-ready datasets, protocol, case report forms, statistical analysis plan, dataset specifications and clinical study report. Before access is granted, proposals must be approved by an independent review panel at www.clinicalstudydatarequest.com and a signed data sharing agreement must be in place. All documents are available for a pre-specified time, typically 12 months, on a password-protected portal.
Results
SV2A occupancy in humans
Twelve healthy volunteers took part in the [11C]UCB-J PET study (eight males, four females; mean age 28.2 ± 11.3 years), and data were available from 11. In two volunteers receiving the first tested dose (100 mg), almost complete SV2A occupancy (>98%) was observed 2 h following PSL administration (Fig. 3); therefore, doses >100 mg were not required to explore the full relationship. Overall, five volunteers received 100 mg PSL, three 25 mg and two each 12.5 mg or 6.25 mg.
Figure 3.

Pharmacokinetic/pharmacodynamic receptor occupancy modelling. Direct relationship between PSL plasma concentration and SV2A occupancy, as observed in PET imaging study (left panel) and predicted and observed SV2A occupancy versus PSL plasma concentration (right panel).
Eleven of the 12 volunteers (91.7%) reported 27 TEAEs, which were all mild in intensity and resolved spontaneously. The most frequently reported TEAEs were dizziness (n = 6, 50%), feeling drunk (4, 33.3%) and blurred vision (2, 16.7%).
A direct relationship between PSL plasma concentrations and SV2A occupancy was observed, best described by an Emax model (Fig. 3). By fixing the Emax to 100% (assuming full occupancy could occur) and the Hill coefficient value to 1, the EC50 was estimated to be 3.1 ng/mL (95% CI 2.0–4.2) and the EC90 27.9 ng/ml (95% CI 18.1–37.7) (Fig. 3). PSL bid dosing regimens were simulated using a popPK model developed using PSL concentration-time data from the PoC trial. The predicted plasma concentrations were compared with the predicted SV2A EC90 from the PET study. These simulations projected that SV2A occupancy would be >90% over the entire dosing interval for 75% of a population receiving PSL 100 mg bid and for 90% of a population receiving PSL 300 or 400 mg bid (Fig. 4).
Figure 4.
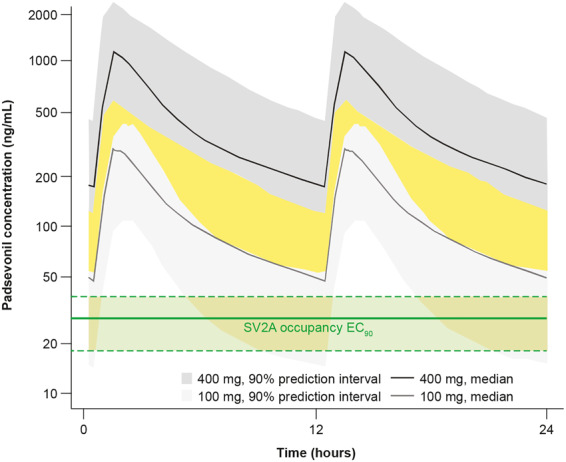
Population pharmacokinetic and receptor occupancy modelling. Estimated PSL concentration leading to 90% SV2A occupancy (median EC90 and 90% confidence interval as shown in the green-shaded area) compared with simulated PSL steady state plasma concentrations following 100 and 400 mg twice daily dosing (median and 90% predicted interval describing the inter-patient variability as shown in areas shaded light and dark grey, respectively). Given the variability in PSL pharmacokinetics following 100 and 400 mg twice daily dosing, some overlap is expected (represented by the yellow-shaded area). (EC90 = concentration producing 90% of maximum effect).
GABAA receptor occupancy in humans
Seven healthy male volunteers took part in the [11C]flumazenil PET study (mean age 29.0 ± 9.40 years). Results showed that PSL binds to GABAARs in multiple brain regions at steady state. For each volunteer, maximum GABAAR occupancy for nine brain regions of interest (ROI) was averaged—occupancy ranged from 1.3% to 9.7% [mean (SD) 6.41% (3.85)] in those who received PSL 200 mg bid, and from 7.6% to 16.8% [mean 13.4% (5.08)] in those who received 400 mg bid. At 2 h and 6 h post-dose, the average occupancy of all ROI was ≤5% for most volunteers. At 4 h post-dose, GABAAR occupancy was measurable in two of four volunteers who received PSL 200 mg and two of three volunteers who received PSL 400 mg, indicating that PSL binds to GABAARs at both dose levels. Mean maximum GABAAR occupancy was ≥5% in both dose groups for all ROI except the parietal cortex in the 200 mg group (3.8%). The mean of minimum values, which were at or near zero, were obtained at 6 h post-dose for most volunteers, indicating that GABAAR binding was transient and returned to baseline during the bid dosing cycle. No clear relationship between PSL PK and GABAAR occupancy was established since a full range of GABAAR occupancy could not be explored.
The seven volunteers reported 35 TEAEs; however, many TEAEs were manifestations of the same event occurring at varying intensities reported separately. All TEAEs were mild or moderate in intensity with the exception of one severe TEAE of somnolence in the PSL 400 mg group. All volunteers reported somnolence, and six (85.7%) reported dizziness. Other TEAEs reported by >1 volunteer were balance disorder (n = 3, 42.9%), decreased appetite (2, 28.6%), and medical device site reaction (2, 28.6%). All TEAEs (except leukocyturia reported 9 days post-treatment, n = 1) were transient and resolved spontaneously.
Proof-of-concept trial
Patient disposition and baseline characteristics
Of 66 patients screened, 55 were randomized to receive either placebo or PSL (1:1) (Fig. 1). PSL dose was titrated up to 400 mg bid over 1 week. All 27 patients randomized to receive placebo transitioned to PSL 400 mg bid during the third week of the inpatient period and continued on PSL during the 8-week open-label maintenance period. Of 28 patients randomized to PSL, 27 completed the inpatient period and continued to the maintenance period. Fifty patients (90.9%) completed the trial and five discontinued. One patient discontinued during the inpatient period due to a TEAE (in the PSL group) while the remaining four discontinued during the outpatient period; three due to lack of efficacy and one due to ‘other’ reason. After an individualized benefit-risk assessment, 42 patients (76%) opted to continue treatment with PSL and entered the OLE (NCT02625090).
Patients’ median age was 36 years (range 18–60), and they had been living with epilepsy for an average of 25 years (Table 1). Epilepsy aetiology was unknown in 36.4% of patients. The most frequently identified aetiology was congenital (30.9%), predominantly due to cortical dysplasia (25.5%), and perinatal events (10.9%). In most patients, localization of the epileptogenic focus was either in the temporal (47.3%) or the frontal lobe (43.6%). Before entering the trial, 74.6% of patients had tried ≥8 AEDs. At baseline, all patients were receiving AEDs, with most (32.7%) being treated with three concomitant AEDs; despite this, median total weekly seizure frequency remained high at 10 (range 3–215). The most frequently used concomitant AEDs were LEV (36.4%), lacosamide, oxcarbazepine (both 30.9%), lamotrigine, valproate (both 27.3%) and perampanel (23.6%).
Table 1.
Demographics and baseline disease characteristics of patients in the proof-of-concept trial
| Placebo (n = 27) | Padsevonil (n = 28) | All patients (n = 55) | |
|---|---|---|---|
| Age (years) | |||
| Mean (SD) | 35.2 (8.7) | 36.2 (11.4) | 35.7 (10.1) |
| Median (range) | 37.0 (21–51) | 35.5 (18–60) | 36 (18–60) |
| Age category (years), n (%) | |||
| ≤18 | 0 | 2 (7.1) | 2 (3.6) |
| 19–<65 | 27 (100) | 26 (92.9) | 53 (96.4) |
| ≥65 | 0 | 0 | 0 |
| Sex, n (%) | |||
| Male | Female | 13 (48.1) | 14 (51.9) | 13 (46.4) | 15 (53.6) | 26 (47.3) | 29 (52.7) |
| Body mass index (kg/m2) | |||
| Mean (SD) | 25.66 (4.82) | 27.20 (4.32) | 26.45 (4.59) |
| Median (range) | 25.30 (16.9–34.9) | 27.55 (18.5–34.7) | 26.60 (16.9–34.9) |
| Race, n (%) | |||
| Black | 0 | 1 (3.6) | 1 (1.8) |
| White | 27 (100) | 25 (89.3) | 52 (94.5) |
| Other/mixed | 0 | 2 (7.1) | 2 (3.6) |
| Age at epilepsy onset (years)a | |||
| Mean (SD) | 13.10 (9.55) | 9.21 (7.70) | 11.08 (8.77) |
| Median (range) | 13.02 (0–45.0) | 8.93 (0–34.8) | 9.71 (0–45) |
| Epilepsy duration (years) | |||
| Mean (SD) | 22.75 (11.27) | 27.19 (11.81) | 25.01 (11.66) |
| Median (range) | 22.09 (1.5–40.6) | 26.02 (12.0–49.7) | 24.24 (1.5–49.7) |
| History of status epilepticus, n (%) | 3 (11.1) | 3 (10.7) | 6 (10.9) |
| Seizure type, n (%)b | |||
| Focal | 27 (100) | 28 (100) | 55 (100) |
| Focal aware | 10 (37.0) | 8 (28.6) | 18 (32.7) |
| Focal impaired awareness | 22 (81.5) | 27 (96.4) | 49 (89.1) |
| Focal to bilateral | 9 (33.3) | 8 (28.6) | 17 (30.9) |
| Generalized | 0 | 1 (3.6) | 1 (1.8) |
| Unclassified | 1 (3.7) | 1 (3.6) | 2 (3.6) |
| Baseline weekly seizure frequency | |||
| Focal seizures, median (range) | 12.60 (3.0–130.6) | 8.00 (3.2–35.7)c | 8.24 (3.0–130.6) |
| All seizures, median (range) | 15.40 (3.0–130.6) | 8.08 (3.2–214.7) | 10.23 (3.0–214.7) |
| Number of prior AEDsd, n (%) | |||
| <4 | 0 | 0 | 0 |
| 4–5 | 3 (11.1) | 5 (17.9) | 8 (14.5) |
| 6–7 | 3 (11.1) | 3 (10.7) | 6 (10.9) |
| 8–10 | 9 (33.3) | 12 (42.9) | 21 (38.2) |
| >10 | 12 (44.4) | 8 (28.6) | 20 (36.4) |
| Number of concomitant AEDs, n (%) | |||
| 1 | 2 (7.4) | 3 (10.7) | 5 (9.1) |
| 2 | 8 (29.6) | 9 (32.1) | 17 (30.9) |
| 3 | 9 (33.3) | 9 (32.1) | 18 (32.7) |
| 4 | 6 (22.2) | 3 (10.7) | 9 (16.4) |
| ≥5 | 2 (7.4) | 4 (14.2) | 6 (10.9) |
| Active vagus nerve stimulation, n (%) | 10 (37.0) | 5 (17.9) | 15 (27.3) |
AED = antiepileptic drug; SD = standard deviation.
Data are from 25 patients in the placebo group and 27 in the padsevonil group (52 patients overall).
Patients could have reported more than one seizure type.
Data are from 27 patients (54 patients overall).
AEDs started before the date of first dose of trial drug.
Efficacy outcomes
Three patients (11.1%) in the placebo group and eight (30.8%) in the PSL group experienced ≥75% reduction from baseline in focal seizure frequency during the inpatient period [odds ratio 4.14 (95% CI 0.90–19.6), P = 0.068]. PSL was associated with a rapid onset of action, as evidenced by a reduction in seizure frequency starting during Week 1 among patients randomized to treatment with PSL, and similarly in the first week of transitioning to PSL among patients initially randomized to receive placebo (Fig. 5). The secondary outcome, median reduction in weekly focal seizure frequency from baseline during the 2-week inpatient period, was 53.7% in the PSL and 12.5% in the placebo groups, a difference of 34.0% (95% CI 3.0, 67.5; unadjusted P = 0.026) (Fig. 5). At the end of the outpatient period, ≥75% RR was 29.2% among patients in the PSL group and 33.3% among those in the placebo group who had transitioned to PSL. For all patients, ≥75% RR was 31.4% and median percent reduction in seizure frequency was 55.2% (n = 51) at the end of the maintenance period (last week of observation) suggesting maintenance of seizure control for ≥8 weeks post-titration. One patient (3.7%) in the placebo group and two (7.4%) in the PSL group were seizure-free during the inpatient period, but none during the outpatient period.
Figure 5.

Median change in seizure frequency throughout the proof-of-concept trial. Treatment with PSL was associated with a rapid and sharp decline in seizure frequency within a week of initiation. No loss of efficacy was observed over the 8-week maintenance period.
Results of the post hoc analysis showed that during the 2-week inpatient period, in the SV2A subgroup (patients on concomitant LEV or BRV), ≥75%RR was 15.4% among patients on placebo (n = 13) and 0 among those on PSL (n = 11); corresponding values in the non-SV2A subgroup were 7.1% (n = 14) and 53.3% (n = 15). In the SV2A subgroup, median percent reduction in seizure frequency was 5.8% and 29.2% among patients on placebo and PSL, respectively, and 23.2% and 75.2%, respectively, in the non-SV2A subgroup. During the last 4 weeks of outpatient maintenance, ≥75% RR was 25.0% among all patients in the SV2A subgroup (n = 24; 20 LEV and 4 BRV) and 37.0% among all patients in the non-SV2A subgroup (n = 27); median percent reduction in seizure frequency was 49.9% and 55.2%, respectively.
Safety outcomes
During the inpatient period, 17 patients (63.0%) on placebo and 24 (85.7%) on PSL reported TEAEs; none were serious (Table 2). During the overall treatment period, 90.9% of patients reported TEAEs; in most (85.5%) these were considered trial drug-related by the investigator. The most frequently reported TEAEs were somnolence (45.5%), dizziness (43.6%), headache (25.5%), fatigue (23.6%) and irritability (14.5%). Most TEAEs were mild or moderate in intensity (84%); the most frequently reported severe TEAE was somnolence (n = 2, 3.6%). Two patients (3.6%), both receiving PSL, experienced serious TEAEs. One patient experienced status epilepticus during the titration period when transitioning from placebo to PSL; the event subsided after 43 min and the patient continued in the trial without any dose change. The second patient experienced impaired judgement, delirium and dysphoria (grouped as ‘acute psychosis’) during the maintenance period; events resolved with dose reduction and the patient continued in the trial and the OLE. One patient (1.8%) discontinued due to concurrent TEAEs of dysphoria and mood swings. These TEAEs were non-serious, moderate in intensity, considered PSL-related and resolved the following day. The overall incidence of TEAEs, as well as incidences of psychiatric and most frequently reported TEAEs, did not differ among patients on concomitant LEV, BRV or non-SV2A AEDs.
Table 2.
Overview of treatment-emergent adverse events (TEAEs) reported in the proof-of-concept trial
| 2-week inpatient period |
All patients when on padsevonil (n = 55) | ||
|---|---|---|---|
| Placebo (n = 27) | Padsevonil (n = 28) | ||
| Any TEAE, n (%) | 17 (63.0) | 24 (85.7) | 50 (90.9) |
| Serious TEAEs, n (%) | 0 | 0 | 2 (3.6) |
| TEAEs necessitating dose change, n (%) | 0 | 1 (3.6) | 18 (32.7) |
| Discontinuations due to TEAEs, n (%) | 0 | 0 | 1 (0.8) |
| Drug-related TEAEs, n (%) | 14 (51.9) | 21 (75.0) | 47 (85.5) |
| Severe TEAEs, n (%) | 0 | 1 (3.6) | 8 (14.5) |
| TEAEs reported for ≥5% of patients when on padsevonil, n (%) | |||
| Somnolence | 4 (14.8) | 5 (17.9) | 25 (45.5) |
| Dizziness | 4 (14.8) | 2 (7.1) | 24 (43.6) |
| Headache | 4 (14.8) | 4 (14.3) | 14 (25.5) |
| Fatigue | 1 (3.7) | 2 (7.1) | 13 (23.6) |
| Irritability | 1 (3.7) | 1 (3.6) | 8 (14.5) |
| Tremor | 0 | 1 (3.6) | 5 (9.1) |
| Insomnia | 0 | 3 (10.7) | 4 (7.3) |
| Gait disturbance | 1 (3.7) | 2 (7.1) | 4 (7.3) |
| Decreased appetite | 1 (3.7) | 2 (7.1) | 4 (7.3) |
| Constipation | 1 (3.7) | 1 (3.6) | 4 (7.3) |
| Dysarthria | 1 (3.7) | 1 (3.6) | 4 (7.3) |
| Memory impairment | 0 | 1 (3.6) | 4 (7.3) |
| Disturbance in attention | 0 | 0 | 4 (7.3) |
| Nystagmus | 0 | 0 | 4 (7.3) |
| Weight increased | 0 | 0 | 4 (7.3) |
| Hyponatremia | 1 (3.7) | 2 (7.1) | 3 (5.5) |
| Paresthesia | 0 | 1 (3.6) | 3 (5.5) |
| Seizure | 0 | 1 (3.6) | 3 (5.5) |
| Hypotension | 0 | 1 (3.6) | 3 (5.5) |
| Dysmenorrhea | 0 | 1 (3.6) | 3 (5.5) |
| Amnesia | 0 | 0 | 3 (5.5) |
| Focal aware seizure | 0 | 0 | 3 (5.5) |
| Depressed mood | 0 | 0 | 3 (5.5) |
| Nervousness | 0 | 0 | 3 (5.5) |
| Diplopia | 0 | 1 (3.6) | 3 (5.5) |
Mean changes in Brief Psychiatric Rating total scores and Mini-Mental State Examination throughout the trial were small and not deemed clinically relevant. TEAEs in the class of psychiatric disorders were reported for 22 patients (40.0%) throughout the overall treatment period; most frequently irritability (n = 8, 14.5%), insomnia (4, 7.3%), depressed mood (3, 5.5%) and nervousness (3, 5.5%) and mostly mild or moderate in intensity. Two patients reported severe TEAEs; delirium and dysphoria reported by one, as described above, and nightmare reported by another. Overall, there was no evidence for consistent PSL effects on laboratory parameters, vital signs, weight, ECG parameters, echocardiography evaluations, or physical and neurological examinations.
Of the 13 patients who did not enter the OLE, and tapered off PSL, one experienced moderate-to-severe withdrawal symptoms, while the remaining 12 experienced only mild symptoms based on the CIWA–B.
Pharmacokinetic and exposure–response analysis
Based on the popPK model, predicted PSL concentrations following the 400 mg bid dosing regimen would achieve the desired target occupancy for both SV2A and GABAARs (Fig. 6).
Figure 6.

Predicted versus observed PSL plasma concentrations. Predicted values were obtained from a population pharmacokinetic model built on data from the proof-of-concept trial and the PET studies, and indicated that PSL concentrations following the 400 mg bid dosing regimen would allow desired target occupancy for both SV2A and GABAA receptors to be achieved.
The variability in PSL’s PK profile allowed exploration of a PK–response relationship (% of seizure reduction), even though a single PSL dose was tested in the trial. Steady state Cmax (maximum drug concentration during the dosing period), Ctrough (minimum concentration at the end of the dosing period) and AUC (area under the concentration–time curve to the end of the dosing period) were considered in the analysis without showing a clear PK–response relationship (Supplementary Fig. 1). Response variability and small sample size may have precluded detection of a correlation between PSL plasma concentration and seizure reduction.
Discussion
Many AEDs introduced over the past two decades display improved PK and safety/tolerability profiles compared with older AEDs; however, the proportion of treatment-resistant patients appears unchanged (Chen et al., 2018; Devinsky et al., 2018). Patients with treatment resistance account for most of the burden of epilepsy in the population, whether medical, psychosocial or economic (Laxer et al., 2014), representing the population with the largest unmet need. Therefore, a novel approach to AED development is warranted to ensure discovery of AEDs with differentiated efficacy compared with the current standard of care.
Unlike many available AEDs, PSL was developed in a target-based, rational drug design program. Target occupancy and therapeutic dose range, determined in the murine amygdala kindling model, indicated that >90% SV2A and approximately 10% GABAAR occupancy were required to achieve an optimal efficacy/tolerability ratio (Leclercq et al., 2020; Wood et al., 2020). Given the availability of radiotracers, it was possible to establish the PSL dose that would provide similar optimal target engagement in humans though objective measurements in imaging studies. A direct, dose-dependent relationship was observed between PSL plasma concentration and SV2A occupancy. The EC50 was estimated to be 3.1 ng/ml, which is over 10-fold higher than that of BRV (0.46 μg/ml) and over 100-fold higher than that of LEV (4.0 μg/ml) (Finnema et al., 2019). Further simulation projected SV2A occupancy over the entire dosing interval would be >90% for 90% of patients receiving PSL 300 or 400 mg bid. As expected, based on target affinity difference (Wood et al., 2020) there was transient and comparatively low GABAAR coverage in the PET study, indicating that with doses <200 mg GABAAR occupancy may not be quantifiable using conventional detection methods. This does not however, rule out a pharmacological effect; for example, only 3–5% GABAAR occupancy is observed at pharmacologically active doses of lorazepam in humans (Sybirska et al., 1993; Lingford-Hughes et al., 2005), while higher occupancy (20–30%) is typically associated with significant drowsiness (Abadie et al., 1996; Malizia et al., 1996). The lack of relationship between PSL plasma concentration and GABAAR occupancy was most likely due to the limited range explored in the study.
Overall, results of the PET studies showed that optimal PSL occupancy at SV2A and GABAARs associated with efficacy in the kindling model was translatable to humans for both molecular targets, and that the differential proportionality was retained. The 400 mg bid dose was selected for the Phase IIa PoC trial, as it was expected to achieve occupancy levels similar to those associated with robust efficacy in the kindling model; high (>90%), sustained SV2A and 10–15% transient GABAAR occupancy. The latter is in line with clinical experience and results of PET studies showing GABAAR occupancy by benzodiazepines and established tolerability thresholds (Abadie et al., 1996; Malizia et al., 1996). Rational dose selection allowed testing the efficacy of PSL in an efficient PoC trial with a single-dose arm.
The design of the PoC trial differed from most previous Phase II AED trials and included several innovative features to maximize rapid safety and efficacy signal detection in a small population. The trial was conducted in a small number of specialized epilepsy centres to ensure diagnostic accuracy and to minimize variance. Patients were required to have failed ≥4 AED regimens of adequate dose and duration, and to be experiencing high baseline seizure frequency (≥4 focal or focal-to-bilateral tonic-clonic seizures per week), allowing for detection of a meaningful reduction in seizure frequency in a short, 3-week, double-blind, placebo-controlled period. The a priori primary efficacy outcome was the ≥75% RR, which is more stringent than the ≥50% RR typically used in AED trials. The high seizure frequency of the patient population meant that a larger reduction in seizure frequency was deemed necessary for the response to be considered clinically meaningful. Conducting the initial double-blind period of the trial in an inpatient setting allowed drug titration in a controlled environment. The subsequent open-label period, where all patients received PSL, was designed to evaluate maintenance of PSL efficacy and any potential for the development of tolerance to its therapeutic effect. Considering the early stage of development, recruiting an appropriate number of patients that would provide sufficient statistical power to detect efficacy was also a key element. Analysis of previous trials has suggested that AED effects detected within the first weeks of treatment in a relatively small sample are informative and tend to predict outcomes with longer treatment duration (French et al., 2013). The trial was powered to detect a level of efficacy suggestive of superior efficacy vs established AEDs; assumptions on expected response rate with current AEDs and placebo were based on observations from previous UCB clinical trials with patients with similar characteristics as those to be enrolled in the PoC trial.
Patients in the PoC trial were relatively young, had a long disease history and most had been assessed for epilepsy surgery. Compared with previous AED trials, more patients in the current trial had cerebral malformations such as cortical dysplasia or epilepsies arising from frontal than from temporal lobes. Disease severity is also reflected by the large number of failed AEDs (75% of patients had tried ≥8) and high seizure frequency at baseline. Response to therapy has been shown to be inversely proportional to the number of previously tested AEDs (Kwan and Brodie 2000; Schiller and Najjar 2008; Chen et al., 2018), while greater seizure frequency at treatment initiation is associated with poor outcomes and is a strong predictor of drug-resistant epilepsy (Mohanraj and Brodie 2006; Hitiris et al., 2007; Schiller 2009). Patients’ median baseline seizure frequency in the current trial was 40 per 28 days, or 32 for observable focal seizures only, approximately three times higher than that seen in a typical trial population—in the perampanel and BRV trials, the two most recently approved AEDs, corresponding values for focal seizures were 11.3 and 9.1, respectively (Steinhoff et al., 2013; Ben-Menachem et al., 2016). Modelling based on data pooled from BRV Phase III trials indicated that patients with high baseline seizure frequency had a much lower probability of being responders (≥50% RR), estimated to be 54.8% at 0.14 seizures/day (1 seizure/week), and 29.3% at 0.32 seizures/day, corresponding to the median baseline seizure frequency in the trials (Schoemaker et al., 2016). In this model, the probability of response would be four times less likely if the median baseline seizure frequency was 1.14 (as in the current trial) compared with 0.32 seizures/day. This observation highlights the significant impact of baseline seizure frequency on efficacy outcomes and should be taken into account when interpreting the results of the current trial. Based on their clinical characteristics—disease severity, ≥4 AED failures and the large number of concomitant AEDs, including vagus nerve stimulation—many of the patients in the current trial would most likely be excluded from other AED trials.
Results of the PoC trial showed that in this population of patients with severe disease and multiple AED failures, adjunctive therapy with PSL was associated with a clinically meaningful improvement. The difference in ≥75% RR between the PSL and placebo groups approached statistical significance, and there was a notable, nominally significant reduction in weekly seizure frequency. Specific design features of the trial, notably the inpatient setting, could have resulted in a substantial placebo effect (Riley et al., 1981). However, patients who had initially received the placebo also showed improvement after switching to PSL, suggesting that the placebo effect may have been limited. Maintenance of efficacy throughout the 8-week, open-label outpatient period indicated that tolerance to the therapeutic effect of PSL was not observed. Development of tolerance is one of the main limitations associated with long-term exposure to some benzodiazepines—not only is there a progressive reduction in efficacy, necessitating higher dosages to obtain the same therapeutic effects, but discontinuation after prolonged treatment can induce withdrawal (Vinkers and Olivier 2012; Gravielle 2016). In epilepsy, tolerance can result in increased seizure frequency and severity in the presence of a constant maintenance dose, and increased risk of withdrawal seizures (Riss et al., 2008). Therefore, while benzodiazepines such as lorazepam, midazolam and diazepam have potent antiseizure effects, their use is limited to the emergency setting for patients presenting with status epilepticus or seizure clusters who require acute therapeutic intervention (Riss et al., 2008; Ochoa and Kilgo 2016). PSL was designed specifically to bind with low-to-moderate affinity to the benzodiazepine site, displaying a partial agonist profile—such properties can help minimize the potential for tolerance (Serra et al., 1994; Bateson 2002; Rudolph and Knoflach 2011; Vinkers and Olivier 2012). Patients who opted not to enter the OLE were monitored throughout the safety follow-up after having tapered off PSL; 12 of the 13 patients experienced mild-to-moderate withdrawal symptoms based on the CIWA–B, and one experienced moderate-to-severe symptoms. Therefore, the possibility of withdrawal effects cannot be ruled be out, and as with most other centrally acting drugs, PSL requires gradual discontinuation by tapering to decrease the risk of rebound seizure activity and withdrawal effects. PSL was well-tolerated and displayed a favourable safety profile; no new, unexpected safety signals were detected. The most common TEAEs, somnolence, dizziness, headache, fatigue and irritability were consistent with PSL’s mechanism of action. Only one patient (1.8%) discontinued from the trial due to a TEAE.
SV2A is the principal therapeutic target of both LEV and BRV (Matagne et al., 2008). While PSL also binds to SV2A, it displays significantly higher affinity for, and markedly slower dissociation from SV2A, and its interaction is not affected by UCB1244283, a positive allosteric modulator that increases LEV and BRV binding (Wood and Gillard 2017; Wood et al., 2020). Furthermore, PSL interacts with SV2B and SV2C with high affinity (Wood et al., 2020); however, their role in the pathophysiology of epilepsy remains unknown. In effect, it is also not clearly understood how binding to SV2A translates into antiseizure activity; as the proteins are located in synaptic vesicles in neurons, this is likely to be mediated via modulation of neurotransmitter release. Given these current knowledge gaps, the clinical consequences of PSL’s high affinity and long-lasting interaction with SV2A, and with the other two SV2 isoforms, remain to be determined. While results of the post hoc analysis indicated that responder numbers were lower among patients who took LEV or BRV compared with those on other AEDs, it is important to note that this observation was based on very small patient numbers.
In further post hoc analyses, concomitant use of oxcarbazepine, lacosamide, lamotrigine, or valproate, specific clinical features (seizure type, semiology, focus location), disease aetiology and use of benzodiazepines as rescue medication did not appear to influence response to PSL (Van Paesschen et al., 2018). Pharmacokinetic and exposure–response relationship analyses confirmed that the predicted PSL concentrations and related SV2A and GABAAR occupancy were achieved during the trial, validating the selection of the 400 mg bid dose, as a single, optimal dose for hypothesis testing. No concentration response with regard to efficacy and tolerability was detected; however, the small sample size limits the interpretation of the results.
In conclusion, the rational, target-driven approach for designing PSL allowed the adoption of a clinical development program based on target engagement, as determined in non-clinical models predictive of drug-resistant epilepsy, and clinical dose selection based on PET target occupancy studies. The ultimate goal of the drug discovery and development program was to offer patients with severe, treatment-resistant epilepsy—the population with the greatest unmet need—a differentiated therapeutic option in an efficient and expedited manner, circumventing some of the limitations of traditional drug discovery programs.
Funding
All studies included in this report were funded by UCB Pharma.
Supplementary Material
Acknowledgements
The authors express their profound gratitude to the patients who participated in the PoC trial and their families. They also thank Henrik Klitgaard (UCB Pharma, Braine l’Alleud) for his contribution to the overall design and development program. The authors acknowledge the contribution of Jacqueline French (New York University School of Medicine, New York, NY, USA), Joop van Gerven (Centre for Human Drug Research, Leiden, the Netherlands), and Paul Boon (Ghent University Hospital, Ghent, Belgium) to the design of the Phase II trial. They also acknowledge Barbara Pelgrims and Fabien Debailleul (UCB Pharma, Brussels, Belgium) for overseeing the development of the manuscript, and Azita Tofighy (Synthesis, London, UK) for providing writing support, funded by UCB Pharma.
Competing interests
S.D., J.H., R.M.K., B.L., P.M., C.O., D.S., E.W. and K.J.W. are current or former employees of UCB Pharma. CB has received personal compensation from Arvelle, Desitin, Eisai, GW, Idorsia and UCB Pharma for consulting services or speaking activities. M.M. has received financial compensation through the epileptology academic centre of Maastricht UMC for industry-sponsored research activities. BJS has received speaker’s honoraria from Al-Jazeera, Desitin, Eisai, GW, Hikma, Novartis, and UCB Pharma, and has participated in advisory boards for, or has consultancy agreements with Arvelle, B. Braun, Eisai, GW, Idorsia, and UCB Pharma. M.T. has received grants and consultation honoraria from UCB Pharma, EISAI Inc, BIAL laboratories and ESTEVE. K.V.L. received contract research grants through KU Leuven for the conduct of the PET studies reported here. W.V.P. received non-financial support from UCB Pharma during the conduct of the Phase II trial, as well as personal fees and non-financial support for other activities from UCB Pharma, grants from EIT Health and personal fees from Idorsia.
Supplementary material
Supplementary material is available at Brain Communications online.
Glossary
- AED =
antiepileptic drug
- BRV =
brivaracetam
- GABAAR =
GABAA receptor
- LEV =
levetiracetam
- OLE =
open-label extension
- PoC =
proof-of-concept
- PSL =
padsevonil
- RR =
responder rate
- SV2 =
synaptic vesicle protein 2
- TEAE =
treatment-emergent adverse event
References
- Abadie P, Rioux P, Scatton B, Zarifian E, Barré L, Patat A, et al. Central benzodiazepine receptor occupancy by zolpidem in the human brain as assessed by positron emission tomography. Eur J Pharmacol 1996; 295: 35–44. [DOI] [PubMed] [Google Scholar]
- Abadie P, Baron JC, In vivo studies of the central benzodiazepine receptors in the human brain with positron emission tomography In: Diksic M, Reba RC, editors. Radiopharmaceuticals and brain pathology studied with PET and SPECT. Boca Raton: CRC Press; 1990. p. 357–80. [Google Scholar]
- Bateson AN. Basic pharmacologic mechanisms involved in benzodiazepine tolerance and withdrawal. Curr PD 2002; 8: 5–21. [DOI] [PubMed] [Google Scholar]
- Ben-Menachem E, Mameniškienė R, Quarato PP, Klein P, Gamage J, Schiemann J, et al. Efficacy and safety of brivaracetam for partial-onset seizures in 3 pooled clinical studies. Neurology 2016; 87: 314–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurol 2018; 75: 279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denayer T, Stöhr T, van Roy M. Animal models in translational medicine: validation and prediction. New Hor Transl Med 2014; 2: 5–11. [Google Scholar]
- Devinsky O, Vezzani A, O'Brien TJ, Jette N, Scheffer IE, de Curtis M, et al. Epilepsy. Nat Rev Dis Primers 2018; 4: 18024. [DOI] [PubMed] [Google Scholar]
- Engel J. What can we do for people with drug-resistant epilepsy? The 2016 Wartenberg Lecture. Neurology 2016; 87: 2483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnema SJ, Nabulsi NB, Eid T, Detyniecki K, Lin SF, Chen MK, et al. Imaging synaptic density in the living human brain. Sci Transl Med 2016; 8: 348ra96. [DOI] [PubMed] [Google Scholar]
- Finnema SJ, Rossano S, Naganawa M, Henry S, Gao H, Pracitto R, et al. A single‐center, open‐label positron emission tomography study to evaluate brivaracetam and levetiracetam synaptic vesicle glycoprotein 2A binding in healthy volunteers. Epilepsia 2019; 60: 958–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA, Cabrera J, Emir B, Whalen E, Lu F. Designing a new proof-of-principle trial for treatment of partial seizures to demonstrate efficacy with minimal sample size and duration – a case study. Epilepsy Res 2013; 106: 230–6. [DOI] [PubMed] [Google Scholar]
- Gillard M, Fuks B, Leclercq K, Matagne A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties. Eur J Pharmacol 2011; 664: 36–44. [DOI] [PubMed] [Google Scholar]
- Gravielle MC. Activation-induced regulation of GABAA receptors: is there a link with the molecular basis of benzodiazepine tolerance? Pharmacol Res 2016; 109: 92–100. [DOI] [PubMed] [Google Scholar]
- Hitiris N, Mohanraj R, Norrie J, Sills GJ, Brodie MJ. Predictors of pharmacoresistant epilepsy. Epilepsy Res 2007; 75: 192–6. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Gillard M, Leclercq K, Hanon E, Lorent G, Dassesse D, et al. Proepileptic phenotype of SV2A-deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia 2009; 50: 1729–40. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Matagne A, Leclercq K, Gillard M, Michel P, Kenda B, et al. SV2A protein is a broad-spectrum anticonvulsant target: functional correlation between protein binding and seizure protection in models of both partial and generalized epilepsy. Neuropharmacology 2008; 54: 715–20. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000; 342: 314–9. [DOI] [PubMed] [Google Scholar]
- Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav 2014; 37: 59–70. [DOI] [PubMed] [Google Scholar]
- Leclercq K, Matagne A, Provins L, Klitgaard H, Kaminski RM. Pharmacological profile of the antiepileptic drug candidate padsevonil – characterization in rodent seizure and epilepsy models. J Pharmacol Exp Ther 2020; 372: 11–20. [DOI] [PubMed] [Google Scholar]
- Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 2005; 79: 241–57. [DOI] [PubMed] [Google Scholar]
- Lingford-Hughes A, Wilson SJ, Feeney A, Grasby PG, Nutt DJ. A proof-of-concept study using [11C]flumazenil PET to demonstrate that pagoclone is a partial agonist. Psychopharmacology (Berl) 2005; 180: 1–91. [DOI] [PubMed] [Google Scholar]
- Löscher W, Klitgaard H, Twyman RE, Schmidt D. New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov 2013; 12: 757–76. [DOI] [PubMed] [Google Scholar]
- Löscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011; 20: 359–68. [DOI] [PubMed] [Google Scholar]
- Löscher W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res 2016; 126: 157–84. [DOI] [PubMed] [Google Scholar]
- Löscher W. The search for new screening models of pharmacoresistant epilepsy: is induction of acute seizures in epileptic rodents a suitable approach? Neurochem Res 2017; 42: 1926–38. [DOI] [PubMed] [Google Scholar]
- Malizia AL, Gunn RN, Wilson SJ, Waters SH, Bloomfield PM, Cunningham VJ, et al. Benzodiazepine site pharmacokinetic/pharmacodynamic quantification in man: direct measurement of drug occupancy and effects on the human brain in vivo. Neuropharmacology 1996; 35: 1483–91. [DOI] [PubMed] [Google Scholar]
- Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H. Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol 2008; 154: 1662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melhem M. Translation of central nervous system occupancy from animal models: application of pharmacokientic/pharmacodynamic modeling. J Pharmacol Exp Ther 2013; 347: 2–6. [DOI] [PubMed] [Google Scholar]
- Mohanraj R, Brodie MJ. Diagnosing refractory epilepsy: response to sequential treatment schedules. Eur J Neurol 2006; 13: 277–82. [DOI] [PubMed] [Google Scholar]
- Nabulsi NB, Mercier J, Holden D, Carre S, Najafzadeh S, Vandergeten M-C, et al. Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med 2016; 57: 777–84. [DOI] [PubMed] [Google Scholar]
- Ochoa JG, Kilgo WA. The role of benzodiazepines in the treatment of epilepsy. Curr Treat Options Neurol 2016; 18: 18. [DOI] [PubMed] [Google Scholar]
- Otoul C, Watling M, Tytgat D, Colzi A, Andersen G, Bani M, Padsevonil Single and Multiple Rising Dose Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics in Healthy Volunteers. American Epilepsy Society (AES) Annual Meeting Abstract Database. AESnet.org; 2017.
- Riley TL, Porter RJ, White BG, Penry JK. The hospital experience and seizure control. Neurology 1981; 31: 912–5. [DOI] [PubMed] [Google Scholar]
- Riss J, Cloyd J, Gates J, Collins S. Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurol Scand 2008; 118: 69–86. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. Brivaracetam: a rational drug discovery success story. Br J Pharmacol 2008; 154: 1555–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABA receptor subtypes. Nat Rev Drug Discov Disc 2011; 10: 685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundfeldt C, Löscher W. The pharmacology of imepitoin: the first partial benzodiazepine receptor agonist developed for the treatment of epilepsy. CNS Drugs 2014; 28: 29–43. [DOI] [PubMed] [Google Scholar]
- Schiller Y, Najjar Y. Quantifying the response to antiepileptic drugs: effect of past treatment history. Neurology 2008; 70: 54–65. [DOI] [PubMed] [Google Scholar]
- Schiller Y. Seizure relapse and development of drug resistance following long-term seizure remission. Arch Neurol 2009; 66: (:1233–9. [DOI] [PubMed] [Google Scholar]
- Schoemaker R, Wade JR, Stockis A. Brivaracetam population pharmacokinetics and exposure-response modeling in adult subjects with partial-onset seizures. J Clin Pharmacol 2016; 56: 1591–1602. [DOI] [PubMed] [Google Scholar]
- Serra M, Ghiani CA, Motzo C, Luisa Perceddu M, Biggio G. Long-term treatment with abecarnil fails to induce tolerance in mice. Eur J Pharmacol 1994; 259: 1–6. [DOI] [PubMed] [Google Scholar]
- Steinhoff BJ, Ben-Menachem E, Ryvlin P, Shorvon S, Kramer L, Satlin A, et al. Efficacy and safety of adjunctive perampanel for the treatment of refractory partial seizures: a pooled analysis of three phase III studies. Epilepsia 2013; 54: 1481–9. [DOI] [PubMed] [Google Scholar]
- Strzelczyk A, Griebel C, Lux W, Rosenow F, Reese JP. The burden of severely drug-refractory epilepsy: a comparative longitudinal evaluation of mortality, morbidity, resource use, and cost using German Health Insurance Data. Front Neurol 2017; 8: 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sybirska E, Seibyl P, Bremner JD, Baldwin RM, Al-Tikriti MS, Bradberry C, et al. [123I]iomazenil SPECT imaging demonstrates significant benzodiazepine receptor reserve in human and nonhuman primate brain. Neuropharmacology 1993; 32: 671–80. [DOI] [PubMed] [Google Scholar]
- Thijs RD, Surges R, O'Brien TJ, Sander JW. Epilepsy in adults. Lancet 2019; 393: 689–701. [DOI] [PubMed] [Google Scholar]
- Van Paesschen W, et al. Dissecting the Heterogeneous Clinical Response to Padsevonil in Adults with Drug-resistant Focal Seizures: Post Hoc Responder Analysis of a Proof-of-Concept Trial. American Epilepsy Society (AES) Annual Meeting Abstract Database. AESnet.org; 2018.
- Vinkers CH, Olivier B. Mechanisms underlying tolerance after long-term benzodiazepine use: a future for subtype-selective GABAA receptor modulators? Adv Pharmacol Sci 2012; 2012: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver DF, Pohlmann Eden B. Pharmacoresistant epilepsy: unmet needs in solving the puzzle (s). Epilepsia 2013; 54: 80–5. [DOI] [PubMed] [Google Scholar]
- Wood MD, Gillard M. Evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia 2017; 58: 255–62. [DOI] [PubMed] [Google Scholar]
- Wood M, Daniels V, Provins L, Wolff C, Kaminski RM, Gillard M. Pharmacological profile of the antiepileptic drug candidate padsevonil – interactions with synaptic vesicle 2 proteins and GABAA receptors. J Pharmacol Exp Ther 2020; 372: 1–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Due to the small volunteer numbers in the PET studies, Individual Patient Data (IPD) cannot be adequately anonymized, and therefore, cannot be shared. Data from the Phase II trial may be requested by qualified researchers 6 months after product approval in the USA and/or Europe, or global development is discontinued. Investigators may request access to anonymized IPD and redacted documents including raw and analysis-ready datasets, protocol, case report forms, statistical analysis plan, dataset specifications and clinical study report. Before access is granted, proposals must be approved by an independent review panel at www.clinicalstudydatarequest.com and a signed data sharing agreement must be in place. All documents are available for a pre-specified time, typically 12 months, on a password-protected portal.