

Abstract
Amyotrophic lateral sclerosis is characterized by the degeneration of upper and lower motor neurons, yet an increasing number of studies in both mouse models and patients with amyotrophic lateral sclerosis suggest that altered metabolic homeostasis is also a feature of disease. Pre-clinical and clinical studies have shown that modulation of energy balance can be beneficial in amyotrophic lateral sclerosis. However, the capacity to target specific metabolic pathways or mechanisms requires detailed understanding of metabolic dysregulation in amyotrophic lateral sclerosis. Here, using the superoxide dismutase 1, glycine to alanine substitution at amino acid 93 (SOD1G93A) mouse model of amyotrophic lateral sclerosis, we demonstrate that an increase in whole-body metabolism occurs at a time when glycolytic muscle exhibits an increased dependence on fatty acid oxidation. Using myotubes derived from muscle of amyotrophic lateral sclerosis patients, we also show that increased dependence on fatty acid oxidation is associated with increased whole-body energy expenditure. In the present study, increased fatty acid oxidation was associated with slower disease progression. However, within the patient cohort, there was considerable heterogeneity in whole-body metabolism and fuel oxidation profiles. Thus, future studies that decipher specific metabolic changes at an individual patient level are essential for the development of treatments that aim to target metabolic pathways in amyotrophic lateral sclerosis.
Keywords: amyotrophic lateral sclerosis, hypermetabolism, skeletal muscle, glucose oxidation, fatty acid oxidation
In superoxide dismutase 1, glycine to alanine substitution at amino acid 93 (SOD1G93A) mice, increased whole-body metabolism occurs alongside fat depletion and increased fatty acid oxidation in glycolytic muscle. Myotubes from patients with amyotrophic lateral sclerosis have increased fatty acid oxidation, and this is associated with increased whole-body energy expenditure. Increased fatty acid oxidation may sustain energy supply to slow disease.
Graphical Abstract
Graphical Abstract.

Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that is characterized by the degeneration of motor neurons in the brain, brainstem and spinal cord. The progressive loss of neurons in ALS results in muscle denervation, weakness and paralysis. Death usually occurs within 3–5 years from diagnosis (Brown and Al-Chalabi, 2017). ALS is variable in terms of age of onset, site of symptom onset and rate and pattern of disease progression. Underlying the clinical variability of ALS is complexity of genetic contribution and the pathogenic pathways that lead to the death of neurons (Brown and Al-Chalabi, 2017; Ghasemi and Brown, 2018).
While multiple pathogenic mechanisms, including neuronal hyperexcitability (Vucic et al., 2008; Kiernan, 2009), glutamate excitotoxicity (Rothstein et al., 1992), protein aggregation (Arai et al., 2006) and oxidative stress (Blasco et al., 2017), are proposed to contribute to the pathogenesis of ALS, studies also indicate that metabolic dysfunction is associated with disease progression (Dupuis et al., 2004; Ahmed et al., 2016; Ioannides et al., 2016). In human ALS, an increase in resting energy expenditure (i.e. hypermetabolism) has been observed in approximately one-third to more than half of all patients (Desport et al., 2001, 2005; Bouteloup et al., 2009; Funalot et al., 2009; Vaisman et al., 2009; Jesus et al., 2018; Steyn et al., 2018a). More recently, we have shown that hypermetabolism in patients with ALS is associated with lower motor neuron dysfunction, more rapid disease progression and increased risk of earlier death (Steyn et al., 2018a). Despite this evidence of the importance of metabolism, the capacity to target metabolism to slow disease progression is restricted by our limited understanding of the underlying biochemistry of altered metabolism in ALS.
Previous studies in superoxide dismutase 1, glycine to alanine substitution at amino acid 93 (SOD1G93A) and superoxide dismutase 1, glycine to arginine substitution at amino acid 86 (SOD1G86R) mouse models of ALS suggest that altered metabolic balance is a feature of the disease (Dupuis et al., 2004), with early and persistent perturbations in the blood levels of metabolic hormones such as leptin, the presence of circulating ketone bodies and increased energy expenditure (Dupuis et al., 2004). Reports of increased peripheral clearance of circulating lipids (Fergani et al., 2007) and increased expression of fatty acid metabolism genes in glycolytic muscle (Palamiuc et al., 2015) suggest that altered skeletal muscle metabolism could contribute to the altered metabolism observed in ALS. However, whether this altered profile of gene expression is associated with functional changes in glucose–fatty acid metabolism in the muscle of ALS mice, and how this might relate to metabolic changes at the whole-body level remains to be determined. Moreover, whether similar metabolic changes occur in the skeletal muscle of ALS patients and the relevance of these changes to the clinical and metabolic presentations in human ALS is unknown.
In this study, we aimed to address this gap in knowledge by characterizing whole-body metabolism in the SOD1G93A mouse model of ALS throughout the course of disease, and by assessing energy substrate utilization in intact glycolytic muscle fibres isolated from SOD1G93A mice when compared to wild-type (WT) controls. We also assessed fatty acid and glucose oxidation in myotubes derived from well characterized ALS patients and healthy controls to determine whether substrate metabolism in ALS muscle is linked to the energy expenditure profiles and clinical features. We report evidence of a functional alteration in glucose–fatty flux in glycolytic muscle of SOD1G93A mice. In ALS patient-derived myotubes, changes in glucose and fatty acid oxidation are associated with whole-body energy expenditure and disease progression, but not hypermetabolism.
Materials and methods
Animal studies
Mice
Experiments at the University of Queensland were approved by the University of Queensland Animal Ethics Committee and conducted in accordance with the Queensland Government Animal Care and Protection Act 2001, associated Animal Care and Protection Regulations (2002 and 2008) and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (2004). Experiments at the University of Notre Dame were conducted in accordance with the Freimann Life Science Center Guidelines and the Notre Dame Institutional Animal Care and Use Committee Policy on Humane Endpoints for animal use.
Transgenic mice overexpressing the human SOD1 G93A mutation [B6-Cg-Tg (SOD1-G93A) 1Gur/J] (Gurney et al., 1994) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and bred on a C57 black 6 (C57BL/6J) background. Male SOD1G93A mice and litter- or age-matched WT control mice (n = 3–26/group) were randomly assigned for experiments at ages that correspond to pre-defined stages of disease in SOD1G93A mice; presymptomatic (5 weeks of age, no symptoms), onset (9–11 weeks of age, early signs of hindlimb tremor and weakness), mid-stage (16–19 weeks of age, pronounced hindlimb weakness) and end-stage (21–25 weeks of age, significant hindlimb weakness leading to paralysis and euthanasia due to loss of the righting reflex) (Ngo et al., 2012; Lee et al., 2013). In line with ethical requirements, SOD1G93A mice were monitored twice daily after the onset of pronounced hindlimb weakness (i.e. mid-stage of disease). All mice were group-housed (3–4 mice per cage) in filter top cages, or in individually ventilated cages when maintained in a specific pathogen-free environment. For the assessment of energy expenditure, mice were single-housed in cages designed specifically for the Phenomaster metabolic phenotyping system (TSE-Systems, Bad Homburg, Germany). Mice were maintained on a 12 h light, 12 h dark cycle (ON at 0600 h and OFF at 1800 h) and had free access to food (20% protein, 4.8% fat; Specialty Feeds, WA, Australia) and water. Room temperature was maintained at 22 ± 2°C. Assessments in live mice or collection of tissue from mice occurred between 1000 and 1400 h on any given day. Prior to tissue collection, all mice were anaesthetized with an intraperitoneal injection of sodium pentobarbitone (32.5 mg/kg, Virbac Animal Health, NSW, Australia). Following complete loss of the pedal withdrawal reflex and eye-blink reflex, mice were killed by cervical dislocation. All animal work was conducted in accordance with the Animal Research: Reporting of In Vivo Experiments guidelines (Kilkenny et al., 2010).
EchoMRI assessment of fat and fat free mass
Imaging took place in the Notre Dame Integrated Imaging Facility Friemann Life Sciences Center. Whole-body fat and fat free mass was measured with an EchoMRI-130™ Quantitative Magnetic Resonance (EchoMRI, TX, USA) (Metzinger et al., 2014). Two X-ray images were taken at different energy levels to assess soft tissue density and bone. Mass measurements for fat and fat free tissue were produced for all scans in the EchoMRI Body Composition Analyzer EMR-184 software (EchoMRI).
Indirect calorimetry
Energy expenditure was measured with a Phenomaster open-circuit indirect calorimetry system housed within a temperature (22°C) and 12 h light, 12 h dark cycle (ON at 0600 h and OFF at 1800 h) controlled chamber (TSE-Systems), as we have done previously (Steyn et al., 2018b). All mice were acclimated for single-housing before collection of individualized measures using the Phenomaster system. Experimental cages (n = 16) were sampled at 60 min intervals for 3.5 min/cage, with concentrations of O2 and CO2 in the outgoing air being measured sequentially within each interval. One vacant cage was included to obtain a reference concentration for ambient gas. Activity (x- and y-plane), food intake and body weight were recorded synchronously with metabolic data. Measurements were performed continuously over 72 h, with analysis restricted to the final 24 h assessment period (allowing 48 h of acclimation). For data analysis, measures of total energy expenditure and food intake were adjusted for body weight. Food intake measures are representative of hourly food intake for each mouse.
18F-deoxyglucose PET/single-photon emission computed tomography/CT imaging
Animals were anaesthetized using isoflurane inhalation for 2–5 min in a vaporized-controlled tank to allow sufficient anaesthesia followed by recovery. Animal breathing was checked once every minute by visual inspection. 18F-deoxyglucose at a dose of 0.200mCI activity was administered to mice intravenously through the tail vein (volume < 100 μl). PET images were acquired on a trimodal Alibra PET/single-photon emission computed tomography/CT image station (Carestream Health, Woodbridge, CT, USA) to produce high‐resolution PET images that were reconstructed for analysis. 18F-deoxyglucose uptake in brown adipose tissue (BAT) was quantified as the mean voxel value within a visually determined volume of interest as described previously (van der Veen et al., 2012).
Ex vivo lipolysis
Epididymal white adipose tissue (WAT) was excised and rinsed in 1× phosphate buffered saline supplemented with 0.1% fatty acid-free bovine serum albumin. All epididymal WAT explants were placed in plastic vials containing 1 ml of modified Krebs–Henseleit buffer (in mM): 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4⋅7H2O, 1.25 CaCl2⋅2H2O, 25 NaHCO3, 5 glucose, 118 NaCl and 4% fatty acid-free bovine serum albumin. Buffer was gassed with 95% O2/5% CO2 for 45 min to reach a pH of 7.4. All procedures were conducted in a shaking water bath at 37°C. For the assessment of non-esterified fatty acids (NEFA), 6 µl of buffer was collected at 0-, 30-, 60-, 90- and 120-min time points, placed immediately on dry ice and stored at −80°C. Samples were assayed on a NEFA-C kit (Wako Chemicals, Osaka, Japan). For glycerol, buffer was collected after 2 h of incubation. Glycerol content was assessed using a free glycerol determination kit with glycerol standard solution (Sigma-Aldrich, MO, USA). Final glycerol content and NEFA-C levels were expressed relative to the weight of the respective epididymal WAT explant.
Plasma NEFA
Following sacrifice, terminal blood samples were collected from mice via cardiac puncture. Samples were transferred into ethylenediaminetetraacetic acid-precoated tubes and centrifuged for 3 min. Plasma was aliquoted and stored at −80°C until use. NEFA levels in plasma were determined using a NEFA-C test kit (Wako Chemicals).
Oil Red O staining
Extensor digitorum longus (EDL) muscles were embedded in optimum cutting temperature compound and rapidly frozen in liquid nitrogen cooled isopentane. Oil Red O (ORO) staining was performed on muscle cryosections (10 μm) to visualize neutral lipids using an ORO kit (Abcam, Cambridge, UK). Briefly, sections were fixed with 10% neutral buffered formalin for 15 min and rinsed three times with distilled water for 30 s at room temperature. ORO solution was added onto the sections and incubated for 10 min, and the slides were differentiated in 85% propylene glycol solution for 1 min at room temperature. After two rinses, slides were air dried and mounted with aqueous mounting agent (Aquatex, EMD Millipore, CA, USA). Bright-field images (20× magnification) were taken with an Aperio ScanScope system (Leica, Mannheim, Germany). ORO labelling was quantified using 10+ randomly selected sections of muscle per animal (n = 5 per group). Representative images of the muscle section were processed using ImageJ to identify the mean ORO intensity for each animal, following thresholding to remove non-specific labelling [threshold set to 225 (0–255)].
Assessment of cellular respiration in muscle fibre bundles
EDL muscle fibre bundles were chemically dissociated as previously described (Li et al., 2016). Muscle fibre bundles were seeded onto Seahorse XFe96 microplates in culture media (low glucose Dulbecco’s Modified Eagle Medium supplemented with 10% foetal bovine serum and 1% Antibiotic-Antimycotic; ThermoFisher, MA, USA) and maintained overnight at 37°C with 5% CO2. Prior to the commencement of metabolic assays, muscle fibre viability was assessed using an alamarBlue cell viability assay (ThermoFisher) and used for the data normalization. Real-time assessment of bioenergetic parameters in EDL fibre bundles was performed on the XFe96 Extracellular Flux Analyzer (Agilent Technologies, CA, USA).
The dependence and capacity of EDL fibres to use glucose and fatty acid as fuel substrates were determined using the Seahorse XF Mito Fuel Flex Test Kit (Agilent Technologies). Prior to the assay, culture media was replaced with pre-warmed assay media (pH 7.4) consisting of extracellular flux base media (Agilent Technologies), 10 mM d-glucose (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich) and 2 mM l-glutamine (ThermoFisher). Carnitine palmitoyltransferase 1A inhibitor etomoxir (ETO, 4.0 μM), mitochondrial pyruvate carrier inhibitor UK5099 (2.0 μM) and glutaminase inhibitor BPTES (3.0 μM) were prepared with assay media and loaded into the XFe96 sensor cartridge following the manufacturer guidelines. Following the first three cycles of baseline measurement of oxygen consumption rate (OCR), the decrease of OCR levels upon inhibition of one or more pathways was continuously recorded for the following six cycles. Each cycle consisted of 3 min mix, 30 s wait and 3 min measurement. The dependence and capacity of each fuel pathway relative to total fuel oxidation were calculated according to the manufacturer guidelines.
Substrate induced maximal respiration was tested in EDL fibre bundles as previously described (Li et al., 2016). Briefly, plates containing muscles fibres were changed into assay media (pH 7.4) containing 120 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgCl2, 2.5 mM d-glucose and 0.5 mM l-carnitine prior to the assay run. OCR was continuously measured for six cycles after sequential injections of sodium pyruvate (10 mM) or palmitate-bovine serum albumin (100 μM) with carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (0.4 μM), followed with antimycin/rotenone (1 μM).
Patient studies
Subjects
Eighteen ALS patients who met the revised El-Escorial criteria for ALS (Brooks et al., 2000) were enrolled from the Royal Brisbane and Women’s Hospital ALS clinic for the collection of skeletal muscle biopsies. Eleven healthy control participants were also enrolled. These control individuals were the spouses, friends or family members of ALS participants. For all participants, exclusion criteria were history of a metabolic condition (e.g. Hashimoto’s disease) and diabetes mellitus. Participant details are shown in Table 1 and Supplementary Table 1. For ALS patients, the ALS functional rating scale-revised (ALSFRS-R) score, King’s stage and deltaFRS were obtained from clinical records. All participants provided written informed consent; participant consent was obtained according to the Declaration of Helsinki. Work performed in this study was approved by the Royal Brisbane and Women’s Hospital and University of Queensland human research ethics committees.
Table 1.
Characteristics of ALS patients and healthy controls at the time of muscle biopsy collection
| Control (n = 11) | ALS (n = 18) | P | |
|---|---|---|---|
| Demographics | |||
| Age (years) | 58.9 ± 10.1 | 55.4 ± 7.2 | 0.277 |
| Sex, female | 3 (27.27) | 4 (22.2) | 0.758 |
| Anthropometric and metabolic measures | |||
| Body mass index (kg/m2) | 25.9 ± 2.7 | 26.6 ± 4.2 | 0.644 |
| Fat free mass (kg) | 53.4 ± 10.5 | 54.7 ± 10.8 | 0.745 |
| Fat mass (%) | 31.1 ± 9.4 | 32.4 ± 11.5 | 0.759 |
| EEkc (kcal/day) | 1596 ± 331.1 | 1809 ± 336.2 | 0.107 |
| MI (%) | 108.4 ± 15.41 | 119.5 ± 9.6 | 0.025 |
| Clinical scores | |||
| Time since onset (months) | 29.2 ± 21.5 | ||
| ALSFRS-R | 36.7 ± 6.1 | ||
| deltaFRS | −0.5 ± 0.3 | ||
| King’s stage | 2.4 ± 1.0 | ||
| FVC (% of predicted) | 88.5 ± 22.1 | ||
Data presented as mean (SD) and n (%) for categorical data. Means were compared by independent t-test and proportions with the chi-square test.
ALSFRS-R = ALS functional rating scale-revised; deltaFRS = change in ALSFRS-R since symptom onset; EEkc = energy expenditure kilocalories; FVC = forced vital capacity; MI = metabolic index (measured EEkc/predicted EEkc).
Assessment of body composition and energy expenditure
Body composition (fat mass and fat free mass) was determined by whole-body air displacement plethysmography using the BodPod system (Cosmed USA, Rome, Italy) (Ioannides et al., 2017). Values of fat mass and fat free mass were used to predict resting energy expenditure (Steyn et al., 2018a). Energy expenditure in fasting and alert ALS and control participants at rest (i.e. resting energy expenditure) was then measured by indirect calorimetry using a Quark RM respirometer (Cosmed) as per our established methodology (Steyn et al., 2018a). Controls were matched to patients with ALS by age, sex, weight, body mass index and body composition. The metabolic index (MI) of each individual was derived by calculating measured resting energy expenditure as a percentage of predicted resting energy expenditure. An MI (i.e. measured energy expenditure relative to predicted resting energy expenditure) ≥120% was defined as hypermetabolism, as we have done previously (Steyn et al., 2018a).
Muscle biopsy and culture
Muscle biopsies were collected from the vastus lateralis of one leg. Lignocaine (1%; 5 ml) was injected to anaesthetize the skin and underlying fat and muscle tissue. A 10-mm incision was made and advanced through the fascia of the muscle. A ∼200 mg sample of muscle was collected using a sterile 6-mm hollow Bergstrom biopsy needle (Pelomi, Albertslund, Denmark) modified for suction (Tarnopolsky et al., 2011) and placed in holding media (Dulbecco’s Modified Eagle Medium/Ham’s F-12 with 0.5% gentamicin; ThermoFisher).
Primary myoblasts were isolated and cultured using a modified muscle tissue explant method (Tarnopolsky et al., 2011) and frozen as low passage cell stocks. Primary myogenic cells were maintained in Dulbecco’s Modified Eagle Medium/Ham’s F-12 medium supplemented with 20% foetal bovine serum, 10% AmnioMAX C-100 and 0.5% gentamicin (ThermoFisher). Culture media was changed every second day and cells were passaged when they reached ∼70% confluence. For experiments, primary myogenic cells were seeded at a density of 15 000 cells/well into Seahorse XFe96 cell culture microplates. At 80% confluence, cells were differentiated into myotubes by replacing maintenance media with differentiation media consisting of Dulbecco’s Modified Eagle Medium/Ham’s F-12 medium with 2% non-inactivated horse serum and 0.5% gentamicin (ThermoFisher). Fresh differentiation medium was fed every 2 days until mature multinucleated myotubes formed. Primary myotubes underwent assessment of key parameters of glucose and fatty acid oxidation dependency and capacity using the Seahorse Mito Fuel Flex Test Kit (Agilent Technologies). Parameters of glycolytic flux (glycolysis, glycolytic capacity and glycolytic reserve) were determined using the Seahorse XF Glycolysis Stress Test Kit (Agilent Technologies).
Statistical analysis
Statistical differences were assessed using Prism 8.0a. (GraphPad Software Inc., La Jolla, CA, USA). Statistical comparisons were performed following testing for normal distribution, and unless otherwise indicated, outcomes were compared using unpaired Student’s t-test, non-parametric t-test or two-way ANOVA followed by Bonferroni multiple comparison. Percentage data were log transformed prior to statistical comparison. Linear relationships were assessed by Pearson correlation. All graphical data are presented as mean ± SD. Values of P < 0.05 were considered to be statistically significant.
Data availability
The data from this study are available from the corresponding author, upon reasonable request.
Results
Disease progression in SOD1G93A mice is associated with a decline in both fat free mass and fat mass
To determine the impact of disease progression on body composition and total body weight in SOD1G93A mice, we conducted a serial assessment of body weight, and whole-body fat free mass and fat mass by EchoMRI (Fig. 1A–C). Total body weight in SOD1G93A mice was significantly lower than that of litter-matched WT controls by 19 weeks of age (Fig. 1A). Loss of body weight was due, in part, to a loss in total fat free mass (Fig. 1B). Tibialis anterior and gastrocnemius weight were reduced by disease onset (Fig. 1D and E), and EDL mass was reduced by the mid-stage of disease (Fig. 1F). When compared to age-matched WT controls, fat accumulation in SOD1G93A mice slowed following disease onset, and whole-body fat mass was significantly lower from 15 weeks of age (Fig. 1C). Epididymal and inguinal fat mass was lower in SOD1G93A mice from the mid-stage of disease (Fig. 1G and H). Consistent with this, circulating levels of leptin were stable in SOD1G93A mice throughout the disease course while the WT controls showed increased levels of leptin with time (Fig. 1I).
Figure 1.

Body weight, fat free mass and fat mass decreases in SOD1G93A mice over the course of disease. (A) Total body weight, (B) fat free mass and (C) fat mass in SOD1G93A mice and WT age-matched controls (n = 9–10/group). Total (D) tibialis anterior (TA), (E) gastrocnemius (Gastroc) and (F) EDL muscle mass in SOD1G93A mice and age-matched WT controls (n = 10–26/group). Total weight of (G) epididymal white adipose tissue (eWAT) and (H) inguinal white adipose tissue (ingWAT) in SOD1G93A mice and age-matched WT controls (n = 5–12/group). (I) Circulating levels of leptin in SOD1G93A mice and age-matched WT controls (n = 5–6/group). White circles represent WT mice; teal circles represent SOD1G93A transgenic mice. All data presented as mean ± SD. *P < 0.05, two-way ANOVA with Bonferroni’s post hoc test. ES = end-stage; MS = mid-stage; OS = onset; PS = presymptomatic.
Total energy expenditure in SOD1G93A mice increases by the mid-stage of disease
We next sought to determine whether energy expenditure in SOD1G93A mice increases relative to disease progression. When compared to WT controls, SOD1G93A mice at the mid-stage of disease had higher levels of energy expenditure during the light and dark cycle, resulting in an overall increase in 24 h total energy expenditure (Fig. 2A and B). SOD1G93A mice had decreased activity by disease onset (Fig. 2C and D).
Figure 2.

Increased energy expenditure occurs in parallel with increased food intake in symptomatic SOD1G93A mice. (A) Representative data trace of oxygen consumption over 24 h in SOD1G93A mice and WT litter-matched controls. (B) Average VO2 consumption in these mice during the light and dark cycle, and over a 24-h period. (C) Representative data trace of daily activity over 24 h. (D) Average total activity during the light and dark cycle, and over a 24-h period in SOD1G93A mice and WT litter-matched controls. (E) Representative data trace of food intake over 24 h. (F) Average food intake during the light and dark cycle, and over a 24-h period in SOD1G93A mice and WT litter-matched controls. Black lines and white circles represent WT mice; teal lines and circles represent SOD1G93A transgenic mice. Data presented as mean ± SD for n = 8/group. *P < 0.05, two-way ANOVA with Bonferroni’s post hoc test. MS = mid-stage; OS = onset; PS = presymptomatic.
We also investigated whether reductions in body weight and fat mass in SOD1G93A mice were due to a decline in food intake. We observed no change in food intake between SOD1G93A mice at disease onset and litter-matched WT controls. SOD1G93A mice at the mid-stage of disease consumed more food than litter-matched WT controls (Fig. 2E and F). Thus, reductions in weight are not associated with decreased food consumption, and despite an increase in food intake and a decline in activity-dependent energy expenditure, SOD1G93A mice are unable to offset increased energy expenditure to prevent the depletion of energy stores.
Lipolytic rate is maintained in SOD1G93A mice
BAT regulates non-shivering thermogenesis, which itself can contribute to total energy expenditure in mice (Even and Blais, 2016). We found no difference in BAT weight (Fig. 3A) or glucose uptake in BAT between SOD1G93A and WT control mice (Fig. 3B and C). Thus, the increase in energy expenditure in SOD1G93A at the mid-stage of disease is unlikely to be due to increased non-shivering thermogenesis.
Figure 3.

Lipolysis is maintained throughout disease course in SOD1G93A mice. (A) BAT weight (n = 5–12/group) and (B, C) glucose uptake in BAT in SOD1G93A mice and age-matched WT controls (n = 9–11/group). (D) Levels of NEFA-C in Krebs–Henseleit buffer as an indicator of lipolytic rate of epididymal white adipose tissue explants from SOD1G93A mice and WT controls (n = 6–17/group). (E) Lipolytic rate expressed as a per cent of that observed at PS stage. (F) Cumulative NEFA (n = 6–17/group) and glycerol (n = 8–17/group) in Krebs–Henseleit buffer. (G) Circulating plasma NEFA in SOD1G93A mice and WT age-matched controls (n = 12/group). White circles represent WT mice; teal circles represent SOD1G93A transgenic mice. All data presented as mean ± SD. *P < 0.05, two-way ANOVA with Bonferroni’s post hoc test. ES = end-stage; MS = mid-stage; OS = onset; PS = presymptomatic.
WAT stores lipids, providing energy reserves that are available during periods of increased energy demand. Given the slowing of accumulation and eventual reduction of fat mass in SOD1G93A mice, we assessed the release of lipids (a proxy measure for lipolysis) from epididymal WAT explants. The lipolytic rate of ex vivo explants of epididymal WAT decreased over the lifespan of WT mice, while the lipolytic rate in SOD1G93A mice was maintained throughout the course of disease. The lipolytic rate in SOD1G93A mice was significantly higher by the mid-stage of disease when compared to WT controls (Fig. 3D and E). This corresponded to a sustained rate of appearance of cumulative NEFA and glycerol from WAT explants (Fig. 3F). Circulating plasma NEFA in WT and SOD1G93A mice did not differ (Fig. 3G). Overall, results suggest that the rate of lipolysis in epididymal WAT explants from SOD1G93A is sustained throughout disease, and that it does not decrease with age, as is seen in WT mice.
SOD1G93A mice exhibit a functional preference for fat oxidation in glycolytic EDL muscle
Previously, a decrease in the expression of glucose handling genes and an increase in the expression of lipid handling genes in glycolytic muscle of SOD1G86R mice have been suggested to drive a switch towards the use of lipid as an energy substrate (Palamiuc et al., 2015). Thus, sustained lipolysis in SOD1G93A mice could serve to mobilize fatty acids for use as an energy source in skeletal muscle. We used ORO staining to quantify intramuscular lipid accumulation in the glycolytic EDL muscle and conducted real-time assessment of substrate utilization in EDL muscle fibre bundles isolated from SOD1G93A and age-matched WT mice. There was no difference in intramuscular lipid content in the EDL muscle of SOD1G93A mice when compared to WT controls (Fig. 4A and B). We observed no difference in glucose oxidation dependence (Fig. 4C and D), and following the inhibition of mitochondrial fatty acid and glutamine uptake, EDL muscle fibres from SOD1G93A mice exhibited similar levels of glucose oxidation capacity across all disease stages (Fig. 4E and F). In contrast to the decline in fat oxidation and dependence in WT mouse skeletal muscle fibres with age, we observed sustained dependence on fat oxidation in isolated EDL muscle fibre bundles from SOD1G93A mice by the mid-stage of disease (Fig. 4G and H). Moreover, the capacity for fat oxidation to sustain mitochondrial respiration after inhibition of pyruvate and glutamine entry into mitochondria was also increased in EDL muscle fibres bundles from SOD1G93A mice at the mid-stage of disease when compared to WT controls, which exhibited a decline in fat oxidation capacity with age (Fig. 4I and J).
Figure 4.

A functional shift in mitochondrial fuel preference from glucose to fat occurs in glycolytic EDL muscle of SOD1G93A mice. (A) ORO staining and (B) quantification of intramuscular lipid in the glycolytic EDL muscle of SOD1G93A and age-matched WT mice (n = 5/group), scale bar = 50 μm. (C) Data trace of mitochondrial OCR (% baseline) contributed to by glucose oxidation dependence. (D) Quantification of glucose oxidation dependence in isolated EDL muscle fibre bundles from SOD1G93A mice and WT controls. (E) Data trace of mitochondrial OCR (% baseline) contributed to by glucose oxidation capacity. (F) Quantification of glucose oxidation capacity in isolated EDL muscle fibre bundles from SOD1G93A mice and WT controls. (G) Data trace of mitochondrial OCR (% baseline) contributed to by fat oxidation dependence. (H) Quantification of fat oxidation dependence in SOD1G93A mice and WT control mice. (I) Data trace of mitochondrial OCR (% baseline) contributed to by fat oxidation capacity. (J) Quantification of fat oxidation capacity in SOD1G93A mice and WT controls. White circles represent WT mice; teal circles represent SOD1G93A transgenic mice. All data presented as mean ± SD for n = 3–13/group. *P < 0.05, two-way ANOVA with Bonferroni’s post hoc test. MS = mid-stage; OS = onset; PS = presymptomatic.
To determine the capacity of glucose and fatty acid oxidation pathways to sustain mitochondrial respiration in the absence of substrate competition, and to study substrate utilization in the presence of increased energy demand, we measured the capacity of mitochondria in EDL fibre bundles to oxidize pyruvate or palmitate in the presence of the mitochondrial uncoupler, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (Fig. 5A and B). The basal OCR of EDL muscles from WT and SOD1G93A mice was similar (Fig. 5C). In the presence of pyruvate, we observed a significant elevation in maximal peak OCR in EDL muscle fibre bundles from SOD1G93A mice at the mid-stage stage of disease (Fig. 5A). In the presence of palmitate, we observed no difference in maximal OCR in EDL muscle fibre bundles between SOD1G93A and WT controls (Fig. 5B). Compared to WT mice, total oxygen consumption in the EDL muscle fibres from SOD1G93A mice was significantly higher at the mid-stage of disease when pyruvate was provided as the external energy substrate (Fig. 5D). However, oxygen consumption between EDL muscle fibres from SOD1G93A mice and WT mice was comparable when palmitate-bovine serum albumin was provided as the external energy substrate (Fig. 5E). Thus, despite exhibiting a functional preference towards fatty acid oxidation, glycolytic EDL muscle fibres from SOD1G93A mice are capable of utilizing glucose metabolism pathways to sustain mitochondrial function when there are no competing fatty acid substrates.
Figure 5.

Glycolytic EDL muscle fibre bundles from SOD1G93A mice are capable of utilizing glucose metabolism pathways. (A) Data traces of mitochondrial OCR (% of baseline) when carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) was used to induce maximal mitochondrial respiration in the presence of (A) pyruvate (PY) or (B) palmitate (PA), followed by inhibition of the mitochondrial electron transport chain complexes III and I by antimycin a (AA) and rotenone (ROT). (C) Quantification of basal OCR in EDL muscle fibre bundles from SOD1G93A mice and WT age-matched control mice. (D) Quantification of area under the curve (AUC) of OCR in muscle fibre bundles from SOD1G93A and WT control mice in the presence of FCCP and pyruvate. (E) Quantification of AUC of OCR in muscle fibre bundles from SOD1G93A and WT controls in the presence of FCCP and palmitate. White circles represent WT mice; teal circles represent SOD1G93A transgenic mice. All data presented as mean ± SD for n = 5–11/group. *P < 0.05, two-way ANOVA with Bonferroni’s post hoc test. MS = mid-stage; OS = onset; PS = presymptomatic.
ALS patient-derived myotubes have increased dependence on fat oxidation
We next aimed to determine whether the preferential use of lipids by mitochondria of SOD1G93A mice was also present in human ALS subjects. We generated primary myotubes from skeletal muscle from ALS patients and age-matched healthy controls and conducted real-time assessment of substrate utilization. Demographics of our study population are detailed in Table 1. Sex, age and clinical demographics of ALS patients are detailed in Supplementary Table 1. When compared to myotubes derived from healthy controls, myotubes from patients with ALS had similar levels of glucose oxidation dependency and capacity (Fig. 6A). ALS patient-derived myotubes also had similar levels of fat oxidation capacity compared to myotubes derived from healthy controls. However, they exhibited an increased dependence on fat oxidation (Fig. 6B).
Figure 6.

Myotubes derived from patients with ALS have increased dependence on fat oxidation. (A) Data traces and quantification of mitochondrial OCR (% baseline) contributed to by glucose oxidation dependency and capacity in ALS and control myotubes. (B) Data traces and quantification of mitochondrial OCR (% baseline) contributed to by fat oxidation dependency and capacity in ALS and control myotubes. (C) Data trace of extracellular acidification rate, and quantification of glycolysis, glycolytic capacity and glycolytic reserve in ALS and control myotubes. All data presented as mean ± SD for n = 11 control and n = 18 ALS individuals. *P < 0.05, Unpaired t-test. Individuals with a C9orf72 expansion are noted in grey, and an individual with a SOD1 mutation is noted in blue.
The transition between glucose and fatty acid oxidation can ultimately impact the flux of metabolic substrates through their respective pathways. We therefore sought to determine whether the increase in fat oxidation dependence in ALS patient-derived myotubes was associated with altered glycolytic function. We found no difference in glycolysis, glycolytic capacity or glycolytic reserve between myotubes derived from control individuals and myotubes derived from patients with ALS (Fig. 6C).
We next assessed the relationships between substrate oxidation and glycolytic function in myotubes and the metabolic and clinical characteristics of our ALS cohort. The metabolic measures were resting energy expenditure and the MI, which we have previously used to define hypermetabolism in ALS patients (Supplementary Table 2) (Steyn et al., 2018a). We found that glucose oxidation capacity and glycolytic reserve were negatively correlated with resting energy expenditure, and that fat oxidation dependency and capacity, as well as glycolysis were greater in myotubes derived from ALS patients with higher resting energy expenditure. Substrate oxidation was not correlated with the MI.
We observed no relationship between substrate oxidation or glycolytic function in ALS patient-derived myotubes relative to the severity of disability as determined by the ALS functional rating scale-revised at the time of metabolic assessment (Supplementary Table 2). However, glucose oxidation capacity was greater, and fat oxidation dependence was lower in patients with a more rapidly progressing disease [indicated by a faster decline in ALS functional rating scale-revised scores (deltaFRS); Supplementary Table 2 and Fig. 7]. Collectively, these data indicate that substrate utilization in myotubes is not related to hypermetabolism but is linked to the resting energy expenditure of ALS patients. Moreover, our data also suggest that substrate utilization in myotubes is associated with the rate of functional decline in patients with ALS.
Figure 7.
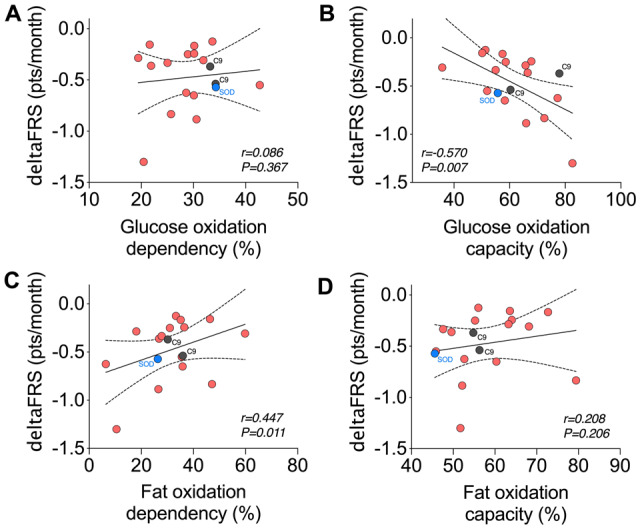
Rate of functional decline in patients with ALS is linked to substrate utilization in myotubes derived from respective donors. Correlation analyses between rate of disease progression (deltaFRS) and (A) glucose oxidation dependency, (B) glucose oxidation capacity, (C) fat oxidation dependency and (D) fat oxidation capacity. DeltaFRS, rate of functional decline as defined by (ALS function rating scale-revised score-48)/disease duration (in months) from symptom onset. Data presented as Pearson correlations for n = 18 ALS patients. Individuals with a C9orf72 expansion are noted in grey, and an individual with a SOD1 mutation is noted in blue.
Discussion
The primary aim of this study was to explore the potential mechanisms underpinning altered whole-body metabolic balance in ALS (Ahmed et al., 2018; Steyn et al., 2018a; Vandoorne et al., 2018). We show that an increase in whole-body energy expenditure in symptomatic SOD1G93A mice is associated with a decline in both fat mass and fat free mass. We also find that glycolytic muscle from symptomatic SOD1G93A mice exhibit an increased dependence on fatty acids as an energy substrate, as well as an increased capacity to utilize fatty acids. In myotubes derived from patients with ALS, we show a similar association between dependence on fatty acid oxidation and resting energy expenditure. Further there was an association between muscle cellular metabolism and rate of disease progression.
Compared to WT littermates, SOD1G93A mice exhibited an increase in total whole-body oxygen consumption (a proxy for energy expenditure) alongside a concomitant decline in body mass, fat mass and fat free mass over the course of disease. These observations are in line with the notion that increased energy expenditure in ALS contributes to weight loss (Dupuis et al., 2011). However, given that hypermetabolism has not been shown to be associated with weight loss in human ALS (Steyn et al., 2018a), these divergent observations between ALS mice and patients suggest that multiple factors are likely to contribute to weight loss in patients with ALS. These include reduced capacity to meet energy requirements (Ngo et al., 2017) [including loss of appetite resulting in loss of fat mass (Ngo et al., 2019; Mezoian et al., 2020)], as well as neurogenic wasting (Al-Sarraj et al., 2014).
A failure to accumulate fat mass was an early feature of disease in SOD1G93A mice. When compared to WT mice, where the lipolytic rate of WAT explants decreased with age, the rate of lipolysis of WAT explants from SOD1G93A mice was sustained over the course of disease. The sustained rate of lipolysis by disease mid-stage corresponded to greater energy needs, as indicated by the higher total energy expenditure of SOD1G93A mice at this time. That circulating levels of fatty acids (NEFA) were not increased in SOD1G93A mice, even though WAT explants were primed for lipolysis, would suggest that lipids that entered circulation were rapidly utilized. It is well established that lipolysis and fatty acid mobilization are upregulated in response to increased muscle energy requirements, for example, during exercise (Goodpaster and Sparks, 2017). Thus, increased mobilization of lipids in ALS mice presumably occurs as a means to sustain metabolic requirements in peripheral skeletal muscle, as has been suggested previously (Dupuis et al., 2004; Fergani et al., 2007). Expanding on previous reports of increased expression of genes associated with fatty acid oxidation in skeletal muscle of SOD1G86R mice (Palamiuc et al., 2015), we have now generated the first evidence to show that there is a greater functional dependence and capacity of glycolytic skeletal muscle from symptomatic SOD1G93A mice to utilize fatty acids as a fuel substrate. This is in contrast to what is observed in WT mice, where fat oxidation declined from 5 weeks of age through to 20–25 weeks of age. This decline in fat oxidation is in line with, and likely to be secondary to our observations of decreased lipolytic rate of WT epididymal WAT explants and stable levels of plasma NEFA, indicating that the metabolic demand of skeletal muscle in WT mice is possibly lower than that of SOD1G93A mice. Although the increased dependence and capacity for fatty acid oxidation in SOD1G93A mice could be an adaptive response to muscle metabolic demand, it might also occur due to reduced muscle glucose uptake and glucose intolerance (Pradat et al., 2010; Desseille et al., 2017). Yet, we found that glycolytic muscle fibres were able to utilize pyruvate, a product of glucose oxidation, following inhibition of fatty acid and glutamine pathways. While providing evidence to suggest that glucose oxidation mechanisms remain intact in glycolytic SOD1G93A muscle, our data lend further support to the notion that increased utilization of fatty acids could inhibit the use of glucose in ALS (Palamiuc et al., 2015), presumably via the Randle cycle; a biochemical process that is characterized by the competition of fatty acids and glucose as cellular energy substrates (Hue and Taegtmeyer, 2009). Indeed, under uncoupling, we found that EDL muscle fibres from both WT and SOD1G93A mice were capable of using either pyruvate or palmitate to sustain energy demand. Interestingly, maximal respiration through consumption of pyruvate was higher in SOD1G93A mice at mid-stage of disease when compared to WT mice. These data demonstrate that pyruvate oxidation also appears to remain intact in glycolytic muscle of SOD1G93A mice, and that when in the absence of a competing fatty acid substrate (i.e. palmitate), glycolytic muscle fibres from SOD1G93A mice will utilize pyruvate to sustain function under increased energy demand.
In order to understand whether the metabolic changes observed in isolated SOD1G93A mouse muscle fibres occur in human ALS, we investigated substrate oxidation capacity and dependence in myotubes derived from patients with ALS. We also extended our studies to assess glycolytic function in human myotubes. Similar to SOD1G93A mouse muscle, we observed an increase in fatty acid oxidation dependence in ALS patient myotubes, although fatty acid oxidation capacity remained unchanged. Myotubes with higher fat oxidation dependence and capacity were derived from ALS patients with higher resting energy expenditure. These results are congruent with the known ability for skeletal muscle to adapt to changes in energy supply and requirement to maintain energy homeostasis (Horowitz, 2003). It is well known that fatty acid metabolism provides more ATP when compared to glucose metabolism (Turner et al., 2014). As such, an increase in the need for fatty acid oxidation in myotubes obtained from patients with higher resting energy expenditure may function to increase ATP availability. Moreover, the correlation observed between resting energy expenditure and fatty acid oxidation dependence aligns with physiological processes whereby continued dependence on fatty acid metabolism requires greater levels of oxygen consumption when compared to glucose metabolism (Turner et al., 2014). Interestingly, myotubes derived from ALS patients with higher resting energy expenditure also had higher glycolysis, but lower glucose oxidation capacity. The finding of increased glycolysis is in agreement with previous studies demonstrating higher activity of phosphofructokinase, the rate limiting enzyme of glycolysis, in skeletal muscles from individuals with higher resting energy expenditure (Zurlo et al., 1994). This higher level of glycolysis likely explains the negative correlation between resting energy expenditure and glycolytic reserve. High levels of glycolysis indicate that the glycolytic activity of a cell is close to the theoretical maximum, and therefore, there is reduced capacity to respond to increased energy demand through this pathway. The negative correlation between resting energy expenditure and glucose oxidation capacity is paradoxical, given that glycolysis is the pathway through which glucose is oxidized. However, this observation can be explained by the known biochemical interplay of the Randle cycle. Fatty acid oxidation inhibits glucose utilization through strong inhibition of pyruvate dehydrogenase, and to a lesser extent, phosphofructokinase (Hue and Taegtmeyer, 2009). Pyruvate dehydrogenase inhibition leads to pyruvate accumulation, which in turn activates lactate dehydrogenase to produce increased levels of lactate. As measurements of extracellular acidification rate via the Seahorse are mainly attributed to the production of lactic acid per unit time following its conversion from pyruvate (TeSlaa and Teitell, 2014), increased glycolysis could represent a higher degree of pyruvate dehydrogenase inhibition by the increase in fatty acid oxidation.
We did not observe a relationship between the MI or any substrate oxidation parameters in ALS patient myotubes. We assume that the findings in myotubes reflect the situation in intact muscle. Hypermetabolism in ALS could arise from other tissues such as the brain. Previous investigations using [18F]fluorodeoxyglucose positron emission tomography supports such a hypothesis, as increased energy use has been observed across a number of brain regions in ALS patients (Cistaro et al., 2012). In ALS patients who show hypermetabolism, the metabolic changes could reflect a whole-body response to disease. Indeed, hypermetabolism is a feature of critical illness, wherein the degree of hypermetabolism varies with the severity and duration of illness (McClave and Snider, 1994).
We found that myotubes with higher fatty acid oxidation dependence were derived from patients with slower clinical decline, indicating that substrate availability and/or use could be a factor that determines disease progression in ALS. Previous studies linking higher levels of serum lipids and fat mass with longer survival (Marin et al., 2011; Lindauer et al., 2013; Huang et al., 2015) provide some evidence to suggest that lipids are beneficial in ALS. This is further supported by recent clinical observations demonstrating that high fat dietary supplementation is able to slow disease progression and extend survival in patients with more rapid progressing disease (Ludolph et al., 2020).
The cause for the change in fuel substrate preference towards fatty acids in skeletal muscle remains unknown. An interesting observation in this study was the functional change in fuel oxidation preference at the symptomatic stage of disease in SOD1G93A mice—this finding is in contrast to work published by Palamiuc et al. who identified gene expression changes suggestive of a similar preference for fat oxidation in asymptomatic SOD1G86R mice (Palamiuc et al., 2015). The differences observed between the two SOD1 mouse models are curious and could be due to the more aggressive disease course in SOD1G86R mice. It is plausible that the timing in the change in preference for fatty acid oxidation is associated with alterations in global metabolic homeostasis (which are apparent in asymptomatic SOD1G86R mice) that results in cellular bioenergetic deficit (Ngo and Steyn, 2015; Loeffler et al., 2016; Vandoorne et al., 2018), and this in turn might subsequently lead to the utilization of fatty acids as an initial adaptive response to yield more ATP than glucose to sustain cellular function and survival. As disease progresses, persistent fatty acid oxidation would likely become maladaptive, with increased production of reactive oxygen species and lipotoxicity exacerbating the degenerative process (Aon et al., 2014; Tracey et al., 2018). If this were the case, could modulation of glucose–fatty acid flux offer therapeutic benefit in ALS? It has previously been shown that dichloroacetate, a synthetic halogenated organic acid that promotes glucose oxidation and pyruvate oxidation, improves function and survival in SOD1 mouse models of ALS (Miquel et al., 2012; Palamiuc et al., 2015). Moreover, the direct targeting of glycolysis or glycolysis associated pathways in skeletal muscle might also show promise. This approach is supported by observations that promotion of glycolytic function in motor neurons improves locomotor function in a transactive response DNA binding protein 43 (TDP-43) fly model of ALS (Manzo et al., 2019), and that pre-supplementation of chromosome 9 open reading frame 72 (C9orf72) induced astrocytes with inosine improves survival of motor neurons by increasing glycolytic flux (Allen et al., 2019).
The biochemical mechanisms that underpin the increased dependence for fatty acid oxidation in skeletal muscle also remain to be determined. It is well known that substrate competition between glucose and fatty acids at the mitochondrial outer membrane is dictated by the activities of the pyruvate dehydrogenase complex and the carnitine palmitoyl transferase system/complexes, and that the interaction between these two complexes is influenced by nutrient availability (Sugden and Holness, 1994). In ALS, an increase in energy expenditure that outstrips nutrient supply might result in negative energy balance, and a switch in substrate preference from glucose to fatty acids through altered pyruvate dehydrogenase and carnitine palmitoyl transferase activity. Future, in-depth and longitudinal assessment of the complex interactions between glucose and fatty acid metabolism pathways is needed to decipher the mechanisms that modulate the adaptation of ALS skeletal muscle towards higher fatty acid oxidation.
Although we have used both mouse and human-derived models to investigate aspects of muscle metabolism in ALS, there are some limitations to our study. First, the SOD1G93A mouse, albeit an accepted pre-clinical model of ALS, is representative of only a small proportion of ALS patients. However, we found similar alterations in substrate oxidation in the myotubes derived from some ALS patients, indicating that muscle metabolic changes are possibly present widely in the disease. Second, we matched measures in SOD1G93A mice with measures in WT control mice rather than measures in mice overexpressing wild-type human SOD1 (WTSOD1). Similar to mutant human SOD1G93A, the overexpression of WTSOD1 in motor neuron-like cells is associated with altered glycolytic function (Richardson et al., 2013; Valbuena et al., 2016), although, to our knowledge, the impact of WTSOD1 on fatty acid metabolism remains to be investigated. Thus, detailed metabolic characterization of WTSOD1 mice is needed if we are to generate further insight into the degree to which mutant human SOD1 alters glucose–fatty acid flux when compared to WTSOD1. Third, while we conducted assessment of energy expenditure in ALS patients using indirect calorimetry (Haugen et al., 2007), and assessed substrate oxidation in real-time using Seahorse technology, our use of primary human myotubes is a caveat. Previously, it has been shown that human primary myotubes mirror the metabolic phenotypes of their donors (Ukropcova et al., 2005). Regardless, as primary myotubes are grown from cells, which have been isolated from muscle biopsies that were removed from their physiological milieu, future studies assessing substrate oxidation in muscle in vivo are needed.
In summary, we demonstrate that an increase in fatty acid oxidation dependence and capacity occurs in glycolytic muscle of SOD1G93A mice during the symptomatic stages of disease when they exhibit increased energy expenditure. In myotubes derived from ALS patients, a similar increase in fatty acid oxidation dependence occurs. While this change in fatty acid oxidation appears to be associated with the progression of disease, it is not linked to hypermetabolism in human ALS. Given the heterogeneity of disease in ALS, there remains an important need for further studies that delineate mechanisms of metabolic imbalance and the link between substrate utilization and energy expenditure throughout the course of the disease. A comprehensive understanding of specific metabolic changes at an individual patient level will be essential for the development of treatments that aim to target metabolic pathways in ALS.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
The authors would like to thank all ALS patients and control participants who took part in the study. We gratefully acknowledge the assistance and support of staff at the University of Queensland Biological Resources (UQBR) and The Centre for Integrated Physiology at the School of Biomedical Sciences, University of Queensland. The authors acknowledge the valuable funding of Wesley Medical Research (Project No. 2016-32).
Funding
This work was funded by grants from The University of Queensland (NSRFS 2012002126 to S.T.N.), Motor Neurone Disease Research Australia (Grant-in-aid, Graham Smith MND Research Grant to F.J.S., P.A.M. and S.T.N. and Charcot MND Research Grant to F.J.S., J.S.C., P.A.M., R.D.H. and S.T.N.), the National Health and Medical Research Council (1101085 and 1185427 to F.J.S. and S.T.N. and 1121962 to F.C.G.) and Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica and Association Française contre les Myopathies-Telethon (HyperALS and AFM-Telethon #22509 to A.F., J.-P.L., F.R. and C.V.). R.L. and T.Y.X. were supported by international postgraduate research scholarships from the Australian Government and University of Queensland centennial scholarships. T.J.T. was funded via a University of Queensland Research Training Program Scholarship and a Motor Neurone Disease Research Australia PhD Scholarship Top-up Grant. S.T.N. acknowledges funding via a Motor Neurone Disease Research Australia Bill Gole Fellowship (2012–15) and the Scott Sullivan MND Research Fellowship (Queensland Brain Institute, the Royal Brisbane & Women’s Hospital Foundation, The MND and Me Foundation; 2015–20). S.T.N. acknowledges current support through a FightMND Mid-Career Research Fellowship and the Australian Institute for Bioengineering and Nanotechnology.
Competing interests
The authors have no conflicts of interest to declares.
Glossary
- ALS =
amyotrophic lateral sclerosis
- BAT =
brown adipose tissue
- C9orf72 =
chromosome 9 open reading frame 72
- EDL =
extensor digitorum longus
- MI =
metabolic index
- NEFA =
non-esterified fatty acids
- OCR =
oxygen consumption rate
- ORO =
oil Red O
- SOD1G86R =
superoxide dismutase 1, glycine to arginine substitution at amino acid 86
- SOD1G93A =
superoxide dismutase 1, glycine to alanine substitution at amino acid 93
- WAT =
white adipose tissue
- WT =
wild-type
- WTSOD1 =
wild-type human SOD1
References
- Ahmed RM, Dupuis L, Kiernan MC. Paradox of amyotrophic lateral sclerosis and energy metabolism. J Neurol Neurosurg Psychiatry 2018; 89: 1013–4. [DOI] [PubMed] [Google Scholar]
- Ahmed RM, Irish M, Piguet O, Halliday GM, Ittner LM, Farooqi S, et al. Amyotrophic lateral sclerosis and frontotemporal dementia: distinct and overlapping changes in eating behaviour and metabolism. Lancet Neurol 2016; 15: 332–42. [DOI] [PubMed] [Google Scholar]
- Allen SP, Hall B, Castelli LM, Francis L, Woof R, Siskos AP, et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019; 142: 586–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sarraj S, King A, Cleveland M, Pradat PF, Corse A, Rothstein JD, et al. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol Commun 2014; 2: 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Bhatt N, Cortassa SC. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol 2014; 5: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006; 351: 602–11. [DOI] [PubMed] [Google Scholar]
- Blasco H, Garcon G, Patin F, Veyrat-Durebex C, Boyer J, Devos D, et al. Panel of oxidative stress and inflammatory biomarkers in ALS: a pilot study. Can J Neurol Sci 2017; 44: 90–5. [DOI] [PubMed] [Google Scholar]
- Bouteloup C, Desport JC, Clavelou P, Guy N, Derumeaux-Burel H, Ferrier A, et al. Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol 2009; 256: 1236–42. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 293–9. [DOI] [PubMed] [Google Scholar]
- Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med 2017; 377: 162–72. [DOI] [PubMed] [Google Scholar]
- Cistaro A, Valentini MC, Chio A, Nobili F, Calvo A, Moglia C, et al. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging 2012; 39: 251–9. [DOI] [PubMed] [Google Scholar]
- Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrere B, et al. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr 2001; 74: 328–34. [DOI] [PubMed] [Google Scholar]
- Desport JC, Torny F, Lacoste M, Preux PM, Couratier P. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis 2005; 2: 202–7. [DOI] [PubMed] [Google Scholar]
- Desseille C, Deforges S, Biondi O, Houdebine L, D’Amico D, Lamaziere A, et al. Specific Physical exercise improves energetic metabolism in the skeletal muscle of amyotrophic-lateral- sclerosis mice. Front Mol Neurosci 2017; 10: 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Oudart H, Rene F, de Aguilar J-LG, Loeffler J-P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A 2004; 101: 11159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Pradat PF, Ludolph AC, Loeffler JP. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol 2011; 10: 75–82. [DOI] [PubMed] [Google Scholar]
- Even PC, Blais A. Increased cost of motor activity and heat transfer between non-shivering thermogenesis, motor activity, and thermic effect of feeding in mice housed at room temperature - implications in pre-clinical studies. Front Nutr 2016; 3: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergani A, Oudart H, Gonzalez De Aguilar J-L, Fricker B, René F, Hocquette J-F, et al. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J Lipid Res 2007; 48: 1571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funalot B, Desport JC, Sturtz F, Camu W, Couratier P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2009; 10: 113–7. [DOI] [PubMed] [Google Scholar]
- Ghasemi M, Brown RH., Jr Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med 2018; 8: a024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab 2017; 25: 1027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994; 264: 1772–5. [DOI] [PubMed] [Google Scholar]
- Haugen HA, Chan LN, Li F. Indirect calorimetry: a practical guide for clinicians. Nutr Clin Pract 2007; 22: 377–88. [DOI] [PubMed] [Google Scholar]
- Horowitz JF. Fatty acid mobilization from adipose tissue during exercise. Trends Endocrinol Metab 2003; 14: 386–92. [DOI] [PubMed] [Google Scholar]
- Huang R, Guo X, Chen X, Zheng Z, Wei Q, Cao B, et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: a study from south-west China and a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener 2015; 16: 359–65. [DOI] [PubMed] [Google Scholar]
- Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 2009; 297: E578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannides ZA, Ngo ST, Henderson RD, McCombe PA, Steyn FJ. Altered metabolic homeostasis in amyotrophic lateral sclerosis: mechanisms of energy imbalance and contribution to disease progression. Neurodegener Dis 2016; 16: 382–97. [DOI] [PubMed] [Google Scholar]
- Ioannides ZA, Steyn FJ, Henderson RD, McCombe PA, Ngo ST. Anthropometric measures are not accurate predictors of fat mass in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2017; 18: 486–491. [DOI] [PubMed] [Google Scholar]
- Jesus P, Fayemendy P, Nicol M, Lautrette G, Sourisseau H, Preux PM, et al. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur J Neurol 2018; 25: 97–104. [DOI] [PubMed] [Google Scholar]
- Kiernan MC. Hyperexcitability, persistent Na+ conductances and neurodegeneration in amyotrophic lateral sclerosis. Exp Neurol 2009; 218: 1–4. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010; 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, et al. Dysregulation of the complement cascade in the hsod1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 2013; 10: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Steyn FJ, Stout MB, Lee K, Cully TR, Calderon JC, et al. Development of a high-throughput method for real-time assessment of cellular metabolism in intact long skeletal muscle fibre bundles. J Physiol 2016; 594: 7197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindauer E, Dupuis L, Muller HP, Neumann H, Ludolph AC, Kassubek J. Adipose tissue distribution predicts survival in amyotrophic lateral sclerosis. PLoS One 2013; 8: e67783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler JP, Picchiarelli G, Dupuis L, Gonzalez De Aguilar JL. The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol 2016; 26: 227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludolph AC, Dorst J, Dreyhaupt J, Weishaupt JH, Kassubek J, Weiland U, et al. Effect of high-caloric nutrition on survival in amyotrophic lateral sclerosis. Ann Neurol 2020; 87: 206–16. [DOI] [PubMed] [Google Scholar]
- Manzo E, Lorenzini I, Barrameda D, O’Conner AG, Barrows JM, Starr A, et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife 2019; 8: e45114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin B, Desport JC, Kajeu P, Jesus P, Nicolaud B, Nicol M, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry 2011; 82: 628–34. [DOI] [PubMed] [Google Scholar]
- McClave SA, Snider HL. Understanding the metabolic response to critical illness: factors that cause patients to deviate from the expected pattern of hypermetabolism. New Horiz 1994; 2: 139–46. [PubMed] [Google Scholar]
- Metzinger MN, Miramontes B, Zhou P, Liu Y, Chapman S, Sun L, et al. Correlation of X-ray computed tomography with quantitative nuclear magnetic resonance methods for pre-clinical measurement of adipose and lean tissues in living mice. Sensors 2014; 14: 18526–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezoian T, Belt E, Garry J, Hubbard J, Breen CT, Miller L, et al. Loss of appetite in amyotrophic lateral sclerosis is associated with weight loss and decreased calorie consumption independent of dysphagia. Muscle Nerve 2020; 61: 230–4. [DOI] [PubMed] [Google Scholar]
- Miquel E, Cassina A, Martinez-Palma L, Bolatto C, Trias E, Gandelman M, et al. Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PLoS One 2012; 7: e34776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo ST, Steyn FJ. The interplay between metabolic homeostasis and neurodegeneration: insights into the neurometabolic nature of amyotrophic lateral sclerosis. Cell Regen 2015; 4: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo ST, Baumann F, Ridall PG, Pettitt AN, Henderson RD, Bellingham MC, et al. The relationship between Bayesian motor unit number estimation and histological measurements of motor neurons in wild-type and SOD1G93A mice. Clin Neurophysiol 2012; 123: 2080–91. [DOI] [PubMed] [Google Scholar]
- Ngo ST, Mi JD, Henderson RD, McCombe PA, Steyn FJ. Exploring targets and therapies for amyotrophic lateral sclerosis: current insights into dietary interventions. Degener Neurol Neuromuscul Dis 2017; 7: 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo ST, van Eijk RPA, Chachay V, van den Berg LH, McCombe PA, Henderson RD, et al. Loss of appetite is associated with a loss of weight and fat mass in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2019; 20: 497–505. [DOI] [PubMed] [Google Scholar]
- Palamiuc L, Schlagowski A, Ngo ST, Vernay A, Grosch S, Henriques A, et al. A metabolic switch towards lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med 2015; 7: 526–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradat PF, Bruneteau G, Gordon PH, Dupuis L, Bonnefont-Rousselot D, Simon D, et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2010; 11: 166–71. [DOI] [PubMed] [Google Scholar]
- Richardson K, Allen SP, Mortiboys H, Grierson AJ, Wharton SB, Ince PG, et al. The effect of SOD1 mutation on cellular bioenergetic profile and viability in response to oxidative stress and influence of mutation-type. PLoS One 2013; 8: e68256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 1992; 326: 1464–8. [DOI] [PubMed] [Google Scholar]
- Steyn FJ, Ioannides ZA, van Eijk RPA, Heggie S, Thorpe KA, Ceslis A, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry 2018. a; 89: 1016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyn FJ, Ngo ST, Chen VP, Bailey-Downs LC, Xie TY, Ghadami M, et al. 17alpha-estradiol acts through hypothalamic pro-opiomelanocortin expressing neurons to reduce feeding behavior. Aging Cell 2018. b; 17: e12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Interactive regulation of the pyruvate dehydrogenase complex and the carnitine palmitoyltransferase system. FASEB J 1994; 8: 54–61. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Pearce E, Smith K, Lach B. Suction-modified Bergstrom muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve 2011; 43: 716–25. [DOI] [PubMed] [Google Scholar]
- TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Meth Enzymol 2014; 542: 91–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci 2018; 11: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Cooney GJ, Kraegen EW, Bruce CR. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J Endocrinol 2014; 220: T61–79. [DOI] [PubMed] [Google Scholar]
- Ukropcova B, McNeil M, Sereda O, de Jonge L, Xie H, Bray GA, et al. Dynamic changes in fat oxidation in human primary myocytes mirror metabolic characteristics of the donor. J Clin Invest 2005; 115: 1934–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman N, Lusaus M, Nefussy B, Niv E, Comaneshter D, Hallack R, et al. Do patients with amyotrophic lateral sclerosis (ALS) have increased energy needs? J Neurol Sci 2009; 279: 26–9. [DOI] [PubMed] [Google Scholar]
- Valbuena GN, Rizzardini M, Cimini S, Siskos AP, Bendotti C, Cantoni L, et al. Metabolomic analysis reveals increased aerobic glycolysis and amino acid deficit in a cellular model of amyotrophic lateral sclerosis. Mol Neurobiol 2016; 53: 2222–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen DR, Shao J, Chapman S, Leevy WM, Duffield GE. A diurnal rhythm in glucose uptake in brown adipose tissue revealed by in vivo PET-FDG imaging. Obesity (Silver Spring) 2012; 20: 1527–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandoorne T, De Bock K, Van Den Bosch L. Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol 2018; 135: 489–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008; 131: 1540–50. [DOI] [PubMed] [Google Scholar]
- Zurlo F, Nemeth PM, Choksi RM, Sesodia S, Ravussin E. Whole-body energy metabolism and skeletal muscle biochemical characteristics. Metabolism 1994; 43: 481–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data from this study are available from the corresponding author, upon reasonable request.