

Abstract
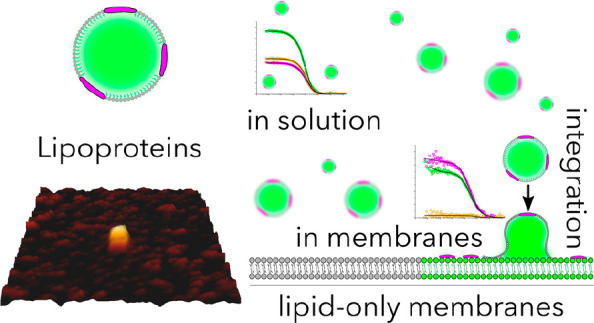
Lipid transfer from lipoprotein particles to cells is essential for lipid homeostasis. High-density lipoprotein (HDL) particles are mainly captured by cell membrane-associated scavenger receptor class B type 1 (SR-B1) from the bloodstream, while low-density and very-low-density lipoprotein (LDL and VLDL, respectively) particles are mostly taken up by receptor-mediated endocytosis. However, the role of the target lipid membrane itself in the transfer process has been largely neglected so far. Here, we study how lipoprotein particles (HDL, LDL, and VLDL) interact with synthetic lipid bilayers and cell-derived membranes and transfer their cargo subsequently. Employing cryo-electron microscopy, spectral imaging, and fluorescence (cross) correlation spectroscopy allowed us to observe integration of all major types of lipoprotein particles into the membrane and delivery of their cargo in a receptor-independent manner. Importantly, the biophysical properties of the target cell membranes change upon delivery of cargo. The concept of receptor-independent interaction of lipoprotein particles with membranes helps us to better understand lipoprotein particle biology and can be exploited for novel treatments of dyslipidemia diseases.
Cholesterol is a major structural element in cell membranes.1 Thus, a steady supply of cholesterol is of utmost importance for cell membrane integrity. Its levels are tightly controlled by homeostatic mechanisms balancing pathways of cholesterol uptake, biosynthesis, and release.2 Specialized cargo vehicles, called lipoprotein particles, are necessary to solubilize their share of nonpolar cargo. Several pathways are operative for cholesterol uptake, the majority via receptor-mediated endocytosis, in which the low-density lipoprotein (LDL) receptor binds apoB- and apoE-containing lipoprotein particles3,4 that are subsequently endocytosed. In addition, selective lipid uptake via scavenger receptor class B family (SR-B) receptors, in which core lipids of the lipoprotein particles are transferred to cells and tissues, has been proposed.5 Furthermore, endocytosis and subsequent transcytosis of lipoprotein particles are operative at least in endothelial cells.6,7 In addition to these well-described pathways, direct transfer of cholesterol from lipoprotein particles to the cell membrane may also occur.8−12 Here, using advanced imaging techniques, we show that cholesterol is transferred from all lipoprotein particles to lipid-only (protein-free) large and giant unilamellar vesicles (LUVs and GUVs, respectively), supported lipid bilayers (SLBs), and cell-derived giant plasma membrane vesicles (GPMVs). Upon delivery, the rigidity of the target membrane increases as expected due to the stiffening effect of cholesterol and other saturated lipids.
Materials and Methods
Reagents
Atto647N NHS ester was obtained from Atto-Tec. Sephadex G-25 fine resin, sodium cyanoborohydride (NaCNBH3), triethylamine (TEA), 3-(aminopropyl)triethoxysilane (APTES), ethanolamine (ETA), sodium deoxycholate, sucrose, glucose, and HEPES were from Sigma. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholesterol linked to BodipyFL (TopFluor-Cholesterol, Bd-Chol) were obtained from Avanti Polar Lipids. C-Laurdan was purchased from 2pprobes. NR12S was provided by A. Klymchenko (University of Strasbourg, Strasbourg, France). Abberior Star Red DOPE was purchased from Abberior.
Lipoprotein Particle Isolation and Labeling
Blood donations, obtained from normolipidemic healthy volunteers, were approved by the Ethics Committee, Medical University of Vienna (EK-Nr. 511/2007, EK-Nr. 1414/2016). Lipoprotein particles were isolated as previously described via sequential flotation ultracentrifugation.13,14 Its proteins were covalently linked to Atto647N at pH 8.3 according to the manufacturer’s instructions. Bd-Chol was incorporated into the lipid leaflet of lipoprotein particles via incubation at 37 °C for 2 h. Free dye and excessive cholesterol were removed via extensive dialysis.
Preparation of Giant Unilamellar Vesicles (GUVs)
GUVs with varying sizes from 10 to 100 μm were prepared by electroformation.15,16 POPC was dissolved in chloroform (1 mg/mL) and deposited on Pt electrodes. The solvent was evaporated by a constant N2 flow for 5 min. Then, 370 μL of 300 mM sucrose was added in a self-made chamber. On the cap of this chamber, we placed two holes with a distance of 5 mm for the electrodes. Then, the electrodes with dried lipids were incubated in the sucrose solution, and a voltage of 2 V at 10 Hz for 1 h and for an additional 30 min at 2 Hz was applied at room temperature (≈23 °C). A fluorescently labeled lipoprotein (HDL, LDL, or VLDL) particle solution was added to the GUV solution (final concentration of lipoproteins of 0.3 mg/mL). Confocal microscopy images were acquired 20 min after the addition of lipoproteins. For imaging, we added 100 μL of the GUV suspension to 100 μL of PBS.
Preparation of Large Unilamellar Vesicles (LUVs)
LUVs were prepared by extrusion (Avanti Mini Extruder, Avanti Polar Lipids). DOPC was dissolved in a chloroform/methanol mixture (2:1, 10 mg/mL), and 10 μL was dried by evaporation. Subsequently, lipids were hydrated using PBS and kept above the phase transition temperature of the lipid during hydration and extrusion. Once the sample was fully hydrated, the mixture was placed into one end of the Mini-Extruder. The plunger of the filled syringe was pushed gently until the lipid solution was completely transferred to the alternate syringe, and then the plunger of the alternate syringe was pushed to transfer the solution back to the original syringe. This process was repeated until the lipid suspension was clear. The lipid solution was stored at 4 °C.
Preparation of Giant Plasma Membrane Vesicles (GPMVs)
CHO cells were grown in DMEM/F12 medium supplemented with 10% FBS and 1% l-glutamine. GPMVs were prepared as previously described.16 Briefly, cells seeded out on a 35 mm Petri dish (≈70% confluent) were washed twice with GPMV buffer [150 mM NaCl, 10 mM Hepes, and 2 mM CaCl2 (pH 7.4)]. One mL of GPMV buffer was added to the cells. Twenty-five mM paraformaldehyde and 2 mM dithiothreitol (final concentrations) were added in GPMV buffer. Cells were incubated for 2 h at 37 °C. Then, GPMVs were collected by collecting the supernatant.
Preparation of Supported Lipid Bilayers (SLBs) on a Mica Substrate [for atomic force microscopy (AFM) force spectroscopy]
SLBs were formed on freshly cleaved (∼1 μm thick) mica, glued onto a glass coverslip using an optically transparent UV glue (optical adhesive 88, Norland Products Inc.). Glass slides (d = 22 mm; Menzel) were incubated in a freshly prepared mixture of sulfuric acid and hydrogen peroxide (3:1) for 20 min, rinsed with deionized water and ethanol, and dried under a N2 flow. Thirty μL of a DOPC solution (10 mg/mL in a 3:1 chloroform/methanol mixture) was evaporated under a N2 flow (20 min) and resuspended in 300 μL of PBS. Vesicles were prepared by sonication for 20 min and applied to the mica. After 20 min, the bilayer was formed and slides were washed with PBS.
Preparation of Supported Lipid Bilayers (SLBs) on a Glass Substrate (for fluorescence microscopy)
POPC was dissolved in a 2:1 chloroform/methanol mixture (1 mg/mL final concentration) with or without 0.01 mol % Abberior Star Red DOPE. Glass coverslips (25 mm in diameter, #1.5) were incubated in a freshly prepared mixture of sulfuric acid and hydrogen peroxide (3:1) for 30 min, rinsed with deionized water, and dried under a N2 flow. The glass coverslip was mounted on the spin-coater, spinning started at 3000 rpm, 25 μL of a lipid solution applied, and spinning continued for 30 s. The glass coverslip was immediately mounted on the metal Attofluor Cell Chamber, and the lipid film was hydrated with SLB buffer [150 mM NaCl and 10 mM HEPES (pH 7.4)].
Confocal and Spectral Microscopy
GUVs and GPMVs were imaged with a laser scanning confocal microscope (LSM 780, Zeiss). The microscope was equipped with a 40×/1.20 NA water immersion objective; 488 and 633 nm lasers were used to excite BodipyFL and Atto647N, respectively. Spectral imaging of C-Laurdan and NR12S was performed on a Zeiss LSM 780 confocal microscope equipped with a 32-channel GaAsP detector array. Laser light at 405 nm was used for fluorescence excitation of Laurdan. The λ detection range was set to 415–691 nm for Laurdan. Laser light at 488 nm was used for excitation of NR12S. The λ detection range was set to 498–691 nm for NR12S. Images were saved in.lsm file format and then analyzed by using a freely available plug-in compatible with Fiji/ImageJ, as described previously.17
Fluorescence Correlation Spectroscopy
For FCS measurements, membranes were incubated with fluorescently labeled lipoproteins for 20 min. GUVs and GPMVs were measured in Ibidi eight-well glass-bottom chambers with a thickness of 0.17 mm. SLBs were measured on 25 mm diameter, 0.17 mm thick glass cover slides.
FCS measurements were carried out using a Zeiss LSM 780 microscope. A 488 nm argon ion laser and a 633 nm He–Ne laser were used for Bd and Atto647N, respectively. A 40×/1.2 NA water immersion objective was used to focus the light. Five curves were taken per spot (5 s each). The laser power was set to 0.1–0.5% of the total laser power that corresponds to 2–10 μW. Curves were then fitted with FoCuS-point software to extract diffusion coefficients with the following two-dimensional diffusion model.18
where N represents the number of fluorescent species within the beam’s focal volume. Next, the diffusion coefficients were calculated as follows:
where w corresponds to the full width of half-maximum of the point spread function, tD is the diffusion time, and D is the diffusion coefficient.
Force Spectroscopy and Bilayer Indentation Experiments
Force measurements were performed on a PicoPlus
AFM instrument (Agilent
Technologies) operated under PicoView 1.6.8 (Agilent Technologies)
in solution (PBS). Force distance cycles were acquired using silicon
cantilevers with a spring constant of 0.01 or 0.02 N/m (Veeco) at
pulling velocities of 0.1–5 μm/s and contact times (hold
times) of 0.1–5 s. Empirical force distributions of the rupture
forces of the last unbinding event (pdf) were calculated as described
previously.19 pdfs were fitted with the
equation 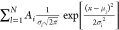 , including the boundary condition
, including the boundary condition 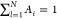 taking the probability density
property
of the pdfs into account, where Ai is a prefactor, μi is the position of the peak, and σi is the width of the peak (see Figure S1). For force spectroscopy, a sweep range
of 3 μm and a sweep rate of 0.2–2 Hz were used. Silicon-nitride
AFM cantilevers with silicon tips (MSNL-10, Bruker AFM Probes) were
amine-functionalized as described previously.20 Briefly, silicon cantilevers were amine-functionalized via gas-phase
silanization with aminopropyl-triethoxysilane (APTES),21 and a heterobifunctional (aldehyde-NHS) linker
was chemically connected. Subsequently, the tips were washed with
chloroform and dried with N2 gas. Tips were incubated with
100 μL of lipoproteins (0.06 mg/mL in PBS), to which 2 μL
of NaCNBH3 (1 M, freshly prepared in 10 mM NaOH) was added
for irreversible binding. Afterward, 5 μL of ethanolamine hydrochloride
(1 M, adjusted to pH 9.6) was added to block nonreacted linker groups,
and incubation was continued for 10 min. This chemical modification
was used to covalently link lipoprotein particles on cantilevers.
The effective spring constant was determined via thermal noise analysis22 before and after chemical modification.
taking the probability density
property
of the pdfs into account, where Ai is a prefactor, μi is the position of the peak, and σi is the width of the peak (see Figure S1). For force spectroscopy, a sweep range
of 3 μm and a sweep rate of 0.2–2 Hz were used. Silicon-nitride
AFM cantilevers with silicon tips (MSNL-10, Bruker AFM Probes) were
amine-functionalized as described previously.20 Briefly, silicon cantilevers were amine-functionalized via gas-phase
silanization with aminopropyl-triethoxysilane (APTES),21 and a heterobifunctional (aldehyde-NHS) linker
was chemically connected. Subsequently, the tips were washed with
chloroform and dried with N2 gas. Tips were incubated with
100 μL of lipoproteins (0.06 mg/mL in PBS), to which 2 μL
of NaCNBH3 (1 M, freshly prepared in 10 mM NaOH) was added
for irreversible binding. Afterward, 5 μL of ethanolamine hydrochloride
(1 M, adjusted to pH 9.6) was added to block nonreacted linker groups,
and incubation was continued for 10 min. This chemical modification
was used to covalently link lipoprotein particles on cantilevers.
The effective spring constant was determined via thermal noise analysis22 before and after chemical modification.
AFM Imaging and Particle Analysis
AFM measurements were performed with an atomic force microscope (JPK BioAFM-NaonWizard 4, JPK, Berlin, Germany). AFM probes made of silicon nitride with a nominal spring constant of 0.3 N/m and a nominal tip radius of 20–60 nm (MLCT-BIO-F, Bruker Nano Inc., Camarillo, CA) were used for the measurements. The exact sensitivity and spring constant of each cantilever were determined on a cleaned coverslip in 300 μL of PBS from a force–displacement experiment and a thermal noise spectrum measurement. All samples (HDL, LDL, and VLDL) were diluted to 1:1000. A volume of 300 μL of the diluted HDL and LDL solution was incubated on the cleaned glass coverslip for at least 5 min and subsequently imaged. Because of the low density of VLDL particles, a volume of 30 μL of the diluted VLDL sample was incubated on the glass coverslip upside down for 5 min. Afterward, AFM images were recorded by using an advanced imaging software (Quantitative Imaging mode QI-mode) from Bruker. A maximal set point force of 500 pN was used.
Particle analysis [full width at half-maximum (fwhm) and height of probe molecules] was performed with JPK Data Processing software (version 6.1.163, JPK). Convolutions of tip artifacts were corrected as described in Figure S2. For each individual particle, the aspect ratio (AR in percent) was calculated. An AR of 100% represents a perfect spherical shape; lower values represent prone discs (Figure S2). The values were later fit with a Gaussian distribution to calculate the mean height and width.
Cryo-electron Microscopy
LUVs (100 μL) were incubated with the respective lipoprotein solutions (5 μL) for 2 min at room temperature. Immediately after incubation, samples were stored on ice and applied to the cryo-grids (2 nm precoated Quantifoil R3/3 holey carbon-supported grids) at a concentration of 10 μM and vitrified using a Vitrobot Mark IV (FEI). Data were collected on a TEM microscope FEI Tecnai F20 equipped with a 4K CCD camera and two side-entry cryo-holders. The data set was collected using a Tecnai F20 instrument (FEI, Eindhoven, The Netherlands) operated at 200 kV and equipped with a 4K charge-coupled device detector FEI Eagle. Micrographs were collected with a pixel size of 1.79 Å and a total dose of 20 e–/Å2. Frames were aligned using MotionCor2,23 and CTF parameters were estimated using Gctf.24
Results and Discussion
First, we imaged lipoprotein particles via AFM to confirm intact lipoprotein particles and to determine their size (Figure 1A). For this purpose, we incubated lipoprotein particles on clean glass for immobilization and performed AFM measurements. From these measurements, we calculated the lateral and axial size for each particle (Figure 1B–D and Figures S2 and S3). The sizes (lateral × axial) were 9.4 ± 2.1 nm × 8.3 ± 3.0 nm (AR = 92%) for HDL particles, 28.8 ± 8.7 nm × 22.5 ± 4.3 nm (AR = 84%) for LDL particles, and 64.6 ± 5.1 nm × 48.8 ± 6.0 nm (AR = 76%) for VLDL particles, in accordance with the literature.25Figure 1D shows the spherical reconstruction of the particles according to the size calculations from AFM images, which confirms the particle structure and integrity. Next, we set out to study the interaction of lipoprotein particles with membranes.
Figure 1.

Topographical characterization of the lipoprotein particles via AFM. (A) AFM images of lipoprotein particles. (B) Height and (C) width probability density function (pdf) (mean ± standard deviation) of 10 analyzed lipoprotein particles. (D) Spherical reconstruction of lipoprotein particles according to the AFM data.
Recently, by using cryo-EM, we studied the interaction of LDL particles with membranes.26 This prompted us to study whether a similar interaction pattern exists with all lipoprotein particles and with lipid-only membranes. We applied lipoprotein particles to LUVs and observed clear interactions between the lipoprotein particles and the LUVs (Figure 2A). Interaction of different lipoprotein particles with LUV membranes (white arrows) was confirmed through recording data under different electron-beam incident angles, thus excluding an accidental overlay of signals originating from different layers of the vitrified ice.
Figure 2.

Lipoprotein particles interact with biomimetic membranes and transfer their cargo. (A) Cryo-EM images of single HDL and VLDL particles (top images) and lipoprotein particle-decorated LUVs (DOPC, bottom images). Images were acquired under low-dose conditions (20 e/Å2). The scale bar is 10 nm. (B) Membrane tethers are formed during the retraction of HDL-modified AFM tips from supported lipid bilayers (DOPC). A representative retraction curve (black) is shown for a functionalized HDL tip on a DOPC membrane. A cantilever with a spring constant of 0.01 N/m was used. The applied pulling velocity was 1 μm/s. For experiments, a maximum contact force of 500 pN was set to prevent penetration of the membrane. During retraction, membrane tethers are formed between the HDL particle on the tip and the bilayer with typical rupture forces of ∼50 pN. Confocal images of (C) SLBs (POPC), (D) GUVs (POPC), and (E) GPMVs incubated with 0.05 mg/mL fluorescently labeled HDL (left), LDL (middle), and VLDL (right) particles. Bd-Chol is colored green, and the Atto647N-labeled protein is colored magenta. Scale bars are 10 μm. (F) Fluorescence cross-correlation spectroscopy of Bd-Chol and proteins measured in solution (intact VLDL) as well as in target membranes [GUVs, GPMVs, and SLBs (see Figure S5 for HDL and LDL)]. In solution, the strong cross correlation of Bd-Chol and protein signals is detected, which suggests co-diffusion. In the target membrane, the cross-correlation curve amplitude is close to zero, which suggests that Bd-Chol and protein molecules diffuse in the target membrane independently.
To further investigate the interaction of lipoprotein particles with membranes, we recorded the interaction force as a function of the distance from the surface via AFM (i.e., force–distance cycles). This technique is used to determine the interactions between molecular compounds (e.g., ligand–receptor). For this purpose, one binding partner is covalently bound to the AFM tip and the other is immobilized on the surface or located in the cell membrane. During the approach of the AFM tip, binding of molecules on the tip to molecules on the surface is facilitated. Retracting the tip breaks the bond. During this process, the strength and kinetics of these interactions can be determined. In our case, lipid-only membranes were used as interaction partners for lipoprotein particles, which were attached to the AFM tip. The maximum force was set below an actual penetration threshold of the membrane. Interestingly, by performing force–distance cycles with an HDL-functionalized tip on a fluid supported lipid bilayer, we detected interaction forces that resembled lipid tube formation27 (see Figure 2B and Figure S4). These membrane tubes (or tethers) are nanocylinders made of lipid bilayers. Here, multiple tether formations were observed on a supported lipid bilayer. In particular, the average force required to form a single tether was found to be ∼50 pN, at different cantilever retraction speeds of 0.5–5 μm/s (Figure S4). Forces were calculated by fitting a Gaussian profile to the force probability density function (pdf). Plotting force versus tether length yields well-defined equidistant force steps (Figure S4). Up to four steps (i.e., unbinding events) were observed, indicating formation of multiple tethers. This result strongly suggests interaction of lipoprotein particles with target membranes, yielding tether formation.
Next, to directly visualize whether interaction of lipoprotein particles led to the transfer of cargo from VLDL, LDL, and HDL particles to biomembrane systems, we applied fluorescence imaging. We used SLBs as supported and GUVs as free-standing lipid-only membrane systems. We labeled lipoprotein particles with cholesterol-BodipyFL (Bd-Chol) and their proteins with Atto647N. Then, we incubated fluorescently labeled lipoprotein particles with synthetic membrane systems (unlabeled GUVs and SLBs). After incubation with all types of lipoprotein particles, we detected the fluorescence signal of both BodipyFL and Atto647N in SLBs and GUVs (Figure 2C,D). This suggests that upon interaction with the target membrane, lipoprotein particles fuse with the target membranes and transfer their cargo. To verify this observation in a more complex membrane system, we prepared GPMVs from Chinese hamster ovary (CHO) cells, which comprise a biological membrane system consisting not only of lipids but also of proteins. Similarly, we observed the transfer of cholesterol and protein from lipoprotein particles to the GPMV membrane (Figure 2E). To unequivocally verify the cargo transfer, we applied fluorescence cross-correlation spectroscopy (FCCS). FCCS is a fluctuation-based method that measures the interaction of two different fluorescently labeled molecules.28 It yields an autocorrelation curve for both differently (e.g., green and red) labeled molecules, providing information about their diffusion, concentration, and molecular brightness. Additionally, it yields a non-zero amplitude cross-correlation curve if molecules co-diffuse (i.e., diffuse as a single entity). Co-diffusion usually means direct interaction or association with the same nanoscale entity (such as a domain, vesicles, etc.) that moves through the focal spot. The amplitude of the cross-correlation curve is proportional to the fraction of molecules that co-diffuse; perfect co-diffusion gives near 100% cross correlation, while no codiffusion yields 0% cross correlation. In principle, intact lipoprotein particles where cholesterol and proteins are both labeled should show perfect co-diffusion with a high amplitude as both molecules move in and out of the focal volume together as one particle unit. We indeed observe a very high cross correlation (nearly 100%) for all types of lipoprotein particles in solution (Figure 2F and Figure S5). This means not only that all lipoprotein particles contain labeled proteins and cholesterol but also that all of the fluorescence signal we record originates from doubly labeled lipoprotein particles. If the content of the lipoprotein particles is released into their target membrane upon interactions, fluorescently labeled proteins and cholesterols should move separately in the membrane (unlike in solution). Thus, cross correlation should disappear. To test this, we measured the cross correlation in SLBs, GUVs, and GPMVs incubated with labeled lipoprotein particles and indeed observed no cross correlation in these samples [near zero amplitude (Figure 2F and Figure S5)]. Moreover, in solution, diffusion coefficients of cholesterol and protein are identical because they move together in the same particle. However, once lipoprotein particles fuse with the target membranes, diffusion of protein is always slower than diffusion of cholesterol due to its larger size (Figure S6). These data confirm that lipoprotein particles interact with synthetic membranes and subsequently release their cargo to the target membranes.
The cholesterol content in the plasma membrane is crucial for membrane biophysical properties such as rigidity, stiffness, elasticity, etc. Therefore, it is necessary to reveal how interaction and cargo transfer of lipoprotein particles alter the biophysical properties of target membranes. We measured the rigidity of target membranes by using two environmentally sensitive probes NR12S29 and C-Laurdan.30 The fluorescence emission of these probes is sensitive to the rigidity of the lipid environment; they demonstrate a red shift in their emission maxima in more fluid membranes. This spectral shift can be used to report on the molecular ordering of the membrane, utilizing an empirical lipid packing parameter, generalized polarization (GP).31 GP is an indirect but robust way to infer lipid packing with values varying between 1 (for very ordered) and −1 (very disordered).32 Confocal spectral imaging can conveniently be used to measure GP values of membranes.17 To this end, we incorporated NR12S in GUVs and C-Laurdan in GPMVs and imaged them with spectral imaging before and after incubation with lipoprotein particles. A higher cholesterol and saturated lipid content yields more rigid membranes, and thus higher GP values. While control GUVs (not treated with any lipoprotein particles) yielded GP values of −0.24 ± 0.07, GUVs incubated with lipoprotein particles showed GP values of −0.19 ± 0.05 (HDL), −0.08 ± 0.08 (LDL), and −0.1 ± 0.07 (VLDL) (Figure 3A,B). Similarly, GPMVs that are not treated with lipoprotein particles showed a value of −0.07 ± 0.02, while lipoprotein-treated ones showed values of 0.05 ± 0.02 (HDL), 0.04 ± 0.01 (LDL), and 0.0 ± 0.01 (VLDL) (Figure 3C,D). These data show that upon cargo transfer, cholesterol and saturated lipids in lipoprotein particles are incorporated into the membrane and rigidify it.
Figure 3.

Changes in rigidity upon interactions of the lipoprotein particle with target membranes. (A) GP images and (B) GP values of NR12S in GUVs (POPC) incubated with HDL, LDL, and VLDL particles compared to control GUVs (no incubation). (C) GP images and (D) GP values of C-Laurdan in GPMVs incubated with HDL, LDL, and VLDL particles compared to control GPMVs. (E) Representative FCS curves and (F) diffusion coefficients for ASR-PE in SLBs (POPC) treated with lipoprotein particles compared to control SLBs. (G) Representative FCS curves and (H) diffusion coefficients for ASR-PE in GUVs (POPC) treated with lipoprotein particles compared to untreated GUVs. Graphs show the mean and the standard deviation; numbers of data points are indicated in graphs in parentheses.
To further confirm this, we also measured the diffusion of a fluorescently labeled lipid analogue [Abberior Star Red-labeled DPPE (ASR-PE)] in the membrane of SLBs and GUVs before and after incubation with unlabeled lipoprotein particles with FCS. Diffusion of lipids in a rigid membrane is slower than in a more fluid membrane.33 If cholesterol and other saturated lipids are indeed transferred to the target membrane, it would become more rigid and diffusion would become slower. We observe this trend with all lipoprotein particles (Figure 3E–H); the diffusion coefficient of ASR-PE was 4.5 ± 1.4 μm2/s in untreated SLBs, while in lipoprotein-treated bilayers, it was 3.9 ± 0.7 μm2/s (HDL), 4.4 ± 0.8 μm2/s (LDL), and 4.4 ± 0.9 μm2/s (VLDL) (Figure 3E,F). The transfer of cargo from freely diffusing lipoprotein particles to a planar supported bilayer on a glass surface may be inefficient. Moreover, in SLBs, the manifestation of compositional changes in diffusion is largely masked by the support effect.33 In contrast, free-standing membranes better reflect the compositional changes.33 Thus, we also tested the diffusion of a lipid analogue in GUVs, where the diffusion coefficient of ASR-PE was 6.6 ± 1.2 μm2/s in untreated GUVs, while in lipoprotein-treated vesicles, it was 4.7 ± 1.2 μm2/s (HDL), 6.1 ± 2.1 μm2/s (LDL), and 5.6 ± 1.8 μm2/s (VLDL) (Figure 3G,H). These data together with GP measurements show that lipoprotein particles transfer cholesterol and possibly saturated lipids to target membranes and thus increase their rigidity.
Conclusions
In this work, we showed that all major types of lipoprotein particles (HDL, LDL, and VLDL), regardless of their size and their lipid and protein composition, integrate with lipid membranes and transfer their lipid cargo (exemplified by Bd-Chol) after integration. One possible explanation is the spontaneous transfer of lipoprotein components. While this type of transfer is possible for hydrophobic lipids,34 it is unlikely for proteins. Moreover, our previous results showed that the height of the lipoprotein particles20,26 decreases upon interaction with target membranes, pointing toward a fusion-based mechanism rather than spontaneous transfer.
This mechanism adds a new aspect to the picture of cholesterol homeostasis where direct cargo transfer of lipoprotein particles might occur in a receptor-independent manner. This will be the first step of further work to elucidate the exact contribution of the direct delivery mechanism in vivo. It is plausible to assume that the fusion-based receptor-independent mechanism is faster in comparison to receptor-mediated process35 because it does not involve complicated receptor coupling and endocytic uptake. However, it is expected that receptor-independent cholesterol transfer will be less efficient as it is limited (and potentially driven) by a preexisting concentration gradient. Thus, we can assume that the receptor-independent process is more immediate, while receptor-driven uptake is more efficient. These two mechanisms are not mutually exclusive, and both could be exploited under physiological conditions; while receptor-independent delivery can supply the basal level of cholesterol cargo quickly, the receptor-dependent mechanism can work to achieve higher concentrations above the equilibrium concentrations. Depending on the target cell type and membrane properties, the efficiency and speed of receptor-independent delivery might vary. Future work on the speed and efficiency of receptor-independent cholesterol transfer compared to receptor-mediated transfer will shed new light on the dynamics of receptor-independent transfer. Despite these unknowns, the ability of lipoprotein particles to directly deliver their cargo to the target membrane can still potentially be exploited for therapeutic approaches against diseases such as familial hypercholesterolemia where the LDL receptor cannot optimally fulfill its function. Therefore, we believe this mechanism may potentially be important for future therapies against dyslipidemia diseases. Elaborated work with cells as well as lipoprotein particles from dyslipidemia patients will also be crucial to see how receptor-independent cargo transfer is affected by the metabolic state of the donors. Finally, it will be crucial to elucidate whether the target membrane properties influence the lipoprotein particle interactions, particularly as a function of lipoprotein particle type.
Acknowledgments
The authors thank Dr. Andrey Klymchenko for providing NR12S.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00748.
Additional figures demonstrating the evaluation of the force steps for AFM force spectroscopy (Figure S1), details of AFM measurements and calculations (Figures S2 and S3), dependence of the force of individual steps on the pulling velocity (Figure S4), and fluorescence cross-correlation spectroscopy of all lipoprotein particles in all target membranes (Figures S5 and S6) (PDF)
Author Contributions
∇ B.P. and T.S. contributed equally to this work.
E.S. is funded by SciLifeLab and the Wellcome Trust Institutional Strategic Support Fund (ISSF). This work has been supported by Austrian Science Fund Projects P22838-B13 and P29110-B21 and the Austrian Research Promotion Agency Innovatives Oberösterreich 2020-851455, the European Fund for Regional Development (EFRE, IWB 2020), the Federal State of Upper Austria, and “Land OÖ Basisfinanzierung”. The authors acknowledge funding by the Wolfson Foundation, the Medical Research Council (MRC, Grant MC_UU_12010/unit programmes G0902418 and MC_UU_12025), MRC/BBSRC/EPSRC (Grant MR/K01577X/1), and the Wellcome Trust (Grant 104924/14/Z/14).
The authors declare no competing financial interest.
Supplementary Material
References
- Ikonen E. (2008) Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 125–138. 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- Brown M. S.; Goldstein J. L. (2009) Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J. Lipid Res. 50, S15–S27. 10.1194/jlr.R800054-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. S.; Goldstein J. L. (1986) A receptor-mediated pathway for cholesterol homeostasis. Science 232, 34–47. 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Mahley R. W.; Ji Z. S. (1999) Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 40, 1–16. [PubMed] [Google Scholar]
- Meyer J. M.; Graf G. A.; Van Der Westhuyzen D. R. (2013) New developments in selective cholesteryl ester uptake. Curr. Opin. Lipidol. 24, 386–392. 10.1097/MOL.0b013e3283638042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Sessa W. C.; Fernández-Hernando C. (2018) Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front. Cardiovasc. Med. 5, 130. 10.3389/fcvm.2018.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung K. Y. Y.; Lee W.; Fairn G. (2020) Inhibition of Low density Lipoprotein Internalization and Transcytosis by HDL; an alternative role for “good” cholesterol. FASEB J. 34, 1. 10.1096/fasebj.2020.34.s1.00598. [DOI] [Google Scholar]
- Robins S. J.; Fasulo J. M. (1999) Delineation of a novel hepatic route for the selective transfer of unesterified sterols from high-density lipoproteins to bile: studies using the perfused rat liver. Hepatology 29, 1541–1548. 10.1002/hep.510290518. [DOI] [PubMed] [Google Scholar]
- Wüstner D. (2005) Mathematical analysis of hepatic high density lipoprotein transport based on quantitative imaging data. J. Biol. Chem. 280, 6766–6779. 10.1074/jbc.M413238200. [DOI] [PubMed] [Google Scholar]
- Wüstner D.; Mondal M.; Huang A.; Maxfield F. R. (2004) Different transport routes for high density lipoprotein and its associated free sterol in polarized hepatic cells. J. Lipid Res. 45, 427–437. 10.1194/jlr.M300440-JLR200. [DOI] [PubMed] [Google Scholar]
- Robins S. J.; Fasulo J. M.; Leduc R.; Patton G. M. (1989) The transport of lipoprotein cholesterol into bile: a reassessment of kinetic studies in the experimental animal. Biochim. Biophys. Acta, Lipids Lipid Metab. 1004, 327–331. 10.1016/0005-2760(89)90080-5. [DOI] [PubMed] [Google Scholar]
- Bravo E.; Botham K. M.; Mindham M. A.; Mayes P. A.; Marinelli T.; Cantafora A. (1994) Evaluation in vivo of the differential uptake and processing of high-density lipoprotein unesterified cholesterol and cholesteryl ester in the rat. Biochim. Biophys. Acta, Lipids Lipid Metab. 1215, 93–102. 10.1016/0005-2760(94)90096-5. [DOI] [PubMed] [Google Scholar]
- Axmann M.; Karner A.; Meier S. M.; Stangl H.; Plochberger B. (2019) Enrichment of Native Lipoprotein Particles with microRNA and Subsequent Determination of Their Absolute/Relative microRNA Content and Their Cellular Transfer Rate. J. Visualized Exp. 147, e59573 10.3791/59573. [DOI] [PubMed] [Google Scholar]
- Schumaker V. N., and Puppione D. L. (1986) [6] Sequential flotation ultracentrifugation. In Methods in Enzymology, pp 155–170, Academic Press. [DOI] [PubMed] [Google Scholar]
- Bak P.; Timonen J. (1978) Coupling between phase solitons and strain near the commensurate- incommensurate transition. J. Phys. C: Solid State Phys. 11, 4901–4905. 10.1088/0022-3719/11/24/019. [DOI] [Google Scholar]
- Sezgin E.; Kaiser H.-J.; Baumgart T.; Schwille P.; Simons K.; Levental I. (2012) Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat. Protoc. 7, 1042–1051. 10.1038/nprot.2012.059. [DOI] [PubMed] [Google Scholar]
- Sezgin E.; Waithe D.; Bernardino de la Serna J.; Eggeling C. (2015) Spectral imaging to measure heterogeneity in membrane lipid packing. ChemPhysChem 16, 1387–1394. 10.1002/cphc.201402794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waithe D.; Clausen M. P.; Sezgin E.; Eggeling C. (2016) FoCuS-point: Software for STED Fluorescence Correlation and Time-Gated Single Photon Counting. Bioinformatics 32, 958–960. 10.1093/bioinformatics/btv687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner W.; Hinterdorfer P.; Schindler H. (2000) Data analysis of interaction forces measured with the atomic force microscope. Ultramicroscopy 82, 85–95. 10.1016/S0304-3991(99)00154-0. [DOI] [PubMed] [Google Scholar]
- Plochberger B.; Röhrl C.; Preiner J.; Rankl C.; Brameshuber M.; Madl J.; Bittman R.; Ros R.; Sezgin E.; Eggeling C.; Hinterdorfer P.; Stangl H.; Schütz G. J. (2017) HDL particles incorporate into lipid bilayers-a combined AFM and single molecule fluorescence microscopy study. Sci. Rep. 7, 15886. 10.1038/s41598-017-15949-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner A.; Hinterdorfer P.; Gruber H. J. (2007) Comparison of different aminofunctionalization strategies for attachment of single antibodies to AFM cantilevers. Ultramicroscopy 107, 922–927. 10.1016/j.ultramic.2007.02.035. [DOI] [PubMed] [Google Scholar]
- Stark R. W.; Drobek T.; Heckl W. M. (2001) Thermomechanical noise of a free v-shaped cantilever for atomic-force microscopy. Ultramicroscopy 86, 207–215. 10.1016/S0304-3991(00)00077-2. [DOI] [PubMed] [Google Scholar]
- Zheng S. Q.; Palovcak E.; Armache J.-P.; Verba K. A.; Cheng Y.; Agard D. A. (2017) MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. (2016) Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12. 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojczynski M. K.; Glasser S. P.; Oberman A.; Kabagambe E. K.; Hopkins P. N.; Tsai M. Y.; Straka R. J.; Ordovas J. M.; Arnett D. K. (2011) High-fat meal effect on LDL HDL, and VLDL particle size and number in the Genetics of Lipid-Lowering Drugs and Diet Network (GOLDN): an interventional study. Lipids Health Dis. 10, 181. 10.1186/1476-511X-10-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axmann M.; Sezgin E.; Karner A.; Novacek J.; Brodesser M. D.; Rohrl C.; Preiner J.; Stangl H.; Plochberger B. (2019) Receptor-Independent Transfer of Low Density Lipoprotein Cargo to Biomembranes. Nano Lett. 19, 2562–2567. 10.1021/acs.nanolett.9b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumí-Audenis B.; Costa L.; Ferrer-Tasies L.; Ratera I.; Ventosa N.; Sanz F.; Giannotti M. I. (2018) Pulling lipid tubes from supported bilayers unveils the underlying substrate contribution to the membrane mechanics. Nanoscale 10, 14763–14770. 10.1039/C8NR03249A. [DOI] [PubMed] [Google Scholar]
- Bacia K.; Schwille P. (2007) Practical guidelines for dual-color fluorescence cross-correlation spectroscopy. Nat. Protoc. 2, 2842–2856. 10.1038/nprot.2007.410. [DOI] [PubMed] [Google Scholar]
- Kucherak O. A.; Oncul S.; Darwich Z.; Yushchenko D. A.; Arntz Y.; Didier P.; Mely Y.; Klymchenko A. S. (2010) Switchable Nile Red-Based Probe for Cholesterol and Lipid Order at the Outer Leaflet of Biomembranes. J. Am. Chem. Soc. 132, 4907–4916. 10.1021/ja100351w. [DOI] [PubMed] [Google Scholar]
- Kim H. M.; Choo H.-J.; Jung S.-Y.; Ko Y.-G.; Park W.-H.; Jeon S.-J.; Kim C. H.; Joo T.; Cho B. R. (2007) A two-photon fluorescent probe for lipid raft imaging: C-laurdan. ChemBioChem 8, 553–559. 10.1002/cbic.200700003. [DOI] [PubMed] [Google Scholar]
- Parasassi T.; De Stasio G.; Ravagnan G.; Rusch R. M.; Gratton E. (1991) Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys. J. 60, 179–189. 10.1016/S0006-3495(91)82041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez S. A., Tricerri M. A., Gunther G., and Gratton E. (2007) Laurdan Generalized Polarization: from cuvette to microscope. In Modern Research and Educational Topics in Microscopy (Méndez-Vilas A., and Díaz J., Eds.) Formatex, Badajoz, Spain. [Google Scholar]
- Beckers D.; Urbancic D.; Sezgin E. (2020) Impact of nanoscale hindrances on the relationship between lipid packing and diffusion in model membranes. J. Phys. Chem. B 124, 1487–1494. 10.1021/acs.jpcb.0c00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund-Katz S.; Phillips M. C. (2010) High Density Lipoprotein Structure–Function and Role in Reverse Cholesterol Transport. Subcell. Biochem. 51, 183–227. 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangl H.; Hyatt M.; Hobbs H. H. (1999) Transport of Lipids from High and Low Density Lipoproteins via Scavenger Receptor-BI. J. Biol. Chem. 274, 32692–32698. 10.1074/jbc.274.46.32692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.