

There is a Blood Commentary on this article in this issue.
Key Points
Differential spatial expression of the chemokine CXCL4/platelet factor-4 marks the progression of fibrosis.
The absence of hematopoietic CXCL4 ameliorates the MPN phenotype and reduces stromal cell activation and BM fibrosis.
Abstract
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm (MPN) that leads to progressive bone marrow (BM) fibrosis. Although the cellular mutations involved in the pathogenesis of PMF have been extensively investigated, the sequential events that drive stromal activation and fibrosis by hematopoietic–stromal cross-talk remain elusive. Using an unbiased approach and validation in patients with MPN, we determined that the differential spatial expression of the chemokine CXCL4/platelet factor-4 marks the progression of fibrosis. We show that the absence of hematopoietic CXCL4 ameliorates the MPN phenotype, reduces stromal cell activation and BM fibrosis, and decreases the activation of profibrotic pathways in megakaryocytes, inflammation in fibrosis-driving cells, and JAK/STAT activation in both megakaryocytes and stromal cells in 3 murine PMF models. Our data indicate that higher CXCL4 expression in MPN has profibrotic effects and is a mediator of the characteristic inflammation. Therefore, targeting CXCL4 might be a promising strategy to reduce inflammation in PMF.
Visual Abstract

Introduction
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm (MPN) that arises from clonal proliferation of hematopoietic stem cells (HSCs) and leads to progressive bone marrow (BM) fibrosis resulting in extramedullary hematopoiesis (typically in the spleen), BM failure, and ultimately death. Although cellular mutations involved in PMF development have been extensively investigated,1-6 the sequential events leading to the transformation of stromal cells to fibrosis-driving cells remain elusive.
It has become increasingly clear over recent years that 2 distinct pathogenic processes contribute to the initiation and progression of PMF: (1) stem cell–derived clonal myeloproliferation; and (2) a reactive cytokine- and chemokine-driven inflammatory fibrosis. On a cellular level, this means that HSCs acquire mutations that lead to increased proliferation of HSCs and the eventual replacement of normal blood formation, whereas nonmutated, non-hematopoietic stromal cells transform into fibrosis-driving cells. The biology of this cross-talk between malignant hematopoietic cells and a normal (non-hematopoietic) stromal cell that transforms into a fibrosis-driving cell is incompletely understood.
Recent research has shown that Gli1+ and LepR+ mesenchymal stromal cells (MSCs) are progenitors of fibrosis-causing myofibroblasts in the BM.7,8 Genetic ablation of Gli1+ MSCs or pharmacologic targeting of Hedgehog-Gli signaling ameliorated fibrosis in mouse models of myelofibrosis. Moreover, pharmacologic or genetic intervention in platelet-derived growth factor receptor α (Pdgfrα) signaling in Lepr+ stromal cells suppressed their expansion and ameliorated myelofibrosis. Together, these 2 recent studies confirmed the longstanding hypothesis that MSCs are indeed the cellular origin of BM fibrosis. However, the precise mechanisms that drive the activation of the stroma by the malignant hematopoietic clone are not well understood. Dissecting the underlying mechanisms has the potential to identify new molecular targets and develop urgently needed novel therapeutics because the only cure for myelofibrosis is an allogeneic BM transplant, which remains a high-risk procedure with high morbidity and high mortality.
We therefore applied a nonhypothesis-driven approach and questioned if the activation of the stroma occurs after short exposure to fibrosis-inducing hematopoietic stem and progenitor cells (HSPCs), and whether we can identify genes that mark the progression of the fibrotic transformation. Using patient samples, we validated and confirmed that the temporospatial distribution of the chemokine CXCL4/platelet factor-4 (PF4) marks progression of fibrosis and dissected the role of CXCL4 knockout in HSPCs in murine models of PMF.
CXCL4, a chemokine synthesized by megakaryocytes (MKs) with key roles in platelet physiology, is known to be secreted by a variety of immune cells such as activated T cells and monocytes.9-11 CXCL4 drives a broad spectrum of immune-modulatory effects in hematopoiesis and angiogenesis and has also been implicated in the pathology of a variety of inflammatory diseases (eg, atherosclerosis, inflammatory bowel disease, rheumatoid arthritis, systemic sclerosis) as well as hematologic diseases.12-15 Although CXCL4 has been under intense investigation for >30 years, its cellular functions, receptors, and their corresponding signaling pathways are still not fully understood and might even be cell type specific.16 At least 2 structurally different receptors, CXCR3-B and a chondroitin sulfate proteoglycan, are capable of binding CXCL4 and inducing specific intracellular signaling machinery. The current study sought to dissect the role of CXCL4 in MPN pathogenesis with specific emphasis on the MK–stromal interaction using genetic fate tracing, RNA sequencing, and pathway identification based on available data and bioinformatics pathway analysis. Our results indicate that CXCL4 plays a significant role in the induction of inflammation in MPN and has profibrotic effects, whereas hematopoietic loss of CXCL4 ameliorates the MPN phenotype and fibrosis.
Methods
Mouse experiments
Gli1CreERT2;tdTomato mice received tamoxifen injections (3×) before lethal irradiation and transplantation of ckit+ cells from wild-type (WT) or Cxcl4−/− expressing either thrombopoietin (ThPO) or empty vector (EV) control (n = 5 per group). For JAK2, CXCL4 overexpression and MPLW515L studies, WT or Cxcl4−/− ckit+ cells transduced with JAK2(V617F), MPLW515L, or pMIG control retrovirus (control: JAK2 EV) were transplanted into lethally irradiated B6.SJL recipients (n = 5-6 mice per group). At the end of the experiment, BM cells were isolated from crushed pelvis and hind legs. Cells were labeled with directly conjugated antibodies, anti-mouse: Gr1, Ter119, CD3, B220, CD11b, ckit, CD41, F4/80, CD48, CD41, Sca1, CD45.2, and CD150 (all 1:100). All samples were analyzed by using an FACSCantoII or FACS Aria (BD Biosciences, San Jose, CA). Hoechst solution was added (1:10 000) to exclude dead cells, and data were analyzed by using FlowJo software version 10 (Tree Star Inc, Ashland, OR).
In vitro experiments, RNA sequencing, and bioinformatics
Gli1+ stromal cells isolated from Gli1CreERt2;tdTomato mice were cultured in αMEM with 20% MSC fetal calf serum, 1% penicillin-streptomycin, 5 ng/mL endothelial growth factor, and 1 ng/mL fibroblast growth factor. For recombinant cytokine stimulation, Gli1+ cells were stimulated with recombinant human transforming growth factor-β (TGF-β; 10 ng/mL, InvivoGen, San Diego, CA) or recombinant murine interleukin-6 (IL-6; 100 ng/mL, PeproTech, Rocky Hill, NJ) for 72 hours. For coculture experiments, ckit+ BM from WT mice transduced with ThPO or EV virus were maintained in Cellgro media (Cellgenix, Freiburg, Germany) containing m-Scf (50 ng/mL) and m-Tpo (50 ng/mL). Transduced ckit+ cells were cocultured with 40 000 primary Gli1+ tdTomato+ cells for 72 hours and sorted on an FACS Aria for Gli1+ tdTomato+ and GFP+ ckit+ cells into TRIzol LS. Complementary DNA (cDNA) libraries were generated by using the Smart-Seq V4 Ultra-Low Input RNA Kit (Clontech Laboratories, Mountain View, CA), and amplified cDNA was further processed to generate Illumina compatible sequence-ready libraries (using the TruSeq Nano DNA Sample Prep Guide [Illumina, San Diego, CA]) that were pair-end sequenced (2 × 75 cycles) on a Hiseq2500 platform (Illumina). RNA data analysis is described in detail in the Extended Methods in the supplemental Files (available on the Blood Web site).
Statistical analysis
Statistical analysis was performed by using GraphPad Prism version 8 software (GraphPad Software Inc, San Diego, CA). Comparisons between 2 groups were performed by using an unpaired Student t test or Mann-Whitney U test as described in the figure legends. For multiple group comparisons, an analysis of variance with post hoc Tukey correction or a Kruskal-Wallis test was applied. Data are shown as mean ± standard error of the mean, and a value of P < .05 was considered significant.
Extended methods are available in the supplemental Files.
Results
Gli1+ stromal cells show fibrotic transformation and functional reprogramming after short exposure to fibrosis-inducing HSPCs
Our previous work showed that Gli1+ stromal cells are completely transcriptionally reprogrammed in progressed BM fibrosis as indicated by upregulation of a “matrisome signature” and significantly decreased expression of genes that are necessary for hematopoiesis support.7 Here, we hypothesized that fibrosis-inducing HSPCs in PMF induce the reprogramming of the stromal cell transcriptome, and we questioned if this occurs after short exposure of fibrosis-inducing HSPCs with Gli1+ stromal cells in vitro. To test this hypothesis, we used a model system in which ThPO is overexpressed in HSPCs to induce BM fibrosis, as it represents a robust, proof-of-principle model, and all mice develop fibrosis 8 to 10 weeks after transplantation.
To trace the fate of Gli1+ stromal cells, bigenic Gli1CreERt2; tdTomato mice received tamoxifen to induce cell-specific expression of the tdTomato fluorochrome. For coculture experiments, Gli1+ stromal cells isolated from bigenic Gli1CreERt2; tdTomato mice after tamoxifen pulse were cocultured with c-kit+ HSPCs transduced with a ThPO-overexpressing vector or its EV control. TdTomato+Gli1+ cells and GFP+ ckit+ HSPCs were separated for subsequent RNA isolation by sort-purifying single, viable GFP+ and tdTomato+ cells using fluorescence-activated cell sorting. As expected, sorted HSPCs expressing ThPO clustered distinctly from control HSPCs in principal component analysis and hierarchical cluster analysis (Figure 1A). Interestingly, the analysis indicated that Gli1+ cells exposed to ThPO-expressing ckit+ HSPCs for only 72 hours were already drastically distinct from Gli1+ cells exposed to control ckit+ HSPCs (Figure 1B). These data suggest that exposure to fibrosis-inducing HSPCs indeed leads to early transcriptional reprogramming of Gli1+ stromal cells.
Figure 1.
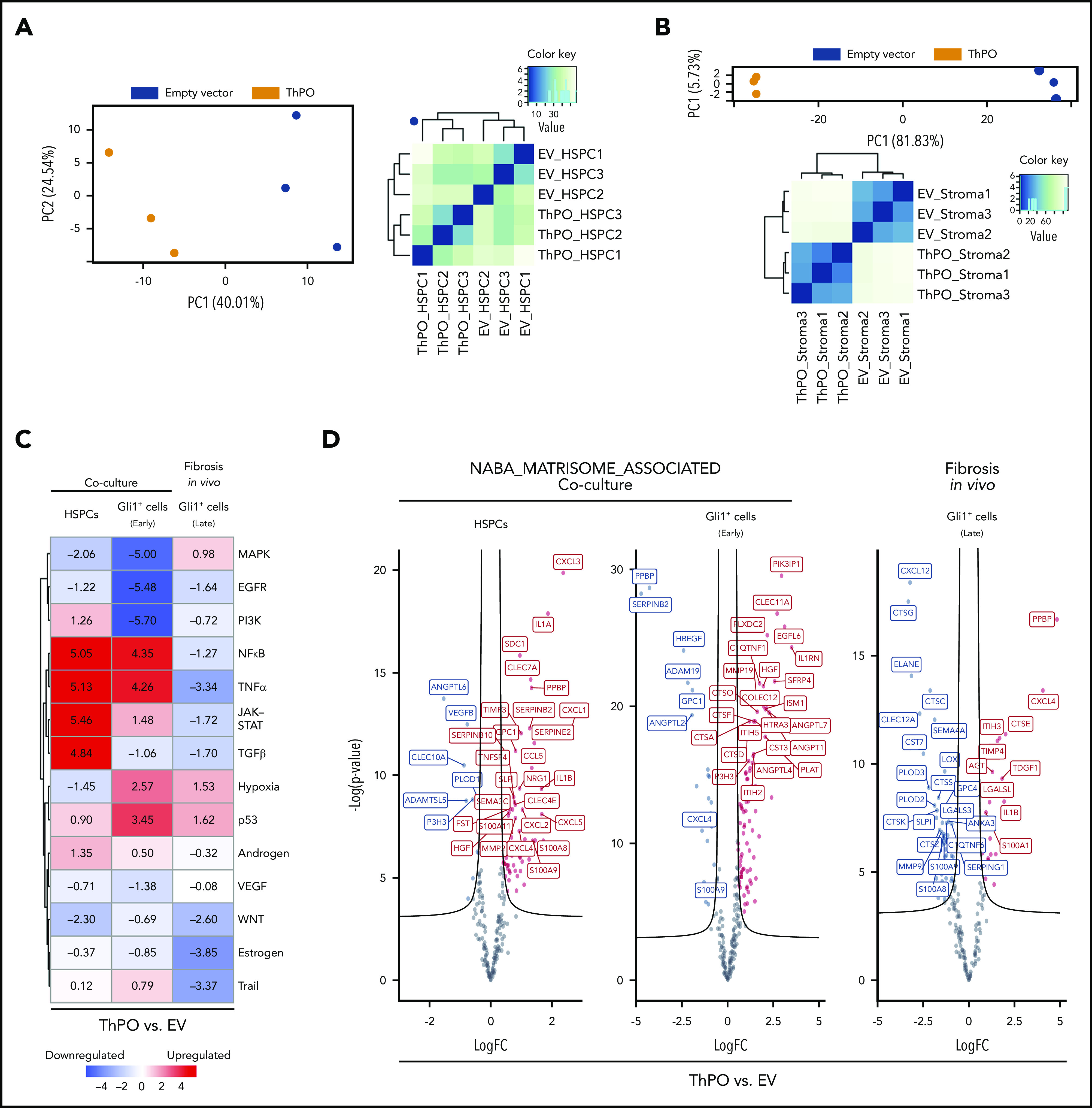
Fibrosis-driving cells are transcriptionally reprogrammed in early stages of BM fibrosis and exhibit induction of inflammatory pathways. HSPCs (ckit+) were transduced with ThPO-overexpression cDNA or EV and cocultured for 72 hours with primary Gli1+ stromal cells (TdTomato+; n = 3 per group). Ckit+ HSPCs and Gli1+ stromal cells were then sort-purified as GFP+ and lin–GFP-tdTomato+, respectively. To analyze fibrosis in vivo, bigenic Gli1CreERt2;tdTomato mice were lethally irradiated 10 days after the last tamoxifen dose and received c-kit–enriched HSPCs from WT littermates expressing either ThPO cDNA (n = 5, 3 male mice) or control cDNA (control, n = 5, 3 male mice). Mice were euthanized at 70 days after transplantation. Gli1+ cells were sort-purified as lin–GFP+ tdTomato+ and subjected to RNA sequencing. (A) Principal component analysis and heatmap representation with hierarchical clustering of HSPCs transduced with either ThPO or EV control. (B) Principal component analysis and heatmap representation with hierarchical clustering of sort-purified Gli1+ stromal cells previously cocultured with either ThPO- or EV-transduced HSPCs. (C) PROGENy pathway analysis of sort-purified HSPCs transduced with ThPO-overexpressing vector (BM fibrosis induction) in a coculture setting with Gli1+ stromal cells (coculture HSPCs, left panel), sort-purified Gli1+ stromal cells with previous short exposure to ThPO (or EV) HSPCs (Gli1+ cells “early,” middle panel), and sort-purified Gli1+ stromal cells isolated from a ThPO-overexpression murine model with severe BM fibrosis (fibrosis in vivo, Gli1+ cells, right panel). All samples are compared with their respective EV control. (D) NABA-matrisome associated genes in HSPCs (left panel), primary Gli1+ stromal cell from coculture (middle panel), and primary Gli1+ stromal cells from murine model of ThPO-induced BM fibrosis (right panel). Curved lines on the volcano plots are asymptotic to a value of P = .05 (-log) and a fold-change = .10 of the maximum observed fold change in the signature.
We performed a Pathway RespOnsive GENes (PROGENy) analysis17 to understand which signaling pathways are mostly affected in: (1) fibrosis-driving (ThPO) HSPCs (compared with control); (2) Gli1+ stroma exposed to fibrosis-inducing HSPCs (early); and (3) Gli1+ stromal cells in progressed ThPO-induced BM fibrosis (late; both compared with Gli1+ cells exposed to HSPCs expressing an EV control). By comparing the differential expression signature of our data with 14 signatures of pathway perturbation,18 we noticed that the second most extreme estimated deregulation was an increased activity of the JAK-STAT pathway in fibrosis-inducing ThPO HSPCs compared with control HSPCs (activity z score >5, uncorrected P value = 1.9e-08) (Figure 1C; supplemental Figure 1). This validates our model, as binding of the ThPO ligand to the MPL receptor activates the JAK/STAT pathway. In addition, profibrotic TGF-β signaling was significantly induced in fibrosis-driving ThPO HSPCs compared with control HSPCs. Gli1+ cells exposed to ThPO-expressing HSPCs were characterized by a hypoxic signature, which is a common hallmark of fibrosis and tissue repair.19 Interestingly, Gli1+ cells in the early condition as well as fibrosis-inducing HSPCs exhibited a significant induction of inflammatory pathways such as NF-κb and tumor necrosis factor-α (TNF-α), which were downregulated in the late condition, suggesting that inflammation is a hallmark feature of stroma in BM fibrosis initiation and is induced by HSPCs.
We then wondered how genes that encode ECM-associated proteins, such as ECM regulators and secreted factors that are crucial for the fibrotic transformation, are expressed in ckit+ HSPCs and stromal cells in both “early” and “late” conditions.
We used a published matrisome-associated gene signature20 and found significant enrichment (using piano in R [Väremo et al21]) in early HSPCs and Gli1+ stromal cells (consensual P value <1e-4) (Figure 1D). In particular, CXC chemokines such as CXCL1, CXCL3, and CXCL4, all known to play a role in fibrosis,22 were significantly upregulated in HSPCs (respective P values of 6e-6, 2e-9, and 6e-4). The strong upregulation of matrisome-associated genes in “early” Gli1+ stromal cells indicated that the fibrotic transformation is likely initiated after short exposure to fibrosis-inducing HSPCs, in line with functional analysis of the stromal cells in culture showing upregulation of α smooth muscle actin (αSMA), collagen 1α1, and fibronectin transcripts after 72 hours (supplemental Figure 1B).
CXCL4 intensity and spatial localization marks the progression of matrix remodeling and BM fibrosis in patients with MPN
Our data confirmed that CXCL4 was significantly upregulated in fibrosis-driving HSPCs and in “late” but not “early” Gli1+ stromal cells. Thus, we wondered whether: (1) CXCL4 in HSPCs is an initiator of the fibrotic transformation; (2) CXCL4 expression in stromal cells is a marker for fibrosis progression; and (3) CXCL4 plays a role in the inflammation seen in the initiation phase. We stained BM biopsy specimens from control patients and patients with MPN and varying grades of fibrosis for CXCL4. We defined 4 grades of CXCL4 expression in the BM (Figure 2A-B). Grade 1 CXCL4 staining is MK specific. In grade 2, MKs are strongly stained, and faint staining is also detected in endosteal and perivascular stromal cells. In grade 3, strong CXCL4 positivity is detected in endosteal and perivascular stromal cells scattered throughout the BM. In the highest grade (4), CXCL4 staining is observed in dense stromal cell patches. Significantly higher grades of CXCL4 staining were observed in patients with MPN compared with control subjects (Figure 2C), together with an increase in CXCL4 grade with increasing myelofibrosis (reticulin) grades (Figure 2D). Interestingly, intermediate grades were specific to MPN patients without fibrosis in the reticulin staining, suggesting that CXCL4 marks remodeling of the microenvironment even in the absence of fibrosis. We observed decreased hemoglobin levels and increases in the number of white blood cells (WBCs) correlating with increases in CXCL4 grade (Figure 2E-F), suggesting that CXCL4 grade progression marks fibrotic progression and the progressive loss of normal hematopoiesis. Together, this shows that CXCL4 is first detected in hematopoietic cells (early stage) such as MKs, with a subsequent shift of localization to stromal cells as fibrosis progresses (late stage), indicating a significant interaction between MKs and stromal cells of the BM during fibrotic transformation.
Figure 2.
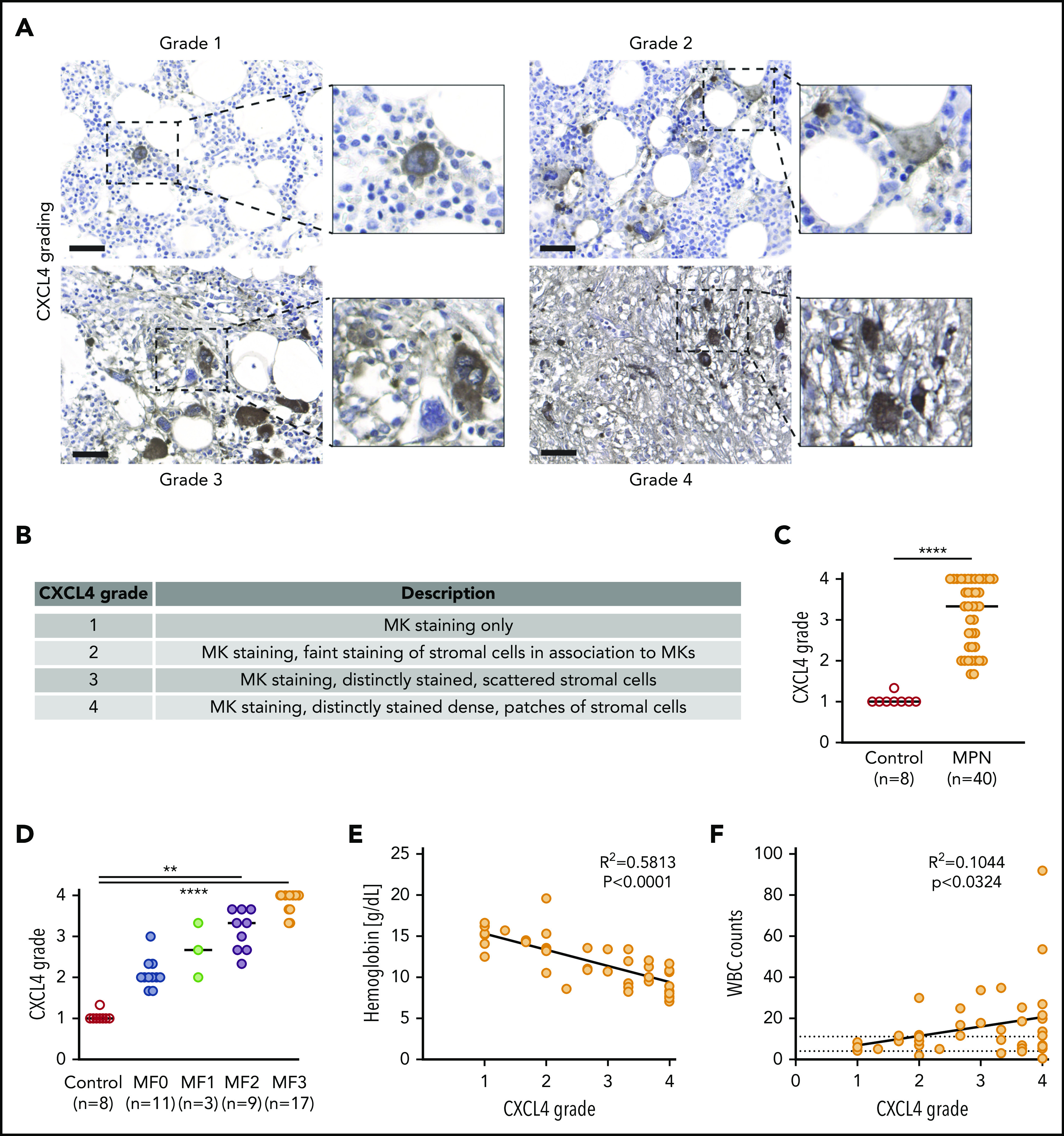
The spatial expression of CXCL4 marks the progression of BM fibrosis in patients. (A) Representative images of CXCL4 grading (1-4) in human BM biopsy samples. Original magnification ×20. Scale bar, 50 μm. (B) Description of the main features of CXCL4 grading scale in human BM biopsy samples. (C) Quantification of the CXCL4 grade in MPN and controls. Data shown as median; Mann-Whitney U test. (D) Quantification of the CXCL4 grade compared with myelofibrosis grade (reticulin staining). Kruskal-Wallis test followed by Dunn’s post hoc test. (E) Comparison of hemoglobin levels vs CXCL4 grade. (F) Comparison of WBCs vs CXCL4 grade. Controls, n = 8; MPN, n = 40. Patient characteristics are shown in supplemental Tables 1 and 2. **P < .01, ****P < .0001.
Knockout of CXCL4 in HSPCs ameliorates the MPN phenotype
Given that the CXCL4 expression is first significantly increased in HSPCs (and MKs) and in progressed disease also in the stroma, we next questioned whether the absence of CXCL4 from the HSPC compartment affects the MPN phenotype. We thus combined genetic fate tracing experiments in a murine model of ThPO-induced myelofibrosis with knockout of Cxcl4 in HSPCs. Bigenic Gli1CreERt2+/−;tdTomato+/− recipient mice were pulsed with tamoxifen to induce expression of the tdTomato fluorochrome in Gli1+ cells, and subsequently BM-transplanted with either WT or Cxcl4−/− ckit+ HSPCs overexpressing ThPO or its EV control (supplemental Figure 2A-C).
CXCL4 knockout in HSPCs in ThPO-induced fibrosis partially restored the progressive anemia with decreased hemoglobin levels commonly observed in ThPO-induced BM (Figure 3A), a measure that often correlates with the degree of fibrosis and the progressive replacement of erythropoiesis in mice.7 In addition, CD71+ erythroid cells in peripheral blood and BM were also partially normalized, suggesting that the absence of CXCL4 preserves erythropoiesis in ThPO-induced fibrosis.
Figure 3.
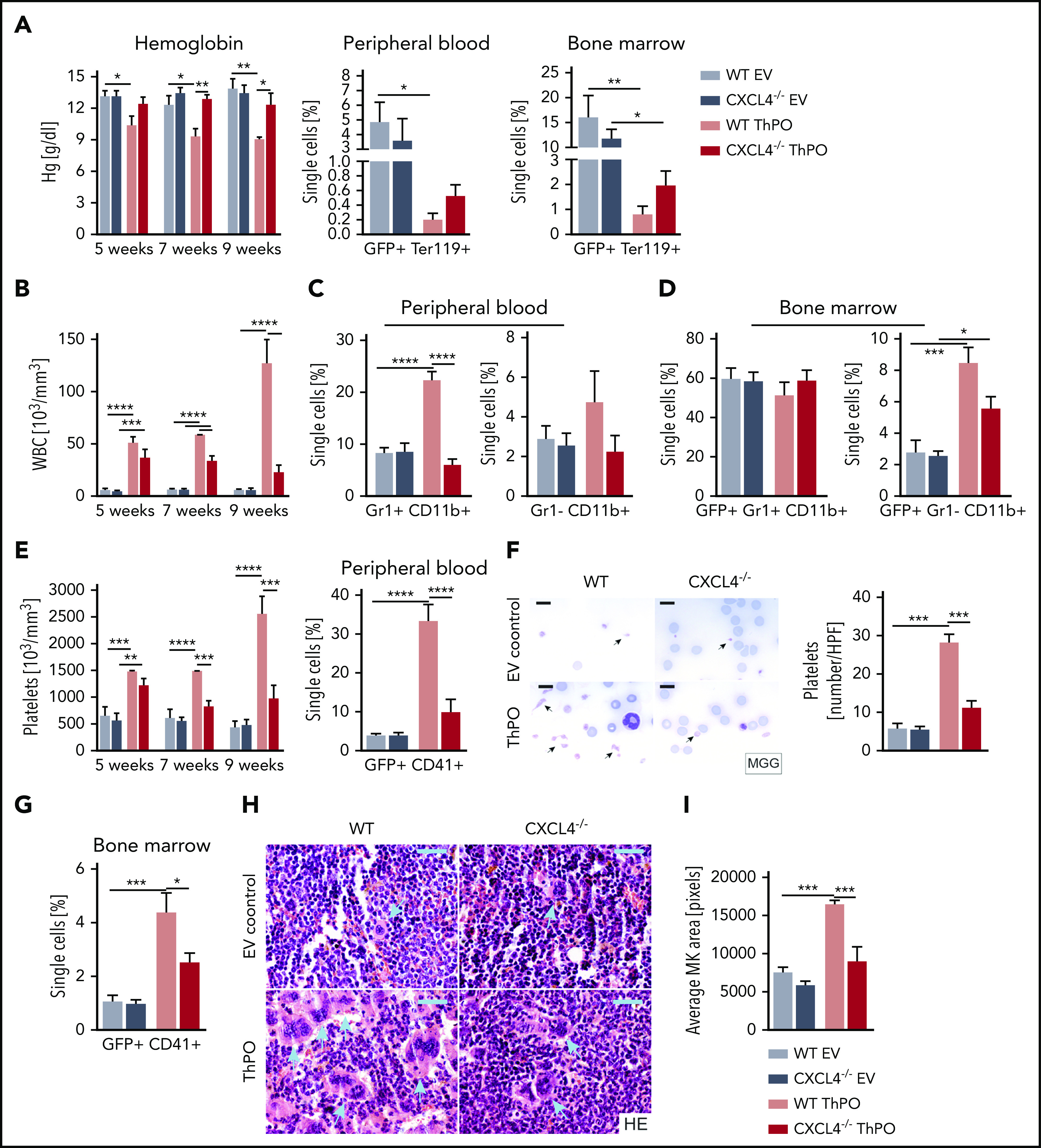
Loss of Cxcl4 in hematopoietic cells ameliorates the MPN phenotype and restores MK and platelet abnormalities. (A) Hemoglobin (Hg) counts monitored over time, together with the frequency of GFP+ Ter119+ cells in peripheral blood and BM 9 weeks’ posttransplant (euthanasia). (B) WBC counts are monitored over time from peripheral blood. (C) Flow cytometric quantification of Gr1+ CD11b+ (granulocytes) and Gr1– CD11b+ (monocytes) at euthanasia in peripheral blood. (D) Flow cytometric quantification of GFP+ Gr1+ CD11b+ (granulocytes) and GFP+ Gr1– CD11b+ (monocytes) at euthanasia in BM. (E) Platelet counts are monitored at 5, 7, and 9 weeks’ posttransplant. Flow cytometric quantification of GFP+ CD41+ cells in peripheral blood. (F) May-Grünwald-Giemsa (MGG) staining and quantification of the number of platelets per high-powered field in murine blood smears. Original magnification ×100. Scale bar, 10 μm. (G) Flow cytometric quantification of GFP+ CD41+ (MKs) in BM. (H) Hematoxylin and eosin (HE) staining of 4-μm BM sections (femur) with particular focus on the size and morphology of MKs (blue arrows). Original magnification ×40. Scale bar, 50 μm. (I) Quantification of the mean area of MKs in BM. n = 5 mice/group, 3 males. Data are shown as mean ± standard error of the mean, 1-way analysis of variance followed by Tukey’s post hoc test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
The typical myeloproliferation seen in ThPO-induced BM fibrosis, as indicated by significantly increased WBC counts, was further completely normalized in the absence of CXCL4 in HSPCs (Figure 3B). Specifically, flow cytometric analysis revealed a significant reduction in the frequency of Gr1+CD11b+ granulocytes and Gr1–CD11b+ monocytes in the peripheral blood (Figure 3C) and Gr1–CD11b+ monocytes in the BM in the absence of CXCL4 in ThPO-induced fibrosis (Figure 3D).
A hallmark feature in ThPO-induced BM fibrosis is significant thrombocytosis. CXCL4 knockout in HSPCs in ThPO-induced fibrosis normalized increased platelet levels and reduced the frequency of CD41+ cells in the peripheral blood (Figure 3E). Interestingly, blood smears showed dysplastic platelets in ThPO-induced fibrosis that were eliminated in the absence of CXCL4 (Figure 3F). In line with the presence of dysplastic platelets in the blood, enlarged, dysplastic, and hyperlobulated MKs, which are pathognomonic for MPN, were also normalized in their frequency, morphology, and size in the BM in the absence of CXCL4 in HSPCs (Figure 3G-I). In summary, the absence of CXCL4 in HSPCs in ThPO-induced BM fibrosis almost completely ameliorated the MPN phenotype and indicated more preserved hematopoiesis in the BM.
Knockout of CXCL4 in HSPCs decelerates the progression of BM fibrosis
Because the analysis of hematopoiesis suggested that the absence of CXCL4 ameliorates hallmark features of the MPN phenotype and preserves hematopoiesis in the BM, we next questioned whether BM fibrosis was reduced. Total cell counts of the BM revealed significantly reduced cell numbers in ThPO-induced BM fibrosis in the presence and absence of CXCL4, although the numbers were slightly higher in the absence of CXCL4 (Figure 4A), in line with decreased fibrosis. This notion was supported by reticulin staining, which revealed severe, grade 2 to 3 fibrosis in WT ThPO mice, characterized by abundant, thick fiber crossing and severe osteosclerosis (Figure 4B). In the absence of CXCL4, reticulin fibers were less densely packed and more diffuse, with less intercrossing and more parallel fibers, characteristic of a less severe, grade 1 to 2 reticulin fibrosis. Importantly, the cellular composition of the BM was normalized in the absence of CXCL4 in ThPO-induced BM fibrosis as seen in the hematoxylin and eosin staining, in line with reduced bone marrow fibrosis and reduced MPN-characteristic myeloproliferation. In addition, CXCL4 immunohistochemistry of femurs revealed strong CXCL4 positivity in dysplastic MKs, in the endosteal stroma and in stromal patches scattered throughout the BM, exactly as seen in patients with progressed fibrosis (Figure 2). In contrast, the CXCL4 expression was markedly reduced in MKs and associated stromal cells in Cxcl4−/− ThPO animals (supplementary figure 3).
Figure 4.
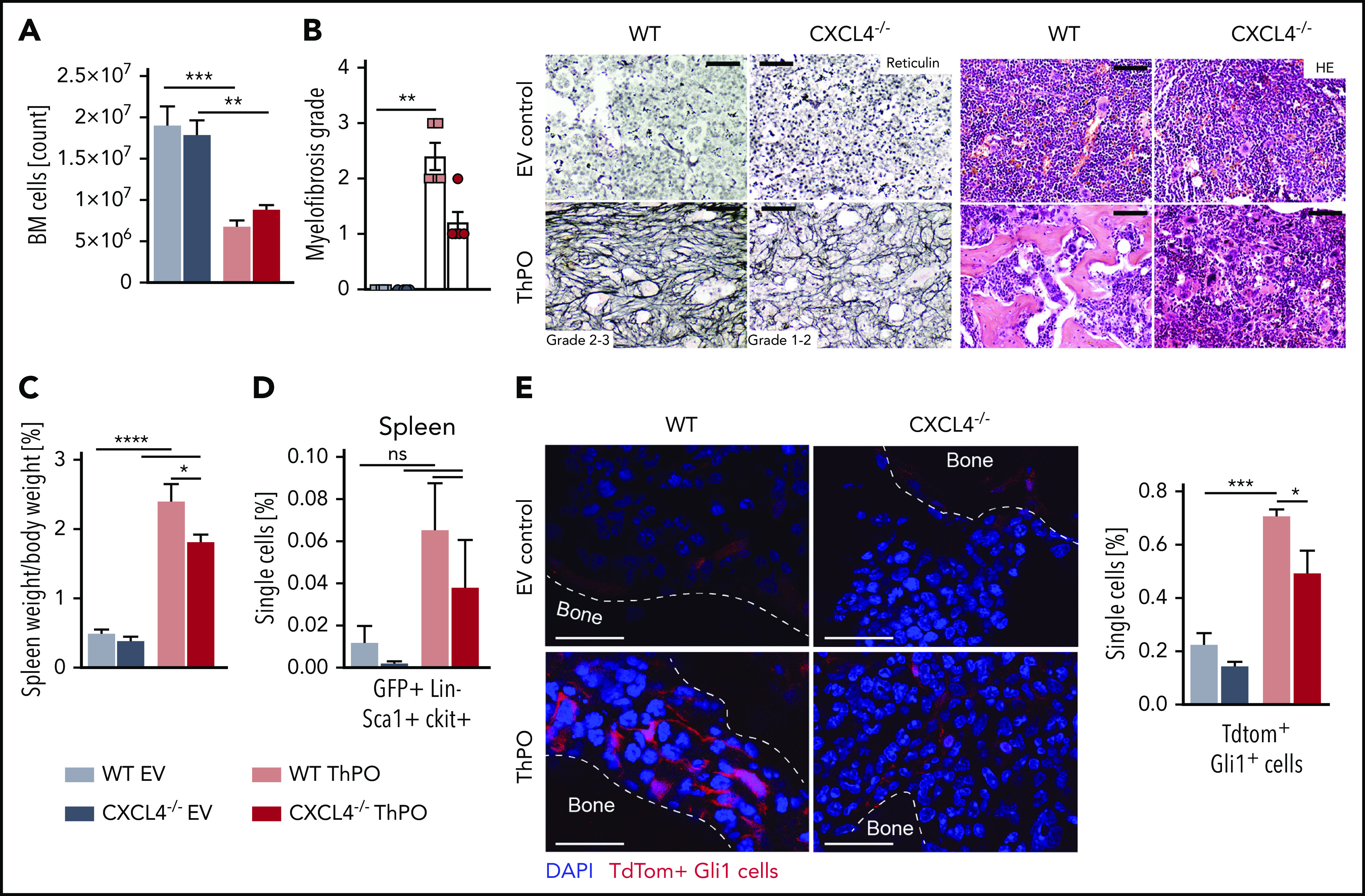
CXCL4 knockout in hematopoietic cells in ThPO-induced PMF decelerates the progression of fibrosis. (A) BM cellularity at 9 weeks’ posttransplantation. (B) Myelofibrosis grading and representative images of reticulin staining of the BM in ThPO-induced and control mice, and hematoxylin and eosin (HE) staining highlighting osteosclerosis in WT ThPO mice. Kruskal-Wallis multiple comparison test. Scale bar, 100 μm. (C) Spleen weight relative to body weight at 9 weeks’ posttransplantation. (D) Flow cytometric analysis of the frequency of GFP+LinlowSca1+ ckit+ cells in the spleen. (E) Representative immunofluorescence images and flow cytometric analysis and quantification of Gli1+tdTomato+ fibrosis-driving stromal cells in the BM. Scale bar, 50 μm. Data are shown as mean ± standard error of the mean, n = 5 per group, 1-way analysis of variance followed by Tukey’s post hoc test. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant.
In line with the severe BM fibrosis in WT ThPO mice, the spleen weight significantly increased (Figure 4C), and mice exhibited extramedullary hematopoiesis indicated by detection of GFP+ lineagelow Sca1+ckit+ HSPCs in the spleen (Figure 4D). Both spleen weight and extramedullary hematopoiesis were reduced in the absence of CXCL4 in ThPO-induced BM fibrosis, further suggesting a reduction although not complete resolution of BM fibrosis.
Importantly for therapeutic purposes, the knockout of CXCL4 in HSPCs in control mice did not have (negative) effects on erythropoiesis, myelopoiesis, or megakaryopoiesis. Only within the HSPC compartment in BM was an increase observed in the frequency of GFP+linlowSca1+ckit+ (LSK), GFP+linlowSca1+ckit+CD48+CD150– (multipotent progenitor cells), GFP+linlowSca1+ckit+CD48–CD150– (short-term HSCs) and GFP+linlowSca1+ckit+CD48–CD150+ (long-term HSCs) in Cxcl4−/− EV mice compared with WT EV (supplemental Figure 4A). This is consistent with data from Bruns et al,23 who highlighted the role of CXCL4 as a regulator of HSC quiescence and HSC cell cycle activity, while its absence leads to HSC cycling and HSC proliferation.
We therefore wondered if the improved MPN phenotype observed in CXCL4 knockout conditions was due to decreased quiescence and exhaustion of HSCs over time. We thus re-transplanted whole BM cells from primary WT ThPO or EV or Cxcl4−/− ThPO or EV animals into lethally irradiated secondary recipients for an additional 32 weeks. Importantly, we observed no difference in BM engraftment or repopulation in either CXCL4−/− control or fibrosis condition (compared with WT; CXCL4+/+) for the complete duration of the study, indicating that CXCL4−/− cells perform equally well and that these cells do not exhaust over time, although CXCL4 affects proliferation. These data indicate that HSC exhaustion is not the primary reason for the amelioration of the MPN phenotype observed in Cxcl4−/− ThPO mice (supplemental Figure 4B-C).
We previously showed that Gli1+ stromal cells are key drivers of BM fibrosis as they differentiate toward myofibroblasts and deposit reticulin fibers and ECM. We questioned whether the absence of CXCL4 in HSPC affects the activation of the genetically tagged Gli1+ cells. Importantly, we observed a significant decrease in the frequency of tdTomato+Gli1+lineagelow stromal cells invading the marrow space in the absence of CXCL4 in HSPCs, indicating a decrease in Gli1+ cell activation, recruitment, and migration (Figure 4E), in line with the decelerated BM fibrosis.
Hematopoietic CXCL4 loss ameliorates the disease phenotype in the clinically relevant JAK2(V617F) and MPLW515L models of MPN
Several somatic mutations responsible for clonal stem cell expansion have been described in MPN. The 3 currently recognized “driver” mutations in PMF are JAK2, CALR, and MPL, and they are typically found to be mutually exclusive.24 Because ∼60% of patients with PMF harbor a JAK2(V617F) mutation, we wondered if we could recapitulate these findings in a JAK2(V617F)-induced murine model of MPN/PMF. We introduced the JAK2(V617F) mutation or JAK2WT control into WT or Cxcl4−/− ckit+-enriched HSPCs and transplanted them into lethally irradiated WT recipients (supplemental Figure 5A). Interestingly, the most significant effect of CXCL4 knockout was also observed in platelets and MKs. WT mice transplanted with JAK2(V617F) progressively developed thrombocytosis over a period of 26 weeks, whereas Cxcl4−/− JAK2(V617F) mice only exhibited a mild elevation in platelet counts (Figure 5A). Importantly, the MK morphology was completely restored in the absence of CXCL4 in the JAK2(V617F)-induced model of MPN (Figure 5B), suggesting that the increased CXCL4 expression in MPN has a strong effect on the pathognomonic MK phenotype and thus positively influences fibrosis, as we again observed a reduction of the reticulin grade (Figure 5C-E).
Figure 5.
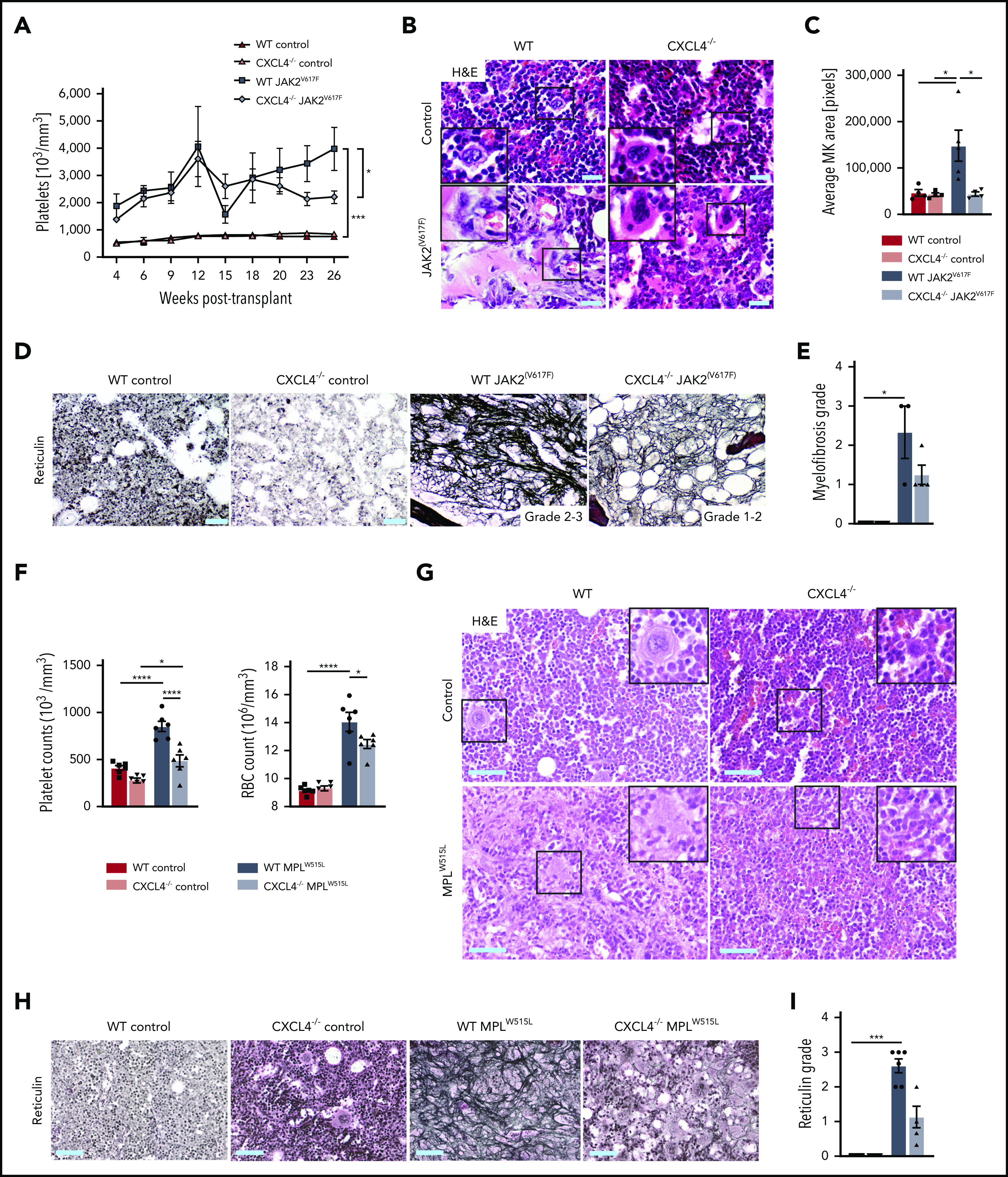
CXCL4 knockdown reduces thrombocytosis and fibrosis grade in JAK2(V617F) and MPLW515L murine models of PMF. C57BL/6 recipient mice received lethal irradiation at 10 weeks of age followed by either WT or Cxcl4−/− HSPCs transduced with JAK2(V617F), MPLW515L, or WT control retroviral vectors (n = 5 per group for JAK2(V617F) experiment; n = 6 per group for MPLW515L experiment). Mice were euthanized at 182 days after transplantation for the JAK2(V617F) experiment and 21 days’ posttransplantation for the MPLW515L experiment. (A) Platelet counts from peripheral blood. (B) Representative images of hematoxylin and eosin (H&E) staining in femurs. Original magnification ×20; scale bar, 100 μm. (C) Quantification of the mean area of MKs in control or JAK2(V617F)-driven BM. (D) Representative images of reticulin staining in control or JAK2(V617F)-driven BM. Original magnification ×20; scale bar, 100 μm. (E) Quantification of myelofibrosis grade based on reticulin staining in control or JAK2(V617F)-mutated BM (Kruskal-Wallis multiple comparisons test). (F) Platelet and red blood cell (RBC) counts in peripheral blood. (G) Representative images of H&E staining in femurs. Original magnification ×20; scale bar, 100 μm.(H) Representative images of reticulin staining in femurs. Original magnification ×20; scale bar, 100 μm. (I) Quantification of myelofibrosis grade based on reticulin staining in control or MPLW515L-mutated BM (Kruskal-Wallis multiple comparisons test). Data are shown as mean ± standard error of the mean, 1-way analysis of variance followed by Tukey’s post hoc test unless otherwise indicated. *P < .05, ***P < .001, ****P < .0001.
To further confirm the involvement of CXCL4 in MPN/PMF, we performed additional experiments in a MPLW515L-induced murine model of PMF. We introduced the MPLW515L mutation or its EV (pMIG EV) in WT or Cxcl4−/− ckit+-enriched HSPCs and transplanted into lethally irradiated recipients (supplemental Figure 5D). At the time of euthanasia at 21 days’ posttransplantation, we observed a significant induction of thrombocytosis and increased red blood cell counts in WT MPLW515L, which were normalized in the absence of Cxcl4 (Figure 5F). Our data showed a significant increase in BM remodeling and reticulin fiber deposition in both BM and spleen in WT MPLW515L, whereas Cxcl4−/− MPLW515L mice showed no apparent MK pathology and a significant reduction in reticulin grade (Figure 5G-I; supplemental Figure 5G). Taken together, these 3 PMF models highlight the importance of CXCL4 in the initiation of BM fibrosis and suggest that targeting CXCL4 in MPN might be an attractive strategy to ameliorate the MPN phenotype.
Our data indicate that CXCL4 plays an important role in the MPN pathogenesis but may not be the sole driver of BM fibrosis. Consequently, we wondered whether increased CXCL4 expression in HSPCs was sufficient to induce fibrosis. We thus transduced WT ckit+ HSPCs with a retroviral vector overexpressing Cxcl4 or EV control (supplemental Figure 6A-B) and transplanted into lethally irradiated WT recipients (supplemental Figure 6C). Critically, CXCL4 overexpression in HSPCs led to an increase in the number of platelets, with no marked signs of fibrosis at 26 weeks’ posttransplant (supplemental Figure 6D). We observed hyperlobulated MKs with CXCL4 overexpression but did not detect an increase in their frequency in the BM (supplemental Figure 6E-F). This outcome suggests that the primary effect of CXCL4 overexpression after 26 weeks is on MKs, acting as a mediator, although we cannot exclude that the kinetics of fibrosis development are slower than in other models.
Hematopoietic CXCL4 knockout in MPN resulted in decreased JAK/STAT activity in MKs and stromal cells and downregulation of interferon target genes in MKs
To determine the exact role of CXCL4 in MKs and their effect on stromal cells, we performed transcriptional analysis in both cell types in fibrosis and control conditions. Because hematopoietic loss of CXCL4 resulted in significantly reduced Gli1+ stromal cell activation and a significantly ameliorated phenotype of MKs in ThPO-induced fibrosis, we sort-purified tdTom+ Gli1+ stromal cells and GFP+ CD41+ cells (MKs) from the BM of control (EV) and fibrotic (ThPO) mice; we then performed RNA sequencing to dissect the interaction between Gli1+ stromal cells exposed to CXCL4 WT or knockout MKs.
We performed pathway analysis with PROGENy and transcription factor analysis with DoRothEA25 to gain a better understanding of the fibrosis-inducing effect of WT or CXCL4−/− MKs on stromal cells (supplemental Figure 7A). Loss of Cxcl4 in HSPCs/MKs resulted in decreased JAK/STAT pathway activity in both MKs and stromal cells in fibrosis (ThPO overexpression compared with EV) (Figure 6A). Activation of JAK/STAT signaling is one of the mechanistic hallmarks of MPN and is observed in every patient with MPN, regardless of founding driver mutation or clinical diagnosis.26 Specifically, in MKs, the knockout of CXCL4 led to the downregulation of interferon-inducible target genes such as Ifi27, Ifi35, Usp18, and Ifitm1 (Figure 6B). In particular, Stat1 was significantly downregulated (Figure 6C). Previous research by Chen et al27 showed that STAT1, a known effector of interferon signaling, promotes MK differentiation and polyploidization, particularly in patients with essential thrombocythemia, and they are primarily characterized by megakaryocytic and platelet abnormalities that are reflected in our ThPO-induced BM fibrosis model. Our data thus suggest that the amelioration of MK abnormalities and dysplasia observed in the absence of CXCL4 can be attributed to a decreased interferon response and decreased Stat1 activation.
Figure 6.
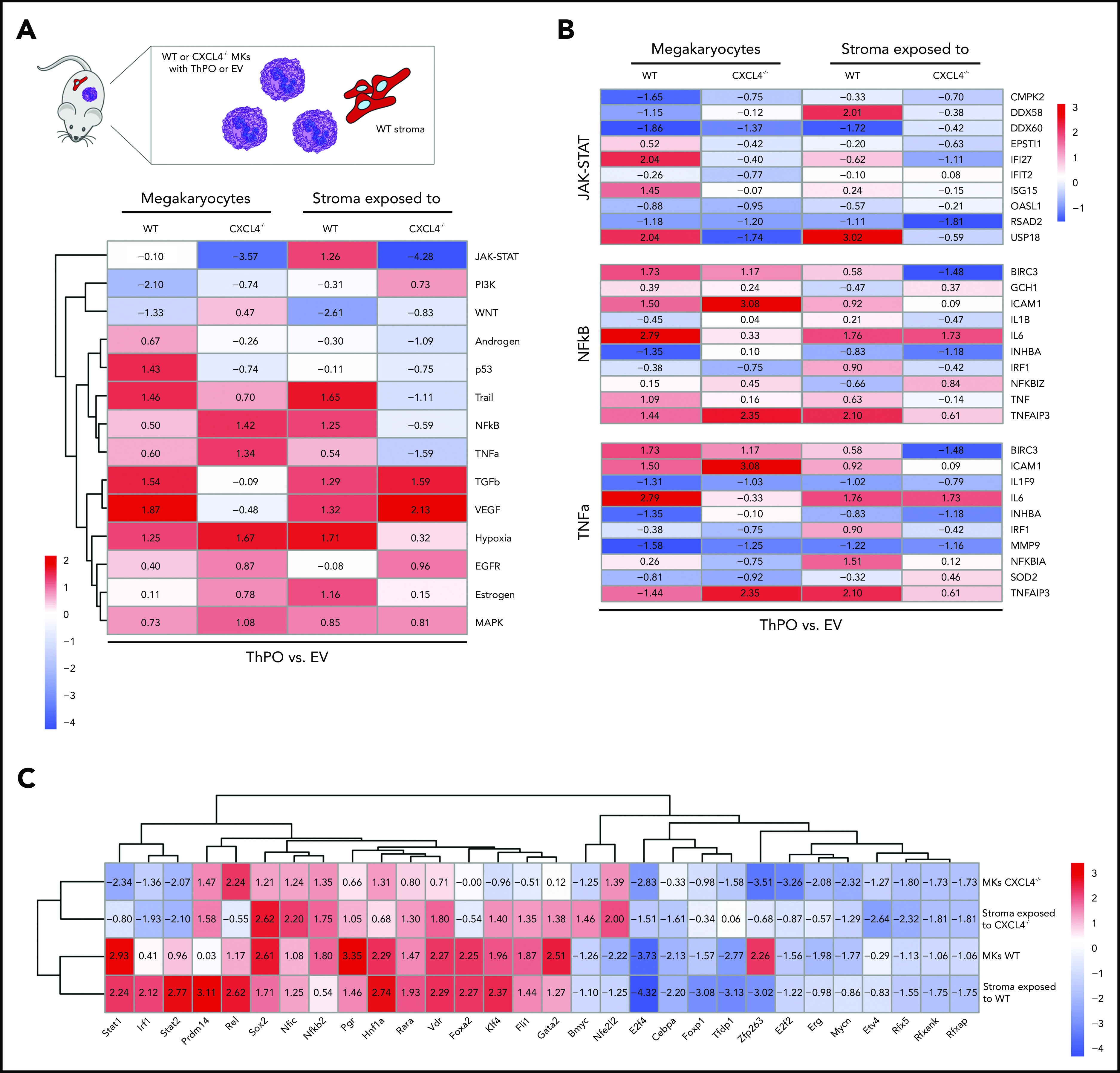
Hematopoietic knockout of CXCL4 leads to reduced expression of profibrotic pathways and reduced inflammation. (A) Schematic of cell populations analyzed. CD41+ MKs are either WT or CXCL4 knockout (GFP+) and transduced with either ThPO-inducing fibrosis or EV as a control. tdTom+ stromal cells were exposed to CXCL4 expressing or knockout hematopoietic cells/MKs in control or fibrosis. Heatmap representation of PROGENy analysis of sort-purified MKs and Gli1+ stromal cells from WT ThPO or Cxcl4−/− ThPO mice compared with EV control mice. (B) Heatmap representation of specific PROGENy pathways such as JAK-STAT, NF-κB, and TNF-α. (C) Heatmap representation of Discriminant Regulon Enrichment Analysis of transcription factors of sort-purified MKs and Gli1+ stromal cells from WT ThPO or Cxcl4−/− ThPO mice compared with EV controls.
Historically, MKs were believed to be fibrosis-inducing cells in PMF/BM fibrosis, and we observed an amelioration of the fibrosis grade in the absence of CXCL4. These observations were confirmed as the hallmark profibrotic pathway TGF-β was upregulated in WT MKs in ThPO-induced fibrosis but not in the absence of CXCL4 (supplemental Figure 7B). Moreover, interleukin-6 (IL-6), a cytokine previously associated with PMF,28,29 was upregulated in WT but not in Cxcl4−/− MKs.
The absence of CXCL4 in hematopoietic cells/MKs resulted in downregulation of inflammatory pathways such as Trail, NF-κb, and TNF-α in stromal cells in BM fibrosis (ThPO compared with EV) (Figure 6A). Interestingly, stromal cells exposed to CXCL4−/− and WT hematopoietic cells were comparable in their TFG-β pathway activities. However, a closer look at the responsive genes of TGF-β pathway activity (supplemental Figure 7B) showed that Col4a1 was overexpressed in stromal cells exposed to WT MKs in ThPO-induced fibrosis but not in the absence of CXCL4. Col4a1, the collagen, type IV, α1 fiber subtype, has previously been identified as a key factor of fibrosis progression.30
To validate the inflammatory landscape in fibrotic mice, we analyzed blood plasma of JAK2(V617F) and MPLW515L mice for common inflammatory cytokines using a bead-based immunoassay (supplemental Figure 5B-C). WT JAK2(V617F) mice showed a global increase in cytokines, specifically increases in IL-1α, monocyte chemotactic protein 1, and TNF-α protein levels, which were reduced in Cxcl4−/− JAK2(V617F) mice (supplemental Figure 5C). Similarly, we observed a significant reduction in IL-1α protein levels in the plasma of Cxcl4−/− MPLW515L mice compared with WT MPLW515L animals, as well as decreased WBC counts (supplemental Figure 5E-F), further confirming that CXCL4 acts as an inflammatory “mediator” in BM fibrosis initiation in these models.
The increased hypoxic signature in stromal cells in ThPO-induced fibrosis was only observed when exposed to WT MKs and is consistent with the severe reticulin fibrosis observed in the BM in the presence of CXCL4. Interestingly, mesenchymal stromal cells in hypoxic conditions survive and proliferate at greater rates than in normoxic conditions, with additional hypoxic preconditioning leading to the release of proangiogenic factors such as vascular endothelial growth factor and IL-6, which may further contribute to the fibrotic transformation.31-33
To better characterize the effect of the CXCL4 knockout itself, we also performed transcription factor analysis on a differential expression signature comparing CXCL4 knockout and WT MKs and stromal cells exposed to those for each of the conditions. Remarkably, this comparison showed again that the JAK-STAT pathway activity, and related transcription factors, was strongly reduced in the absence of CXCL4 in ThPO-induced BM fibrosis in both stromal cells and MKs (Figure 6C).
CXCL4 knockout ameliorates increased expression of the profibrotic IL-6
Because IL-6 was one of the highest upregulated cytokines in MKs in the WT ThPO condition, and significantly reduced by CXCL4 knockout, we hypothesized that IL-6 plays a CXCL4-dependent role in the fibrotic transformation and inflammation in stromal cells. We questioned whether exogenous IL-6 had a direct effect on fibrosis, specifically on the activation and differentiation of Gli1+ stromal cells into ECM-producing myofibroblasts. Sustained in vitro stimulation of primary Gli1+ stromal cells with TGF-β (strong inducer of profibrotic phenotype and reduced in MKs in ThPO-induced fibrosis after CXCL4 knockout) or IL-6 significantly increased αSMA expression (Figure 7A). Critically, IL-6– and TGF-β–stimulated cells displayed myofibroblast morphology and αSMA+ stress fibers (Figure 7B). Together, these data suggest an involvement of CXCL4 in a profibrotic IL-6 axis in which proinflammatory IL-6 is highly upregulated in MK during fibrosis and induces myofibroblast differentiation of Gli1+ stromal cells. Hematopoietic loss of CXCL4 resulted in reduced IL-6 expression by MKs with subsequently reduced myofibroblast differentiation and fibrosis.
Figure 7.
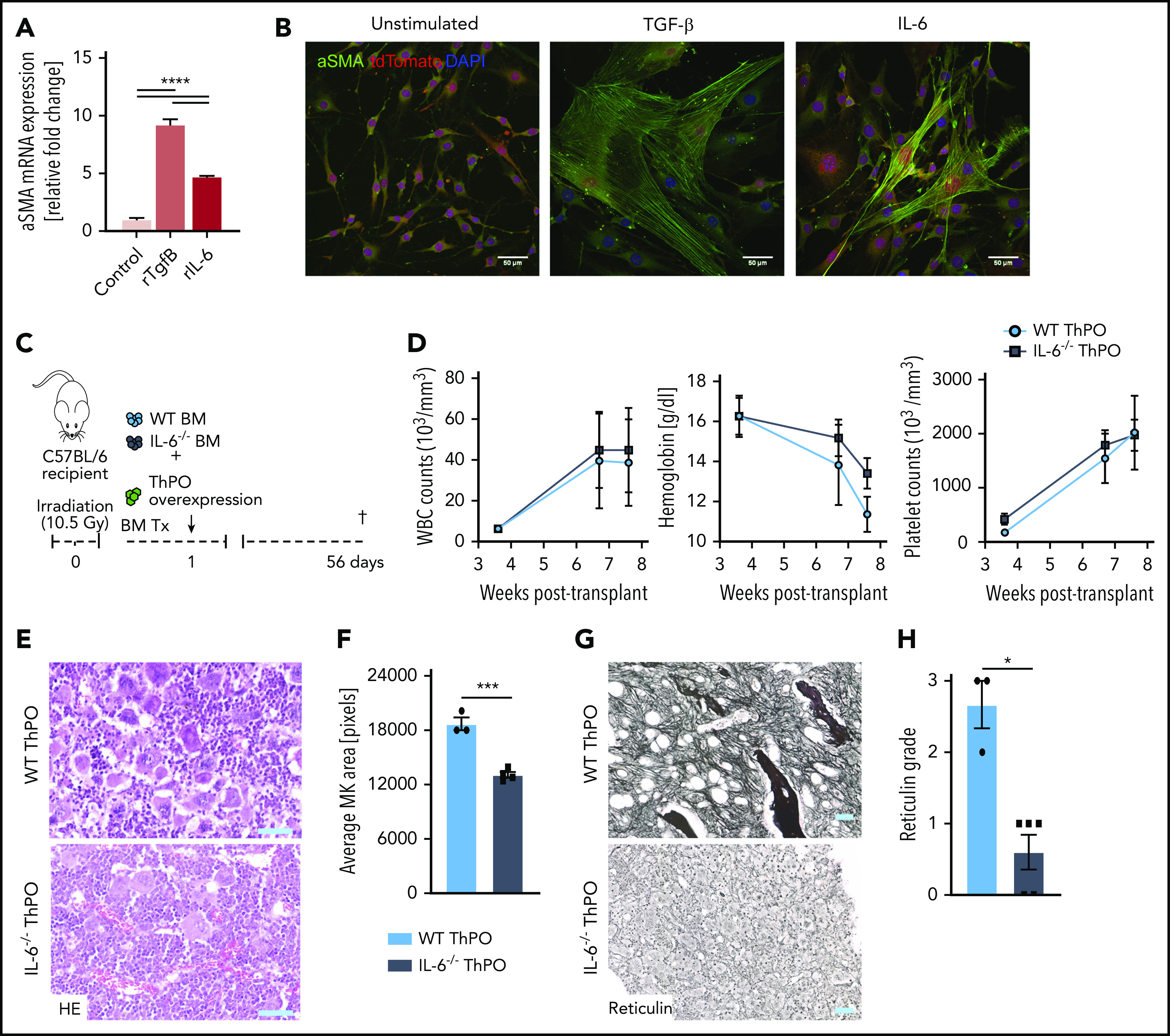
IL-6 knockout in BM reduces fibrosis in a ThPO-induced model of PMF. (A) Quantitative polymerase chain reaction of αSMA and collagen1α1 in primary murine Gli1+ stromal cells stimulated with recombinant cytokines for 72 hours. n = 3. (B) Representative immunofluorescence images of primary Gli1+ stromal cells stimulated with recombinant cytokines for 72 hours and stained for αSMA. Scale bar, 50 μm. (C) Schematic representation of transplantation of WT or IL-6−/− ckit+ cells in a model of ThPO-induced fibrosis (n = 3, WT ThPO; n = 5, IL-6−/− ThPO). (D) WBC, hemoglobin, and platelet parameters in weeks posttransplantation. (E) Representative images of hematoxylin and eosin (HE) staining of WT ThPO or IL-6−/− ThPO femurs. Original magnification ×20; scale bar, 100 μm. (F) Average pixel size of MKs in 3 randomly selected panels from WT ThPO or IL-6−/− ThPO femurs. Original magnification ×10; scale bar, 100 μm. (G) Representative images of reticulin staining (fibrosis) in WT ThPO or IL-6−/− ThPO femurs. (H) Quantification of myelofibrosis grade based on reticulin staining in WT ThPO or IL-6−/− ThPO femurs (Kruskal-Wallis multiple comparisons test). Data are shown as mean ± standard error of the mean, 1-way analysis of variance followed by Tukey’s post hoc test. *P < .05, ***P < .001, ****P < .0001. mRNA, messenger RNA.
To further elucidate whether the absence of IL-6, downstream of CXCL4, in hematopoietic cells ameliorates fibrosis, we transplanted WT or IL-6−/− ckit+ HSPCs transduced with ThPO into lethally irradiated recipients (Figure 7C). Although MPN features, including blood counts and hemoglobin (Figure 7D) as well as MK dysplasia (Figure 7E-F), were only mildly reduced and ameliorated in the absence of IL-6, the grade of fibrosis was significantly reduced in IL-6−/− ThPO animals (Figure 7G-H). This outcome further confirms the profibrotic role of IL-6 in the activation of the BM stroma and in the production of reticulin fibers.
Discussion
Our studies converged on a critical role of CXCL4 in the pathogenesis and progression of MPN and specifically PMF. Using genetic knockout of CXCL4 in hematopoietic cells in murine models of BM fibrosis/PMF, we found that the absence of CXCL4 leads to: (1) an ameliorated MPN phenotype; (2) decreased severity of BM fibrosis; (3) reduced Gli1+ cell activation including reduced inflammation; and (4) decreased JAK/STAT activation as a hallmark feature of MPN.
We showed that the spatial distribution of CXCL4 marks the progression of BM fibrosis. This is of particular interest as previous studies found that CXCL4 levels in patients with systemic sclerosis seem to predict progression in systemic sclerosis/fibrosis phenotypes.34 The identification of CXCL4 as a marker for fibrosis may be helpful (together with conventional reticulin staining) in early diagnosis and risk assessment, an important factor in patients who require aggressive treatment.
Accumulating evidence suggests a role for CXCL4 in chronic fibroproliferative and inflammatory conditions.14,35 However, in systemic sclerosis, CXCL4 induced skin inflammation but did not induce fibrosis. This is in line with our studies as we observed the most significant effects of CXCL4 on inflammation. It is thus tempting to speculate that although CXCL4 may sensitize cells to inflammatory stimuli, culminating in fibrosis, the presence of CXCL4 alone is not sufficient.
Inflammation is a hallmark feature of MPN and PMF, and MPN driver mutations constitutively activate the JAK/STAT signaling pathway.36-38 We observed a strong upregulation of inflammatory pathways and cytokines such as IL-1α and the alarmins S100A8/S100A9 in ThPO-overexpressing HSPCs. Interestingly, CXCL4 knockout led to decreased activation of JAK/STAT and inflammation, mainly reflected by a decrease in IL-6 levels in MKs. Our data thus link CXCL4 in hematopoietic cells in MPN to increased JAK/STAT activation and increased inflammation, partially through IL-6 activation. Importantly, we showed that IL-6 can induce a myofibroblast phenotype in Gli1+ fibrosis-driving cells, highlighting the cascade and vicious cycle of increased inflammation and fibrosis induction. The central role of IL-6 was shown in previous studies in MPN/PMF and increased levels of IL-6 cytokines correlate to shorter survival in patients with PMF.28,38 All IL-6–type cytokines strongly activate STAT3, and to a lesser extent STAT1.39 Kleppe et al37 showed that pan-hematopoietic STAT3 deletion resulted in improved survival and decreased cytokine secretion in a similar fashion to ruxolitinib therapy. We report increased expression of IL-6 not only in hematopoietic cells but also in the stroma. IL-6 secretion from stromal cells was shown to protect JAK2(V617F)-mutated cells from JAK2 inhibitor therapy.40 Thus, combined therapeutic strategies targeting the malignant clone but also the inflammatory environment (eg, by targeting CXCL4) have the potential to eliminate the disease-causing cells and also their maintaining environment. We here highlight an important crosstalk between mutated hematopoietic progenitors and nonmutated MSCs, which are eventually activated and subsequently contribute to the formation of a malignant niche that supports the neoplastic clone and the development of fibrosis.
In line with significantly reduced platelets, CXCL4 knockout had the strongest effect on the pathognomonic MK dysplasia, in both ThPO and JAK2(V617F)-induced PMF. MKs showed a strong upregulation of profibrotic pathways such as vascular endothelial growth factor and TGF-β signaling in fibrosis, which were normalized comparably to control MKs by CXCL4 knockdown. Interestingly, the profibrotic cytokine TGF-β, which is also overexpressed in patients with PMF and MPLW515L mice, is additionally linked to inflammation, in particular to the NF-κB pathway that was also upregulated in stromal cells in PMF in our models and normalized by CXCL4 knockdown.41 In sum, these data highlight the vicious cycle of inflammation, JAK/STAT activation, and activation of profibrotic pathways.
The only current treatment option that is able to prolong survival or provide a potential cure for PMF is an allogeneic stem transplant, which is associated with a 50% rate of transplant-related deaths or morbidities.42 The JAK1/2 inhibitor ruxolitinib was shown to reduce spleen size and JAK2(V617F) allele burden, and it suppressed cytokine secretion both in a murine model of PMF and in patients.43-45 CXCL4 inhibition alone was not sufficient to entirely rescue fibrosis but had a strong effect on the MPN phenotype, inflammation, MK dysplasia, and JAK/STAT activation. Our work implies that other chemokines or cytokines continue to drive the fibrotic transformation even in the absence of CXCL4. This was particularly obvious in the model of JAK2(V617F)-induced PMF, in which the absence of CXCL4 mainly affected the MK dysplasia and had a weaker effect on fibrosis, most likely because the development of fibrosis takes longer and other drivers can take over. Thus, combinatorial strategies might be optimal to block the vicious cycle of inflammation and activation of profibrotic pathways. Potential targets to combine with ruxolitinib, for example, include inhibition of the MK–stroma crosstalk through CXCL4 inhibition, inhibition of TGF-β signaling, or direct inhibition of inflammation (in particular IL-6).41,46-50
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors kindly thank the staff of the Erasmus Laboratory Animal Science Center (EDC) facility at Erasmus MC for their help with experiments.
H.F.E.G. was supported by the European Union’s Horizon 2020 Research and Innovation Programme under a Marie Sklodowska grant (LEaDing Fellows Programme, 707404) and is a Marie-Curie LEaDing Fellow at Erasmus MC. A.J.F.D. was supported by the European Union’s Horizon 2020 Research and Innovation Program (675585 Marie-Curie ITN “SymBioSys," awarded to J.S.-R.) and was a Marie-Curie early stage researcher at RWTH (Aachen, Germany); he was also supported by the JRC for Computational Biomedicine, which is an institute partially funded by Bayer AG. N.B.L. was funded by a postdoctoral fellowship through the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, BA 6349/1-1). R.K. was supported by grants of the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, SCHN1188/5-1, CRU344, Subproject 1), a grant of the European Research Council (ERC-StG 677448), and a grant from the Interdisciplinary Centre for Clinical Research within the Faculty of Medicine at the RWTH Aachen University (O3-11). R.K.S. was supported by grants from the European Hematology Association (EHA, John Goldman Clinical Research grant), the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, SCHN1188/6-1, within CRU344), the MPN foundation (2017 MPNRF/LLS Award), a KWF Kankerbestrijding young investigator grant (11031/2017–1, Bas Mulder Award, Dutch Cancer Foundation) and a grant of the European Research Council (deFIBER, ERC-StG 757339). R.K. and R.K.S. received funding in the BMBF-funded eMed consortium FibroMap.
Footnotes
The RNA sequencing data have been deposited with the European Nucleotide Archive (accession number PRJEB39607).
Authors will make renewable materials, data sets, and protocols available to other investigators upon e-mails to the corresponding author. For correspondence regarding the bioinformatics analysis please contact J.S.-R. (julio.saez@bioquant.uni-heidelberg.de).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H.F.E.G. designed and conducted experiments, analyzed results, and wrote the manuscript; A.J.F.D. analyzed data and wrote the manuscript; N.B.L. performed experiments, analyzed data, and wrote the manuscript; S.M., S.Z., B.B., and J.E.P. performed experiments; I.A.M.S. and S.N.R.F. performed experiments and analyzed data; N.K., I.T., G.B., and H.K. selected patient specimens and reviewed the manuscript; R.H. analyzed data and reviewed the manuscript; E.M.B. performed experiments and reviewed the manuscript; N.S., S.R.-J., and S.E. provided reagents and reviewed the manuscript; J.S.-R. obtained funding, supervised the bioinformatics analysis of the data by A.J.F.D., and reviewed the manuscript; R.K. and R.K.S. obtained funding, designed the study, performed experiments, analyzed data, and wrote the manuscript; and all authors provided critical analysis of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rebekka K. Schneider, RWTH Aachen University Hospital, Pauwelsstrasse 30, 52074 Aachen; e-mail: reschneider@ukaachen.de.
REFERENCES
- 1.Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416-1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baxter EJ, Scott LM, Campbell PJ, et al. ; Cancer Genome Project . Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054-1061. [DOI] [PubMed] [Google Scholar]
- 3.James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148. [DOI] [PubMed] [Google Scholar]
- 4.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790. [DOI] [PubMed] [Google Scholar]
- 5.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7(9):673-683. [DOI] [PubMed] [Google Scholar]
- 6.Thorsten K, Gisslinger H, Harutyunyan AS, et al. Frequent mutations in the calreticulin gene CALR in myeloproliferative neoplasms. Blood. 2013;122(21):LBA-1. [Google Scholar]
- 7.Schneider RK, Mullally A, Dugourd A, et al. Gli1+ mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell Stem Cell. 2017;20(6):785-800.e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Decker M, Martinez-Morentin L, Wang G, et al. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat Cell Biol. 2017;19(6):677-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine SP, Wohl H. Human platelet factor 4: purification and characterization by affinity chromatography. Purification of human platelet factor 4. J Biol Chem. 1976;251(2):324-328. [PubMed] [Google Scholar]
- 10.Schaffner A, Rhyn P, Schoedon G, Schaer DJ. Regulated expression of platelet factor 4 in human monocytes—role of PARs as a quantitatively important monocyte activation pathway. J Leukoc Biol. 2005;78(1):202-209. [DOI] [PubMed] [Google Scholar]
- 11.Vandercappellen J, Van Damme J, Struyf S. The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev. 2011;22(1):1-18. [DOI] [PubMed] [Google Scholar]
- 12.Affandi AJ, Silva-Cardoso SC, Garcia S, et al. CXCL4 is a novel inducer of human Th17 cells and correlates with IL-17 and IL-22 in psoriatic arthritis. Eur J Immunol. 2018;48(3):522-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vrij AA, Rijken J, Van Wersch JW, Stockbrügger RW. Platelet factor 4 and beta-thromboglobulin in inflammatory bowel disease and giant cell arteritis. Eur J Clin Invest. 2000;30(3):188-194. [DOI] [PubMed] [Google Scholar]
- 14.Pitsilos S, Hunt J, Mohler ER, et al. Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb Haemost. 2003;90(6):1112-1120. [DOI] [PubMed] [Google Scholar]
- 15.Aivado M, Spentzos D, Germing U, et al. Serum proteome profiling detects myelodysplastic syndromes and identifies CXC chemokine ligands 4 and 7 as markers for advanced disease. Proc Natl Acad Sci U S A. 2007;104(4):1307-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasper B, Petersen F. Molecular pathways of platelet factor 4/CXCL4 signaling. Eur J Cell Biol. 2011;90(6-7):521-526. [DOI] [PubMed] [Google Scholar]
- 17.Schubert M, Klinger B, Klünemann M, et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun. 2018;9(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holland CH, Szalai B, Saez-Rodriguez J. Transfer of regulatory knowledge from human to mouse for functional genomics analysis. Biochim Biophys Acta Gene Regul Mech. 2020;1863(6):194431. [DOI] [PubMed] [Google Scholar]
- 19.Darby IA, Hewitson TD. Hypoxia in tissue repair and fibrosis. Cell Tissue Res. 2016;365(3):553-562. [DOI] [PubMed] [Google Scholar]
- 20.Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11(4):014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Väremo L, Nielsen J, Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013;41(8):4378-4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahin H, Wasmuth HE. Chemokines in tissue fibrosis. Biochim Biophys Acta. 2013;1832(7):1041-1048. [DOI] [PubMed] [Google Scholar]
- 23.Bruns I, Lucas D, Pinho S, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20(11):1315-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tefferi Ayalew, Nicolosi Maura, Mudireddy Mythri, et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am. J. Hematol. 2018;93(3):348-355. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, Saez-Rodriguez J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29(8):1363-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rampal R, Al-Shahrour F, Abdel-Wahab O, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123(22):e123-e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen E, Beer PA, Godfrey AL, et al. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell. 2010;18(5):524-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Čokić VP, Mitrović-Ajtić O, Beleslin-Čokić BB, et al. Proinflammatory cytokine IL-6 and JAK-STAT signaling pathway in myeloproliferative neoplasms. Mediators Inflamm. 2015;2015:453020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sollazzo D, Forte D, Polverelli N, et al. Circulating calreticulin is increased in myelofibrosis: correlation with interleukin-6 plasma levels, bone marrow fibrosis, and splenomegaly. Mediators Inflamm. 2016;2016:5860657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alavi MV, Mao M, Pawlikowski BT, et al. Col4a1 mutations cause progressive retinal neovascular defects and retinopathy. Sci Rep. 2016;6(1):18602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mylotte LA, Duffy AM, Murphy M, et al. Metabolic flexibility permits mesenchymal stem cell survival in an ischemic environment. Stem Cells. 2008;26(5):1325-1336. [DOI] [PubMed] [Google Scholar]
- 32.Hu X, Yu SP, Fraser JL, et al. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J Thorac Cardiovasc Surg. 2008;135(4):799-808. [DOI] [PubMed] [Google Scholar]
- 33.Grayson WL, Zhao F, Bunnell B, Ma T. Hypoxia enhances proliferation and tissue formation of human mesenchymal stem cells. Biochem Biophys Res Commun. 2007;358(3):948-953. [DOI] [PubMed] [Google Scholar]
- 34.van Bon L, Affandi AJ, Broen J, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. 2014;370(5):433-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaldivar MM, Pauels K, von Hundelshausen P, et al. CXC chemokine ligand 4 (Cxcl4) is a platelet-derived mediator of experimental liver fibrosis. Hepatology. 2010;51(4):1345-1353. [DOI] [PubMed] [Google Scholar]
- 36.Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30(2):88-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleppe M, Kwak M, Koppikar P, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015;5(3):316-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10):1356-1363. [DOI] [PubMed] [Google Scholar]
- 39.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374(Pt 1):1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manshouri T, Estrov Z, Quintás-Cardama A, et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011;71(11):3831-3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yue L, Bartenstein M, Zhao W, et al. Efficacy of ALK5 inhibition in myelofibrosis. JCI Insight. 2017;2(7):e90932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ballen KK, Shrestha S, Sobocinski KA, et al. Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2010;16(3):358-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. ; COMFORT-II investigators . Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis [published correction appears in Blood. 2016;128(25):3013]. Blood. 2013;122(25):4047-4053. [DOI] [PubMed] [Google Scholar]
- 44.Tefferi A. Challenges facing JAK inhibitor therapy for myeloproliferative neoplasms. N Engl J Med. 2012;366(9):844-846. [DOI] [PubMed] [Google Scholar]
- 45.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ceglia I, Dueck AC, Masiello F, et al. Preclinical rationale for TGF-β inhibition as a therapeutic target for the treatment of myelofibrosis. Exp Hematol. 2016;44(12):1138-1155.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kampan NC, Xiang SD, McNally OM, Stephens AN, Quinn MA, Plebanski M. Immunotherapeutic interleukin-6 or interleukin-6 receptor blockade in cancer: challenges and opportunities. Curr Med Chem. 2018;25(36):4785-4806. [DOI] [PubMed] [Google Scholar]
- 48.Huselton E, Cashen AF, Jacoby M, Pusic I, Romee R, Uy GL. CX-01, an inhibitor of CXCL12/CXCR4 axis and of platelet factor 4 (PF4), with azacitidine (AZA) in patients with hypomethylating agent (HMA) refractory AML and MDS. J Clin Oncol. 2018;36(15):7027. [Google Scholar]
- 49.Kovacsovics TJ, Mims A, Salama ME, et al. Combination of the low anticoagulant heparin CX-01 with chemotherapy for the treatment of acute myeloid leukemia. Blood Adv. 2018;2(4):381-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. 2018;17(6):395-412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.