

Abstract
Analyzing the genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from clinical samples is crucial for understanding viral spread and evolution as well as for vaccine development. Existing RNA sequencing methods are demanding on user technique and time and, thus, not ideal for time-sensitive clinical samples; these methods are also not optimized for high performance on viral genomes. We developed a facile, practical, and robust approach for metagenomic and deep viral sequencing from clinical samples. We demonstrate the utility of our approach on pharyngeal, sputum, and stool samples collected from coronavirus disease 2019 (COVID-19) patients, successfully obtaining whole metatranscriptomes and complete high-depth, high-coverage SARS-CoV-2 genomes with high yield and robustness. With a shortened hands-on time from sample to virus-enriched sequencing-ready library, this rapid, versatile, and clinic-friendly approach will facilitate molecular epidemiology studies during current and future outbreaks.
Keywords: COVID-19, SARS-CoV-2, whole-genome sequencing, metagenomics
Graphical Abstract
The novel coronavirus disease 2019 (COVID-19) pandemic poses a serious public health risk. Chen et al. develop a facile and robust approach for transcriptomic sequencing of COVID-19 samples that will facilitate molecular epidemiology studies during current and future outbreaks.
Introduction
As of November 4, 2020, the ongoing coronavirus disease 2019 (COVID-19) viral pandemic has affected more than 46 million people in over 200 countries and territories around the world and has claimed more than 1,204,000 lives (WHO, 2020). Closely monitoring the genetic diversity and distribution of viral strains at the population level is essential for epidemiological tracking and for understanding viral evolution and transmission; additionally, examining the viral heterogeneity within a single individual is imperative for diagnosis and treatment (Wölfel et al., 2020). The disease-causing pathogen, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was identified from early disease cases, and its draft genome was sequenced within weeks because of rapid responses from researchers around the world (Lu et al., 2020c; Ren et al., 2020; Wu et al., 2020; Zhou et al., 2020b). The initial SARS-CoV-2 draft genome was obtained independent from the same early COVID-19 patient samples using various conventional RNA sequencing (RNA-seq) library construction methods. Although these library construction methods successfully generated a draft genome, several drawbacks hinder use of these methods for routine viral genome sequencing from the surge of clinical samples during an outbreak.
One direct library construction approach used to generate the SARS-CoV-2 draft genome (Lu et al., 2020c; Ren et al., 2020; Wu et al., 2020; Zhou et al., 2020b) essentially captures each sample’s entire metatranscriptome, in which SARS-CoV-2 is just one species among many. The abundance of SARS-CoV-2 in clinical swabs, sputum, and stool samples is often low (Wang et al., 2020; Wölfel et al., 2020); therefore, this catch-all method requires deeper sequencing of each sample to obtain sufficient coverage and depth of the whole viral genome, which increases the time and cost of sequencing. Target enrichment with spiked-in primers can improve SARS-CoV-2 genome coverage (Deng et al., 2020), but reliance on specific primers inherently limits this approach for profiling of evolving viruses. The same limitation applies to multiplex RT-PCR-based strategies (Xiao et al., 2020). Additionally, when the sample is subjected to targeted amplification during the initial RT steps, its metatranscriptomic information is lost forever.
Currently, the most comprehensive strategy is a combination of metatranscriptomics profiling with post-library SARS-CoV-2 target enrichment (Xiao et al., 2020). However, in most conventional RNA-seq methods, the double-strand DNA ligation (dsDL) portion of the protocol is usually the most demanding regarding hands-on time and user technique (Di et al., 2020). When superimposed on the target enrichment process, these labor-intensive and lengthy protocols become impractical for routine use in the clinic and for timely monitoring of viral genetics and evolution on large volumes of samples during an outbreak. Furthermore, because of the low molecular efficiency of dsDL, these protocols also require a high amount of input material, further restricting their application to clinical samples.
Although next-generation sequencing platforms are high throughput and have a short turn-around time, library construction from samples, including targeted enrichment or not, remains a major bottleneck. To broadly apply viral sequencing to clinical samples, especially during outbreaks, when biomedical resources are already limited, a rapid, simple, versatile, and scalable sample library construction method that does not compromise performance is urgently needed.
Recently, we reported a new RNA-seq library construction strategy that aims to address some of these challenges: sequencing hetero RNA-DNA-hybrid (SHERRY) avoids the problematic dsDL step in library construction by taking advantage of the newly discovered Tn5 tagmentation activity on RNA/DNA hybrids to directly tag RNA/cDNA fragments with sequencing adapters (Di et al., 2020). Therefore, SHERRY has minimal sample transfer and greatly reduced hands-on time, making it simple, robust, and suitable for input ranging from single cells to 200 ng total RNA. We now combine the advantages of a tailored SHERRY protocol, which improves coverage of whole metatranscriptomes, with a simplified post-library target enrichment protocol. MetagenomIc RNA enrichment viral sequencing (MINERVA) is an easy-to-use, versatile, scalable, and cost-effective protocol that yields a high-coverage, high-depth SARS-CoV-2 genome while preserving the sample’s rich metatranscriptomic profile. The hands-on time required from clinical sample to sequencing-ready library using conventional approaches without enrichment is 190 min; MINERVA requires only 100 min hands-on time, and when deep viral coverage is desired, an additional 90 min for post-library enrichment, totaling 190 min for the entire workflow (Figure S1A). MINERVA also costs less than dsDL because of its simpler procedure and much lower sequencing depth (Figure S1A). These features make MINERVA practical for high-volume, routine clinical use. We applied MINERVA to various types of COVID-19 samples and successfully obtained up to 10,000-fold SARS-CoV-2 genome enrichment. This strategy will facilitate all studies regarding SARS-CoV-2 genetic variations in the current pandemic and can also be applied to other pathogens of interest.
Results
Minerva
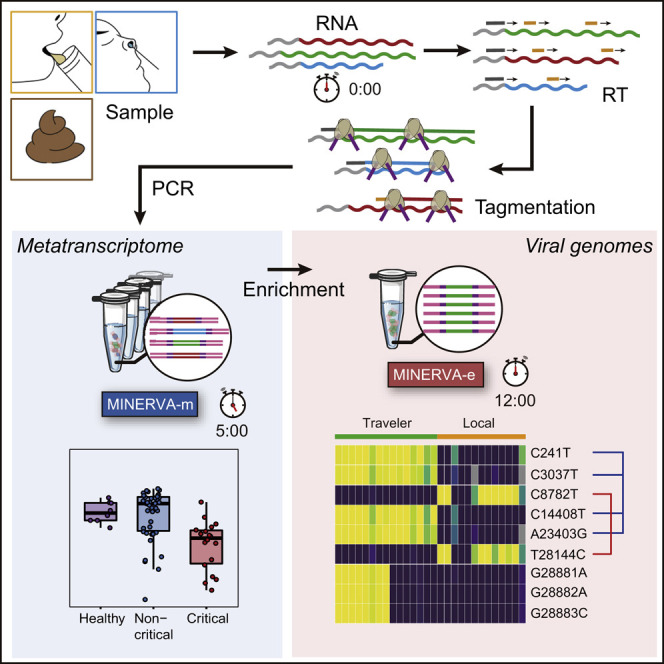
To analyze metagenomics and SARS-CoV-2 genetics from COVID-19 patient samples, we developed a two-stage metagenomic RNA enrichment viral sequencing strategy named MINERVA (Figure 1 A and Methods S1). First, we employed a SHERRY-based RNA-seq pipeline for metagenomic analysis (MINERVA-m). Because clinical samples may contain DNA, RNA, and possibly carrier RNA, MINERVA starts with ribosomal RNA (rRNA) removal and optional simultaneous carrier RNA removal, followed by DNase I treatment. The remaining RNA is then subject to standard SHERRY. Previously, we observed 3′ bias in SHERRY libraries; to address this, we used 10 ng mouse 3T3 cell total RNA as starting material and tested whether adding a random decamer (N10) during reverse transcription could improve coverage evenness (Figures 1B, 1C, and S1B–S1E). Compared with the standard SHERRY protocol, which uses 1 μM T30VN primer during reverse transcription, supplementation with 1 μM N10 in MINERVA indeed improves gene body coverage evenness, presumably by improving the reverse transcription efficiency. When the N10 concentration was increased further to 10 μM, we observed almost no coverage bias in the gene body. The high N10 concentration can result in an increased rRNA ratio in the product, sometimes as high as 90%, but MINERVA employs rRNA removal as the first step prior to reverse transcription, negating this problem. We also performed enzyme titration with homemade and commercial Tn5 transposomes. Based on these N10 and Tn5 titration results, we used 10 μM N10 during reverse transcription and 0.5 μL V50 for each 20-μL tagmentation reaction in all following experiments. The whole procedure, from nucleic acid to metagenomic sequencing-ready library, including wait time, takes 5.5 h (Figure S1A).
Figure 1.

Scheme and Development of MINERVA
(A) RNA extracted from pharyngeal swabs and sputum and stool samples undergo rRNA and DNA removal before metagenomic sequencing library construction (MINERVA-m). Multiple libraries were then pooled for SARS-CoV-2 sequence enrichment.
(B) Effect of the N10 primer during reverse transcription and Tn5 amount on detected gene number.
(C) Effect of the N10 primer during reverse transcription and Tn5 amount on gene body coverage evenness.
For target enrichment (MINERVA-e), we first quantified SARS-CoV-2 abundance in each metagenomic sequencing library using an N gene qPCR assay and pooled eight libraries based on quantification results. Then we performed standard in-solution hybridization on the pooled library with biotinylated RNA probes covering the whole viral genome. The enrichment procedure takes ~7–13 h; the entire MINERVA pipeline can be completed within ~12–18 h.
MINERVA Is Compatible with COVID-19 Samples
To evaluate its performance on clinical samples, we applied MINERVA to 136 samples collected from 91 individuals with COVID-19, with samples types including pharyngeal swabs, sputum, and stool. These individuals were admitted to Ditan Hospital within a 3-month period from January to April 2020, presenting different symptom severity (Figure 2 A; Tables S1 and S2). Some individuals were re-sampled longitudinally to investigate temporal and intra-host viral heterogeneity. In addition to the samples from individuals with COVID-19, we also included different sample types from eight healthy individuals as well as non-template control (NTC) samples. We first tested the effect of sample input volume on MINERVA results. Using just 2.7-μL of rRNA- and DNA-depleted sample led to satisfactory SARS-CoV-2 coverage, and scaling up the reaction volume and sample input further improved the MINERVA data quality (Figure 2B). Using the same samples and the same sequencing depth, more input in a higher reaction volume generated deeper SARS-CoV-2 genome coverage.
Figure 2.

Optimization of MINERVA for Clinical Samples
(A) COVID-19 sample profiles, showing the age group, sex, severity, and re-sampling status of each individual.
(B) Effect of sample input and reaction volume on sequencing depth of the SARS-CoV-2 genome.
(C) Mapping ratios of human, fungus, bacterium, and virus reads showed good performance of carrier RNA removal. “No carrier RNA” refers to samples with no carrier RNA during extraction; “with post-added carrier RNA” refers to samples with post-added carrier RNA after RNA extraction. “RNA extraction with carrier RNA” refers to samples with carrier RNA during extraction. All samples with carrier RNA went through a carrier RNA removal step.
(D) SARS-CoV-2 genome coverage and depth of MINERVA-m and -e for “RNA extraction with carrier RNA” samples.
Carrier RNA, which is widely used in viral DNA/RNA extraction before qRT-PCR assays, severely affects high-throughput sequencing analysis. Therefore, most qRT-PCR-positive clinical samples are not amenable to further viral genetic studies. We explored the effect of adding polyT oligos during the rRNA removal step to simultaneously remove spike-in poly(A) RNA and carrier RNA. By incorporating this step in MINERVA, we successfully avoided overwhelming representation of unwanted RNA sequences while retaining desired metagenomic and SARS-CoV-2 information (Figures 2C and 2D).
MINERVA Captures Metagenomic Signatures of COVID-19 Samples
We benchmarked MINERVA against conventional dsDL strategies in head-to-head comparisons of the first 79 clinical samples sequenced. On average, we sequenced 2–5 gigabase pairs (Gbp) for each MINERVA-m and MINERVA-e library, and nearly 100 Gbp for each dsDL library (Figure S2A). The MINERVA-m and dsDL libraries were comparable: bacterial heterogeneity as measured by species richness and Shannon index was correlated between the two (Figures S2B and S2C). To ensure the accuracy of our metagenomic computational pipeline, we employed metagenomics and metagenome assembly strategies to profile microbial compositions of different types of samples. For assembly, we combined reads from samples of the same type for co-assembly, obtaining contigs for each sample type. After taxonomy assignment at the contig level, reads from each individual sample were mapped back to the co-assembled contigs to obtain the microbial composition of each individual sample. Overall, we found the results of microbial compositions profiled from metagenomic and assembly strategies to be comparable (Figures S2D–S2F). There were slightly more taxa identified with the metagenomic strategy, especially those with relatively lower abundance. Detailed metagenome assemblies and mapping statistics of each sample are outlined in the STAR Methods and listed in Tables S3 and S4.
To study the metagenomic signature of individuals with COVID-19, we first analyzed the data from NTC samples, which showed distinct microbial compositions and significantly lower species richness, suggesting that NTC samples contain very low-level biomass and have little influence on real samples (Figures S3A and S3B). We then assessed potential factors that may be associated with microbial compositions in all types of samples. Permutational multivariate analysis of variance (PERMANOVA) revealed significant association with sample types as well as disease severity (Figure S3C). To exclude the influence from sample type, we performed further analyses on separated sample types. Disease severity was found to have significant associations in pharyngeal and sputum samples, and no factors were found with significant effect on stool samples (Figure 3 A).
Figure 3.

Metagenomics Analysis of COVID-19 Samples Using MINERVA
(A) PERMANOVA analysis highlights factors associated with microbial compositions in different sample types, including pharyngeal (n = 68), sputum (n = 59), and stool (n = 33). (PERMANOVA test, *p < 0.05, **p < 0.01, ***p < 0.001). Permutation was constrained within the same time point to account for repeated measures.
(B) PCoA analysis of pharyngeal samples based on Bray-Curtis distance, calculated at the bacterial genus, fungal genus, and viral family levels. Samples are colored according to different disease groups, including healthy controls (n = 8), mild (n = 10), moderate (n = 23), severe (n = 9), critical (n = 18), and non-template controls (NTCs; n = 2).
(C) Comparison of alpha diversity, including species richness (left panel) and Shannon index (right panel), among different groups of pharyngeal samples. Groups were segmented as healthy controls (n = 8), non-critical (including mild, moderate, and severe; n = 42) and critical (n = 18). Kruskall-Wallis test and Wilcoxon rank-sum test were used for multi-group and two-group comparisons, respectively.
(D) Analysis of association of microbial taxa with disease severity using pharyngeal samples. The generalized estimating equation (GEE) model was applied. Results were filtered based on significance (BH-adjusted p < 0.05) and effect size (absolute coefficient > 0.1). Taxa found to be significantly associated with disease are shown in the left panel, and their abundance in different groups of samples is shown in the right panel.
We performed various analyses to explore the metagenomic signatures that correlate with disease severity. Principal coordinates analysis (PCoA) analysis based on Bray-Curtis distance among pharyngeal samples indicates differences between samples from individuals with COVID-19 and healthy control subjects as well as a difference between critical samples and samples from individuals with less severe disease status. NTC samples were also separated from other samples on the PCoA plot (p < 0.001 by PERMANOVA test) (Figure 3B). Based on this result, we segmented pharyngeal samples into healthy, non-critical (including “mild,” “moderate” and “severe” samples) and critical groups. Decreased alpha diversities were observed in samples from individuals with COVID-19. A lower Shannon index was observed in the non-critical group compared with healthy control subjects. The most strongly and significantly decreased alpha diversity was observed in critical samples in terms of species richness and Shannon index (Figure 3C).
Furthermore, we applied multivariate analysis by using a generalized estimating equation (GEE) model to explore microbes associated with the difference among these three groups. To avoid introducing noise, especially for relatively low-abundance microbes, we set stringent filtering based on effect size and significance for the results, and only microbes with an absolute coefficient of more than 0.1 and Benjamini-Hochberg (BH) adjusted p value of less than 0.05 were kept. Streptococcus, Rothia, Acinetobacter, and Acidovorax were found to be associated with non-critical disease, whereas Halomonas, Campylobacter, Prevotella, and Veillonella were associated with critical disease (Figure 3D, left panel). Among these, Streptococcus and Rothia were highly enriched in the non-critical disease group, and Prevotella and Veillonella were abundant and prevalent in the healthy and non-critical groups. Acinetobacter and Acidovorax were enriched in the healthy and critical groups, whereas Halomonas was only highly enriched in critical samples (Figure 3D, right panel). Although some of these microbes were also detected in one of the NTC samples, their abundance was quite low compared with samples from individuals with COVID-19 (Figure S3I). They are also known to be abundant commensal microbes in human oral-pharyngeal samples (de Lastours et al., 2015; Human Microbiome Project Consortium, 2012; Zaura et al., 2009) and, therefore, were not removed as contaminants.
For sputum samples, PCoA analysis also showed separation between samples from individuals with COVID-19 and healthy control samples (p < 0.001 by PERMANOVA test), whereas samples from individuals with COVID-19 were not separated by disease severity (Figure S3D). The Shannon index was lower in samples from individuals with COVID-19 compared with healthy control samples, whereas no significant difference of species richness was observed (Figure S3E). GEE analysis found that Streptococcus, Rothia, and Veillonella were associated with disease (Figure S3F): Streptococcus and Rothia were enriched in in the disease group, whereas Veillonella was enriched and more prevalent in healthy samples. For stool samples, no difference was observed between samples from individuals with COVID-19 and healthy control subjects in terms of PCoA on Bray-Curtis distance (Figure S3G) and alpha diversities, including species richness and Shannon index (Figure S3H). These results were also consistent with PERMANOVA analysis (Figure 3A).
By surveying the metagenomic landscape of these samples, we also observed several patient samples with exceptionally high abundance of additional known pathogens, including Candida albicans, Staphylococcus aureus, Corynebacterium jeikeium, Corynebacterium striatum, and Klebsiella aerogenes (Figure 4 A, left panels). To validate these results, we directly mapped the reads to the individual genome of each species and assessed the genome coverage of each sample, normalized by sequencing depth (Figure 4A, right panel). Generally, the metagenomics results were consistent with the direct mapping results and also consistent with the assembly results (Figure S4). The occurrence rate of these species with high abundance identified by metagenomics and direct mapping is also associated with disease severity (Figure 4B). For severe viral pneumonia, co-infections can greatly affect patient outcome; one recent study showed that 50% of patients with COVID-19 who died in this pandemic had secondary bacterial infections (Cox et al., 2020; Crotty et al., 2015; Shah et al., 2016). We were unable to retrospectively confirm clinical co-infection in the cases identified by MINERVA; however, the sparse occurrence of these pathogenic microbes, combined with their high abundance, raises concerns and can be investigated in future studies.
Figure 4.

Co-existence of Additional Pathogens in Samples from Individuals with COVID-19
(A) Abundance of other potential pathogens. Sparse occurrence of high abundance of several pathogens with the potential to cause secondary infections was identified by metagenomics analysis (presented as relative abundance, left panels) and validated by direct mapping to their genomes (presented as coverage normalized by sequencing depth, right panels). Identified pathogens include Candida albicans, Staphylococcus aureus, Corynebacterium jeikeium, Corynebacterium striatum, and Klebsiella aerogenes.
(B) Occurrence rate of high-risk pathogens in different severity groups (samples with one or several high-risk pathogens identified were all considered). Only samples with a high abundance of these pathogens identified by metagenomics analysis and direct mapping were considered here and are also labeled in (A). The occurrence rate was associated with disease severity.
MINERVA Achieves Better SARS-CoV-2 Genome Coverage Compared with Conventional dsDL Strategies
In MINERVA-m and dsDL data, we detected low but significant levels of SARS-CoV-2 sequences. The viral ratio is between 10−7 and 10−1. It is worth noting that the SARS-CoV-2 sequence ratio is higher in MINERVA-m data than in dsDL data (Figures 5A and 5B), suggesting that MINERVA-m libraries capture more SARS-CoV-2 sequences. Although SARS-CoV-2 genome coverage and depth were not high in MINERVA-m results because of low viral ratios and low sequencing depth, performing MINERVA-e subsequently can enrich the SARS-CoV-2 sequence ratio up to 10,000-fold (Figures 5C and S5A–S5C). To evaluate potential false positive SARS-CoV-2 signals from targeted enrichment, we performed MINERVA-e on several 3T3 total RNA samples and three types of samples from eight healthy donors. The results showed extremely low coverage of the SARS-CoV-2 genome from a few duplicated amplicons in 3T3 data (Figure S5D). We observed low-level false-positive SARS-CoV-2 signals in a few healthy donor samples, likely because of sequencing index hopping, but the coverage depth is generally lower than in patient samples (Figure S5E). In summary, MINERVA gives more complete and deeper coverage of SARS-CoV-2 genomes (Figures 5D and 5E), despite sequencing dsDL libraries to two orders of magnitude more depth (Figure S2A).
Figure 5.

Direct Comparison of Sequencing Libraries Constructed from MINERVA and Conventional dsDL Strategies
(A) SARS-CoV-2 mapping ratio statistics of the MINERVA-m and dsDL libraries.
(B) Comparison of SARS-CoV-2 mapping ratios between the MINERVA-m and dsDL libraries.
(C) Comparison of SARS-CoV-2 mapping ratios between the MINERVA-m and MINERVA libraries.
(D and E) SARS-CoV-2 genome coverage and depth statistics of the MINERVA-e and dsDL libraries.
(F and G) Comparison of SARS-CoV-2 sequencing results between the MINERVA-e and dsDL libraries.
(H) Metagenomic sequencing and qPCR result features of samples with poor SARS-CoV-2 genome coverage.
The superior quality of MINERVA data became clearer when we included clinical qRT-PCR results. The dsDL and MINERVA libraries detect SARS-CoV-2 sequences for samples with various cycle threshold (Ct) values, but MINERVA produced more complete and deeper genome coverage than dsDL methods (Figures 5F and 5G), and this advantage is more pronounced for low-viral-load samples, including two samples with negative qPCR results, and stool samples. By studying the relationship between SARS-CoV-2 qPCR results and read ratio, we identified two groups of samples that resulted in low SARS-CoV-2 genome coverage when processed using dsDL (Figure 5H). The first group had a low SARS-CoV-2 read ratio, which prohibited acquisition of enough SARS-CoV-2 sequencing reads. The second group, which included most stool samples, had relatively high SARS-CoV-2 Ct values and read ratios, suggesting that these samples had low total nucleic acid amounts. Because dsDL approaches are less sensitive and require more input, this may explain why MINERVA outperforms dsDL most evidently in stool samples.
MINERVA Can Facilitate Multiple Facets of COVID-19 Research
As a novel virus, little is known about the evolutionary features of SARS-CoV-2. Using 136 samples, we constructed a SARS-CoV-2 mutational profile (Figure 6 A) that was distinct from the Guangdong profile (Lu et al., 2020b). A few mutation sites, including the two linked to the S and L strains (Lu et al., 2020a), were found in multiple samples. Aided by the deep genome coverage in MINERVA data, we not only detected strong linkage between position 8,782 and 28,144 but also observed high concordance of allele frequencies between these two positions. Furthermore, we detected strong linkage and high allele frequency concordance among four other positions: 241, 3,037. 14,408, and 23,403. Such allele frequency information offers additional layers of evidence supporting co-evolution of positions within the SARS-CoV-2 genome in two distinct groups of samples. It is worth noting that, in some samples, not all linked alleles are detected simultaneously because of low coverage at some positions; these alleles can indeed be observed at low coverage in the raw data for these samples but are missing from the post-processing data because they do not pass the stringent quality filtering steps. Nonetheless, the linkage was established by observing such linkage over many samples.
Figure 6.

MINERVA Could Facilitate COVID-19 and SARS-CoV-2 Research through Accurate and Sensitive Identification of Viral Mutations
(A) SARS-CoV-2 mutation profile obtained from 136 samples.
(B) SARS-CoV-2 mutation profiles of asymptomatic individuals with COVID-19 and their infected family members. Individual origin is labeled in red (local) or blue (international traveler).
(C) Longitudinal SARS-CoV-2 mutation analysis of individuals with COVID-19.
Apart from its high infectiousness, containment of SARS-CoV-2 transmission is challenging because of the existence of asymptomatic infected individuals (Bai et al., 2020). Although qRT-PCR can be used to identify these individuals, elucidation of the chain of transmission requires complete SARS-CoV-2 sequences. To evaluate the performance of MINERVA when tracking SARS-CoV-2 transmission, we sequenced samples from several asymptomatic individuals and infected family members. SARS-CoV-2 SNV analysis could separate local patients from international travelers. Asymptomatic individuals each harbor viral sequences with unique signatures, and these individuals are clustered by viral SNV signature with their respective family members rather than other individuals with COVID-19, which indicates that viral SNVs within infected families are similar to each other and unique from other families (Figure 6B). Summarily, despite the asymptomatic phenotype of some infected individuals, the viral SNV signature generated by MINERVA can be used to accurately place these individuals in the chain of transmission, enabling better epidemiological tracking.
Recent studies have identified genetic variations of SARS-CoV-2 and raised the possibility that multiple variants could co-exist in the same host individual. The intra-host SNVs (iSNVs) detected in many samples (Figure 6A) suggest that SARS-CoV-2 is evolving within hosts (Wölfel et al., 2020). Through longitudinal sampling, we confirmed that iSNVs were generally relatively stable across time and body sites (Figure S6) but found that some patients harbored greater variations in iSNVs (Figure 6C). In P40 and P41, iSNVs were stable within the same sample type across time but varied across different sample types. These results support co-existence of multiple SARS-CoV-2 variants in the same individual, and further investigation is warranted to understand this phenomenon.
In summary, MINERVA effectively converts metagenomes and SARS-CoV-2 sequences into sequencing libraries with a simple and quick experimental pipeline, and subsequent target enrichment can further improve SARS-CoV-2 genome coverage and genetic variation detection. MINERVA can facilitate study of SARS-CoV-2 genetics and can be implemented easily to fight future RNA pathogen outbreaks.
Discussion
As of today, our knowledge of SARS-CoV-2 is still preliminary, and much of it is extrapolated from past studies of other betacoronaviruses such as SARS-CoV and MERS-CoV. However, the epidemiology, physiology, and biology of COVID-19 are evidently unique (Fauci et al., 2020). To speed up our investigation of this virus and the disease it causes, a practical protocol for viral genome research of clinical samples is urgently needed. Currently, methods for transforming clinical samples into sequencing libraries are laborious and painstaking, and clinical personnel at the frontlines are already strained for time and energy. MINERVA minimizes the need for expert technique and hands-on operation; we believe it will be pivotal in accelerating clinical research of SARS-CoV-2.
Recent evolutionary tracing studies suggest emergence of multiple novel, evolved subtypes of SARS-CoV-2 (Gudbjartsson et al., 2020), such as the S/L subtypes (Lu et al., 2020a). New variants will likely continue to emerge as the virus mutates, and to uncover them requires deep, complete coverage of viral genomes from a large number of patients. With the existence of asymptomatic carriers (Bai et al., 2020) and possible recurrent infections in the same individual (An et al., 2020), longitudinal re-sampling of individuals with COVID-19 is also important to uncover intra-host viral heterogeneity, but as viral load decreases with time (He et al., 2020), the sensitivity of the sample processing method becomes critical. These studies require processing large volumes of clinical samples with a highly robust and scalable method that does not compromise sensitivity. We demonstrated that MINERVA libraries from clinical samples can generate deep and complete coverage of SARS-CoV-2 genomes that can be used for evolutionary tracing and variant characterization research. Furthermore, the high sensitivity, high coverage, and high depth of the SARS-CoV-2 viral genomes obtained by MINERVA can reveal unique viral SNV signatures in individuals with COVID-19, even when they are asymptomatic. We showed that these viral SNVs allow families of infected individuals to be co-clustered but are unique between families, which enables each individual to be accurately placed in the chain of transmission. Because MINERVA is easily scalable and implementable in a clinical lab setting, it can serve as a robust strategy for timely and critical epidemiological tracking and monitoring during a pandemic.
It is well established that SARS-CoV-2 can infect multiple organ systems, tissue compartments, and cell types (Chen et al., 2020; Wang et al., 2020; Wölfel et al., 2020; Young et al., 2020). In our profiling of COVID-19 clinical samples from multiple body sites of the same individual, we found that the viral load and viral subtypes vary across different body sites, possibly affected by interactions between microbial and other viral species as well as overall metagenomic diversity in different microenvironments of each body site. The effects of metagenomic diversity and inter-compartment heterogeneity on SARS-CoV-2 biology and COVID-19 symptom severity are also not understood. The biomass of different sample types varies, which requires library construction methods compatible with various sample content and different RNA quality. It is difficult to obtain high-quality, unbiased metagenomic data using conventional library construction methods from low-quantity samples as well as samples such as stool, in which bacteria dominate the metagenomes, because conventional methods are not sufficiently sensitive. Our previous study (Di et al., 2020) demonstrated a wide tolerance of sample input amount by our DNA/RNA hybrid tagmentation strategy. Our method showed the strongest data improvement in stool samples, likely because of better performance with low-input samples.
The versatility of MINERVA as a two-part protocol integrating MINERVA-m and MINERVA-e makes it possible to use one standard sample pipeline for highly sensitive metagenomic analysis and targeted deep sequencing of specific transcripts. To further improve the integration level of MINERVA, we evaluated the possibility of analyzing the merged datasets of MINERVA-m and MINERVA-e. It was revealed that the merged datasets (MINERVA-m+e) had highly concordant metagenomic compositions as the MINERVA-m datasets alone (Figures 7A and 7B), demonstrating the feasibility to perform metagenomic analysis and deep SARS-CoV-2 sequencing in a single sequencing run by MINERVA. The MINERVA-m+e and dsDL libraries were also compatible in terms of alpha diversities, species richness, and Shannon index (Figures S7A and S7B) and microbial compositions of different sample types (Figures 7C and S7C).
Figure 7.

Evaluation of Microbial Profiles by Merged MINERVA Datasets
(A and B) Correlation of alpha diversity, including species richness (A) and Shannon index (B), between MINERVA-m and merged (m+e) datasets.
(C) Comparison of bacterial composition of pharyngeal samples between dsDL and merged MINERVA (m+e) datasets. Genera with a relative abundance over 1% are shown here.
Using MINERVA, we demonstrated the first large-scale profiling of metagenomic composition of different body sites in the context of COVID-19. Several studies have investigated the relationship between gut microbes with SARS-CoV-2 infection and COVID-19 severity (Gou et al., 2020; Gu et al., 2020; Zuo et al., 2020); however, there is no discussion of metagenomic composition of other body sites. As we show here with MINERVA data from a wide range of sample types, there are large body site-specific differences, and our data suggest that microbial composition in pharyngeal swab samples also correlates significantly with disease severity. The metagenomic profile of these other body sites, which are arguably more directly involved in viral infection, have not been reported or investigated elsewhere with such a large sample size (Shen et al., 2020). Using MINERVA, we highlight several new directions of clinical and basic research, and with further investigation, these could shed light on the complex interactions between SARS-CoV-2 pathology, host microbial communities, host immunity, and disease progression. We also showed that MINERVA metagenomic profiles can identify highly abundant pathogenic species in samples from individuals with COVID-19 in a non-targeted fashion, which is challenging with conventional approaches. In our samples, we found ~13.3% (6 of 45) of individuals with COVID-19 whose samples contained a high relative abundance of a pathogenic microbe other than SARS-CoV-2. Although this could indicate colonization or co-infection, it raises concerns regarding potentially missed co-infections. One secondary study found 8% of patients experiencing bacterial/fungal co-infection, but the rate of broad-spectrum antibiotics use for COVID-19 patients is much higher (72%) (Rawson et al., 2020). It is well known that co-infections in severe pneumonia can greatly affect patient outcome (Crotty et al., 2015; Shah et al., 2016), and it is estimated that 50% of patients with COVID-19 who died in this pandemic had secondary bacterial infections (Zhou et al., 2020a). Further primary studies using MINERVA could help to elucidate true co-infection rates to better guide strategies for antibiotics use.
Limitations
MINERVA was not created to be a rapid diagnostic assay; rather, we hope that its ease of use, versatility, scalability, sensitivity, and cost effectiveness will drive adoption of routine sequencing of COVID-19 clinical samples and facilitate multiple areas of much-needed SARS-CoV-2 and COVID-19 research for clinicians and researchers.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| clinical samples | Beijing Ditan Hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DNase I (RNase-free) | NEB | Cat#M0303 |
| Superscript II reverse transcriptase | Invitrogen | Cat#18064014 |
| N,N-Dimethylformamide | Sigma | Cat#D4551 |
| Recombinant RNase Inhibitor | Takara | Cat#2313 |
| Deoxynucleotide (dNTP) Solution Set | NEB | Cat#N0446S |
| Betaine solution | Sigma | Cat#B0300 |
| PEG8000 | VWR Life Science | Cat#97061 |
| ATP | NEB | Cat#P0756 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| RNA Clean & Concentrator-5 kit | Zymo Research | Cat#R1015 |
| QIAamp Viral RNA Mini Kit | QIAGEN | Cat#52906 |
| MGIEasy rRNA removal kit | BGI | Cat#1000005953 |
| TruePrep DNA Library Prep Kit V2 for Illumina | Vazyme | Cat#TD501 |
| TargetSeq One Cov Kit | iGeneTech | Cat#502002-V1 |
| Q5 High-Fidelity 2x Master Mix | NEB | Cat#M0492 |
| VAHTS DNA Clean Beads | Vazyme | Cat#N411 |
| ChamQ SYBR qPCR master mix | Vazyme | Cat#Q311-02 |
| Deposited Data | ||
| The sequencing data generated during this study | This paper | Genome Sequencing Archive: PRJCA002533 |
| Experimental Models: Cell Lines | ||
| NIH/3T3 | ATCC | CRL-1658 |
| Oligonucleotides | ||
| Decamer(N10): NNNNNNNNNN |
Sangon | N/A |
| T30VN: TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN |
Sangon | N/A |
| SARS-CoV-2 qPCR N gene forward primer: GGGGAACTTCTCCTGCTAGAAT |
sangon | N/A |
| SARS-CoV-2 qPCR N gene reverse primer: CAGACATTTTGCTCTCAAGCTG |
sangon | N/A |
| xGen Universal Blockers | IDT | Cat#1079586 |
| Software and Algorithms | ||
| BBmap | Bushnell, 2014 | https://www.osti.gov/biblio/1241166 |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Centrifuge | Kim et al., 2016 | https://codeload.github.com/infphilo/centrifuge/zip/centrifuge-genome-research |
| MEGAHIT | Li et al., 2015 | https://github.com/voutcn/megahit |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml |
| Bedtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/# |
| vegan | Oksanen et al., 2019 | https://cran.r-project.org/web/packages/vegan/index.html |
| MaAsLin | (Morgan et al., 2012) | https://github.com/pooranis/maaslin/ |
| Picard Tools | (Broad Institute, 2019) | http://broadinstitute.github.io/picard/ |
| Other | ||
| bench protocol | This paper | Methods S1 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jianbin Wang (jianbinwang@tsinghua.edu.cn)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The sequencing data generated during this study have been uploaded to Genome Sequencing Archive (PRJCA002533). Detailed bench protocol is available from Mendeley Data at https://doi.org/10.17632/45w7hv53yr.1.
Experimental Model and Subject Details
Cell Lines
The NIH/3T3 cell line was purchased from ATCC. The complete growth medium was made using DMEM (cat. No.11965-092; Life Technologies), 10% fetal bovine serum (cat. No. 16000-044; Life Technologies), and 1% penicillin and streptomycin. The cell line was incubated with 5% carbon dioxide at 37°C in a culture flask.
Ethics Approval
This study was approved by the Ethics Committee of Beijing Ditan Hospital, Capital Medical University (No. KT2020-006-01).
Patients and Clinical Samples
From January 23, 2020 to April 20, 2020, 91 patients were enrolled in this study according to the 7th guideline for the diagnosis and treatment of COVID-19 from the National Health Commission of the People’s Republic of China. All patients, diagnosed with COVID-19, were hospitalized in Beijing Ditan Hospital and classified into four severity degrees, mild, moderate, severe, and critical illness, according to the 7th guideline for the diagnosis and treatment of COVID-19 from the National Health Commission of the People’s Republic of China (https://www.chinadaily.com.cn/pdf/2020/1.Clinical.Protocols.for.the.Diagnosis.and.Treatment.of.COVID-19.V7.pdf). Briefly, mild cases are those with mild clinical symptoms, and there was no sign of pneumonia on imaging. Moderate cases are those showing fever and respiratory symptoms with radiological findings of pneumonia. Severe cases include the adult cases meeting any of the following criteria: (1) respiratory distress (≥30 breaths/min); (2) oxygen saturation ≤ 93% at rest; (3) arterial partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ≤ 300 mmHg. We collected 136 samples (60 pharyngeal swab samples, 51 sputum samples, and 25 stool samples) from these patients. Pharyngeal swab samples were collected into the viral sampling medium (Yocon, MT0301-1), sputum samples were collected directly into sterile containers, and stool samples were collected in the storage buffer using the stool sampling kit (Longsee, LS-R-P-003). All samples were stored at −80 °C.
Method Details
Optimization of MINERVA Protocol
We used the total RNA extracted from 3T3 cells to optimize experimental protocols. RNA extraction was performed using RNeasy Mini Kit (QIAGEN, Cat.No.74104). DNA was then removed through DNase I (NEB, Cat.No.M0303) digestion. The resulting total RNA was concentrated by RNA Clean & Concentrator-5 kit (Zymo Research, Cat R1015), and its quality was assessed by the Fragment Analyzer Automated CE System (AATI). Its quantification was done by Qubit 2.0 (Invitrogen). To optimize the MINERVA protocol, different amount of random decamer (N10) (0, 1, or 10 μM) was used to set up reverse transcription reactions. Titration of Tn5 transposome (0.2, 0.5, or 1.0 μl Vazyme V50; 0.05 or 0.25 μl home-made pTXB1) was performed in tagmentation procedure. In all tests, 10 ng 3T3 total RNA was used, and all reagents except for N10 or Tn5 transposome remain unchanged. All libraries were sequenced on Illumina NextSeq 500 with 2x75 paired-end mode. Clean data was aligned to GRCm38 genome and known transcript annotation using Tophat2 v2.1.1. Ribosome-removed aligned reads were proceeded to calculate FPKM by Cufflinks v2.2.1 and gene body coverage by RSeQC v.2.6.4.
RNA Extraction and rRNA Removal
For all the clinical samples, nucleic acids extraction was performed in a BSL-3 laboratory. Samples were deactivated by heating at 56°C for 30 min before extraction. Total RNA was extracted using QIAamp Viral RNA Mini Kit (QIAGEN) following the manufacturer’s instructions. In most (130 out of 136) samples we specifically omitted the use of carrier RNA due to its interference on the most prevalent sample preparation protocols for high-throughput sequencing. After nucleic acids extraction, rRNA was removed by rDNA probe hybridization and RNase H digestion, followed by DNA removal through DNase I digestion, using MGIEasy rRNA removal kit (BGI, Shenzhen, China). The final elution volume was 12-20 μl for each sample. For carrier RNA removal tests, 1.7 μg polyA carrier RNA was spiked into 18 μl of elute from QIAamp Viral RNA Mini Kit. To remove the carrier RNA from these spike-in samples and other samples extracted with carrier RNA, 2 μg poly(T) 59-mer (T59) oligo was added during the rDNA hybridization step.
dsDL Metagenomic RNA Library Construction and Sequencing
The libraries were constructed using MGIEasy reagents (BGI, China) following manufacture’s instruction. The purified RNA, after rRNA depletion and DNA digestion, underwent reverse transcription, second strand synthesis, and sequencing adaptor ligation. After PCR amplification, DNA was denatured and circularized before being sequenced on DNBSEQ-T7 sequencers (BGI, China).
MINERVA Library Preparation
A step-by-step protocol is available (Methods S1). Briefly, 2.7 μl RNA from rRNA and DNA removal reaction was used for standard SHERRY reverse transcription, with the following modifications: 1) 10 pmol random decamer (N10) was added to improve coverage; 2) initial concentrations of dNTPs and oligo-dT (T30VN) were increased to 25 mM and 100 μM, respectively. For 5.4 μl and 10.8 μl input, the entire reaction was simply scaled up 2 and 4 folds, respectively. The RNA/DNA hybrid was tagmented in TD reaction buffer (10 mM Tris-Cl pH 7.6, 5 mM MgCl2, 10% DMF) supplemented with 3.4% PEG8000 (VWR Life Science, Cat.No.97061), 1 mM ATP (NEB, Cat.No. P0756), and 1U/μl RNase inhibitor (TaKaRa, Cat.No. 2313B). The reaction was incubated at 55°C for 30 min. 20 μl tagmentation product was mixed with 20.4 μl Q5 High-Fidelity 2X Master Mix (NEB, Cat.No. M0492L), 0.4 μl SuperScript II reverse transcriptase, and incubated at 42°C for 15 min to fill the gaps, followed by 70°C for 15 min to inactivate SuperScript II reverse transcriptase. Then index PCR was performed by adding 4 μl 10 μM unique dual index primers and 4 μl Q5 High-Fidelity 2X Master Mix, with the following thermo profile: 98°C 30 s, 18 cycles of [98°C 20 s, 60°C 20 s, 72°C 2 min], 72°C 5 min. The PCR product was then purified with 0.8x VAHTS DNA Clean Beads (Vazyme, Cat. No. N411). These libraries were sequenced on Illumina NextSeq 500 with 2x75 paired-end mode for metagenomic analysis.
For preparing MINERVA-e libraries through SARS-CoV-2 enrichment, 1 μL metagenomic library was first quantified for N gene using quantitative PCR (F: GGGGAACTTCTCCTGCTAGAAT, R: CAGACATTTTGCTCTCAAGCTG) after 1:200 dilution. Then 8~16 libraries were pooled together based on qPCR results to obtain relatively uniform amount of data. Specifically, Ct values were divided into 4 groups and each group corresponded to a pooling volume: Ct above 28 (50ul), Ct of 24-28 (20ul), Ct of 20-24 (8ul), Ct below 20 (3ul). Pooled library was further processed with TargetSeq One Cov Kit (iGeneTech, Cat.No.502002-V1) following manufacturer’s instruction. The iGeneTech Blocker was replaced by the IDT xGen Universal Blockers (NXT). These MINERVA libraries were sequenced on Illumina NextSeq 500 with 2x75 paired-end mode for deep SARS-CoV-2 analysis.
Data Processing
For metagenomic RNA-seq data, raw reads were quality controlled using BBmap (version 38.68) (Bushnell, 2014) and then mapped to the human genome reference (GRCh38) using STAR (version 2.6.1d) (Dobin et al., 2013) with default parameters. All unmapped reads were collected using samtools (version 1.3) (Li et al., 2009) for microbial taxonomy assignment by Centrifuge (version 1.0.4) (Kim et al., 2016). Custom reference was built from all complete bacterial, viral and any assembled fungal genomes downloaded from NCBI RefSeq database (viral and fungal genomes were downloaded on February 4th, 2020, and bacterial genomes were downloaded on November 14th, 2018). There were 11,174 bacterial, 8,997 viral, and 308 fungal genomes respectively. Bacterial Shannon diversity (entropy) was calculated at species level, and the species abundance was measured based on total reads assigned at the specific clade normalized by genome size and sequencing depth. Bacterial genus composition was analyzed based on reads proportion directly assigned by Centrifuge. For dsDL sequencing data, sub-sampling was performed for each sample to obtain ~12M pair-end nonhuman reads, which is the median of MINERVA datasets. The same workflow as above was performed for the removal of human reads and microbial taxonomy assignment.
For metagenome assembly, nonhuman reads from samples of the same sample type were merged first for co-assembly using MEGAHIT (version 1.2.9) (Li et al., 2015) with default parameters. In total there were approximately 200M, 150M and 50M read pairs for pharyngeal (n = 68), sputum (n = 59) and stool (n = 33) samples respectively. Contigs longer than 200nt were kept, resulting in 273,434; 266,932 and 58,836 contigs for each sample type, and the N50 values were 800bp, 689bp and 699bp respectively. Centrifuge was used to assign taxonomies to contigs using the database mentioned above. After taxonomy assignment at contigs level, we mapped reads of each sample back to the co-assembled contigs to assess the microbial composition of each individual sample. Reads mapping to the contigs was done using Bowtie2 (version 2.3.5.1) (Langmead and Salzberg, 2012) with “–very-sensitive” mode. Pileup from BBmap (version 38.68) package were used to calculate the coverage of mapped reads for each individual samples. The overall mapping ratio were 56.3%, 57.4% and 46.2% for each sample type. For species identified with high abundance by metagenomics analysis, we mapped reads back to their genome reference to check the genome coverage of each sample by using Bowtie2 (version 2.3.5.1) with “–very-sensitive” mode. Bedtools (version 2.29.0) (Quinlan and Hall, 2010) was used to calculate the genome coverage, and the genome coverage was then normalized by dividing the sequencing depth of corresponding samples and then multiply a scaling factor of 1,000,000 for comparisons. We calculated the relative abundance of taxa as the ratio of reads assigned to that taxa for the metagenomic strategy, then compared it with the ratio of reads mapped to contigs which were assigned as that taxa for the metagenome assembly strategy.
For SARS-CoV-2 genome analysis, raw reads were trimmed to remove sequencing adaptors and low-quality bases with Cutadapt v1.15. BWA 0.7.15-r1140 was used to align reads to the SARS-CoV-2 reference genome (NC_045512.2). Then we depleted duplicates from the properly-paired alignment with Picard Tools v2.17.6 (Broad Institute, 2019). The resulting bam was further filtered for mutation calling by two criterions: 1) The mapping position was before 29856 in SARS-CoV-2 genome; 2) The soft-clip bases of each read accounted for less than 50%. We used mpileup function in samtools v1.10 to call SNP and InDel with parameter -C 50 -Q 20 -q 15 -E -d 0. We called mutation if the depth ≥ 10 and strand bias ≥ 0.2 or supported by independently distributed reads, which were checked in IGV (Integrative Genome Browser). The strand bias is defined as the value that minimum of positive strand depth and negative strand depth divided by the maximum. For cluster heatmap analysis, we chose samples of high coverage and two asymptomatic family, and included all mutations which variant allele frequency ≥ 0.2 and strand bias ≥ 0.2 occurred in at least one sample. The frequency of mutated allele was simply calculated as observed mutant reads divided by total reads for each site.
Quantification and Statistical Analysis
All statistical analyses were performed in R, version 3.5.1. Permutational multivariate analysis of variance (PERMANOVA) was applied to assess the effect of meta factors, including age, gender and disease status on the Bray-Curtis distance among samples by using adonis2 function in R package vegan (Oksanen et al., 2020). To account for potential effect of repeated-measures of multiple samples from the same subject, permutation was constrained within the same time point during PERMANOVA analysis. PCoA ordination analysis was performed based on Bray-Curtis distance to explore the difference among samples. The Bray-Curtis distance was calculated by vegdist function and PCoA axes were calculated using cmdscale functions from R package vegan. The p values were calculated by PERMANOVA analysis. Generalized Estimating Equations (GEE) was applied to assess associations of microbes with disease status by using gee function in R (Carey, 2006; Carl and Kühn, 2007). Only microbes with relative abundance > 0.01 and prevalence > 0.01 were used in this model. P values were calculated from the estimated robust z-score based on standard Gaussian distribution. The Benjamini-Hochberg procedure was applied for the correction of the p values. Results were filtered based on both significance (BH-adjusted p < 0.05) and effect size (absolute coefficient > 0.1). Kruskall-Wallis test and Wilcoxon rank-sum test were used for other multi-group and two-group comparisons respectively if not specifically stated.
Additional Resources
Detailed Protocol
A detailed bench protocol is available as Methods S1.
Acknowledgments
We thank Chenyang Geng and the BIOPIC sequencing platform at Peking University for assistance with high-throughput sequencing experiments and Amelia Huang for assistance with figure preparation. This work was supported by the Ministry of Science and Technology of China (2018YFA0108100, 2018YFA0800200, and 2018YFC1002300), the National Natural Science Foundation of China (21675098, 21927802, and 21525521), the 2018 Beijing Brain Initiative (Z181100001518004), the Beijing Advanced Innovation Center for Structural Biology, the Beijing Advanced Innovation Center for Genomics, the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20SC01), the Hong Kong Research Grants Council Theme-based Research Scheme (RGC TBRS T12-704/16R-2) and Collaborative Research Fund (RGC CRF C6002-17G), the Hong Kong RGC Early Career Support Scheme (RGC ECS 26101016), the Hong Kong Epigenomics Project (LKCCFL18SC01-E), and HKUST BDBI Labs.
Author Contributions
C.C., Y.C., X.S.X., H.Z., Y.H., and J.W. conceived the project. J.L., P.D., Q.L., and C.S. conducted experiments. C.C., L.D., Q.J., J.L., Y.H., and J.W. analyzed the data. C.C., J.L., L.D., Q.J., A.R.W., Y.H., and J.W. wrote the manuscript with help from all other authors.
Declaration of Interests
The authors declare no competing interests.
Published: November 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.11.030.
Supplemental Information
References
- An J., Liao X., Xiao T., Qian S., Yuan J., Ye H., Qi F., Shen C., Liu Y., Wang L. Clinical characteristics of the recovered COVID-19 patients with re-detectable positive RNA test. Ann. Transl. Med. 2020;8 doi: 10.21037/atm-20-5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Yao L., Wei T., Tian F., Jin D.Y., Chen L., Wang M. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA. 2020;323:1406–1407. doi: 10.1001/jama.2020.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B. Accurate, Splice-Aware Aligner; 2014. BBMap: A Fast. [Google Scholar]
- Carey V.J. Ported to R by Thomas Lumley (versions 3.13, 4.4, version 4.13). R package version. 2006;4:13. B. R. gee: Generalized Estimation Equation solver. https://CRAN.R-project.org/package=gee. 11. [Google Scholar]
- Carl G., Kühn I. Analyzing spatial autocorrelation in species distributions using Gaussian and logit models. Ecol. Modell. 2007;207:159–170. [Google Scholar]
- Chen C., Gao G., Xu Y., Pu L., Wang Q., Wang L., Wang W., Song Y., Chen M., Wang L. SARS-CoV-2-Positive Sputum and Feces After Conversion of Pharyngeal Samples in Patients With COVID-19. Ann. Intern. Med. 2020;172:832–834. doi: 10.7326/M20-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox M.J., Loman N., Bogaert D., O’Grady J. Co-infections: potentially lethal and unexplored in COVID-19. Lancet Microbe. 2020;1:e11. doi: 10.1016/S2666-5247(20)30009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty M.P., Meyers S., Hampton N., Bledsoe S., Ritchie D.J., Buller R.S., Storch G.A., Micek S.T., Kollef M.H. Epidemiology, Co-Infections, and Outcomes of Viral Pneumonia in Adults: An Observational Cohort Study. Medicine (Baltimore) 2015;94:e2332. doi: 10.1097/MD.0000000000002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lastours V., Malosh R.E., Aiello A.E., Foxman B. Prevalence of Escherichia coli carriage in the oropharynx of ambulatory children and adults with and without upper respiratory symptoms. Ann. Am. Thorac. Soc. 2015;12:461–463. doi: 10.1513/AnnalsATS.201412-586LE. [DOI] [PubMed] [Google Scholar]
- Deng X., Gu W., Federman S., du Plessis L., Pybus O.G., Faria N., Wang C., Yu G., Pan C.Y., Guevara H. A Genomic Survey of SARS-CoV-2 Reveals Multiple Introductions into Northern California without a Predominant Lineage. medRxiv. 2020 doi: 10.1101/2020.03.27.20044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L., Fu Y., Sun Y., Li J., Liu L., Yao J., Wang G., Wu Y., Lao K., Lee R.W. RNA sequencing by direct tagmentation of RNA/DNA hybrids. Proc. Natl. Acad. Sci. USA. 2020;117:2886–2893. doi: 10.1073/pnas.1919800117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauci A.S., Lane H.C., Redfield R.R. Covid-19 - Navigating the Uncharted. N. Engl. J. Med. 2020;382:1268–1269. doi: 10.1056/NEJMe2002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou, W., Fu, Y., Yue, L., Chen, G.-d., Cai, X., Shuai, M., Xu, F., Yi, X., Chen, H., Zhu, Y.J., et al. (2020). Gut microbiota may underlie the predisposition of healthy individuals to COVID-19. medRxiv. 10.1101/2020.04.22.20076091. [DOI]
- Gu S., Chen Y., Wu Z., Chen Y., Gao H., Lv L., Guo F., Zhang X., Luo R., Huang C. Alterations of the Gut Microbiota in Patients with COVID-19 or H1N1 Influenza. Clin. Infect. Dis. 2020:ciaa709. doi: 10.1093/cid/ciaa709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson D.F., Helgason A., Jonsson H., Magnusson O.T., Melsted P., Norddahl G.L., Saemundsdottir J., Sigurdsson A., Sulem P., Agustsdottir A.B. Spread of SARS-CoV-2 in the Icelandic Population. N. Engl. J. Med. 2020;382:2302–2315. doi: 10.1056/NEJMoa2006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Lau E.H.Y., Wu P., Deng X., Wang J., Hao X., Lau Y.C., Wong J.Y., Guan Y., Tan X. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020;26:672–675. doi: 10.1038/s41591-020-0869-5. [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan X.C., Tickle T.L., Sokol H. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13 doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J., Blanchet F.G., Friendly M., Kindt R., Legendre P., McGlinn D., Minchin P.R., O’Hara R.B., Simpson G.L., Solymos P. vegan: Community Ecology Package. R package version. 2019;2:5–6. https://cran.r-project.org/web/packages/vegan/index.html [Google Scholar]
- Kim D., Song L., Breitwieser F.P., Salzberg S.L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26:1721–1729. doi: 10.1101/gr.210641.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Liu C.M., Luo R., Sadakane K., Lam T.W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- Lu J., Cui J., Qian Z., Wang Y., Zhang H., Duan Y., Wu X., Yao X., Song Y., Li X. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020;7:1012–1023. doi: 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., du Plessis L., Liu Z., Hill V., Kang M., Lin H., Sun J., Francois S., Kraemer M.U.G., Faria N.R. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell. 2020;181:997–1003.e9. doi: 10.1016/j.cell.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson T.M., Moore L.S.P., Zhu N., Ranganathan N., Skolimowska K., Gilchrist M., Satta G., Cooke G., Holmes A. Bacterial and fungal co-infection in individuals with coronavirus: A rapid review to support COVID-19 antimicrobial prescribing. Clin. Infect. Dis. 2020:ciaa530. doi: 10.1093/cid/ciaa530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L.L., Wang Y.M., Wu Z.Q., Xiang Z.C., Guo L., Xu T., Jiang Y.Z., Xiong Y., Li Y.J., Li X.W. Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin. Med. J. (Engl.) 2020;133:1015–1024. doi: 10.1097/CM9.0000000000000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N.S., Greenberg J.A., McNulty M.C., Gregg K.S., Riddell J., 4th, Mangino J.E., Weber D.M., Hebert C.L., Marzec N.S., Barron M.A. Bacterial and viral co-infections complicating severe influenza: Incidence and impact among 507 U.S. patients, 2013-14. J. Clin. Virol. 2016;80:12–19. doi: 10.1016/j.jcv.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Xiao Y., Kang L., Ma W., Shi L., Zhang L., Zhou Z., Yang J., Zhong J., Yang D. Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020;71:713–720. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Xu Y., Gao R., Lu R., Han K., Wu G., Tan W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA. 2020;323:1843–1844. doi: 10.1001/jama.2020.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . 2020. WHO Coronavirus Disease (COVID-19) Dashboard.https://covid19.who.int/ [Google Scholar]
- Wölfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Müller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M., Liu X., Ji J., Li M., Li J., Yang L., Sun W., Ren P., Yang G., Zhao J. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020;12:57. doi: 10.1186/s13073-020-00751-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B.E., Ong S.W.X., Kalimuddin S., Low J.G., Tan S.Y., Loh J., Ng O.T., Marimuthu K., Ang L.W., Mak T.M., Singapore 2019 Novel Coronavirus Outbreak Research Team Epidemiologic Features and Clinical Course of Patients Infected With SARS-CoV-2 in Singapore. JAMA. 2020;323:1488–1494. doi: 10.1001/jama.2020.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaura E., Keijser B.J., Huse S.M., Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259. doi: 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo T., Zhang F., Lui G.C.Y., Yeoh Y.K., Li A.Y.L., Zhan H., Wan Y., Chung A.C.K., Cheung C.P., Chen N. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology. 2020;159:944–955.e8. doi: 10.1053/j.gastro.2020.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “Picard Toolkit.” 2019. Broad Institute, GitHub Repository. http://broadinstitute.github.io/picard/; Broad Institute.
- 2020. 2.5-7. https://cran.r-project.org/web/packages/vegan/index.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated during this study have been uploaded to Genome Sequencing Archive (PRJCA002533). Detailed bench protocol is available from Mendeley Data at https://doi.org/10.17632/45w7hv53yr.1.