

Abstract
Cyclin-dependent kinases 4 and 6 (CDK4/6) are key regulators of the cell cycle, and CDK4/6 inhibitors are FDA-approved for treating patients with metastatic breast cancer. However, due to conservation of their ATP-binding sites, development of selective agents has remained elusive. Here, we report imide-based degrader molecules capable of degrading both CDK4/6, or selectively degrading either CDK4 or CDK6. We were also able to tune the activity of these molecules against Ikaros (IKZF1) and Aiolos (IKZF3), well-established targets of imide-based degraders. We found that in mantle cell lymphoma cell lines, combined IKZF1/3 degradation with dual CDK4/6 degradation exhibited enhanced anti-proliferative effects compared to CDK4/6 inhibition, CDK4/6 degradation, or IKZF1/3 degradation. In sum, we report here the first compounds capable of inducing selective degradation of CDK4 and CDK6 as tools to pharmacologically dissect their distinct biological functions.
Keywords: CDK4, CDK6, degradation, PROTAC, CRBN
Cyclin-dependent kinases 4 and 6 (CDK4/6) regulate the G1-S cell cycle transition by phosphorylating the tumor suppressor retinoblastoma (Rb), thereby triggering gene expression programs that promote S phase entry[1]. As such, CDK4/6 are attractive targets for cancer therapy, and the dual CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib have been FDA approved for treating patients with advanced or metastatic breast cancer, leading to prolonged progression-free-survival[2]. These agents are also currently under investigation in subsets of lung cancers, sarcomas, and lymphomas, such as mantle cell lymphoma (MCL), that exhibits aberrant cell cycle progression via activation of CDK4/6[3].
Although CDK4 and CDK6 are highly homologous, CDK4- and CDK6-specific functions have been reported. For example, in a mouse model of non-small cell lung carcinoma, genetic ablation of CDK4 but not CDK6 induced senescence in lung cancer cells expressing mutant K-Ras[4]. On the other hand, CDK6-specific functions include participating as part of a transcription complex that regulates expression of p16INK4a and VEGF-A[5] and acting as a co-factor for NFκB-dependent gene expression[6]. We also recently demonstrated that CDK6, but not CDK4, phosphorylates NFAT family transcription factors to promote T cell activation and enhance the anti-tumor immune response[7].
As current CDK4/6 inhibitors target their highly conserved ATP-binding pockets and are approximately equipotent inhibitors of both proteins, we hypothesized that achieving selectivity would require exploiting selectivity determinants outside of the ATP-binding pocket. One approach is to develop small molecule degraders, bifunctional molecules that consist of an E3 ubiquitin ligase-binding moiety and a target-binding moiety connected via an optimizable linker. Successful degradation requires induced heterodimerization between the E3 ligase and the target of interest, resulting in the target’s polyubiquitination and subsequent proteasomal degradation[8]. This strategy has been deployed to achieve selective degradation using non-selective ligands. For example, selective degradation of CDK9 was achieved by converting the non-selective CDK inhibitor SNS-032 into a degrader[9]. We also developed a BRD4 degrader that achieves selectivity versus BRD2 and BRD3 by exploiting structural differences in the bromodomain/E3 ligase protein-protein interface[10].
The small molecule thalidomide, which binds to the ubiquitously expressed E3 ligase substrate adapter Cereblon (CRBN), has often been used as the E3 ligase-binding moiety in degraders[11]. Here, we generated CRBN-recruiting degraders and identified molecules capable of inducing both dual CDK4/6 and selective CDK4 or CDK6 degradation.
High-resolution crystal structures of palbociclib [PDB ID: 5L2I], ribociclib [PDB ID: 5L2T], and abemaciclib [PDB ID: 5L2S] with CDK6 revealed similar binding modes where the aminopyrimidine moiety forms three hydrogen bonds with the backbones of Val101 and Asp163, and the piperazine ring is stabilized via direct interactions with a solvent-exposed ridge consisting of Asp104 and Thr107. We hypothesized that we could attach linkers at the piperazine moiety without compromising affinity to CDK4/6, so we designed palbociclib-, ribociclib-, and abemaciclib-based degraders using thalidomide as the CRBN-recruiting moiety (Figure 1). Compounds were alkylated from the 4-nitrogen of the piperazine, extended with either a polyethylene glycol (PEG) or an alkyl linker, and terminated at either the 4- or 5-position of thalidomide.
Figure 1.
Chemical structures of CDK4/6 degraders derived from palbociclib (A), ribociclib (B), or abemaciclib.
We first assessed the CDK4/6 kinase inhibition activity of these degraders using enzymatic assays. Most compounds maintained a biochemical IC50 ranging from 1 – 50 nM (Table S1), validating our choice of linker attachment site. Next, we evaluated whether biochemically-active compounds could degrade CDK4 or CDK6 in Jurkat cells, which express both proteins, and treated Jurkat cells for 4h with 1 µM of degraders derived from each CDK4/6 inhibitor: BSJ-02–162 (palbociclib), BSJ-01–152 (ribociclib), or BSJ-01–184 (abemaciclib). Immunoblotting for CDK4/6 revealed that each inhibitor could be successfully converted into active CDK4/6 degraders (Figure 2). Interestingly, abemaciclib-based degraders also induced degradation of CDK9, a known off-target[12].
Figure 2.

Palbociclib-, ribociclib-, and abemaciclib-based PROTACs induce CDK4/6 degradation. Immunoblots from Jurkat cells treated with 1 µM of the indicated compound for 4h.
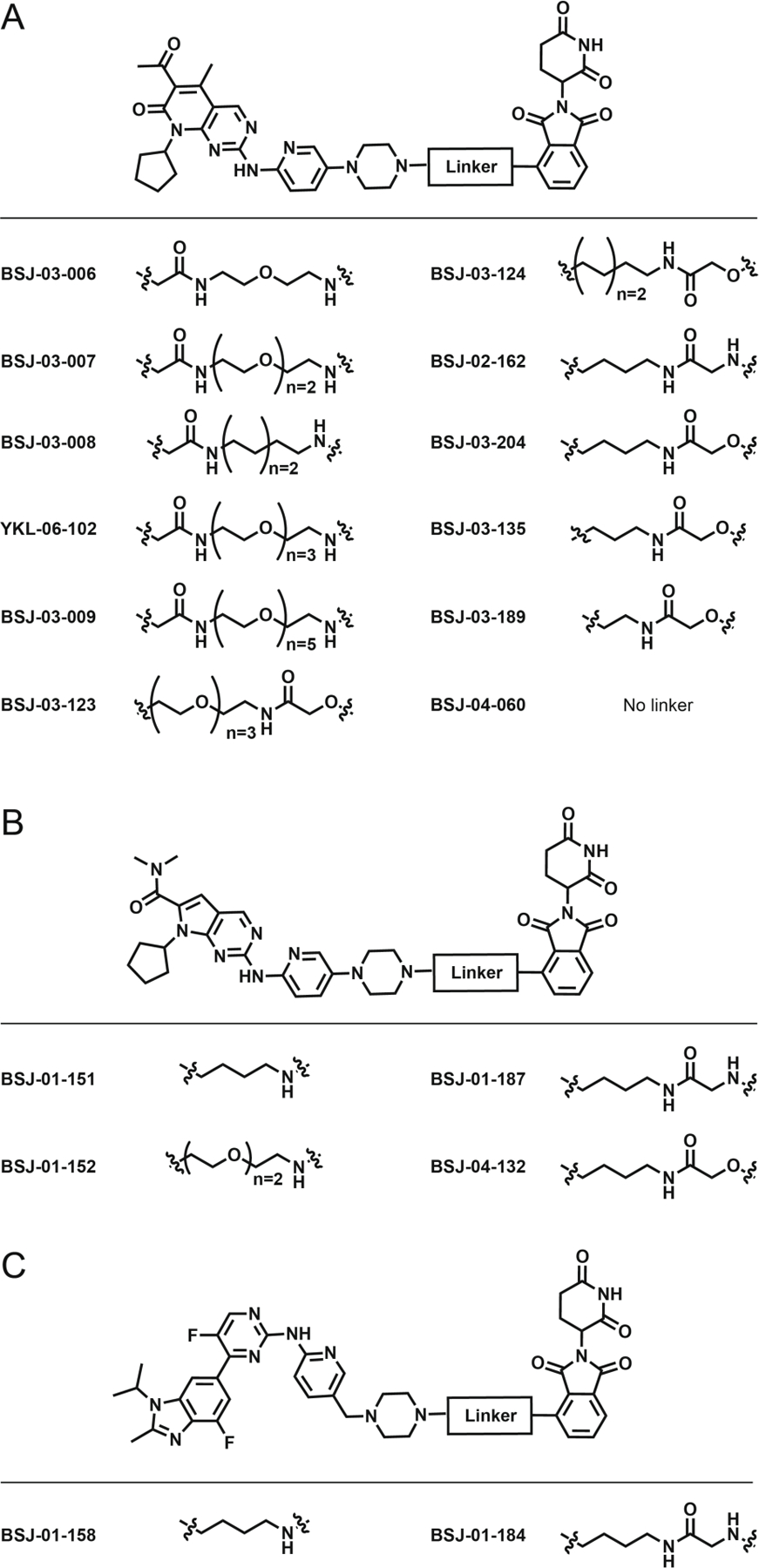
Subsequently, we found that acute exposure to some degraders differentially affected the abundance of CDK4 and CDK6, as a function of inhibitor, linker length, and linker composition. For example, BSJ-02–162, which consists of an alkyl linker conjugated to palbociclib, degraded both CDK4 and CDK6 (Figure 3A). In contrast, BSJ-01–187, a ribociclib-based degrader with a 4-carbon alkyl linker, selectively degraded CDK4 (Figure 3C), while the extended PEG-3 linker of the palbociclib-derived compound YKL-06–102 induced selective degradation of CDK6 (Figure 3E).
Figure 3.
Variations of linker length and composition result in differing selectivity for CDK4/6 and IKZF1/3 degradation. Immunoblots from WT or Crbn−/− Jurkat cells treated for 4h with (A) BSJ-02–162; (B) BSJ-03–204; (C) BSJ-01–187; (D) BSJ-04–132; (E) YKL-06–102; and (F) BSJ-03–123.
We also evaluated compound-induced degradation of IKZF1 and IKZF3, known targets of imides and some imide-based degraders[13], and found that BSJ-02–162, BSJ-01–187, and YKL-06–102 all induced degradation of IKZF1/3 (Figure 3). As previous studies indicated that varying the thalidomide aryl amine nitrogen to an oxylamide could prevent recruitment of IKZF1/3 to CRBN[10, 13c], we investigated whether the equivalent modification would have the same effect here. Thus, we assessed the analogs BSJ-03–204 (Figure 3B; dual CDK4/6 degrader), BSJ-04–132 (Figure 3D; selective CDK4 degrader), and BSJ-03–123[14] (Figure 3F; selective CDK6 degrader), none of which induced IKZF1/3 degradation. Finally, we found that treatment of isogenic Jurkat cells in which CRBN was deleted by CRISPR/Cas9 had no effect on the abundance of CDK4/6 and/or IKZF1/3 protein, demonstrating that that degradation was dependent on CRBN (Figure 3).
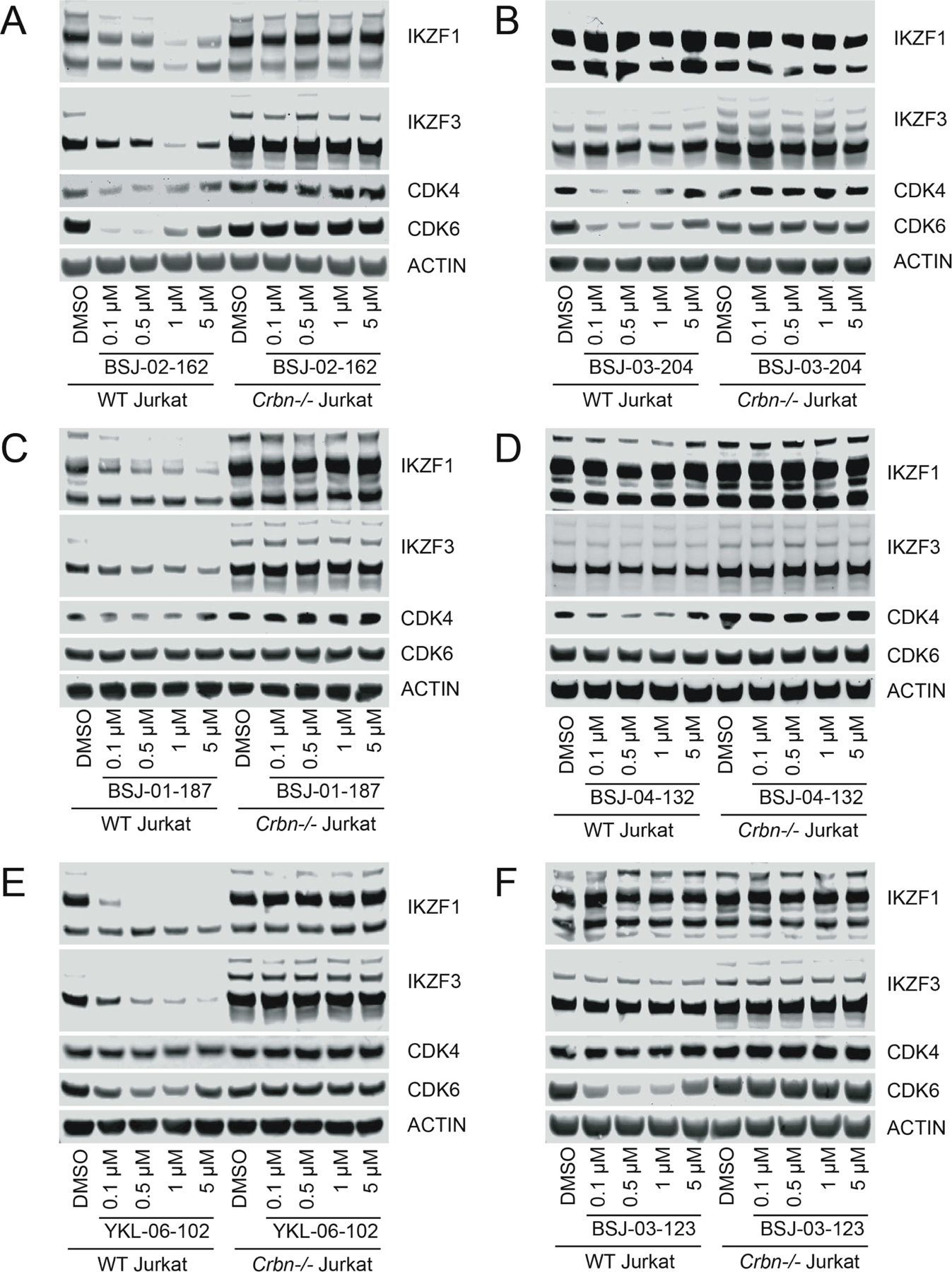
As CDK4/6 inhibition induces cell cycle arrest, we compared the effects of BSJ-02–162 to those of palbociclib or lenalidomide on the cell cycle profile of wildtype and Crbn−/− Jurkat cells by quantifying DNA content via propidium iodide staining (Figure 4). As expected, treatment of wildtype or Crbn−/− Jurkat cells with 100 nM palbociclib for 24h induced profound G1 arrest in both cell lines, while treatment with 100 nM lenalidomide for 24h had no effect. In contrast, treatment with 100 nM of BSJ-02–162 for 24h induced G1 arrest in wildtype but not Crbn−/− Jurkat cells, indicating that the cytostatic effect of CDK4/6 degraders required CRBN-dependent degradation of CDK4/6 (Figure 4).
Figure 4. CDK4/6 degrader-induced cell cycle arrest is dependent on CRBN.
Fluorescence histograms (A) and quantitation (B) of wildtype or Crbn−/− Jurkat cells treated with 100 nM of indicated compound for 24h and stained with PI.
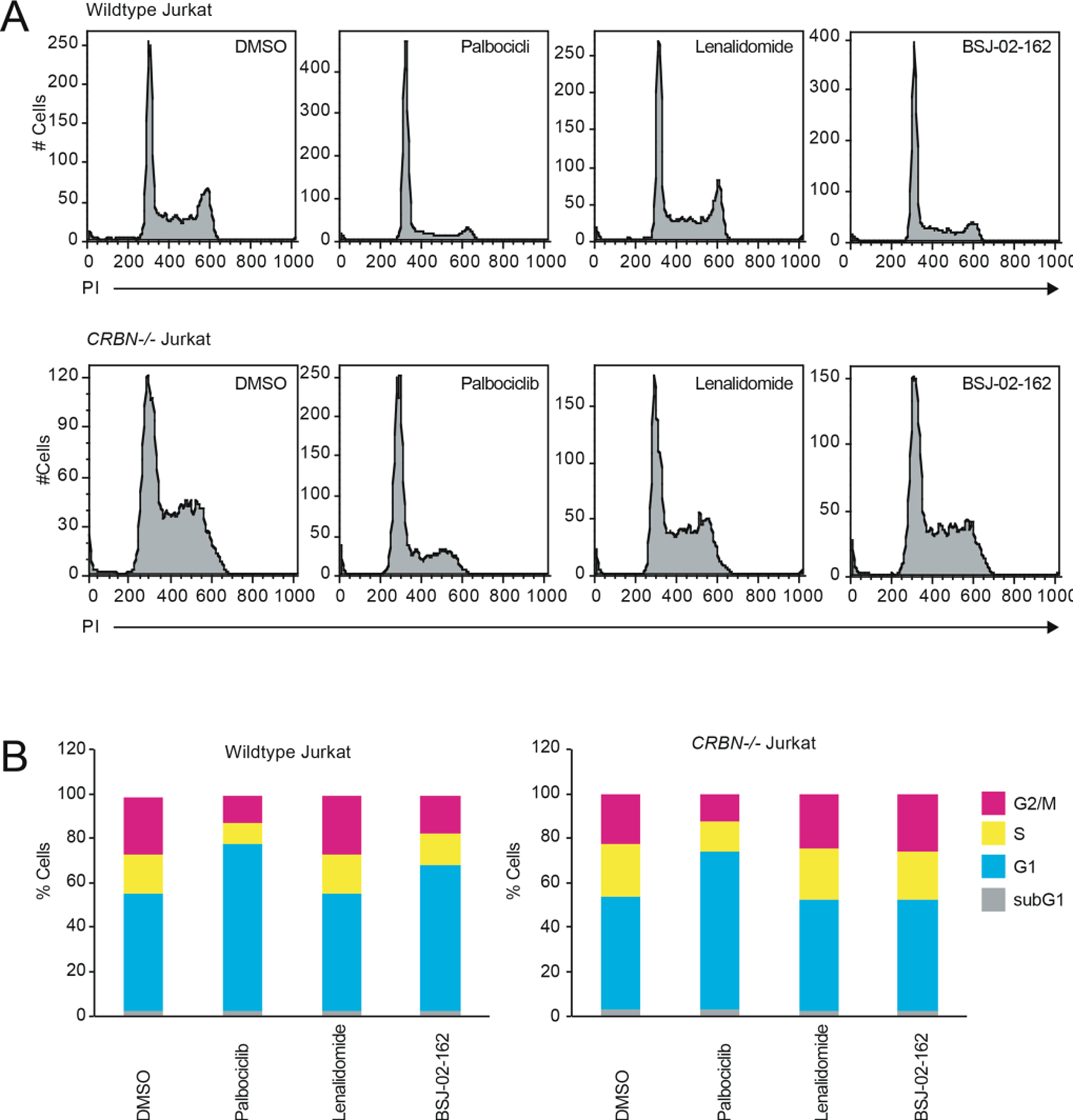
To more broadly assess degrader selectivity, we performed multiplexed mass spectrometry (MS)-based proteomic analysis[15] in Molt4 cells treated with 250 nM of BSJ-02–162, BSJ-03–204, BSJ-04–132 or BSJ-03–123 for 5h. As expected, BSJ-02–162 induced loss of CDK4/6, IKZF1/3, and other zinc finger proteins known to be recruited to CRBN by imides (Figure 5A)[16]. In contrast, its non-IKZF1/3-recruiting analog BSJ-03–204 was verified to be selective for CDK4/6 degradation (Figure 5B). In cells treated with BSJ-04–132, CDK4 (~1.9-fold change) did not meet our minimum 2-fold change threshold to be depicted on the scatterplot (Figure 5C), but its level was reduced to a greater extent than those of CDK6, IKZF1 and IKZF3 (Table S2). Finally, treatment of Molt4 cells with BSJ-03–123 resulted in loss of CDK6, but not CDK4 or IKZF1/3 (Figure 5D). Thus, profiling confirmed that we could achieve both dual and selective degradation of CDK4/6 across the proteome after acute compound treatment, and that we could tune degradation of IKZF1/3.
Figure 5.
Proteome-wide selectivity of CDK4/6 degrader molecules. Quantitative proteomics showing relative abundance of proteins in Molt4 cells treated for 5h with 250 nM of (A) BSJ-02–162; (B) BSJ-03–204; (C) BSJ-04–132; and (D) BSJ-03–123.
CDK4/6 inhibitors are currently being evaluated for MCL, an incurable B cell non-Hodgkin lymphoma whose molecular hallmark is the t(11;14)(q13;q32) chromosomal translocation that juxtaposes CCND1 (encoding cyclin D1) with the Ig heavy chain gene locus, resulting in aberrant cell cycle progression via activation of CDK4/6[17]. As such, we examined the effects of CDK4/6 degraders in several MCL cell lines.
We first tested our CDK4/6 degraders in Granta-519 cells, a MCL cell line that exhibits the characteristic overexpression of cyclin D1. Treatment of Granta-519 cells with 1 µM BSJ-02–162 for 1d resulted in pronounced loss of CDK4/6 and IKZF1/3 protein, while treatment with BSJ-03–204 only resulted in degradation of CDK4/6 (Figure 6A). Similar to palbociclib, these dual CDK4/6 degraders also reduced levels of phosphorylated Rb (Figure 6A). Finally, treatment of Granta-519 cells with these degraders also potently induced a G1 arrest (Figure 6B).
Figure 6. Combined CDK4/6 and IKZF1/3 degradation has potent anti-proliferative effects on MCL cell lines.
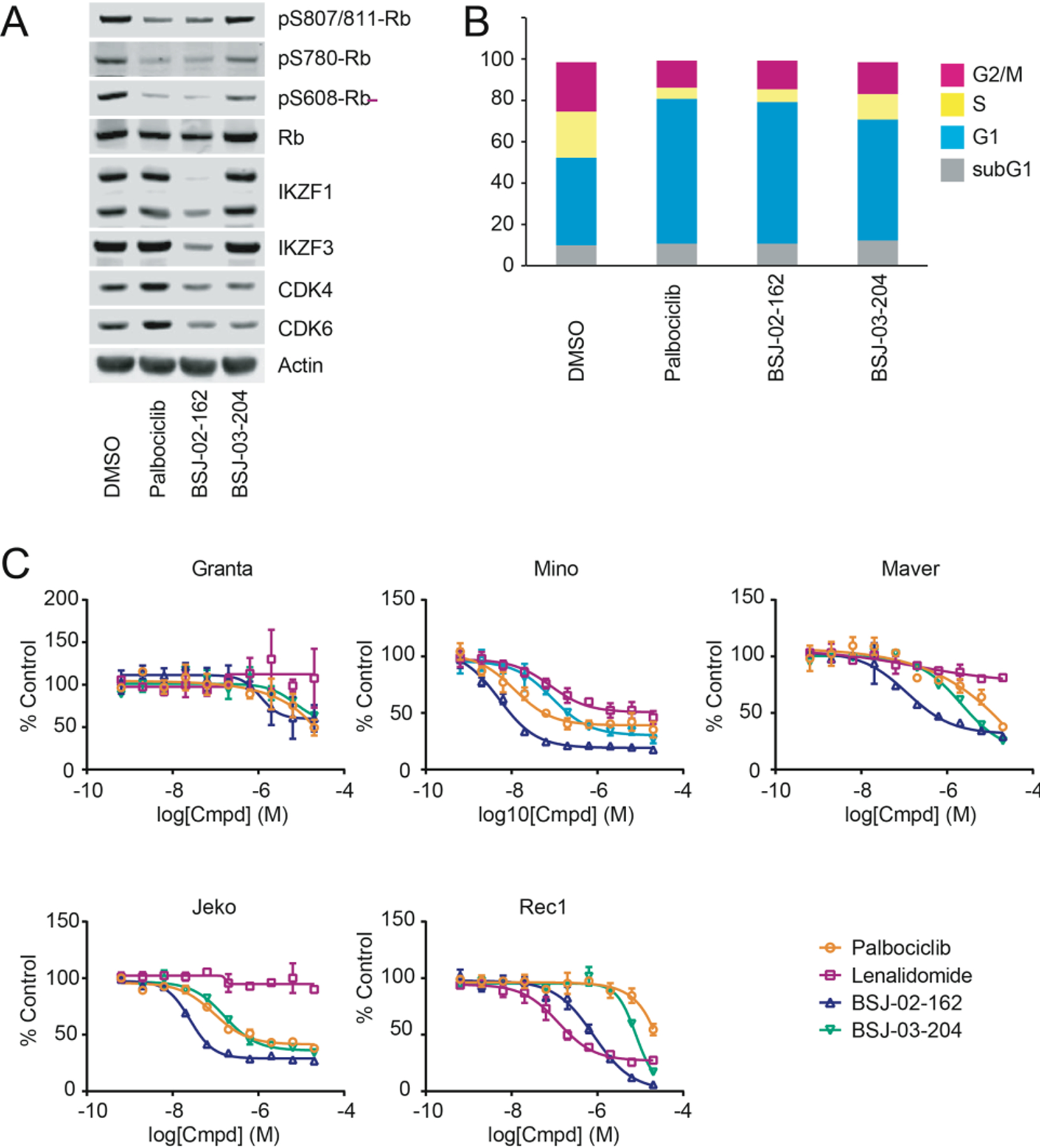
(A) Immunoblots from Granta-519 cells treated with 1 µM of indicated compounds for 1d. (B) Cell cycle analysis of Granta-519 cells by quantitation of DNA content after treatment with 1 µM of indicated compounds for 1d. (C) MCL cells were treated with the indicated compounds and concentrations for 3d (4d for Granta-519). Anti-proliferative effects of compounds were assessed using the CellTiter-Glo assay (Promega), and ED50s were determined using Graphpad Prism nonlinear regression curve fit.
Next, we profiled the anti-proliferative activities of BSJ-02–162 and BSJ-03–204 in comparison with palbociclib and lenalidomide, which itself is FDA-approved for the treatment of patients with relapsed MCL, in a panel of MCL cell lines that exhibit a range of sensitivity to CDK4/6 inhibition. Interestingly, BSJ-02–162, which degrades CDK4/6 and IKZF1/3, had increased anti-proliferative effects in many MCL cell lines in comparison to BSJ-03–204, which only degrades CDK4/6 (Figure 6C), suggesting that co-targeting these two distinct pathways may be a promising strategy for the treatment of MCL.
In sum, we developed a series of CDK4/6 degraders that largely recapitulated the cellular effects of CDK4/6 inhibition, including reduction in pRB levels, G1 arrest, and anti-proliferative activity. As these effects required the presence of CRBN, they were dependent on target degradation rather than inhibition.
We also discovered that simultaneously degrading CDK4/6 and IKZF1/3 demonstrated enhanced anti-proliferation effect in MCL cell lines in comparison to palbociclib, lenalidomide, or a selective CDK4/6 degrader, similar to what was observed with a previously reported BTK degrader that also targeted IKZF1/3[13c]. Thus, compounds that target multiple vulnerabilities may be a promising therapeutic strategy, particularly for B-cell malignancies that are sensitive to IKZF1/3 degradation.
The highly homologous ATP-binding pockets of CDK4 and CDK6 has hindered the development of selective CDK4 or CDK6 probes. Here, by varying the linkers for CDK4/6-targeted degraders, we identified molecules capable of acutely and selectively degrading either CDK4 or CDK6. Structural analysis of kinase-degrader-CRBN ternary complexes will be required to reveal the basis for the observed selectivity. These compounds may serve as selective probes for pharmacological dissection of the distinct CDK4 or CDK6 biological functions and as starting points for the development of therapeutic degrader molecules.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge the generous financial support of the following sources: Damon Runyon Cancer Research Fellowship DRG-2270–16 (E.S.W.). This work was funded by NIH grant R01CA218278 (E.S.F). Eric S. Fischer is a Damon Runyon-Rachleff Innovator supported in part by the Damon Runyon Cancer Research Foundation (DRR-50–18).
Footnotes
Conflicts of Interest
N.S.G. is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, B2S and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield and Sanofi. E.S.F is a member of the scientific advisory board of C4 Therapeutics and is a consultant to Novartis and Deerfield. The Fischer lab receives or has received research funding from Novartis, Deerfield and Astellas. B.J., E.S.W., Y.L., and N.S.G are inventors on patents covering CDK4/6 degraders owned by Dana-Farber.
References
- [1].O’Leary B, Finn RS, Turner NC, Nat Rev Clin Oncol 2016, 13, 417–430. [DOI] [PubMed] [Google Scholar]
- [2].Laderian B, Fojo T, Semin Oncol 2017, 44, 395–403. [DOI] [PubMed] [Google Scholar]
- [3].Sherr CJ, Beach D, Shapiro GI, Cancer Discov 2016, 6, 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G,Guerra C, Santamaria D, Barbacid M, Cancer Cell 2010, 18, 63–73. [DOI] [PubMed] [Google Scholar]
- [5].Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, Ott RG, Schafer M, Fajmann S, Schlederer M, Schiefer AI, Reichart U, Mayerhofer M, Hoeller C, Zochbauer-Muller S, Kerjaschki D, Bock C, Kenner L, Hoefler G, Freissmuth M, Green AR, Moriggl R, Busslinger M, Malumbres M, Sexl V, Cancer Cell 2013, 24, 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Handschick K, Beuerlein K, Jurida L, Bartkuhn M, Muller H, Soelch J, Weber A, Dittrich-Breiholz O, Schneider H, Scharfe M, Jarek M, Stellzig J, Schmitz ML, Kracht M, Molecular cell 2014, 53, 193–208. [DOI] [PubMed] [Google Scholar]
- [7].Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, Chhabra S, Huang W, Liu H, Aref AR, Ivanova E, Paweletz CP, Bowden M, Zhou CW, Herter-Sprie GS, Sorrentino JA, Bisi JE, Lizotte PH, Merlino AA, Quinn MM, Bufe LE, Yang A, Zhang Y, Zhang H, Gao P, Chen T, Cavanaugh ME, Rode AJ, Haines E, Roberts PJ, Strum JC, Richards WG, Lorch JH, Parangi S, Gunda V, Boland GM, Bueno R, Palakurthi S, Freeman GJ, Ritz J, Haining WN, Sharpless NE, Arthanari H, Shapiro GI, Barbie DA, Gray NS, Wong KK, Cancer Discov 2018, 8, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Toure M, Crews CM, Angew Chem Int Ed Engl 2016, 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- [9].Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green JL, Haas W, Nomanbhoy T, Fischer ES, Young RA, Bradner JE, Winter GE, Gray NS, Nat Chem Biol 2018, 14, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, Zhang T, Mancias JD, Gray NS, Bradner JE, Fischer ES, Nat Chem Biol 2018, 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a An S, Fu L, EBioMedicine 2018, 36, 553–562; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cromm PM, Crews CM, Cell Chem Biol 2017, 24, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, Torres R, Ajamie RT, Wishart GN, Flack RS, Neubauer BL, Young J, Chan EM, Iversen P, Cronier D, Kreklau E, de Dios A, Invest New Drugs 2014, 32, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG Jr., Science 2014, 343, 305–309; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, Ebert BL, Science 2014, 343, 301–305; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, Donovan KA, Yang G, Li Z, Fischer ES, Treon SP, Weinstock DM, Gray NS, Blood 2018; [DOI] [PMC free article] [PubMed]; d Huang HT, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho JH, Ko E, Jang J, Shi K, Choi HG, Griffin JD, Li Y, Treon SP, Fischer ES, Bradner JE, Tan L, Gray NS, Cell Chem Biol 2018, 25, 88–99 e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brand M, Jiang B, Bauer S, Donovan KA, Liang Y, Wang ES, Nowak RP, Yuan JC, Zhang T, Kwiatkowski N, Muller AC, Fischer ES, Gray NS, Winter GE, Cell Chem Biol 2018. [DOI] [PMC free article] [PubMed]
- [15].Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL, Fischer ES, Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a An J, Ponthier CM, Sack R, Seebacher J, Stadler MB, Donovan KA, Fischer ES, Nat Commun 2017, 8, 15398; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sievers QL, Petzold G, Bunker RD, Renneville A, Slabicki M, Liddicoat BJ, Abdulrahman W, Mikkelsen T, Ebert BL, Thoma NH, Science 2018, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jares P, Colomer D, Campo E, Nat Rev Cancer 2007, 7, 750–762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.