

Abstract
Ricin toxin A subunit (RTA) removes an adenine from the universally conserved sarcin/ricin loop (SRL) on eukaryotic ribosomes thereby inhibiting protein synthesis. No high affinity and selective small-molecule therapeutic antidotes have been reported against ricin toxicity. RTA binds to the ribosomal P stalk to access the SRL. The interaction anchors RTA to the P protein C-termini at a well-defined hydrophobic pocket, which is on the opposite face relative to the active site. The RTA ribosome binding site has not been previously targeted by small molecule inhibitors. We used fragment screening with surface plasmon resonance to identify small-molecular-weight-lead compounds that bind RTA and defined their interactions by crystallography. We identified five fragments, which bound RTA with mid-micromolar affinity. Three chemically distinct binding fragments were co-crystallized with RTA and crystal structures were solved. Two fragments bound at the P stalk binding site and the third bound to helix D, a motif distinct from the P stalk binding site. All fragments bound RTA remote from the catalytic site and caused little change in catalytic site geometry. Two fragments uniquely bound at the hydrophobic pocket with affinity sufficient to inhibit the catalytic activity on eukaryotic ribosomes in the low micromolar range. The binding mode of these inhibitors mimicked the interaction of the P stalk peptide, establishing that small molecule inhibitors can inhibit RTA binding to the ribosome with the potential for therapeutic intervention.
Keywords: Toxin, ricin, fragments, inhibitor, ribosome
Graphical Abstract

Ricin toxin A subunit binds to the ribosomal P stalk protein C-termini at a well-defined hydrophobic pocket to depurinate the sarcin/ricin loop. We carried out fragment screening with Biacore and identified small-molecular-weight lead compounds that bind to the hydrophobic pocket, which is covered by the B subunit and defined their interactions by crystallography.
INTRODUCTION
Ricin is one of the most potent toxins known and is a worldwide problem as a biothreat agent. It is an AB toxin containing an enzymatically active A subunit (RTA) and a cell binding B subunit (RTB) linked by a disulfide bond. Ricin and related Shiga toxins (Stxs) produced by E. coli are type II ribosome inactivating proteins (RIPs). The B chains of type II RIPs bind receptors to promote endocytosis. A portion of ricin holotoxin undergoes retrograde trafficking to the trans-Golgi network and then to the endoplasmic reticulum (ER)1. Reduction of the disulfide bond releases RTA from the B subunit in the ER2. RTA is thought to exploit the ER-associated degradation (ERAD) pathway to enter the cytosol3. RTA has high catalytic activity to specifically remove a single adenine base from the conserved sarcin/ricin loop (SRL) of the 28S rRNA by hydrolytic depurination, inhibit protein synthesis and cause cell death4. One molecule of ricin toxin delivered to the cytosol is sufficient to kill cultured cells5. RTA-antibody complexes have been explored as immunotoxins against leukemia and lymphoma, but off-target effects, including the vascular leak syndrome have limited their utility to date6–8. Small molecule selective inhibitors against RTA could rescue normal cells after RTA-immunotoxin cancer therapy and might also be used as antidotes against ricin9. At present, there are no high affinity and selective small-molecule therapeutics available against ricin intoxication.
We previously identified the ribosomal P stalk as the binding site for RTA and showed that P stalk binding is critical for catalysis of depurination and toxicity10–12. The P stalk binding site was mapped to the RTA interface with RTB, distant from the catalytic site (Figure 1)13–15. The active site in ricin holotoxin is exposed, but ricin holotoxin cannot depurinate the ribosome because the ribosome binding site is blocked by RTB16. Neither ricin holotoxin nor RTA has detectable enzymatic activity toward the naked rRNA or an RNA mimic of the SRL at physiological pH. However, RTA can depurinate RNA substrates in acidic conditions, a non-physiologic state where ribosome binding is not required for activity. In higher eukaryotes, the P stalk is a pentameric complex containing P1 and P2 proteins, which are attached to the uL10 protein (previously known as P0), in the form of two heterodimers17–21. The last 11 amino acids of the P1-P2 dimers and uL10 have identical sequence in all eukaryotes and are responsible for the recruitment of translational GTPases (trGTPases), including elongation factor 2 (eEF2) and stimulation of factor-dependent GTP hydrolysis17–21. It is not known how the five C-terminal sequences coordinate the interaction with RTA and whether RTA interacts with P proteins in a similar way as the translation factors. eEF2 binding protects the ribosome against RTA by preventing binding of RTA to the ribosome22–23. Mutant ribosomes missing P1-P2 proteins and retaining only uL10 interact with RTA very weakly10–11. Using purified human P1-P2 heterodimer missing the C-terminal residues we confirmed that RTA binds to the C-termini of P1-P2 proteins24. The P1-P2 C-termini do not have an equal role in the interaction with RTA, suggesting that the overall architecture of the P stalk complex is important24–25. The X-ray crystal structure analysis of RTA with a peptide (P11) corresponding to the C-terminal 11 amino acids of P proteins (SDDDMGFGLFD; PDB ID: 5GU4) showed that RTA binds to the last 6 amino acids (P6) at a well-defined hydrophobic pocket remote from the catalytic site created by reductive loss of the B subunit (Figure 1)26–27. Phe111, Leu113 and Phe114 of P6 (GFGLFD) are inserted into a hydrophobic pocket formed by Tyr183, Arg235, Phe240 and Ile251 residues of RTA26–27. Strong electrostatic and hydrophobic interactions contribute to anchoring RTA on the ribosome with nanomolar affinity14–15. Arginine residues at the RTA/RTB interface are important for the electrostatic interactions with the P stalk. Arg235 was identified as the most important arginine residue at the P stalk binding site13–14. The crystal structure showed that Arg235 forms hydrogen bonds with the last two residues (Phe114 and Asp 115) of P626–27. Mutation analysis of residues at the hydrophobic pocket of RTA indicated that Leu232, Tyr183 and Phe240 contribute to toxicity with Leu232 as the most critical residue15. When mutations in critical hydrophobic residues were combined with the R235A mutation, activity and toxicity of RTA were eliminated in mammalian cells without altering the active site15.
Figure 1. Model showing the interaction of RTA with the P stalk to access the SRL.

S. cerevisiae 26 S rRNA (PDB ID:3U5H) and 60 S subunit (PDB ID:3U5I) are indicated as light gray and dark gray colors, respectively. The fitted schematic structure of P0 fragment complexed with the N-terminal domain of P-proteins (PDB ID: 3A1Y) from Archaea is depicted as yellow and green, respectively. One CTD domain of a P-protein is shown as a gray line attached to the RTA molecule with a gray circle at the end. The yeast 60 S subunit is oriented to show A3027 (red) of the SRL (blue) and RTA (PDB ID:1RTC) oriented to show the putative SRL binding site (cyan), and the hydrophobic pocket at the stalk binding site (orange). RTA binds to the C-terminal tail of a P protein using arginine residues and the hydrophobic pocket located on the stalk binding surface. The flexible hinge of the P protein orients the active site of RTA toward the SRL through a conformational change and positions A3027 of the SRL into the active site of RTA. An enlarged image of RTA showing the P stalk binding site, which is distant from the SRL binding site is shown in the inset.
The active site of RTA has been extensively explored as a potential target for inhibitors by structure-based design. Early studies identified substrate analogs that target the active site and inhibit RTA with modest inhibitory activity28–29. However, these compounds had poor solubility and none of them exhibited potent inhibitory activity against ricin due to the large and highly polar active site30. Transition state analogs of the RTA catalytic reaction were powerful inhibitors of the depurination of small stem-loop RNA substrates at low pH, but did not protect eukaryotic ribosomes at physiological pH9, 31–35. High-throughput screening (HTS) of compounds against protein synthesis inhibitory activity or cytotoxicity yielded compounds that target toxin trafficking or cell death pathways, but not the toxin itself35–40.
Modulation of RTA-ribosome interactions by small molecules is a previously unexplored strategy for inhibition of RTA. Peptides mimicking the conserved C-termini of P proteins inhibit the activity of RTA by binding to the ribosome binding site41. Fragment-based lead discovery (FBLD) has become a widely established approach to identify small molecule fragments that directly bind to the target as starting points for chemical optimization42–44. One of the major advantages of FBLD with surface plasmon resonance (SPR) is that it provides kinetic information, which can be a valuable tool during hit to lead optimization. The fragments usually bind with low affinity due to their low molecular mass and have low inhibitory activity. Once target binding is confirmed they can be expanded by growing, merging, or linking to produce a more potent and selective binder with higher affinity for the binding site. Here, we report FBLD with SPR to identify inhibitors that target the hydrophobic pocket at the ribosome binding site of RTA. Lead compounds are identified and the crystal structures of three fragments are solved in complex with RTA. These fragments are chemically diverse and two of them bind at the P stalk binding site, which is remote from the catalytic site and indirectly reduce the catalytic activity of RTA. This structure-function study describes identification of the first small molecule inhibitors that bind at the ribosome binding site of RTA and provides insights for designing small molecule inhibitors against ricin intoxication.
RESULTS
Binding Level and Affinity Screen of the fragment library against RTA.
One thousand fragments from the Maybridge Ro3 core library, which has been prescreened for promiscuous binders using Biacore instruments with different chips, was screened against RTA using Biacore T200 in a Binding Level screen to identify fragments that bind to RTA as indicated in the methods. Fragments were passed over both reference and RTA surfaces at a single concentration of 200 μM and binding levels and sensorgram shapes were analyzed. Since there are no known small molecules that bind at the ribosome binding site of RTA, an 11-mer peptide (P11) mimicking the last 11 amino acids of P proteins (molecular weight 1218.25 Da) was used as a positive control and myo-inositol, which does not bind to chip surface or RTA, has high solubility and has a similar molecular weight as the fragments, was used as a negative control (Figure S1). Because of the low affinity of P11 (80 μM in this buffer) and limited solubility, a concentration (200 μM), which is lower than the saturating concentration, was used. Even at this concentration the surface activity for P11 binding decreased gradually. The PT peptide, which was reported to bind to the active site of RTA45 showed the opposite behavior, with gradual increase of binding activity from an initial very low level. PT did not bind when RTA was freshly immobilized but did show some binding as binding of P11 decreased, indicating PT can bind partially deactivated RTA. Both P11 and PT were used to monitor the surface activity, which is critical for picking reliable binders because once the surface lost activity, it became sticky and generated false positives. Adenine was used as a positive control for the active site of RTA (Figure S1).
A sample result from screening one of the 13 plates is shown in Figure 2. The binding level of each fragment and control was solvent corrected against DMSO, normalized for molecular weight, and shown as the relative response compared to adenine. The binding level of P11 decreased slightly with the cycle time, indicating that RTA gradually lost binding activity to P11. The binding level of adenine was relatively stable during the screen. Promising binders were selected based on the binding strength as well as binding behavior by analyzing the sensorgram shape. The fragments with binding levels higher than 80 response units were analyzed manually for sensorgram shape. The fragments that showed square sensorgrams due to fast on and off rates (Fragment A) were picked as good binders for further testing. The fragments with sloped sensorgrams indicated super stoichiometric sticky binders (Fragment B), which were not tested further.
Figure 2. Results of single-dose screening from one plate.

RTA was immobilized on a CM5 chip to around 6000 RU using amine coupling. The reference surface was activated and blocked. Fragments at 200 μM were passed over both surfaces at 30 μL/min for 30 seconds and dissociation was monitored for another 30 seconds. The running buffer was PBS-P (20 mM phosphate, 2.7 mM KCl, 137 mM NaCl and 0.05% surfactant P20, pH 7.4) with 2% DMSO. Both positive and negative controls were tested repeatedly between the screening cycles. DMSO corrections were run at the beginning, after 50 cycles and at the end of the screening cycles. Interaction sensorgrams from fragments A and B are shown. The binding levels of 80 fragments are shown in green, the positive controls, P11 and adenine are shown as red and magenta, and the negative controls, PT and myo-inositol are shown in black and cyan, respectively.
Seventy-nine fragments were selected from the binding level screen for the affinity screen. The 79 fragments were run at five different concentrations ranging from 65.2 to 1000 μM to verify binding and to determine the equilibrium dissociation constant (KD). An example of this screen is shown in Figure S2. The binding level data up to 500 μM was used in the fitting to avoid the effect of aggregation or sticky fragments. The KD values were obtained by fitting the binding levels at steady state since the kinetic rate constants could not be resolved due to fast on and fast off binding. The binders were ranked from high to low affinity. Three out of 79 fragments either did not show dose-dependent binding (two fragments) or showed no binding (one fragment), indicating that the result of the single dose screening was reliable. The KD values of the fragments are summarized in Table S1. Some fragments showed gradually saturated fitting curves and maximal binding levels with relatively low KD, indicating relatively few binding sites (Figure S2 A–F). Most of the fragments showed a linear increase in the binding level with increasing fragment concentration and projected very high maximal binding, indicating either multiple binding sites or aggregation or both (Figure S2 G–P).
Inhibition of the depurination activity of RTA by the fragment inhibitors.
We measured the depurination inhibition activity of the 79 fragments against yeast and rat liver ribosomes using qRT-PCR46. The qRT-PCR method measures the depurination level directly after isolation of the depurinated rRNA46. Hence, it is subject to less interference by the fragments than the reporter based assays, which add potential sources of interference47. Due to the sensitivity differences between yeast and rat liver ribosomes, different concentrations of RTA were used based on their linear response ranges. The level of depurination inhibition and the affinity data for the 79 fragments are shown in Table S1. Twelve fragments that showed good affinity and consistent inhibition of both yeast and rat liver ribosomes were selected for dose dependent inhibition activity. Five fragments (shown in green in Figure S3 and Table S1) that showed dose dependent inhibitory activity and less than 1 mM affinity, were selected for X-ray crystallography analysis.
Structure of RTA-fragment complexes.
RTA was co-crystallized with CC10501, CC70601 and BTB13068 (Figure 3A) using sitting drop vapor diffusion with varied crystallization conditions as summarized in (Table S2). Both CC10501, CC70601 showed square shaped sensorgrams in the binding level screen and bound RTA better than the positive controls (Figure S4). Molecular replacement using PHASER was done to solve the crystal structures of the complexes (Figure 3B). CC10501 and CC70601 complex structures were solved in the P6322 space group with a monomer in the asymmetric unit. The BTB13068 complex was solved in space group P43212 with a dimer in the asymmetric unit. Except a few N and C terminal residues, the electron density was observed clearly for the entire polypeptide backbone structure. The side chain electron density of few surface residues was also not observed in the structures (Table S3). All amino acid residues were found to be in the most favored or in allowed regions of the Ramachandran plot except for a few residues found in high B-factor loops (Table 1). The electron density corresponding to the inhibitors were well resolved in the structure (Figure 4).
Figure 3. Inhibitory fragments cocrystallized with RTA.

(A) CC10501 is 5-phenylthiophene-2-carboxylic acid, CC70601 is 4-(thien-2-ylmethyl) benzoic acid and BTB13068 is 9-oxofluorene-4-carboxamide. (B) Inhibitor-bound RTA structures (stereoviews). The superposition of RTA structures in complex with CC10501 (blue), CC70601 (cyan) and BTB13068 (red).
Table 1.
Data collection and refinement statistics of RTA complexes.
| Dataseta | Ricin Catalytic Subunit | ||
|---|---|---|---|
| RTA + CC10501 | RTA + CC70601 | RTA + BTB13068 | |
| Unit cell data | |||
| Space group | P6322 | P6322 | P43212 |
| Cell parameters (Å, °) | a = 168.35, b = 168.35, | a = 168.62, b = 168.62, | a = 66.23, b = 66.23, |
| γ = 120.0 | γ = 120.0 | ||
| Vm (Å3/Dalton) | 3.7 | 3.7 | 2.4 |
| Number of subunits in the asymmetric unit | 1 | 1 | 2 |
| Data collection | |||
| Beamline | LRL-CAT | LRL-CAT | LRL-CAT |
| Wavelength (Å) | 0.97931 | 0.97931 | 0.97931 |
| Temperature (K) | 100 | 100 | 100 |
| Resolution range (Å) | 145.80 – 1.99 (2.04 – 1.99) | 146.02 – 2.40 (2.49 – 2.40) | 66.14 – 1.54 (1.57 – 1.54) |
| Total number of observed reflections | 775660 (53962) | 411233 (26050) | 2238366 (46300) |
| Number of unique reflections | 31903 (2190) | 18435 (1891) | 88500 (4211) |
| Rmerge (%)b | 19.9 (222.4) | 13.8 (153.9) | 12.4 (117.4) |
| Rpim (%)c | 4.1 (45.5) | 2.9 (42.9) | 2.5 (35.6) |
| CC1/2 (%) | 0.99 (0.81) | 0.99 (0.75) | 0.99 (0.75) |
| <I/σ(I)>d | 17.6 (2.0) | 20.9 (1.9) | 18.2 (2.0) |
| Completeness (%) | 100 (100) | 100 (100) | 100 (99.4) |
| Multiplicity | 24.3 (24.6) | 22.3 (13.8) | 25.3 (11) |
| Wilson B-factor (Å2) | 25.8 | 33.1 | 12.1 |
| Refinement | |||
| Rwork (%)e | 18.4 | 18.5 | 16.4 |
| Rfree (%)f | 21.8 | 23.5 | 19.5 |
| No. of atoms | 2336 | 2248 | 5083 |
| Protein atoms | 2060 | 2042 | 4202 |
| Ligand atoms | 14 | 15 | 17 |
| Solvent atoms | 262 | 191 | 864 |
| Model quality | |||
| RMS deviation from ideal value | |||
| Bond length (Å) | 0.01 | 0.01 | 0.01 |
| Bond angle (°) | 1.43 | 1.49 | 1.48 |
| Average B-factor | |||
| Protein atoms (Å2) | 34.3 | 45.6 | 16.8 |
| Ligand atoms (Å2) | 42.2 | 88.3 | 10.7 |
| Waters (Å2) | 52.1 | 65.3 | 31.1 |
| Ramachandran plotg | |||
| Most favored regions (%) | 98.1 | 97.7 | 99.6 |
| Allowed regions (%) | 1.5 | 1.9 | 0.4 |
| Outlier regions (%) | 0.4 | 0.4 | 0.0 |
| PDB entries | 6URX | 6URW | 6URY |
Values in parentheses refer to the highest resolution shell.
Rmerge= (ΣhklΣi|li(hkl) - < l(hkl)>|)ΣhklΣi<li(hkl)>, where li(hkl) is the intensity of the ith measurement of reflection (hkl) and < l(hkl) > is its mean intensity.
Rpim = (Σhkl[1/(Nhkl-1)]1/2Σi|li(hkl) - < l(hkl) >|) / ΣhklΣi<li(hkl)>, where li(hkl) is the intensity of the ith measurement of reflection (hkl), < l(hkl) > is its mean intensity and N is the number of measurements.
l is the integrated intensity and σ(l) is its estimated standard deviation.
Rwork = (Σhkl|Fo-Fc|)ΣhklFo where Fo and Fc are the observed and calculated structure factors.
Rfree is calculated as for Rwork but from a randomly selected subset of the data (5%), which were excluded from the refinement calculation.
Calculated by MOLPROBITY.
Figure 4: The omit density map (Fo-Fc) of CC10501, CC70601 and BTB13068 inhibitors bound to the RTA.

The omit map was calculated after 15 cycles of omit refinement by REFMAC5, leaving out the inhibitors. The contour levels are at 2.5σ. The inhibitors CC10501, CC70601 and BTB13068 are shown in panels A, B, and C, respectively.
The crystal structure of RTA with the C-terminal amino acid sequence of P proteins (SDDDMGFGLFD; PDB ID: 5GU4) established that Phe111 and the penultimate Phe114 residues bind to the hydrophobic pocket of the RTA occupied by the B-chain lectin (RTB) in intact ricin26–27. Inhibitors CC10501 and CC70601 bind in the hydrophobic site occupied by Phe114 of the P protein C-termini, thereby precluding the anchoring of RTA to the ribosome (Figure 5). Structural analysis suggests similar binding contacts for CC10501 and CC70601, confirmed by their similar binding constants. CC10501 showed more favorable stacking interactions with the aromatic ring of the inhibitor than CC70601 (Figures 5 and 7). BTB13068 binds close to the helix D of the RTA to interact with Glu146, Ser149, Ala150, Try153, Gly158, Thr159 and Thr163, all within 4 Å contacts (Figures 6 and 7). The position of the BTB13068 binding is 31 Å away from the P6 hydrophobic pocket and on the surface of RTA. The binding of BTB13068 is unlikely to prevent P protein interaction with RTA, and therefore may act by preventing conformational changes related to adenine depurination.
Figure 5. Superposition of RTA-inhibitor complexes with P stalk peptide (stereoview).

(A) Electrostatic surface representation of CC10501 (green), CC70601 (yellow) and C-terminal stalk protein (P2, magenta) in the hydrophobic pocket of RTA. (B) Cartoon representation (zoomed in) of CC10501 (stick, green), CC70601 (stick, yellow) and C-terminal stalk protein (P2, stick, magenta) in the hydrophobic pocket of RTA.
Figure 7. Binding interactions of RTA in complex with inhibitors (stereoview).

Inhibitor complexes of CC10501, CC70601 and BTB13068 (panels A, B, and C, respectively). Amino acids interacting with inhibitors from RTA are highlighted. Selected hydrogen bond interactions are shown in orange dotted lines. The RTA residues interacting with inhibitors within 4Å are highlighted.
Figure 6. Electrostatic surface representation (stereoview) of BTB13068 (green) binding in a remote hydrophobic pocket close to helix-D4.

The binding of BTB13068 is mostly driven by hydrophobic interactions shown in Figure 7.
The RTA complex with CC10501 was determined to 1.99 Å resolution. CC10501 is bound in the hydrophobic pocket of RTA where Phe114 of the P protein also binds (Figure 5). In ricin holotoxin this hydrophobic pocket interacts with Phe262 of RTB (Figure S5). The aromatic ring of CC10501 has π-π stacking with Tyr183 as well as π -T stacking with Phe240 of RTA. Other residues in hydrophobic interaction with the CC10501 aromatic ring are Leu232 and Ile251. The O1 of the inhibitor is in hydrogen bond interaction with Water258 (wat258), NE(Arg235) and backbone N(Arg235) (Figure 7).
The structure of RTA with CC70601 solved to 2.40 Å in the space group P6322. CC70601 is also bound in the same hydrophobic pocket of RTA as CC10501 inhibitor. The binding of CC70601 is stabilized primarily by hydrophobic interactions. The aromatic ring of CC70601 is in offset stacking with Tyr183 and Phe240 whereas the thiophane ring of the inhibitor has a hydrophobic interaction with Ile251. The O1 of the inhibitor has a hydrogen bond interaction with wat127 (Figure 7).
The structure of BTB13068 with RTA was determined at 1.54 Å resolution in space group P43212. There are two monomers of RTA in the asymmetric unit in which BTB13068 is bound between both subunits of RTA. BTB13068 is bound with the 0.5 occupancy at the dimer symmetry interface. One hydrogen bond interaction is observed between O1 of the inhibitor with O1 of an ethylene glycol molecule, also bound in the symmetry interface. The other amino acids, which are within 4 Å of BTB13068 are Glu146, Ser149, Ala150, Try153, Gly158, Thr159 and Thr163 (Figure 7). The same residues from the symmetry related monomer are also interacting with BTB13068.
Structural Comparison.
The crystal structures of RTA and full-length ricin have been determined previously and are similar to the RTA structures reported here, except for the binding of the fragment inhibitors. The RMSD deviation of unliganded and inhibitor-bound RTAs are from 0.662 to 0.798 Å. Crystal structures of RTA in complex with CC10501, CC70601 and BTB13068 have been compared with unliganded RTA (PDB ID: 1RTC), the RTA-RTB complex (PDB ID: 2AAI) and with RTA in complex with the C-terminal peptide of ribosomal P protein (PDB ID: 5GU4). When the catalytic sites of unliganded RTA (1RTC) and known complex structures were compared with the current structures, no major structural changes were observed. In complexes with CC10501 and CC70601 the loop from Gly35 to His40 has moved towards the β-sheet-2. Also, the side chain of Thr159 has flipped out in all the ligand-bound structures. The Arg234 and Arg235 side chains have moved toward the binding pocket for better interactions with the inhibitor (Figure 7). There were no major structural changes upon BTB13068 binding except flipping of Thr159 and a conformational change for Glu146.
The affinity (KD) and the IC50 of the three fragments for RTA.
The affinity of the three fragments for RTA was determined using Biacore T200 at 12.5, 25, 50, 100 and 200 μM in triplicate measurements. The results were fitted globally (Figure 8). Because of their lower affinity and propensity to aggregate at higher concentrations the data for CC10501 and CC70601 were fitted with the “Steady State Affinity Constant Rmax model” using 69% surface activity calculated based on the binding affinity of the P6 peptide41. CC10501 and CC70601 had similar KD values of 270 and 404 μM, respectively, while BTB13068 had slightly lower KD of 153 μM. The 50% of inhibitory activity (IC50) was determined by qRT-PCR46. The data for the percent inhibition at different fragment concentrations were fitted with Michaelis-Menten kinetics using Origin software (Figure 9). CC70601 gave the best inhibitory activity with an IC50 of 32 μM. CC10501 was 5.6-fold weaker with IC50 of 181 μM and BTB13068 gave the weakest IC50, which was greater than 400 μM (Figure 9).
Figure 8. Binding sensorgrams (left) and kinetic fittings (right) of the three fragments to RTA.

Biacore T200 was used for the measurements. The conditions were the same as described in Figure 2. The fragment concentrations were 12.5, 25, 50, 100, and 200 μM. The binding measurements were repeated five different times using four different chips with three replicates each time. The KD values are shown as the mean ± S.D. The Steady State Affinity Constant Rmax model with global fitting was used to determine the KD values for CC10501 and CC70601. The surface activity calculated based on the binding affinity of P6 was 69%. The Steady State Affinity model with global fitting was used to determine the KD value for BTB13068 using the Biacore T200 Evaluation software 3.0.
Figure 9. The 50% inhibitory concentration (IC50) of fragments against RTA.
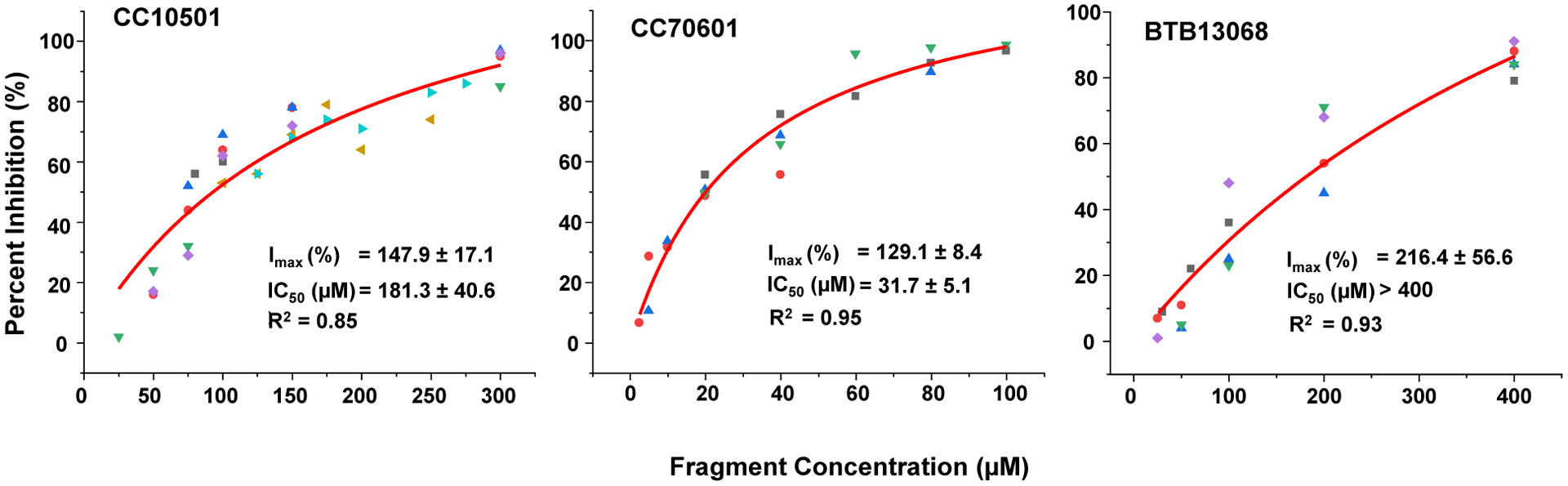
Depurination inhibitory activity of the fragments was determined by qRT-PCR. Fragment concentrations varied depending on the inhibitory activity of each fragment. The highest concentration for CC10501 was 300 μM, for CC70601 was 100 μM, and BTB13068 was used at 400 μM due to the limited solubility of this fragment. The different colors indicate the different measurements which were repeated 4 to 6 times. The Michaelis-Menten model was used to fit the inhibition curves using the Origin software.
DISCUSSION
The effort to develop drugs against ricin has been challenging. Small-molecule inhibitors targeting the active site of RTA have been based on pterins, guanines, and pyrimidines, which inhibited RTA with IC50 values ranging from 200 to 2000 μM35. Peptide conjugated pterins had improved IC50 values (6–115 μM)48. However, pterin derivatives suffered from solubility, toxicity and cell permeability issues35. Transition state analogs in an RNA framework inhibited RTA activity at acidic pH around 49. Only one small molecule, Retro-2 had marginal activity in protecting mice against ricin challenge37. Retro-2 inhibits transport of ricin and Stx from endosomes to Golgi and shows pre-exposure protection of mice from nasal challenge with 1 LD90 ricin37. The structure of Retro-2 is chemically unstable and spontaneously converts to a racemic and cyclic structure of Retro-2cycl 49. Retro-2 not only blocked the cellular traffic of ricin and Stx, but also blocked papillomavirus and polyomavirus infection50. Recent results showed that Retro-2 blocks the delivery of tail-anchored proteins to the ER-targeting factor ASNA151. Retro-2 is challenging to optimize in the absence of structural information about its complex with its target.
A distinct strategy for inhibition of RTA is to prevent the binding of RTA to the ribosomal P stalk. Currently no direct binding screens have been carried out against RTA. The ribosome binding site on RTA is partially defined13–15, 26–27. Peptides mimicking the C-termini of ribosomal P proteins have been shown to block RTA-ribosome binding and inhibit RTA activity41. The lack of promising inhibitor candidates is primarily due to the rapid and strong electrostatic interactions at the interface between RTA and the ribosome which make it difficult to identify drug-like molecules that can compete with the ribosome binding. In this study, we used fragment screening to identify lead compounds. Kinetic and SPR analysis were used to define the binding affinity and crystal structure analysis was used to define candidate interactions with RTA. Structure analysis showed that inhibitors CC10501 and CC70601 are bound in the hydrophobic pocket at the C-terminal domain of RTA, the interface for binding of the hydrophobic motif at the C-termini of P proteins or RTB of the ricin toxin, but the binding models were slightly different. The third inhibitor BTB13068 bound allosterically between the RTA crystallographic dimer symmetry interface. We did not see any major structural changes in the structure with the three different fragments. The structural information from the X-ray crystallography analysis provides a starting point to grow these fragment hits into more potent leads.
The solubility limitations and the low affinity of the fragments forced the use of suboptimal concentrations for the affinity analysis. Therefore, the affinities reported here are projected affinities. When RTA was immobilized on the CM5 chip the surface activity calculated based on the binding affinity of P6 peptide was 69%, indicating that 69% of the RTA molecules were able to interact with P6. The affinities were in the mid micromolar range for CC10501 and CC70601, while the inhibitory activities were greater in the low micromolar range with IC50 of 32 μM and 181 μM for CC70601 and CC10501, respectively. A peptide mimicking the last 8 amino acids of P proteins (P8) bound yeast ribosomes with a similar KD of 439 ± 111 μM in the running buffer as CC10501 and CC70601 and inhibited the activity of RTA with a similar IC50 of 23 ± 4.4 μM as CC7060141. The IC50 values of the fragment inhibitors were lower than the KD values as previously observed with peptides that bind in the hydrophobic pocket41. Although the fragments did not bind RTA very tightly, they inhibited the ability of RTA to inactivate eukaryotic ribosomes. The fragments bind at the P protein binding site, which is distant from the active site (Figure 1). Therefore, they do not compete with the SRL for binding to RTA but compete with binding of RTA to the P stalk. The interaction of the P stalk C-termini with the hydrophobic pocket stimulates the activity of RTA by promoting a conformational change to correctly position the active site residues for catalytic attack on the active site13, 15. Consequently, inhibition of P stalk binding does not lead to a proportional decrease in activity. Even low affinity binding to the fragments leads to a greater level of reduction in depurination activity because the inhibitory effect is amplified.
Peptides corresponding to C-terminal residues of P proteins have been shown to interact with several other RIPs, including trichosanthin (TCS)52, maize RIP (MOD)53, and Stx54–56. Only the last six residues of P11 (SDDDMGFGLFD) bound to a hydrophobic pocket composed of Gln182, Tyr183, Ser203, Leu207, Gln233, Arg234, Arg235, Phe240, Ile247, P250 and Ile251 on RTA26–27. The main chain carbonyl of the penultimate Phe114 of P6 formed a hydrogen bond with the main chain amide of Arg235. The side chain atom OD1 of Asp115 formed another hydrogen bond with the side chain atom NE of Arg235 in RTA26–27. The binding mode of CC70601 and CC10501 mimicked the interaction of the P stalk peptide. They occupied the same binding site as Phe114 of P11. The interaction of CC70601 and CC10501 was mostly hydrophobic in the binding pocket with only few hydrogen bonding interactions (Figures 5 and 7). The O1 of CC10501 formed a hydrogen bond with the side chain atom NE of Arg235, mimicking the interaction of Asp115 of P6 with Arg235 of RTA (Figure 7A).
In the ricin toxin heterodimer structure Cys4 of RTB forms a disulfide bond with Cys259 of RTA. C-terminal residue Phe262 of RTB interacts with RTA in a pocket surrounded by Tyr183, Leu207, Leu232, Phe240 and Ile25127. This hydrophobic interaction facilitates the disulfide bond that normally keeps the two chains aligned. CC10501 and CC70601 bound in the same hydrophobic pocket where Phe262 of RTB binds with RTA (Figure S5).
BTB13068 bound remote from the P protein site and on the surface of helix D. BTB13068 showed the highest affinity, but consistent with its binding away from the P protein site, had the lowest inhibitory activity (IC50 of BTB13068 was >400 μM). These results indicate that the binding site of helix D is not a good target for inhibitor development. We did not see any major structural changes in BTB13068 bound RTA, but this does not exclude the possibility that a conformational change of helix D occurs during depurination by RTA. CC10501 and CC70601 have similar binding affinity but higher inhibitory activity than BTB13068, indicating that the P protein binding site is a better target for inhibition. No fragments were found to bind to the active site of RTA, suggesting that the active site is a less suitable target for inhibition by fragment screening. We identified two fragments that bind at the ribosome binding site after screening 1000 fragments. This relatively high hit rate and the relatively low IC50 with respect to the size of the fragments indicate that there is a possibility to generate more powerful inhibitors to target the ribosome binding site of RTA. The identified fragment hits can be optimized to increase their molecular mass and binding specificity based on their binding environment. An alternative method is to identify fragments that bind to adjacent sites and link them together to make a more potent ligand42–44.
We demonstrate here that FBLD using SPR is a feasible approach to identify new lead compounds that target a small number of residues at the RTA-ribosome binding interface and inhibit the activity of RTA. These compounds will be useful tools for modulating toxin/ribosome interactions as a new target for inhibitor discovery. The structural information from this study can contribute to the design of the next generation of RTA inhibitors. We propose that a similar approach can be used to search for inhibitors of other RIPs such as Stxs, which are the virulence factors of Stx-producing Escherichia coli (STEC) and for inhibitors targeting the bacterial translation factors which interact with the prokaryotic stalk.
CONCLUSION
We screened Maybridge fragment Ro3 core library of 1000 fragments for binding to the ricin toxin A subunit and identified three lead fragments. Two of them bound at a hydrophobic pocket that interacts with the hydrophobic amino acids at the C-termini of ribosomal P proteins. Another fragment bound remotely on the surface of helix D of RTA. All three fragments showed mid-micromolar binding affinity and low micromolar inhibitory activity against ribosome depurination. This study provides a blueprint for designing the next generation inhibitors targeting RTA-ribosome interactions with greater potency and selectivity against ricin toxin action.
METHODS
The fragment library.
The Ro3 core library obtained from Maybridge (Waltham, MA, USA) contains 1000 fragments representing the diversity of their entire 2500 fragment library. A clean screen was performed by GE Life Sciences to remove fragments that show persistent binding and behave promiscuously (GE poster 29155462 AC 06/2016).
Recombinant RTA proteins.
N-terminal 6x-His tagged RTA was obtained from Biresources (Manassas, VA, USA). Untagged RTA was purified by Nexomics (Nexomics Biosciences, New Brunswick, NJ, USA).
Fragment Screening.
A Biacore T-200 (GE Healthcare Life Sciences, Marlborough, Massachusetts, USA) was used for screening 1000 fragments from the Maybridge Ro3 core library and for the affinity measurements. The 6xHis-tagged RTA was immobilized on a carboxymethyl dextran (CM5) chip to around 6000 RU using the amine coupling kit (GE Healthcare). The reference surface was activated and blocked in the same way as the active surface without RTA. A peptide corresponding to the last 11 amino acids of the ribosomal P proteins (P11) was used as a positive control for the ribosome binding site and adenine was used as a positive control for the active site of RTA. Myo-inositol was used as a negative control. PT, a peptide reported to bind to the active site of RTA (Figure S1)45, but does not bind to freshly immobilized RTA, was used to monitor surface activity over time. The structurally diverse 1000 fragment Maybridge Ro3 core library was screened against RTA in Binding Level Screen and Affinity Screen experiments for identification and validation of binders, respectively.
The running buffer was PBS-P (20 mM phosphate, 2.7 mM KCl, 137 mM NaCl and 0.05% Surfactant P20, pH 7.4) with 2% DMSO for both screens. For the Binding Level Screen, the fragments were passed over both reference and RTA surface in phosphate buffer (PBS-P) with 2% DMSO at 200 μM concentration and binding levels and sensorgram shapes were analyzed using Biacore T200 Evaluation Software 3.0. For the Affinity Screen, the fragment concentrations were varied at 62.5, 125, 250, 500 and 1000 μM and the KD of each fragment was determined by fitting the steady state binding level using Biacore T200 Evaluation Software 3.0. Solvent correction against DMSO was run in Binding Level Screen and Affinity Screen. The fragments were ranked based on their affinity.
Depurination inhibition.
Depurination inhibitory activity of the fragments was determined by qRT-PCR at 500, 200, and 100 μM using both yeast and rat liver ribosomes (Table S1). Depurination reaction buffer contained 10 mM Tris-HCl pH 7.4, 60 mM KCl, 10 mM MgCl2, 0.5 % DMSO. RTA was used at 1.0 and 0.2 nM for yeast and rat liver ribosomes, respectively. RTA and fragments were first preincubated at room temperature for 5 min followed by addition of ribosomes (50 nM) to start the reaction. The reaction was incubated at room temperature for 5 min. 100 μL of 2X extraction buffer (240 mM NaCl, 50 mM Tris-HCl pH 8.8, 20 mM EDTA, 2% SDS) was added to stop the reaction. RNA was extracted using phenol/chloroform, precipitated with ethanol, and dissolved in 30 μL RNase-free water. The depurination level was determined using qRT-PCR46. The reaction mixture without RTA was used as no depurination control (0%) and the reaction mixture with RTA, but without the fragment was used as the 100% depurination control. The data were fitted to the Michaelis-Menten equation using Origin (OriginLab, Northampton, Massachusetts, USA). Fragments that showed dose dependent inhibition and affinity lower than 1 mM were selected for X-ray crystallography analysis.
The IC50 was determined for each fragment by qRT-PCR by measuring the percent inhibition at different fragment concentrations. RTA (1nM) and fragments were preincubated for 5 min at room temperature before addition of yeast ribosomes (50 nM). The reaction was incubated at room temperature for 5 min in the depurination buffer (20 mM Tris-HCl pH 7.4, 25 mM KCl, 5 mM MgCl2, 0.5 % DMSO). Fragment concentrations varied depending on the inhibitory activity of each fragment. The highest concentration for CC10501 was 300 μM, for CC70601 was 100 μM, and BTB13068 was used at 400 μM due to the limited solubility of this fragment. The measurements were repeated 4 to 6 times. The Michaelis-Menten model was used to fit the inhibition curves using the Origin software. As shown in Figure S6, the depurination rate was linear over the 8 min time course.
Co-crystallization of RTA with lead fragments.
The co-crystallization of RTA with CC10501, CC70601 and BTB13068 was done using sitting drop vapor diffusion at 22°C. RTA (5 mg/ml) was mixed with inhibitors in a 1:5 molar ratio and incubated for two hours on ice. The incubated samples were screened by using the Microlytic (MCSG1–4) and Hampton (crystal screenHT) crystallization conditions. The crystallization drops were set up in 96-well INTELLI plates (ART ROBBINS) using the CRYSTAL-GRYPHON crystallization robot (ART ROBBINS). Each crystallization drop contained 0.5 μL of enzyme inhibitor mixture and 0.5 μL of well solution. The volume of the well solution was 70 μL. Good quality crystals were obtained in one week. The crystallization and crystal handling process are summarized in Table S2.
Data collection and processing.
The diffraction data were collected from 1.54 to 2.40 Å resolution at the LRL-CAT beam line (Argonne National Laboratory, Argonne, IL) at 0.97931 Å wavelength (Table 1). All diffraction data were processed using the iMOSFLM program and scaled by the AIMLESS program of the CCP4 suite in different space groups as summarized in Table 157–58 The quality of the data was analyzed using the SFCHECK and XTRIAGE58–59. The Matthews coefficient (Vm) calculations was done to calculate the number of monomer molecules present the asymmetric unit58. The data collection and processing statistics are summarized in Table 1.
Structure determination and refinement.
The crystal structures of RTA in complex with fragment inhibitors were solved by molecular replacement using PHASER60. The chain-A of wild-type RTA (PDB ID: 1RTC) structure was used as the initial phasing model. The model obtained from PHASER was manually adjusted and completed using the graphics program COOT61. The structure refinement was performed by REFMAC5 program, using standard protocols for the NCS refinement62. The inhibitor molecules were left out from the models in the beginning of the refinement. After building structural waters into the complexes, inhibitor molecules were fitted into their respective electron densities. The final refinement statistics of the structure is summarized in Table 1.
Structure analysis.
The crystal structure of unliganded wild-type RTA (PDB ID: 1RTC, chain: A) has been used for structure comparisons63. Also, RTA complexes with adenine (adenosine became adenine at the active site, PDB ID: 1IFS, chain: A), RTA-RTB dimer (PDB ID: 2AAI) and RTA in complex with C-terminal peptide of ribosomal P protein (PDB ID: 5GU4) structures were used for structure comparisons27, 64–66. Structural superimpositions were done using the SSM protocol of COOT. The geometry analyses of the final model were done using MolProbity67. Further structure analyses, including the calculation of the B-factor profiles was done using BAVERAGE program of the CCP4 suite58. The Figures were made using the molecular graphics program PyMOL. For RTA structures, subunit-A was used for all the structural analyses and comparisons.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Allergy and Infectious Diseases funding AI072425 to NET and National Institutes of General Medical Sciences funding GM041916 to VLS. The School of Environmental and Biological Sciences (SEBS) Biomolecular Interaction Analysis Core Facility is supported by NIH Shared Instrumentation Grant, S10 OD026750. The Einstein Crystallographic Core X-Ray diffraction facility is supported by NIH Shared Instrumentation Grant S10 OD020068. We gratefully acknowledge them. We thank Drs. David Kimball and Stephen Burley for help with the New Jersey Health Foundation funding for the fragment library. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
ABBREVIATIONS
- FBLD
fragment based ligand discovery
- RTA
ricin toxin A subunit
- RTB
ricin toxin B subunit
- RIP
ribosome inactivating protein
- SPR
surface plasmon resonance
- SRL
sarcin/ricin loop
- STEC
Shiga toxin producing E. coli, Stx, Shiga toxin
Footnotes
Supporting information
The following files are included with this manuscript:
Supplementary Table S1. Affinity and percent inhibition of the 79 fragments with yeast and rat liver ribosomes at 100 μM fragment concentration.
Supplementary Table S2: Crystallization and crystal handling.
Supplementary Table S3: Missing amino acid side chains in the RTA complex structures.
Supplementary Figure S1. Binding controls for the fragment screen with Biacore.
Supplementary Figure S2. Results of kinetic screening from one plate.
Supplementary Figure S3. Inhibition of RTA depurination by fragment inhibitors.
Supplementary Figure S4. Results of a single-dose screening from one plate.
Supplementary Figure S5: Superposition of RTA structure in complex with inhibitors with RTA-RTB complex.
Supplementary Figure S6: Time course of depurination under the same conditions as the IC50 measurements in Figure 9.
Accession codes
The structures generated in this study were deposited in the Protein Data Bank (PDB) under accession numbers 6URX, 6URW and 6URY. Authors will release the atomic coordinates and experimental data upon article publication.
Conflicts of interest
The authors have no conflicts of interest.
REFERENCES
- (1).Sowa-Rogozinska N, Sominka H, Nowakowska-Golacka J, Sandvig K, and Slominska-Wojewodzka M (2019) Intracellular transport and cytotoxicity of the protein toxin ricin. Toxins (Basel) 11 (6). DOI: 10.3390/toxins11060350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Spooner RA, Watson PD, Marsden CJ, Smith DC, Moore KA, Cook JP, Lord JM, and Roberts LM (2004) Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J 383 (Pt 2), 285–93. DOI: 10.1042/BJ20040742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Spooner RA and Lord JM (2012) How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol 357, 19–40. DOI: 10.1007/82_2011_154. [DOI] [PubMed] [Google Scholar]
- (4).Endo Y, Chan YL, Lin A,Tsurugi K, and Wool IG (1988) The cytotoxins alpha-sarcin and ricin retain their specificity when tested on a synthetic oligoribonucleotide (35-mer) that mimics a region of 28 S ribosomal ribonucleic acid. J. Biol. Chem 263 (17), 7917–20. [PubMed] [Google Scholar]
- (5).Eiklid K, Olsnes S, and Pihl A (1980) Entry of lethal doses of abrin, ricin and modeccin into the cytosol of HeLa cells. Exp. Cell Res 126 (2), 321–6. [DOI] [PubMed] [Google Scholar]
- (6).Liu XY, Pop LM, Schindler J, and Vitetta ES (2012) Immunotoxins constructed with chimeric, short-lived anti-CD22 monoclonal antibodies induce less vascular leak without loss of cytotoxicity. mAbs 4 (1), 57–68. DOI: 10.4161/mabs.4.1.18348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Weidle UH, Tiefenthaler G, Schiller C, Weiss EH, Georges G, and Brinkmann U (2014) Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genom. Proteom 11 (1), 25–38. [PubMed] [Google Scholar]
- (8).Polito L, Djemil A, and Bortolotti M (2016) Plant toxin-based immunotoxins for cancer therapy: A short overview. Biomedicines 4 (2). DOI: 10.3390/biomedicines4020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ho MC, Sturm MB, Almo SC, and Schramm VL (2009) Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc. Natl. Acad. Sci. USA 106 (48), 20276–81. DOI: 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chiou JC, Li XP, Remacha M, Ballesta JP, and Tumer NE (2008) The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol 70 (6), 1441–52. DOI: 10.1111/j.1365-2958.2008.06492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li XP, Chiou JC, Remacha M, Ballesta JP, and Tumer NE (2009) A two-step binding model proposed for the electrostatic interactions of ricin a chain with ribosomes. Biochemistry 48 (18), 3853–63. DOI: 10.1021/bi802371h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).May KL, Li XP, Martinez-Azorin F, Ballesta JP, Grela P, Tchorzewski M, and Tumer NE (2012) The P1/P2 proteins of the human ribosomal stalk are required for ribosome binding and depurination by ricin in human cells. FEBS J. 279 (20), 3925–36. DOI: 10.1111/j.1742-4658.2012.08752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li XP, Kahn PC, Kahn JN, Grela P, and Tumer NE (2013) Arginine residues on the opposite side of the active site stimulate the catalysis of ribosome depurination by ricin A chain by interacting with the P-protein stalk. J. Biol. Chem 288 (42), 30270–84. DOI: 10.1074/jbc.M113.510966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhou Y, Li XP, Chen BY, and Tumer NE (2017) Ricin uses arginine 235 as an anchor residue to bind to P-proteins of the ribosomal stalk. Sci. Rep 7, 42912 DOI: 10.1038/srep42912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhou Y, Li XP, Kahn JN, McLaughlin JE, and Tumer NE (2019) Leucine 232 and hydrophobic residues at the ribosomal P stalk binding site are critical for biological activity of ricin. Biosci. Rep 39 (10). DOI: 10.1042/BSR20192022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li XP and Tumer NE (2017) Differences in Ribosome Binding and Sarcin/Ricin Loop Depurination by Shiga and Ricin Holotoxins. Toxins (Basel) 9 (4). DOI: 10.3390/toxins9040133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wahl MC and Moller W (2002) Structure and function of the acidic ribosomal stalk proteins. Curr. Protein Pept. Sci 3 (1), 93–106. [DOI] [PubMed] [Google Scholar]
- (18).Gonzalo P and Reboud JP (2003) The puzzling lateral flexible stalk of the ribosome. Biol. Cell 95 (3–4), 179–93. [DOI] [PubMed] [Google Scholar]
- (19).Ballesta JP and Remacha M (1996)The large ribosomal subunit stalk as a regulatory element of the eukaryotic translational machinery. Prog. Nucleic Acid. Res. Mol. Biol 55, 157–93. [DOI] [PubMed] [Google Scholar]
- (20).Grela P, Gajda MJ, Armache JP, Beckmann R, Krokowski D, Svergun DI, Grankowski N, Tchorzewski M (2012) Solution structure of the natively assembled yeast ribosomal stalk determined by small-angle X-ray scattering. Biochem. J 444 (2), 205–9. [DOI] [PubMed] [Google Scholar]
- (21).Tchorzewski M (2002) The acidic ribosomal P proteins. Int. J. Biochem. Cell. Biol 34 (8), 911–5. [DOI] [PubMed] [Google Scholar]
- (22).Fernandez-Puentes C, Benson S, Olsnes S, and Pihl A (1976) Protective effect of elongation factor 2 on the inactivation of ribosomes by the toxic lectins abrin and ricin. Eur. J. Biochem 64 (2), 437–43. [DOI] [PubMed] [Google Scholar]
- (23).Cawley DB, Hedblom ML, and Houston LL (1979) Protection and rescue of ribosomes from the action of ricin A chain. Biochemistry 18 (12), 2648–54. [DOI] [PubMed] [Google Scholar]
- (24).Grela P, Li XP, Horbowicz P, Dzwierzynska M, Tchorzewski M, and Tumer NE (2017) Human ribosomal P1-P2 heterodimer represents an optimal docking site for ricin A chain with a prominent role for P1 C-terminus. Sci. Rep 7 (1), 5608 DOI: 10.1038/s41598-017-05675-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Grela P, Li XP, Tchorzewski M, and Tumer NE (2014) Functional divergence between the two P1-P2 stalk dimers on the ribosome in their interaction with ricin A chain. Biochem. J 460 (1), 59–67. DOI: 10.1042/BJ20140014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fan X, Zhu Y, Wang C, Niu L, Teng M, and Li X (2016) Structural insights into the interaction of the ribosomal P stalk protein P2 with a type II ribosome-inactivating protein ricin. Sci. Rep 6, 37803 DOI: 10.1038/srep37803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shi WW, Tang YS, Sze SY, Zhu ZN, Wong KB, and Shaw PC (2016) Crystal Structure of ribosome-inactivating protein ricin A chain in complex with the C-terminal peptide of the ribosomal stalk protein P2. Toxins (Basel) 8 (10). DOI: 10.3390/toxins8100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Robertus JD and Monzingo AF (2004) The structure of ribosome inactivating proteins. Mini Rev. Med. Chem 4 (5), 477–86. [DOI] [PubMed] [Google Scholar]
- (29).Wahome PG, Robertus JD, and Mantis NJ (2012) Small-Molecule Inhibitors of Ricin and Shiga Toxins. Curr. Top. Microbiol 357, 179–207. DOI: 10.1007/82_2011_177. [DOI] [PubMed] [Google Scholar]
- (30).Jasheway K, Pruet J, Anslyn EV, and Robertus JD (2011) Structure-based design of ricin inhibitors. Toxins 3 (10), 1233–48. DOI: 10.3390/toxins3101233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chen XY, Link TM, and Schramm VL (1998) Ricin A-chain: kinetics, mechanism, and RNA stem-loop inhibitors. Biochemistry 37 (33), 11605–13. DOI: 10.1021/bi980990pbi980990p [pii]. [DOI] [PubMed] [Google Scholar]
- (32).Roday S, Amukele T, Evans GB, Tyler PC, Furneaux RH, and Schramm VL (2004) Inhibition of ricin A-chain with pyrrolidine mimics of the oxacarbenium ion transition state. Biochemistry 43 (17), 4923–33. DOI: 10.1021/bi0498499. [DOI] [PubMed] [Google Scholar]
- (33).Roday S, Saen-oon S, and Schramm VL (2007) Vinyldeoxyadenosine in a sarcin-ricin RNA loop and its binding to ricin toxin A-chain. Biochemistry 46 (21), 6169–82. DOI: 10.1021/bi0621821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sturm MB, Roday S, and Schramm VL (2007) Circular DNA and DNA/RNA hybrid molecules as scaffolds for ricin inhibitor design. J. Ame. Chem. Soc 129 (17), 5544–50. DOI: 10.1021/ja068054h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wahome PG, Robertus JD, and Mantis NJ (2012) Small-molecule inhibitors of ricin and Shiga toxins. Curr. Top. Microbiol. Immunol 357, 179–207. [DOI] [PubMed] [Google Scholar]
- (36).Saenz JB, Doggett TA, and Haslam DB(2007) Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun 75 (9), 4552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Stechmann B, Bai SK, Gobbo E, Lopez R, Merer G, Pinchard S, Panigai L, Tenza D, Raposo G, Beaumelle B, Sauvaire D, Gillet D, Johannes L, and Barbier J (2010) Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 141 (2), 231–42. [DOI] [PubMed] [Google Scholar]
- (38).Wahome PG, Bai Y, Neal LM, Robertus JD, and Mantis NJ (2010) Identification of small-molecule inhibitors of ricin and Shiga toxin using a cell-based high-throughput screen. Toxicon 56 (3), 313–23. DOI: 10.1016/j.toxicon.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wahome PG, Ahlawat S, and Mantis NJ (2012) Identification of small molecules that suppress ricin-induced stress-activated signaling pathways. PLoS One 7 (11), e49075 DOI: 10.1371/journal.pone.0049075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Redmann V, Gardner T, Lau Z, Morohashi K, Felsenfeld D, and Tortorella D (2013) Novel class of potential therapeutics that target ricin retrograde translocation. Toxins 6 (1), 33–53. DOI: 10.3390/toxins6010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Li XP, Kahn JN, and Tumer NE (2018) Peptide mimics of the ribosomal P stalk inhibit the activity of ricin A chain by preventing ribosome binding. Toxins (Basel) 10 (9). DOI: 10.3390/toxins10090371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hajduk PJ and Greer J (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nature Rev. Drug Discov 6 (3), 211–9. DOI: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- (43).Hubbard RE, and Murray JB (2011) Experiences in fragment-based lead discovery. Methods Enzymol. 493, 509–31. [DOI] [PubMed] [Google Scholar]
- (44).Chessari G and Woodhead AJ (2009) From fragment to clinical candidate--a historical perspective. Drug Discov. Today 14 (13–14), 668–75. [DOI] [PubMed] [Google Scholar]
- (45).Wang S, Feng J, Guo J, Li Y, Sun Y, Qin W, Hu M, and Shen B (2005) Structural-based rational design of an antagonist peptide that inhibits the ribosome-inactivating activity of ricin A chain. Int. J. Peptide Res. Ther 11 (3), 211–218. [Google Scholar]
- (46).Pierce M, Kahn JN, Chiou J, and Tumer NE (2011) Development of a quantitative RT-PCR assay to examine the kinetics of ribosome depurination by ribosome inactivating proteins using Saccharomyces cerevisiae as a model. RNA 17 (1), 201–10. DOI: 10.1261/rna.2375411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhou Y, Li XP, Kahn JN, and Tumer NE (2018) Functional assays for measuring the catalytic activity of ribosome inactivating proteins. Toxins (Basel) 10 (6). DOI: 10.3390/toxins10060240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Saito R, Pruet JM, Manzano LA, Jasheway K, Monzingo AF, Wiget PA, Kamat I, Anslyn EV, and Robertus JD (2013) Peptide-conjugated pterins as inhibitors of ricin toxin A. J. Med. Chem 56 (1), 320–9. DOI: 10.1021/jm3016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Park JG, Kahn JN, Tumer NE, and Pang YP (2012) Chemical structure of Retro-2, a compound that protects cells against ribosome-inactivating proteins. Sci. Rep 2, 631 DOI: 10.1038/srep00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Nelson CD, Carney DW, Derdowski A, Lipovsky A, Gee GV, O’Hara B, Williard P, DiMaio D, Sello JK, and Atwood WJ (2013) A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. mBio 4 (6), e00729–13. DOI: 10.1128/mBio.00729-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Morgens DW, Chan C, Kane AJ, Weir NR, Li A, Dubreuil MM, Tsui CK, Hess GT, Lavertu A, Han K, Polyakov N, Zhou J, Handy EL, Alabi P, Dombroski A, Yao D, Altman RB, Sello JK, Denic V, Bassik MC (2019) Retro-2 protects cells from ricin toxicity by inhibiting ASNA1-mediated ER targeting and insertion of tail-anchored proteins. eLife 8 DOI: 10.7554/eLife.48434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chan DS, Chu LO, Lee KM, Too PH, Ma KW, Sze KH, Zhu G, Shaw PC, and Wong KB (2007) Interaction between trichosanthin, a ribosome-inactivating protein, and the ribosomal stalk protein P2 by chemical shift perturbation and mutagenesis analyses. Nucleic Acid Res. 35 (5), 1660–72. DOI: 10.1093/nar/gkm065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Yang Y, Mak AN, Shaw PC, and Sze KH (2010) Solution structure of an active mutant of maize ribosome-inactivating protein (MOD) and its interaction with the ribosomal stalk protein P2. J. Mol. Biol 395 (5), 897–907. DOI: 10.1016/j.jmb.2009.10.051. [DOI] [PubMed] [Google Scholar]
- (54).McCluskey AJ, Poon GM, Bolewska-Pedyczak E, Srikumar T, Jeram SM, Raught B, and Gariepy J (2008) The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol 378 (2), 375–86. [DOI] [PubMed] [Google Scholar]
- (55).McCluskey AJ, Bolewska-Pedyczak E, Jarvik N, Chen G, Sidhu SS, and Gariépy J (2012) Charged and hydrophobic surfaces on the A chain of Shiga-like toxin 1 recognize the C-terminal domain of ribosomal stalk proteins. PloS one 7 (2), e31191 DOI: 10.1371/journal.pone.0031191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Chiou JC, Li XP, Remacha M, Ballesta JP, and Tumer NE (2011) Shiga toxin 1 is more dependent on the P proteins of the ribosomal stalk for depurination activity than Shiga toxin 2. Int. J. Biochem. Cell. Biol 43 (12), 1792–801. DOI: 10.1016/j.biocel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D. Biol. Crystallogr 67 (Pt 4), 271–81. DOI: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).The CCP4 suite: programs for protein crystallography. (1994) Acta Crystallogr. D. Biol. Crystallogr 50 (Pt 5), 760–3. DOI: 10.1107/s0907444994003112. [DOI] [PubMed] [Google Scholar]
- (59).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr 66 (Pt 2), 213–21. DOI: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40 (Pt 4), 658–674. DOI: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr 66 (Pt 4), 486–501. DOI: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr 53 (Pt 3), 240–55. DOI: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- (63).Mlsna D, Monzingo AF, Katzin BJ, Ernst S, and Robertus JD (1993) Structure of recombinant ricin A chain at 2.3 A. Protein Sci. 2 (3), 429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Rutenber E, Katzin BJ, Ernst S, Collins EJ, Mlsna D, Ready MP, and Robertus JD (1991) Crystallographic refinement of ricin to 2.5 A. Proteins 10 (3), 240–50. [DOI] [PubMed] [Google Scholar]
- (65).Montfort W, Villafranca JE, Monzingo AF, Ernst SR, Katzin B, Rutenber E, Xuong NH, Hamlin R, and Robertus JD (1987) The three-dimensional structure of ricin at 2.8 A. J. Biol. Chem 262 (11), 5398–403. [PubMed] [Google Scholar]
- (66).Weston SA, Tucker AD, Thatcher DR, Derbyshire DJ, and Pauptit RA (1994) X-ray structure of recombinant ricin A-chain at 1.8 A resolution. J. Mol. Biol 244 (4), 410–22. [DOI] [PubMed] [Google Scholar]
- (67).Chen VB, Arendall WB 3rd, Headd JJ, and Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr 66 (Pt 1), 12–21. DOI: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.