

Abstract
Gardnerella spp. are considered a hallmark of bacterial vaginosis, a dysbiosis of the vaginal microbiome. There are four cpn60 sequence-based subgroups within the genus (A, B, C and D), and thirteen genome species have been defined recently. Gardnerella spp. co-occur in the vaginal microbiome with varying abundance, and these patterns are shaped by a resource-dependent, exploitative competition, which affects the growth rate of subgroups A, B and C negatively. The growth rate of rarely abundant subgroup D, however, increases with the increasing number of competitors, negatively affecting the growth rate of others. We hypothesized that a nutritional generalist lifestyle and minimal niche overlap with the other more abundant Gardnerella spp. facilitate the maintenance of subgroup D in the vaginal microbiome through negative frequency-dependent selection. Using 40 whole-genome sequences from isolates representing all four subgroups, we found that they could be distinguished based on the content of their predicted proteomes. Proteins associated with carbohydrate and amino acid uptake and metabolism were significant contributors to the separation of subgroups. Subgroup D isolates had significantly more of their proteins assigned to amino acid metabolism than the other subgroups. Subgroup D isolates were also significantly different from others in terms of number and type of carbon sources utilized in a phenotypic assay, while the other three could not be distinguished. Overall, the results suggest that a generalist lifestyle and lack of niche overlap with other Gardnerella spp. leads to subgroup D being favoured by negative frequency-dependent selection in the vaginal microbiome.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00248-020-01643-1.
Keywords: Gardnerella, Vaginal microbiome, Negative frequency-dependent selection, Pangenome, Competition
Introduction
Gardnerella spp. are an important diagnostic marker of bacterial vaginosis (BV), a dysbiosis of the vaginal microbiome characterized by a shift from the lactobacilli-dominated vaginal microbiome to a more diverse microbiome, containing many aerobic and anaerobic bacterial species, including Gardnerella spp. Gardnerella is a diverse genus and at least four subgroups (A, B, C and D) have been identified using cpn60 universal target barcode sequencing [1], which correspond to four clades defined by Ahmed et al. [2]. Recently, Gardnerella subgroups have been reclassified into thirteen genome species, of which four are now named as G. vaginalis (subgroup C/clade 1), G. swidsinskii and G. leopoldii (subgroup A/clade 4) and G. piotii (subgroup B/clade 2) [3, 4]. These Gardnerella species differ in their phenotypic traits, including sialidase activity and vaginolysin production, which may render some of the subgroups more pathogenic than the others [5–7].
Women with vaginal microbiomes dominated by Gardnerella are usually colonized by at least two Gardnerella spp. [4, 8]. The relative abundances of these co-occurring species, however, are not equal. Subgroup A (G. swidsinskii and G. leopoldii) and subgroup C (G. vaginalis) are most frequently dominant in reproductive-aged women [4, 8]. These two subgroups are also often associated with the clinical symptoms of bacterial vaginosis [4, 9, 10]. Subgroup B has been suggested to be associated with intermediate microbiota [7, 9, 10]. Subgroup D, comprised of several unnamed “genome species”, has only been detected at low prevalence and abundance [4, 10].
Several factors can affect the abundance and co-occurrence of Gardnerella spp. in the vaginal microbiome, including host physiology, host-microbiota interactions, nutrient availability and ecological interactions among bacteria [11, 12]. Ecological interactions are perhaps the most important factors which may affect the co-occurrence and ecological succession of Gardnerella species in the vaginal microbiome. Recently, we demonstrated that an indirect, exploitative competition between subgroups of Gardnerella is prevalent in co-cultures in vitro. While the growth rates of isolates in subgroups A, B and C were negatively affected by competition, growth rates of Gardnerella subgroup D isolates increased with the increasing number of competing subgroups in co-culture communities [12].
The strength of microbial interactions between bacterial species can be affected by niche overlap [13, 14], and species with similar nutritional requirements will naturally compete over the same resources [15]. In addition to competition for nutritional resources, bacteria may also compete for resources essential for colonizing a specific site. Since isolates from Gardnerella subgroups A, B and C are negatively affected by competition, and subgroup D isolates experienced a boost in growth rate, the degree of niche overlap between subgroups A, B and C is presumably higher than between subgroup D and any of the others.
Although the growth rate of subgroup D increases in co-cultures, it does not have an intrinsically high growth rate. In fact, the in vitro growth rate of subgroup D is half of that of subgroup C, which may contribute to its low abundance in the vaginal microbiome [12]. Low-abundance species are often favoured by negative frequency-dependent selection [16, 17], which can be governed by nutritional requirements [18]. Bacteria capable of utilizing relatively few, abundantly available nutrients in a particular environment are nutritional specialists in the context of that environment. Generalists, on the contrary, are bacteria capable of utilizing more nutrient sources than their specialist counterparts. In negative frequency-dependent selection, the resources accessible to rapidly growing specialists will dwindle, reducing the fitness of the specialists as their population increases. As a result, the population of more generalist bacteria capable of utilizing a wider range of nutrient sources will expand in a density-dependent manner [18, 19]. Generalists can also negatively affect the growth of specialists by competing for the resources that can be utilized by both of them [14].
Although the growth of the rarely abundant subgroup D is facilitated in co-cultures, the degree of overlap in nutrient utilization among the subgroups and the range of nutrient utilization by individual subgroups are yet unknown. The objective of our present study was, therefore, to evaluate the amount of genomic and phenotypic overlap in nutrient utilization among the subgroups of Gardnerella and to determine if subgroup D is a nutritional generalist relative to the three other subgroups. Findings are interpreted in relation to the hypothesis that subgroup D is maintained in the vaginal microbiome through negative frequency-dependent selection.
Methods
Bacterial Isolates
Thirty-nine Gardnerella isolates from our culture collection representing all four subgroups (based on cpn60 barcode sequencing) were selected for the study (n = 12 subgroup A, 12 subgroup B, 8 subgroup C and 7 subgroup D isolates) (Table S1). Isolates were streaked on Columbia agar plates with 5% (v/v) sheep blood and were incubated anaerobically at 37 °C for 48 h. For broth culture, colonies from blood agar plates were suspended in BHI broth supplemented with 10% horse serum and 0.25% (w/v) maltose.
Whole-Genome Sequencing
Whole-genome sequences for 10 of the study isolates had been published previously, and the remaining 29 were sequenced as part of the current study (Table S1). DNA was extracted from isolates using a modified salting-out protocol [20] and was stored at −20 °C. DNA was quantified using Qubit dsDNA BR assay kit (Invitrogen, Burlington, Ontario) and the quality of the extracts was assessed by the A260/A280 ratio. Isolate identity was confirmed by cpn60 barcode sequencing as follows. cpn60 barcode sequences were amplified from extracted DNA with the primers JH0665 (CGC CAG GGT TTT CCC AGT CAC GAC GAY GTT GCA GGY GAY GGH CHA CAA C) and JH0667 (AGC GGA TAA CAA TTT CAC ACA GGA GGR CGA TCR CCR AAK CCT GGA GCY TT). The reaction contained 2-μL template DNA in 1× PCR buffer (0.2 M Tris-HCl at pH 8.4, 0.5 M KCl), 2.5 mM MgCl2, 200 μM dNTP mixture, 400 nM of each primer, 2 U AccuStart Taq DNA polymerase and water to bring to a final volume of 50 μL. PCR was carried out with incubation at 94 °C for 30 s, 40 cycles of 94 °C 30 s, 60 °C for 1 min, 72 °C for 1 min and a final extension at 72 °C for 10 min. PCR products were purified and sequenced by Sanger sequencing and compared with the chaperonin sequence database cpnDB [21] to confirm identity.
Following confirmation of the identity of isolates, sequencing libraries were prepared using the Nextera XT DNA library preparation kit according to the manufacturer’s instructions (Illumina, Inc., San Diego, CA). PhiX DNA (15% [vol/vol]) was added to the indexed libraries before loading onto the flow cell. The 500 cycle V2 reagent kit was used for the Illumina MiSeq platform (Illumina, Inc.).
Raw sequences were trimmed using Trimmomatic [22] with a minimum quality score of 20 over a sliding window of 4 and a minimum read length of 40. Trimmed sequences were assembled using SOAPdenovo2 [23] or SPAdes (NR002, NR043, NR044) [24]. Assembled genomes were annotated using the National Center for Biotechnology Information Prokaryotic Genome Annotation Pipeline [25].
Pangenome Analysis
Pangenome analysis of the 39 study isolates and the published genome of G. vaginalis strain ATCC 14019 (Accession number: PRJNA55487) was performed using the micropan R package [26]. We used a “complete” linkage for clustering, and the cutoff value for the generation of clusters was set to 0.75. For initial visualization of the results, the Jaccard index was used to calculate the similarity of patterns of presence and absence of protein clusters among all isolates and a dendrogram was constructed from the results by the unweighted pair group method with arithmetic mean (UPGMA) using DendroUPGMA (http://genomes.urv.cat/UPGMA/).
COG Analysis
Predicted protein sequences from individual genomes were classified into Clusters of Orthologous Groups (COG) categories using WebMEGA (http://weizhonglab.ucsd.edu/webMEGA). Based on the output from this process, the proportion of proteins in each of the COG categories was calculated for each genome. The distributions of proportional abundances of each category were then used to assess the relationships of the four subgroups in terms of COG category representation.
Carbon Source Utilization Assay
Bacterial isolates from freezer stocks were streaked on 5% sheep blood agar plates and were grown for 48 h anaerobically, prior to the inoculation of AN microplates (Biolog Inc, Hayward, CA). Each plate contained 95 carbon sources and one blank well. Colonies of Gardnerella isolates were harvested using a sterile swab and suspended in 14 mL of inoculating fluid supplied by the manufacturer. The cell density was adjusted to 55% T (OD595 approximately 0.25) using a turbidimeter. Each well was filled with 100 μL of culture suspension and was incubated at 35 °C anaerobically for 48 h. All inoculations and incubations were performed in an anaerobic chamber containing 10% CO2, 5% hydrogen and 85% nitrogen. All plates were read visually after 48 h of incubation. If there was no carbon source utilization, the wells remained colourless. A visual change from colourless to purple indicated carbon source utilization. To avoid bias in interpretation, a subset of the plates was read by a second observer who was blinded to the identity of the isolates. There was no disagreement between independent observers. The entire experiment was performed in two biological replicates.
Carbon Source Profiling of Co-cultures
Representative isolates (VN003 of subgroup A, VN002 of subgroup B, NR001 of subgroup C and WP012 of subgroup D) from the four subgroups were co-cultured in the Biolog AN Microplate in a pairwise fashion (n = 6, AB, AC, AD, BC, BD, CD), by combining 50 μL of each isolate suspended in inoculation fluid in each well. The co-cultured AN microplates were incubated at 35 °C for 48 h before being assessed visually for colour change. The experiment was repeated on separate days.
Statistical Analysis
The degree of similarity between the isolates in terms of presence/absence of protein clusters generated in the pangenome analysis, proportional abundance of proteins in various COG categories and carbon source utilization patterns was calculated using the Bray-Curtis dissimilarity matrix. Principle components analysis (PCA) was performed on the distance matrices and the significance of relationships was tested using PERMANOVA with the ADONIS function in the vegan package [27]. The SIMPER function was used to identify variables driving the differences between groups.
One-way ANOVA, student’s t test and chi-square tests were applied to determine if the utilization of particular carbon sources was associated with specific subgroups.
All statistical analyses were performed in RStudio (version 3.5.2). Figures were generated using GraphPad Prism 8.0 and RStudio (version 3.5.2).
Results
Overlap Between the Subgroups Based on Pangenome and COG Analysis
The purpose of our pangenome analysis was to estimate the degree of niche overlap between Gardnerella subgroups based on comparisons of their predicted proteomes. Hierarchical clustering using complete linkage produced 4868 clusters or predicted proteins in the pangenome of the 40 isolates included (rarefaction curve is shown in Fig. S1). The strict core (defined as the protein clusters present in all isolates) included 176 clusters. Most of these core proteins were related to metabolism, transcriptional control, DNA replication and protein synthesis. The clustering of the genomes by subgroup was apparent in a UPGMA dendrogram based on the presence/absence patterns of the 4868 protein clusters (Fig. 1a). PCA was performed to determine the extent of overlap between the subgroups. The amount of variance explained by the two principal components was 19.4%, based on which, the four subgroups were separable (Fig. 1b). The dissimilarity between the four subgroups was significant (pairwise ADONIS, Bonferroni adjusted, p < 0.05, A vs B, R2 = 0.45; A vs C, R2 = 0.48; A vs D, R2 = 0.26; B vs C, R2 = 0.34; B vs D, R2 = 0.45 and C vs D, R2 = 0.55).
Fig. 1.

Comparison of predicted proteomes of study isolates. a UPGMA dendrogram based on the presence/absence of protein clusters in the predicted proteomes of Gardnerella isolates. b Principle components analysis (PCA) of Bray-Curtis dissimilarity matrices calculated from protein cluster distributions. The dissimilarity between the four subgroups is significant (pairwise ADONIS p < 0.05, Bonferroni adjusted). Subgroup affiliations of isolates are indicated by colour as shown in the legend between the panels
Following the identification of core and accessory proteins, we investigated the distribution of functional classifications of proteins encoded by isolates in the four subgroups. COG analysis resulted in the assignment of predicted proteins into 23 functional categories. As expected, the hierarchical clustering of the COG distribution patterns corresponded to subgroup affiliation (Fig. 2a). PCA was performed on the Bray-Curtis dissimilarity matrix and the differences between all subgroups were found to be significant (pairwise ADONIS, Bonferroni adjusted, p < 0.05, A vs B, R2 = 0.31; A vs C, R2 = 0.71; A vs D, R2 = 0.26; B vs C, R2 = 0.48; B vs D, R2 = 0.20 and C vs D, R2 = 0.74) (Fig. 2b).
Fig. 2.

COG analysis of predicted proteomes. a Hierarchical clustering of the study isolates based on the proportional abundance of COG categories in their predicted proteomes. Abundance is indicated by the blue colour intensity in the heat map. b PCA of Bray-Curtis dissimilarity matrices calculated from the proportional abundance data. Dissimilarity between the four subgroups is significant (pairwise ADONIS, p < 0.05, Bonferroni adjusted). Subgroup affiliations of isolates are indicated by colour as shown in the legend between the panels
We identified the variables which caused the four subgroups to diverge in terms of abundance of different COG categories in a multivariate analysis using SIMPER [28]. SIMPER calculates the contribution of each variable to the dissimilarity observed between two groups and relies on the Bray-Curtis dissimilarity matrix for calculating the proportion of the contribution of each variable being tested. Thirty-six percent of the differences between subgroup A and subgroup B were accounted for by amino acid transport and metabolism (COG category E), inorganic ion transport metabolism (category P), translation, ribosomal structure and biogenesis proteins (category J). The proportion of proteins with functions related to carbohydrate transport and metabolism (category G) was the major factor that differentiated subgroup A from C, contributing to 34% of the dissimilarity observed. Carbohydrate transport and metabolism also accounted for 31% of the dissimilarity observed between subgroups B and C and 36% of the dissimilarity between subgroups C and D. The major contributing factors that differentiated subgroups A and D were proportional abundance of proteins assigned to functional categories H (co-enzyme transport and metabolism) and E (amino acids transport and metabolism) (23%). Subgroups B and D were separated primarily based on functional categories P (inorganic ion transport and metabolism), J (translation, ribosomal structure and biogenesis), G (carbohydrate transport and metabolism proteins) and E (amino acid transport and metabolism proteins), which together accounted for 37% of the dissimilarity observed.
Functional Categories of Proteins Differentiating Subgroups of Gardnerella
We tested if the proportions of individual functional categories of proteins that drive the overall separation of the four subgroups in multivariate analysis were significantly different between pairs of Gardnerella subgroups. This analysis revealed that subgroup C has a significantly higher proportion of its encoded proteins associated with carbohydrate transport and metabolism and transport and transcriptional regulation than the other subgroups (unpaired t test, p ≤ 0.01, Bonferroni adjusted, Fig. 3a and d). The proportion of proteins associated with amino acid transport and metabolism is significantly higher in subgroup D than subgroups A, B and C (unpaired t test, p ≤ 0.01, Bonferroni adjusted, Fig. 3b). Proteins involved in co-enzyme transport and metabolism were found in significantly higher proportional abundance in subgroup A than in subgroups B, C and D (unpaired t test, p ≤ 0.0001, Bonferroni adjusted, Fig. 3c). Subgroup B has a significantly higher abundance of proteins associated with inorganic ion transport and metabolism than subgroups A and D (unpaired t test, p ≤ 0.0001, Fig. 3e), but the difference between subgroup B and C was not significant. Subgroup B also has a significantly higher proportion of translation, ribosomal structure and biogenesis proteins (unpaired t test, p ≤ 0.001, Fig. 3f) compared to subgroup C.
Fig. 3.

Differential abundance of six COG categories that were identified by SIMPER analysis as main drivers of subgroup separation. a Carbohydrate metabolism and transport proteins (category G), b amino acid transport and metabolism proteins (category E), c co-enzyme transport and metabolism proteins (category H), d transcription proteins (category K), e inorganic ion transport and metabolism proteins (category P) and f translation, ribosomal structure and biogenesis proteins (category J). Results of unpaired t tests are indicated where * is p ≤ 0.05, ** is p ≤ 0.01, *** is p ≤ 0.001, **** is p ≤ 0.0001 and ns is not significant
Carbon Source Utilization Phenotypes
We hypothesized that subgroup D, a slow growing, rarely detected Gardnerella subgroup is maintained in the vaginal microbiome at a low level and avoids competitive exclusion through negative frequency-dependent selection, made possible by being a nutritional generalist. We performed carbon source utilization profiling of thirty-six representative isolates (n = 12, subgroup A; n = 9, subgroup B; n = 8, subgroup C (including type strain G. vaginalis ATCC 14018) and n = 7, subgroup D). The number of carbon sources utilized by any Gardnerella strain ranged from 5 to 24. Only 25% (9/36) of the isolates utilized more than 17 carbon sources, including two subgroup C (NR001, NR038) and all subgroup D isolates. Twenty isolates utilized at least 13 carbon sources, including three subgroup A (3/12, 25%), four subgroup B (4/8, 50%), six subgroup C (6/7, 86%) and all seven isolates of subgroup D (100%). The average number of carbon sources utilized by isolates in subgroups A, B, C and D was 10.4 ± 3.1, 11.8 ± 1.75, 13.9 ± 3.6 and 20.3 ± 1.9, respectively (Fig. 4). A one-way ANOVA was performed to compare the overall difference in carbon sources utilization among the four subgroups showed significant difference among the subgroups (F(3,32) = 18.15, p < 0.05). A post hoc comparison between the subgroups revealed that the number of carbon sources utilized by subgroup D was significantly higher than subgroups A, B and C (Tukey’s HSD, p < 0.05). All of the tested Gardnerella isolates were able to utilize pyruvic acid, palatinose and l-rhamnose. The next most frequently utilized carbon sources were d-fructose (32/36, 97%) and l-fucose (32/36, 97%).
Fig. 4.
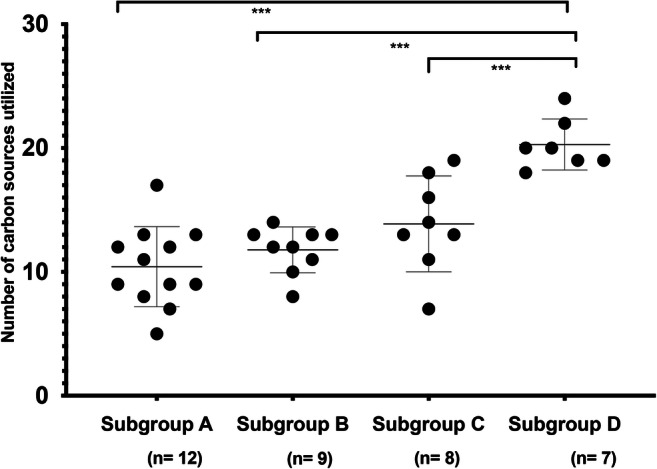
Comparison of numbers of carbon sources utilized by isolates in each subgroup. The number of carbon sources utilized by isolates in subgroup D was significantly higher than those in subgroups A, B and C (Tukey’s HSD, p ≤ 0.001). Only one-fourth (9/36) of the tested isolates utilized more than 17 carbon sources, including all seven tested isolates of subgroup D
Overall, 31/95 carbon sources were utilized by at least one isolate, and the majority (20/31) of these were sugars (mono- or oligosaccharides). Together, subgroup D isolates (n = 7) utilized more of the sugar substrates (18/37 available) than any other subgroup, including subgroup C (n = 8), which utilized 15/37 available sugars. The utilization of any of the 11 available amino acids was rarely observed, with only two of the subgroup C isolates positive for l-methionine or l-valine utilization.
Overlap in Carbon Sources Utilization Among the Subgroups
To determine if subgroups could be distinguished based on carbon source utilization profiles, a principal component analysis was performed (Fig. 5). The overlap between the representative isolates of subgroups A, B and C was significant. Subgroup D was significantly dissimilar to subgroups A and B (Fig. 5, pairwise ADONIS, A vs D, R2 = 0.55; B vs D, R2 = 0.55, p < 0.05). Although the dissimilarity between subgroups C and D was not statistically significant after Bonferroni adjustment, 39% (pairwise ADONIS, C vs D, R2 = 0.39) of the variation in carbon source utilization could be explained by subgroup affiliation of the tested isolates, which was higher than between subgroups A, B and C (A vs B: 13%, A vs. C: 21% and B vs. C: 8%).
Fig. 5.

Subgroup D has minimal overlap with the other subgroups in carbon source utilization. The degree of variation based on carbon source utilization between subgroup D and subgroups A and B was significant (pairwise ADONIS, p < 0.05, after Bonferroni adjustment). The variation in carbon source utilization between subgroups C and D can be explained by subgroup affiliation in 39% of cases. Overall, 42% of differences in carbon sources utilization between subgroups can be explained by their subgroup affiliation (Adonis, R2 = 0.42, p < 0.05)
Association of Carbon Source Utilization Pattern with Subgroups
To identify carbon sources that differentiate the subgroups, we selected twelve substrates that were utilized by more than five isolates but fewer than thirty isolates. Chi-square tests were performed to determine if the subgroups significantly differ in the utilization of those twelve carbon sources. The four Gardnerella subgroups differed in their use of 3 of the 12 carbon sources: turanose, inosine and uridine 5-monophosphate (chi-square test, p < 0.05, Bonferroni adjusted) (Fig. 6). For each of these three carbon sources, subgroups A and B had a low frequency of use (9.5% = 2/21, 0.0% = 0/21, 0.0% = 0/21; subgroups A and B combined) and subgroup C had low or intermediate frequency of use (25.0% = 2/8, 50.0% = 4/8 and 62.5% = 5/8), whereas subgroup D had a high frequency of use (100.0% = 7/7, 100.0% = 7/7, 100.0% = 7/7).
Fig. 6.

Subgroup D can be differentiated from the other subgroups based on its capacity to utilize inosine (a), uridine 5-monophosphate (b) and turanose (c). The percentage of isolates in each subgroup that utilize the indicated carbon source is shown. The utilization of these three carbon sources is significantly associated with subgroup D (chi-square test, p ≤ 0.001)
Carbon Source Utilization by Co-cultured Isolates
Since the four subgroups co-exist in the same ecosystem, it is possible that mixing them might facilitate the utilization of certain carbon sources. To detect any such facilitation in carbon sources utilization, we co-cultured isolates from all four subgroups in six pairwise combinations (A+B, A+C, A+D, B+C, B+D and C+D). The representative isolates of subgroups A–D utilized 11, 13, 19 and 24 carbon sources, respectively, when grown alone while co-cultures utilized from 12 to a maximum of 22 carbon sources (Table 1). In every case, the co-culture utilized fewer carbon sources than the isolate that utilized the most carbon sources on its own.
Table 1.
Carbon source utilization by co-cultured isolates
| Isolate 1 (subgroup) | Isolate 2 (subgroup) | No. carbon sources | ||
|---|---|---|---|---|
| Isolate 1 | Isolate 2 | Co-culture | ||
| VN003 (A) | VN002 (B) | 11 | 13 | 12 |
| VN003 (A) | NR001 (C) | 11 | 19 | 13 |
| VN003 (A) | WP012 (D) | 11 | 24 | 22 |
| VN002 (B) | NR001 (C) | 13 | 19 | 15 |
| VN002 (B) | WP012 (D) | 13 | 24 | 20 |
| NR001 (C) | WP012 (D) | 19 | 24 | 19 |
Discussion
Rarely abundant species can be maintained in the human microbiome through a variety of mechanisms, which include but are not limited to sequestration of essential nutrients from competing species, diversification of phenotype [29], social cheating [30] and negative frequency-dependent selection [17]. Differences in nutrient utilization among community members can be a key factor that sets the stage for negative frequency-dependent selection [31]. The reproductive fitness of nutritional specialist species will remain high as long as the supply of nutrients usable by the specialists is abundant. As soon as the supply of these nutrients drops, slower-growing generalists, by virtue of their greater utilization capacity, will have increased fitness, which will eventually lead to their dominance in the absence of any other negative influences.
Among the four subgroups of Gardnerella spp. that colonize the vaginal microbiome of reproductive-aged women, subgroup D is the rarest in terms of abundance and prevalence among women [4]. Subgroup D is also relatively slow growing, yet shows an increased growth rate when the number of competitors in an in vitro community increases [12]. We have reported previously that resource-based competition is common among Gardnerella spp. and no evidence for contact-dependent interaction was observed. Therefore, we set out to investigate if the negative frequency-dependent selection is responsible for the persistence of subgroup D, which would require relatively small niche overlap and a more generalist lifestyle than the other Gardnerella spp. in the vaginal microbiome.
Predicted Niche Overlap Between the Four Subgroups
Niche overlap may lead to competition for nutrients and space [31–34] and it has been reported that competition is prevalent among metabolically similar bacterial species [14]. Occupying distinct niches can therefore help bacterial species avoid competition for space, growth factors and nutrients, resulting in increased reproductive fitness. Since subgroup D isolates have higher growth rates in vitro in the presence of competitors compared to when grown alone, these isolates presumably occupy a distinct niche. The pangenome analysis showed that the four subgroups differ significantly based on the composition of their predicted proteomes (Fig. 1), with only 176 proteins comprising the strict core of proteins found in all isolates. This finding is not surprising since the genetic diversity among Gardnerella is well established, and genome sequence comparisons formed the basis for the recent reclassification of Gardnerella into 13 genome species [1–3].
Comparisons of the entire predicted proteomes do not, however, focus on the key factors for a resource-based competition: nutrient utilization potential. Proteins involved in nutrient uptake and metabolism account for only a fraction of the 4868 protein clusters comprising the pangenome. Analysis of the distribution of various functional (COG) categories of proteins revealed significant differences among subgroups in their predicted capacity to utilize carbohydrates and amino acids (Fig. 3a and b), with subgroup D having significantly more of its proteome dedicated to amino acid transport and metabolism than any of the other subgroups. Since a resource-based competition encapsulates competition for space, growth factors and nutrients, our findings from the pangenome and COG analyses suggest that the competition among the four subgroups is not spatial but may be primarily for nutrients, a speculation supported by the previous observation that Gardnerella spp. form multi-subgroup biofilms [12].
Subgroup D Is a Nutritional Generalist Relative to Subgroups A, B and C
The diversity of nutrients available to microbiota in the vaginal microbiome is less than in the gastrointestinal microbiome, where food intake provides a constant source of diverse nutrients that affect the assembly of gut microbiota [35, 36]. Vaginal microbiota, on the contrary, are largely dependent upon host-derived nutrients, the most abundant of which is glycogen. Glycogen is deposited in the vaginal lumen by epithelial cells under the influence of estrogen [37] and is digested into maltooligosaccharides, maltodextrins and glucose by the combined activities of host and microbial enzymes prior to uptake and metabolism by the microbiota [38–40]. Given the relatively narrow range of nutrients available in the vaginal microbiome, it is expected that the resident microbiota, including the four subgroups of Gardnerella, overlap to a considerable extent in their nutrient utilization capacity, resulting in some level of competition among them [32, 36, 41]. As discussed earlier, subgroup D is an exception since the growth of these isolates was actually facilitated in co-cultures, suggesting that while it may compete with other Gardnerella spp. over common nutrients like the breakdown products of glycogen, it may be able to utilize a greater overall diversity of nutrients (i.e. it is a generalist).
The AN microplate assay results showed that subgroup D isolates utilized more of the provided carbon sources than isolates in the three other subgroups (Fig. 4). Furthermore, when the patterns of substrate use were considered, subgroups A, B and C were not separable from each other, but subgroup D was significantly different (Fig. 5). The distinct pattern observed in subgroup D was partially driven by the utilization of three particular substrates: turanose, inosine and uridine 5-monophosphate (Fig. 6). Turanose is an isomer of sucrose, known as a non-accumulative osmoprotectant, aiding bacterial growth at high osmolarity [42]. The importance of turanose utilization in the vaginal environment is not known yet, but our observation is an indication that subgroup D isolates can metabolize sucrose-like sugars. The two other carbon sources: inosine and uridine 5-monophosphate are probably used in purine and pyrimidine biosynthesis in Gardnerella spp.
Some findings of the pangenome and COG analyses could not be reconciled with the phenotypic carbon source utilization assay. For example, although subgroups C and D have higher proportions of their proteomes predicted to be involved in transport and metabolism of carbohydrates (category G) and amino acids (category E), respectively, than the other subgroups, subgroup C isolates did not utilize the greatest number of available sugar substrates in the AN microplate and subgroup D isolates did not utilize any of the amino acid substrates available. It is, however, important to consider that the carbon source utilization assay was performed in a plastic environment and included only 95 substrates, many of which are not relevant to the vaginal microbiome. More relevant amino acid sources available in the vagina, including those whose abundance is altered in bacterial vaginosis, such as isoleucine, leucine, proline and tryptophan, are not included [43–45]. Ideally, this study would have involved a vaginal-microbiome specific nutrient panel, but such reagents were not available. Even with this limitation, our results suggest that subgroup D is a nutritional generalist relative to other Gardnerella spp. Most of the ecological studies that have been performed to date to elucidate mechanisms shaping the assembly of bacterial communities have included either environmental bacterial species or well-characterized model organisms [14, 16, 29, 36, 41, 46–50]. There are understandably fewer studies that focus on interactions among host-associated microbiota [51, 52].
Negative Frequency-Dependent Selection in the Vaginal Microbiome
The genomic and phenotypic differences we observed between subgroup D and the three other subgroups, including the potential to utilize more amino acids, use of a greater number of carbon sources and a distinct pattern of substrate utilization, suggest that subgroup D is a candidate for negative frequency-dependent selection. Why then are these Gardnerella spp. only observed rarely and in low abundance in reproductive-aged women? Among 413 vaginal samples from reproductive-aged Canadian women, genome species comprising subgroup D of Gardnerella were detected in <10% of samples and never accounted for more than 5% of the microbiota [4]. Vaginal environmental dynamics and related host factors, such as menstruation, sloughing of epithelial cells and fluctuating pH, contribute to the turnover of bacterial species, shifting the bacterial population density and changing the nutrients available [53]. A decline in population density would reshuffle the vaginal ecosystem, increasing the supply of abundant nutrients accessible to faster growing, specialists, and checking the growth of slower-growing generalist subgroup D.
Although subgroup D is likely rare due to the factors described above, it could still be a major player in ecological succession and transition of vaginal microbiota between a Lactobacillus dominated community and the overgrowth of anaerobes characteristic of bacterial vaginosis. These organisms may also play a particular role in biofilm formation or competition for occupancy of the vaginal mucosa. Rarely abundant species often act as keystone species helping colonization by other bacterial species, which are also essential to maintain homeostasis of an ecosystem [54–57]. The resolution of the role of low abundant Gardnerella spp. will depend on the development and application of experimental systems that more closely model the human vaginal microbiome. Rodent models have shown some promise, especially for studies of specific combinations of organisms [58], but there is also potential in bioreactors [59], and cell and tissue culture systems that attempt to recapitulate many of the environmental and physiological aspects of the vaginal microbiome [60]. Further study of rare Gardnerella spp. will likely also result in the definition of additional species within this diverse genus.
Supplementary Information
(PDF 67 kb)
(PDF 110 kb)
Acknowledgements
The authors are grateful to Champika Fernando for their excellent technical support and to all members of the Hill Lab for their review of the manuscript. Special thanks to Prairie Diagnostic Services for access to their anaerobic chamber. No thanks to COVID-19.
Availability of Data and Material
NCBI Bioproject accession numbers for all genome sequence data are included in Table S1.
Authors’ Contributions
Conceived and designed the study: Salahuddin Khan and Janet E. Hill; performed the experiments: Salahuddin Khan and Sarah J. Vancuren; analysed the data: Salahuddin Khan, Sarah J. Vancuren, Janet E. Hill; wrote and revised the manuscript: Salahuddin Khan, Sarah J. Vancuren, Janet E. Hill.
Funding
The research was supported by a Natural Sciences and Engineering Research Council of Canada Discovery Grant to JEH.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
Contributor Information
Salahuddin Khan, Email: salahuddin.khan@usask.ca.
Sarah J. Vancuren, Email: svancure@uoguelph.ca
Janet E. Hill, Email: Janet.Hill@usask.ca
References
- 1.Paramel Jayaprakash T, Schellenberg JJ, Hill JE. Resolution and characterization of distinct cpn60-based subgroups of Gardnerella vaginalis in the vaginal microbiota. PLoS ONE. 2012;7:e43009. doi: 10.1371/journal.pone.0043009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed A, Earl J, Retchless A, Hillier SL, Rabe LK, Cherpes TL, Powell E, Janto B, Eutsey R, Hiller NL, Boissy R, Dahlgren ME, Hall BG, Costerton JW, Post JC, Hu FZ, Ehrlich GD. Comparative genomic analyses of 17 clinical isolates of Gardnerella vaginalis provide evidence of multiple genetically isolated clades consistent with subspeciation into genovars. J Bacteriol. 2012;194:3922–3937. doi: 10.1128/JB.00056-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaneechoutte M, Guschin A, Van Simaey L, et al. Emended description of Gardnerella vaginalis and description of Gardnerella leopoldii sp. nov., Gardnerella piotii sp. nov. and Gardnerella swidsinskii sp. nov., with delineation of 13 genomic species within the genus Gardnerella. Int J Syst Evol Microbiol. 2019;69:679–687. doi: 10.1099/ijsem.0.003200. [DOI] [PubMed] [Google Scholar]
- 4.Hill JE, Albert AYK. Resolution and co-occurrence patterns of Gardnerella leopoldii, Gardnerella swidsinskii, Gardnerella piotii and Gardnerella vaginalis within the vaginal microbiome. Infect Immun. 2019;87:e00532–e00519. doi: 10.1128/IAI.00532-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janulaitiene M, Gegzna V, Baranauskiene L, Bulavaitė A, Simanavicius M, Pleckaityte M. Phenotypic characterization of Gardnerella vaginalis subgroups suggests differences in their virulence potential. PLOS ONE. 2018;13:e0200625. doi: 10.1371/journal.pone.0200625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeoman CJ, Yildirim S, Thomas SM, Durkin AS, Torralba M, Sutton G, Buhay CJ, Ding Y, Dugan-Rocha SP, Muzny DM, Qin X, Gibbs RA, Leigh SR, Stumpf R, White BA, Highlander SK, Nelson KE, Wilson BA. Comparative genomics of Gardnerella vaginalis strains reveals substantial differences in metabolic and virulence potential. PLoS ONE. 2010;5:e12411. doi: 10.1371/journal.pone.0012411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schellenberg JJ, Paramel Jayaprakash T, Withana Gamage N, Patterson MH, Vaneechoutte M, Hill JE. Gardnerella vaginalis subgroups defined by cpn60 sequencing and sialidase activity in isolates from Canada, Belgium and Kenya. PLoS One. 2016;11:e0146510. doi: 10.1371/journal.pone.0146510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albert AY, Chaban B, Wagner EC, et al. A study of the vaginal microbiome in healthy Canadian women utilizing cpn60-based molecular profiling reveals distinct Gardnerella subgroup community state types. PLoS One. 2015;10:e0135620. doi: 10.1371/journal.pone.0135620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hilbert DW, Schuyler JA, Adelson ME, Mordechai E, Sobel JD, Gygax SE. Gardnerella vaginalis population dynamics in bacterial vaginosis. Eur J Clin Microbiol Infect Dis. 2017;36:1269–1278. doi: 10.1007/s10096-017-2933-8. [DOI] [PubMed] [Google Scholar]
- 10.Shipitsyna E, Krysanova A, Khayrullina G, Shalepo K, Savicheva A, Guschin A, Unemo M. Quantitation of all four Gardnerella vaginalis clades detects abnormal vaginal microbiota characteristic of bacterial vaginosis more accurately than putative G. vaginalis sialidase A gene count. Mol Diagn Ther. 2019;23:139–147. doi: 10.1007/s40291-019-00382-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schellenberg JJ, Patterson MH, Hill JE. Gardnerella vaginalis diversity and ecology in relation to vaginal symptoms. Res Microbiol. 2017;168:837–844. doi: 10.1016/j.resmic.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Khan S, Voordouw MJ, Hill JE. Competition among Gardnerella subgroups from the human vaginal microbiome. Front Cell Infect Microbiol. 2019;9:374. doi: 10.3389/fcimb.2019.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madsen JS, Røder HL, Russel J, Sørensen H, Burmølle M, Sørensen SJ. Coexistence facilitates interspecific biofilm formation in complex microbial communities. Environ Microbiol. 2016;18:2565–2574. doi: 10.1111/1462-2920.13335. [DOI] [PubMed] [Google Scholar]
- 14.Russel J, Røder HL, Madsen JS, Burmølle M, Sørensen SJ. Antagonism correlates with metabolic similarity in diverse bacteria. Proc Natl Acad Sci U S A. 2017;114:10684–10688. doi: 10.1073/pnas.1706016114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darwin C. On the origin of species by means of natural selection, or preservation of favoured races in the struggle for life. London: John Murray; 1859. [PMC free article] [PubMed] [Google Scholar]
- 16.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin BR. Frequency-dependent selection in bacterial populations. Philos Trans R Soc Lond B Biol Sci. 1988;319:459–472. doi: 10.1098/rstb.1988.0059. [DOI] [PubMed] [Google Scholar]
- 18.Kurihara Y, Shikano S, Toda M. Trade-off between interspecific competitive ability and growth rate in bacteria. Ecology. 1990;71:645–650. doi: 10.2307/1940318. [DOI] [Google Scholar]
- 19.Ross-Gillespie A, Gardner A, Buckling A, West SA, Griffin AS. Density dependence and cooperation: theory and a test with bacteria. Evolution. 2009;63:2315–2325. doi: 10.1111/j.1558-5646.2009.00723.x. [DOI] [PubMed] [Google Scholar]
- 20.Martin-Platero AM, Valdivia E, Maqueda M, Martinez-Bueno M. Fast, convenient, and economical method for isolating genomic DNA from lactic acid bacteria using a modification of the protein “salting-out” procedure. Anal Biochem. 2007;366:102–104. doi: 10.1016/j.ab.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Vancuren SJ, Hill JE. Update on cpnDB: a reference database of chaperonin sequences. Database. 2019;2019:baz033. doi: 10.1093/database/baz033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu SM, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam TW, Wang J. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes de novo assembler. Curr Protoc Bioinforma. 2020;70:e102. doi: 10.1002/cpbi.102. [DOI] [PubMed] [Google Scholar]
- 25.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snipen L, Liland KH. micropan: an R-package for microbial pan-genomics. BMC Bioinformatics. 2015;16:79. doi: 10.1186/s12859-015-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oksanen J, Blanchet FG, Kindt R et al (2012) vegan: community ecology package. R package version 2.0-10. Available: http://CRAN.R-project.org/package = vegan
- 28.Whitfield-Cargile CM, Cohen ND, Suchodolski J, Chaffin MK, McQueen CM, Arnold CE, Dowd SE, Blodgett GP. Composition and diversity of the fecal microbiome and inferred fecal metagenome does not predict subsequent pneumonia caused by Rhodococcus equi in foals. PLOS ONE. 2015;10:e0136586. doi: 10.1371/journal.pone.0136586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martín PV, Muñoz MA, Pigolotti S. Bet-hedging strategies in expanding populations. PLOS Comput Biol. 2019;15:e1006529. doi: 10.1371/journal.pcbi.1006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen R, Déziel E, Groleau M-C, Schaefer AL, Greenberg EP. Social cheating in a Pseudomonas aeruginosa quorum-sensing variant. Proc Natl Acad Sci. 2019;116:7021–7026. doi: 10.1073/pnas.1819801116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freter R, Brickner H, Botney M, Cleven D, Aranki A. Mechanisms that control bacterial populations in continuous-flow culture models of mouse large intestinal flora. Infect Immun. 1983;39:676–685. doi: 10.1128/IAI.39.2.676-685.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holt RD. Bringing the Hutchinsonian niche into the 21st century: ecological and evolutionary perspectives. Proc Natl Acad Sci. 2009;106:19659–19665. doi: 10.1073/pnas.0905137106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hutchinson GE. Concluding remarks. Cold Spring Harb Symp Quant Biol. 1957;22:415–427. doi: 10.1101/SQB.1957.022.01.039. [DOI] [Google Scholar]
- 34.Amarasekare P. Interference competition and species coexistence. Proc R Soc Lond B Biol Sci. 2002;269:2541–2550. doi: 10.1098/rspb.2002.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graf D, Di Cagno R, Fåk F, et al. Contribution of diet to the composition of the human gut microbiota. Microb Ecol Health Dis. 2015;26:26164. doi: 10.3402/mehd.v26.26164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CC, Snowberg LK, Gregory Caporaso J, et al. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 2015;9:2515–2526. doi: 10.1038/ismej.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirmonsef P, Hotton AL, Gilbert D, Burgad D, Landay A, Weber KM, Cohen M, Ravel J, Spear GT. Free glycogen in vaginal fluids is associated with Lactobacillus colonization and low vaginal pH. PLoS One. 2014;9:e102467. doi: 10.1371/journal.pone.0102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhandari P, Tingley JP, Abbott DW, Hill JE (2020) Characterization of an α-glucosidase enzyme conserved in Gardnerella spp. isolated from the human vaginal microbiome. bioRxiv 2020.05.11.086124. 10.1101/2020.05.11.086124 [DOI] [PMC free article] [PubMed]
- 39.Nunn KL, Forney LJ. Unraveling the dynamics of the human vaginal microbiome. Yale J Biol Med. 2016;89:331–337. [PMC free article] [PubMed] [Google Scholar]
- 40.Spear GT, French AL, Gilbert D, Zariffard MR, Mirmonsef P, Sullivan TH, Spear WW, Landay A, Micci S, Lee BH, Hamaker BR. Human alpha-amylase present in lower-genital-tract mucosal fluid processes glycogen to support vaginal colonization by Lactobacillus. J Infect Dis. 2014;210:1019–1028. doi: 10.1093/infdis/jiu231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bauer MA, Kainz K, Carmona-Gutierrez D, Madeo F. Microbial wars: competition in ecological niches and within the microbiome. Microb Cell. 2018;5:215–219. doi: 10.15698/mic2018.05.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gouffi K, Blanco C. Is the accumulation of osmoprotectant the unique mechanism involved in bacterial osmoprotection? Int J Food Microbiol. 2000;55:171–174. doi: 10.1016/s0168-1605(00)00192-6. [DOI] [PubMed] [Google Scholar]
- 43.Srinivasan S, Morgan MT, Fiedler TL, Djukovic D, Hoffman NG, Raftery D, Marrazzo JM, Fredricks DN (2015) Metabolic signatures of bacterial vaginosis. mBio 6. 10.1128/mBio.00204-15 [DOI] [PMC free article] [PubMed]
- 44.Vicente-Muñoz S, Cobo T, Puchades-Carrasco L, Sánchez-García AB, Agustí N, Palacio M, Pineda-Lucena A, Gratacós E. Vaginal metabolome: towards a minimally invasive diagnosis of microbial invasion of the amniotic cavity in women with preterm labor. Sci Rep. 2020;10:5465. doi: 10.1038/s41598-020-62542-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vitali B, Cruciani F, Picone G, Parolin C, Donders G, Laghi L. Vaginal microbiome and metabolome highlight specific signatures of bacterial vaginosis. Eur J Clin Microbiol Infect Dis. 2015;34:2367–2376. doi: 10.1007/s10096-015-2490-y. [DOI] [PubMed] [Google Scholar]
- 46.Stubbendieck RM, Straight PD. Escape from lethal bacterial competition through coupled activation of antibiotic resistance and a mobilized subpopulation. PLOS Genet. 2015;11:e1005722. doi: 10.1371/journal.pgen.1005722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonzalez DJ, Haste NM, Hollands A, Fleming TC, Hamby M, Pogliano K, Nizet V, Dorrestein PC. Microbial competition between Bacillus subtilis and Staphylococcus aureus monitored by imaging mass spectrometry. Microbiology. 2011;157:2485–2492. doi: 10.1099/mic.0.048736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Griffin AS, West SA, Buckling A. Cooperation and competition in pathogenic bacteria. Nature. 2004;430:1024–1027. doi: 10.1038/nature02744. [DOI] [PubMed] [Google Scholar]
- 49.Oliveira NM, Martinez-Garcia E, Xavier J, Durham WM, Kolter R, Kim W, Foster KR. Biofilm formation as a response to ecological competition. PLoS Biol. 2015;13:e1002191. doi: 10.1371/journal.pbio.1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rezzoagli C, Granato ET, Kümmerli R. Harnessing bacterial interactions to manage infections: a review on the opportunistic pathogen Pseudomonas aeruginosa as a case example. J Med Microbiol. 2020;69:147–161. doi: 10.1099/jmm.0.001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.García-Bayona L, Comstock LE (2018) Bacterial antagonism in host-associated microbial communities. Science 361. 10.1126/science.aat2456 [DOI] [PubMed]
- 52.Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533:255–259. doi: 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abreu CI, Friedman J, Andersen Woltz VL, Gore J. Mortality causes universal changes in microbial community composition. Nat Commun. 2019;10:2120. doi: 10.1038/s41467-019-09925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee S, Schlaeppi K, van der Heijden MGA. Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol. 2018;16:567–576. doi: 10.1038/s41579-018-0024-1. [DOI] [PubMed] [Google Scholar]
- 55.Herren CM, McMahon KD. Keystone taxa predict compositional change in microbial communities. Environ Microbiol. 2018;20:2207–2217. doi: 10.1111/1462-2920.14257. [DOI] [PubMed] [Google Scholar]
- 56.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. A low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and the complement pathway. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, Brodie EL, Dominguez-Bello MG. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5:574–579. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teixeira GS, Carvalho FP, Arantes RM, et al. Characteristics of Lactobacillus and Gardnerella vaginalis from women with or without bacterial vaginosis and their relationships in gnotobiotic mice. J Med Microbiol. 2012;61:1074–1081. doi: 10.1099/jmm.0.041962-0. [DOI] [PubMed] [Google Scholar]
- 59.Guzman-Rodriguez M, McDonald JAK, Hyde R, et al. Using bioreactors to study the effects of drugs on the human microbiota. Methods. 2018;149:31–41. doi: 10.1016/j.ymeth.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 60.Herbst-Kralovetz MM, Pyles RB, Ratner AJ, Sycuro LK, Mitchell C. New systems for studying intercellular interactions in bacterial vaginosis. J Infect Dis. 2016;214:S6–S13. doi: 10.1093/infdis/jiw130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 67 kb)
(PDF 110 kb)
Data Availability Statement
NCBI Bioproject accession numbers for all genome sequence data are included in Table S1.