

ABSTRACT
Prenatal smoke exposure (PSE) is a risk factor for nicotine dependence. One susceptibility gene for nicotine dependence is Cytochrome P450 (CYP) 2A6, an enzyme responsible for the conversion of nicotine to cotinine and nicotine clearance in the liver. Higher activity of the CYP2A6 enzyme is associated with nicotine dependence, but no research has addressed the PSE effects on the CYP2A6 gene or its mouse homologue Cyp2a5. We hypothesized that PSE affects Cyp2a5 promoter methylation, Cyp2a5 mRNA levels, and nicotine metabolism in offspring. We used a smoke-exposed pregnant mouse model. RNA, DNA, and microsomal protein were isolated from liver tissue of foetal, neonatal, and adult offspring. Enzyme activity, Cyp2a5 mRNA levels, and Cyp2a5 methylation status of six CpG sites within the promoter region were analysed via HPLC, RT-PCR, and bisulphite pyrosequencing. Our data show that PSE induced higher cotinine levels in livers of male neonatal and adult offspring compared to controls. PSE-induced cotinine levels in neonates correlated with Cyp2a5 mRNA expression and promoter methylation at CpG-7 and CpG+45. PSE increased methylation in almost all CpG sites in foetal offspring, and this effect persisted at CpG-74 in male neonatal and adult offspring. Our results indicate that male offspring of mothers which were exposed to cigarette smoke during pregnancy have a higher hepatic nicotine metabolism, which could be regulated by DNA methylation. Given the detected persistence into adulthood, extrapolation to the human situation suggests that sons born from smoking mothers could be more susceptible to nicotine dependence later in life.
KEYWORDS: Prenatal smoke exposure, epigenetics, cotinine, CYP2A5, nicotine dependence, sex difference, mouse model
Introduction
Nicotine is the most prominent and the primary psychoactive component in cigarette smoke, which is primarily metabolized by the enzyme Cytochrome P450 (CYP) 2A6 [1–3]. The encoding gene CYP2A6 has been identified as a susceptibility gene for nicotine dependence [4], and variation in CYP2A6 predicts heavier cigarette consumption [5,6] and delayed smoking cessation [7], which are both related to nicotine dependence [4,8]. Up until today, a number of pharmacogenetics studies in humans have shown that nicotine dependence relies on alterations in nicotine metabolism [9], which in turn has been associated with higher CYP2A6 enzyme activity [10,11].
Cyp2a5, the mouse homologue of CYP2A6, plays a prominent role in the clearance of nicotine in the liver, as CYP2A5 protein converts 80% of nicotine to cotinine [12,13]. This conversion requires the production of nicotine- Δ1ʹ(5ʹ)-iminium ion first, which is then subsequently converted to cotinine by aldehyde oxidase [12]. Similar to CYP2A6, Cyp2a5 is primarily expressed in the liver [14].
Until now, many studies have described the adverse effects of smoking during pregnancy on foetal development [15], metabolic disease [16,17], smoking behaviour [18,19] and the risk to develop the smoking-related diseases such as chronic obstructive pulmonary disease (COPD) [20]. These observations are supported by experimental mouse studies from our own group [15,21,22] and others [23–25].
Despite the above-mentioned plethora of studies, relatively little is known about the underlying mechanisms that contribute to the impact of prenatal smoke exposure (PSE) on smoking behaviour and nicotine dependence. However, following the prenatal programming paradigm regarding prenatal epigenetic changes such as alterations in DNA methylation patterns and their role in onset of diseases, it is suggested that changes in DNA methylation could be one of the contributing mechanisms in dependency establishment. Thus far, a number of birth cohort studies indeed reported aberrantly methylated CpG sites in peripheral blood [26], placenta [27] and buccal cells [28] from children of smoking mothers. Furthermore, in the mouse studies from our group, PSE was shown to induce differential gene regulation and DNA methylation profiles of a variety of genes [29,30], but this effect was not shown by other studies [31].
However, to our knowledge, nothing has been reported on the effect of PSE on nicotine metabolism and possible nicotine dependence. Therefore, we investigated the effect of PSE on hepatic Cyp2a5 gene expression, promoter methylation, and enzyme activity in male and female offspring from smoke exposed pregnant mice. To establish persistence of the PSE effect, this was investigated across three developmental stages, namely foetal (E17.5), neonatal (D3), and adult (D140).
Results
Higher Cyp2a5 mRNA expression and promoter methylation in male PSE offspring
PSE increased Cyp2a5 mRNA levels in the liver of neonates (p = 0.029, Figure 1(c)), which was most prominent in male offspring (p = 0.015, Figure 1(d)). PSE had no effect on Cyp2a5 mRNA steady state in foetal (Figure 1(a,b)) and adult livers of both sexes (Figure 1(e,f)). Interestingly, when comparing control male and female adult groups, females had a higher Cyp2a5 mRNA content than male adult offspring (p < 0.0001, Figure 1(f)).
Figure 1.
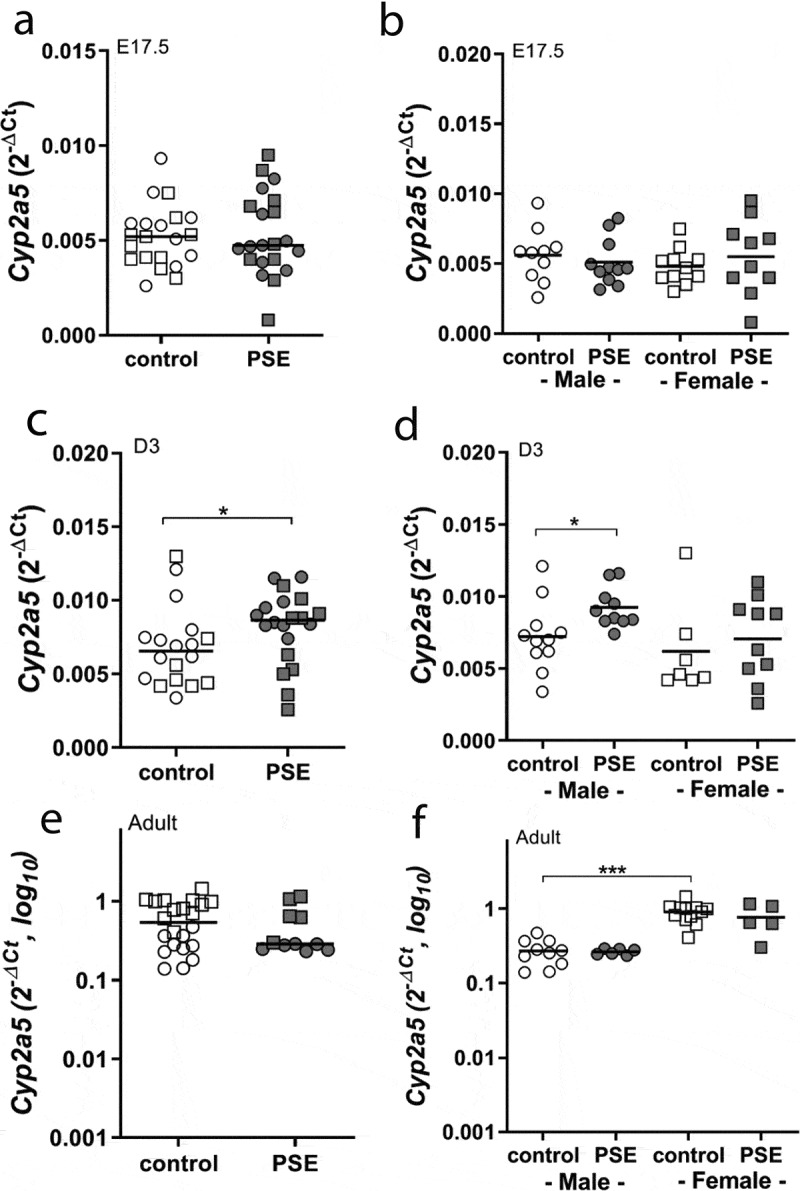
Cyp2a5 expression in liver of PSE and control in E17.5, D3 mice and adult offspring
Cyp2a5 mRNA expression in whole liver of foetal offspring (E17.5, a and b), neonatal offspring (D3, c and d) and adult offspring (e and f) in PSE (closed symbols) and control groups (open symbols). Data are shown as individual values. If not stated otherwise, the comparison of shown groups was not significant. (Mann–Whitney U-test). *p ≤ 0.05, *** p ≤ 0.001 (Mann–Whitney U-test). Circle (○) symbol(s) = male, square (□) symbol(s) = female.
To investigate whether the observed PSE-induced mRNA levels were associated with Cyp2a5 promoter methylation, the percentage of methylation of six CpG sites (CpG-614, CpG-589, CpG-542, CpG-74, CpG-7, and CpG+45) was measured using bisulphite pyrosequencing.
In foetal livers, PSE-induced Cyp2a5 hypermethylation, both in male and female offspring at CpG-614 (male: p = 0.0009, fold-change = 1.06, female: p = 0.001, fold-change = 1.05; Figure 2(a)), CpG-589 (male: p = 0.0005, fold-change = 1.06, female: p = 0.003, fold-change = 1.08; Figure 2(a)), and CpG-542 (male: p = 0.001, fold-change = 1.1, female: p = 0.004, fold-change = 1.1; Figure 2(a)). PSE-induced hypermethylation was found in male foetuses at CpG-74 (p = 0.01, fold-change = 1.1, Figure 2(a)), but not in females. In contrast, in females, PSE-induced hypermethylation at CpG-7 (p = 0.04, fold-change = 1.12, Figure 2(a)) and CpG+45 (p = 0.05, fold-change = 1.17, Figure 2(a)). In neonatal livers, PSE-induced Cyp2a5 hypermethylation was detected in both male and female mice at CpG-74 (male: p = 0.01, fold-change = 1.1, female: p = 0.03, fold-change = 1.07; Figure 2(b)). In male neonates, PSE-induced hypermethylation was found at CpG-614 (p = 0.05, fold-change = 1.03, Figure 2(b)) and CpG-542 (p = 0.04, fold-change = 1.05, Figure 2(b)). In females, PSE-induced hypermethylation was found at CpG+45 (p = 0.04, fold-change = 1.17, Figure 2(b)). All other analysed CpG sites of the Cyp2a5 promoter were not found to be influenced by PSE (Figure 2(b)). In adult livers, higher methylation of CpG-74 was detected in PSE male offspring (p = 0.03, fold-change = 1.21, Figure 2(c)), while no PSE effect was observed in all other analysed CpG sites, both in male and female offspring (Figure 2(c)). In addition, we investigated whether these differentially methylated CpG sites were mediated by DNA methyltransferase (DNMT) enzymes. Dnmt1 expression was lower in PSE male foetal offspring, compared to the control group (Supplementary Figure 4(a,b)), while no differences were detected in the neonates and adult offspring (Supplementary Figure 4(c-f)). Expression of Dnmt3a mRNA and Dnmt3b mRNA was not affected by PSE (Supplementary Figures 5(a-f) and Figure 6(a-f)).
Figure 2.
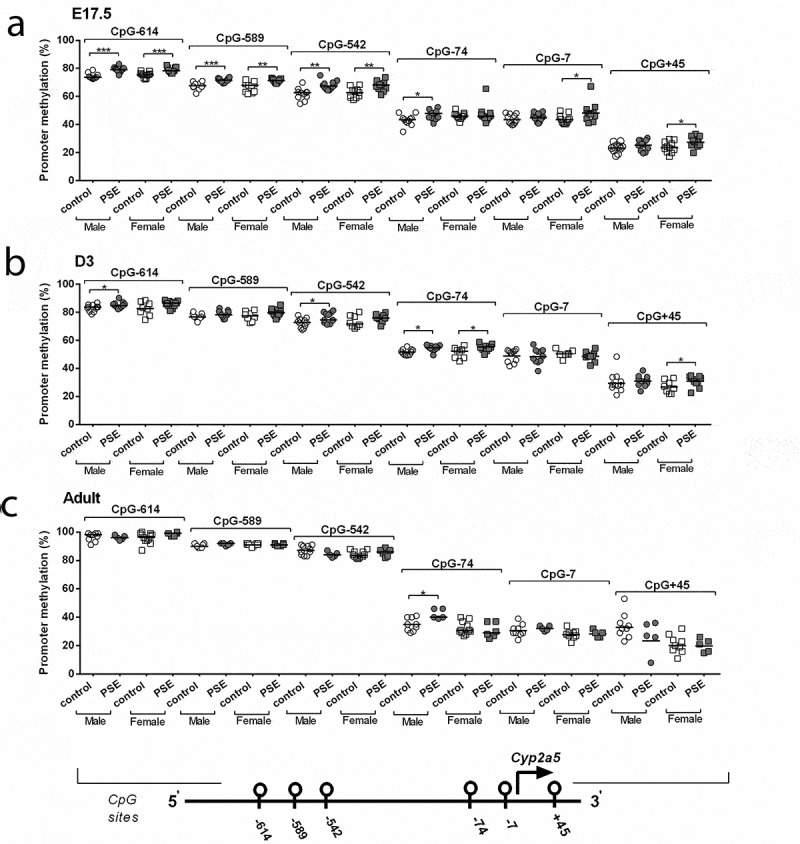
Sex-dependent Cyp2a5 promoter methylation across three time points (E17.5, D3 mice and adult offspring). DNA was isolated from whole liver of foetuses (E17.5, a), neonatal offspring (D3, b) and adult offspring (c) in PSE (closed symbols) and control groups (open symbols). DNA was subjected to bisulphite sequencing-based methylation analysis of the Cyp2a5 promoter region and the percentage of DNA methylation was assessed. Data of the 6 targeted CpG-sites are presented per sex and exposure as individual values with median as a horizontal line. CpG-site annotations are relative to the ATG start codon. If not stated otherwise, the comparison of shown groups was not significant. *p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 (Mann-Whitney U-test). Circle (○) symbol(s) = male, square (□) symbol(s) = female
Linking Cyp2a5 mRNA levels with methylation across three time points, in PSE male neonates, Cyp2a5 mRNA content was positively correlated with methylation at CpG+45 site (r = 0.77, p = 0.009), as shown in Table 2 and Supplementary Figure 1(c). This correlation was not seen when PSE and control male neonates were taken together (Table 2, Supplementary Figure 1(a)), or within male neonate controls only (Table 2, Supplementary Figure 1(b)). For CpG-614, methylation in all male neonatal livers correlated positively with Cyp2a5 mRNA expression (r = 0.48, p = 0.027, Table 2), while no correlations were found in PSE female neonates. In PSE adult offspring, methylation levels of CpG-614 were positively correlated with Cyp2a5 expression (r = 0.83, p = 0.003, Table 2), while no correlations were observed in combined and control adult offspring. Furthermore, negative correlations were observed for CpG-74 (r = −0.81, p = 0.004, Table 2) and CpG-7 (r = −0.84, p = 0.004 Table 2) in PSE adult offspring, while no additional correlations were detected in both adult offspring groups. In foetuses, positive correlations were found at CpG-614 (r = 0.65, p = 0.029, Table 2) and CpG-542 in control female foetal livers (r = 0.7, p = 0.016, Table 2) while no correlation was found in PSE foetal livers.
Table 1.
Sequences of primers used in bisulphite-based methylation analysis
| Gene | Targeted CpG-sites | Sequence 5ʹ-3’ |
|---|---|---|
| Cyp2a5 | CpG-614 to −542 | For: TTTGTGTTTGTTTTGAGTGTTGGGATTA Rev: CCCCATCCACAACCATTCTT Seq: GTTGGGATTATAGGTTTATATTA Sequence to analyse: TTATATTYGATTTTTGGGAGTTTTTTAATGAAGAGGATTTTGAATTTAAGGATGYGAGAA GTGGAGATTT TAGGGTTATYGG |
| CpG-74 to +45 | For: AGTGGATAGTTTGGAGGTGAAAT Rev: ACAACTTTCCTAAAAACTTTCTCTACTTC Seq1: GGAGGTGAAATAGTTGTATAATTAA Seq2: TAGTTATTATTGTTTGTTTATTAT Sequence to analyse: S1: GATTAAAGTTYGTTTTTTTGTTTTTGGATGTATAAAAGTAAGTTAATT S2: TTATYGTTATTATGTTGATTTTAGGATTTTTTTTGGTGGTTGTAGTGGTTTTTTTTAGYGTTTTGGTTTT |
Table 2.
Correlations between Cyp2a5 mRNA expression and promoter methylation in foetal (E17.5), neonatal (D3) and adult offspring liver
| E17.5 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| correlation of / with | Cyp2a5 (2-dCT) | all | all male | all female | all control | all PSE | male control | male PSE | female control | female PSE | |
| Cyp2a5 promoter methylation [%] | CpG-614 | r | -0.02 | -0.23 | 0.17 | 0.17 | -0.29 | -0.23 | -0.45 | 0.65 | -0.13 |
| p-value | ns | ns | ns | ns | ns | ns | ns | 0,02* | ns | ||
| CpG-589 | r | 0.23 | 0.04 | 0.38 | 0.46 | 0,19 | 0.32 | -0.1 | 0,57 | 0.47 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-542 | r | 0.11 | -0.38 | 0.41 | 0.26 | 0.1 | -0.56 | -0.38 | 0.70 | 0.23 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | 0,01* | ns | ||
| CpG-74 | r | 0.21 | 0.19 | 0.25 | 0.35 | 0.05 | -0.05 | 0.13 | 0.48 | 0.06 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-7 | r | 0.02 | -0.04 | 0.01 | 0.19 | -0.12 | 0.07 | -0.15 | 0.40 | -0.14 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG+45 | r | 0.25 | 0.1 | 0.38 | 0.24 | 0.20 | -0.25 | 0.16 | 0.44 | 0.29 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| D3 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| correlation of / with | Cyp2a5 (2-dCT) | all | all male | all female | all control | all PSE | male control | male PSE | female control | female PSE | |
| Cyp2a5 promoter methylation [%] | CpG-614 | r | 0,29 | 0.48 | 0.16 | 0.26 | 0.05 | 0.08 | 0.13 | 0.07 | 0.04 |
| p-value | ns | 0.02* | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-589 | r | 0.28 | 0.38 | 0.27 | 0.21 | 0.15 | 0.12 | 0.31 | 0.04 | 0.26 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-542 | r | 0.24 | 0.33 | 0.23 | 0.48 | 0.12 | 0.52 | 0.33 | 0.32 | 0.08 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-74 | r | 0.35 | 0.17 | 0.02 | 0.37 | 0.24 | -0.19 | 0.50 | -0.39 | -0.02 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-7 | r | 0.12 | 0.17 | 0.00 | 0.03 | 0.37 | 0.23 | 0.51 | -0.61 | 0.47 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG+45 | r | 0.27 | 0.4 | -0.04 | 0.29 | 0.23 | 0.09 | 0.77 | 0.11 | -0.27 | |
| p-value | ns | ns | ns | ns | ns | ns | 0,009** | ns | ns | ||
| Adult |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| correlation of /with | Cyp2a5 (2-dCT) | all | all male | all female | all control | all PSE | male control | male PSE | female control | female PSE | |
| Cyp2a5 promoter methylation [%] | CpG-614 | r | 0.22 | 0.12 | 0.21 | 0.02 | 0.83 | 0.18 | 0.2 | 0.15 | -0.81 |
| p-value | ns | ns | ns | ns | 0.003** | ns | ns | ns | ns | ||
| CpG-589 | r | 0.24 | 0.17 | 0.33 | 0.38 | -0.11 | 0.24 | -0.12 | 0.29 | 0.31 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-542 | r | -0.18 | 0.1 | 0.01 | -0.36 | 0.22 | 0.22 | -0.60 | 0.1 | -0.31 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-74 | r | -0.41 | 0.43 | -0.11 | -0.17 | -0.82 | 0.62 | 0.15 | 0.41 | -0.6 | |
| p-value | ns | ns | ns | ns | 0.004** | ns | ns | ns | ns | ||
| CpG-7 | r | -0.42 | 0.03 | -0.47 | -0.32 | -0.85 | 0.11 | -0.66 | -0.64 | 0.61 | |
| p-value | ns | ns | ns | ns | 0.004** | ns | ns | ns | ns | ||
| CpG+45 | r | -0.44 | 0.12 | 0.11 | -0.57 | -0.57 | 0.1 | -0.1 | -0.28 | 0.4 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
*p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001. P-values were adjusted by the Bonferroni method.
Our next question addresses the potential persistence of DNA methylation levels across three developmental stages, foetal, neonatal, and adult. As shown in Figure 3, all of CpG sites showed a variable pattern at the three different stages (p-values<0.0001, Figure 3). For CpG-614, CpG-589 and CpG-542 sites, basal levels of DNA methylation increased from foetal to adulthood (p-values<0.0001, Figure 3(a-c)) in both sexes, while it was not affected by PSE (Figure 3(a-c)). In control male and female offspring, methylation patterns of CpG-74 and CpG-7 increased from foetal to neonatal stage but reversed to hypomethylated in adulthood (p-values<0.0001, Figure 3(d and e)). Interestingly, PSE effects were detected for CpG-74 across three developmental stages in male offspring, (p-values<0.05, Figure 3(d)), whereas at CpG-74, the PSE effect on methylation disappeared in female foetus and adults (Figure 3(d)). For CpG+45, hypermethylation was observed at the neonatal stage when compared to neonates across foetal to neonatal stage (p < 0.01, Figure 3f), which was only observed in male offspring of both the control and PSE groups (Figure 3(f)). In female controls, methylation at CpG+45 remained stable across three time points, while PSE increased methylation at both the foetal and neonatal stages (p-values<0.05, figure 3f).
Figure 3.
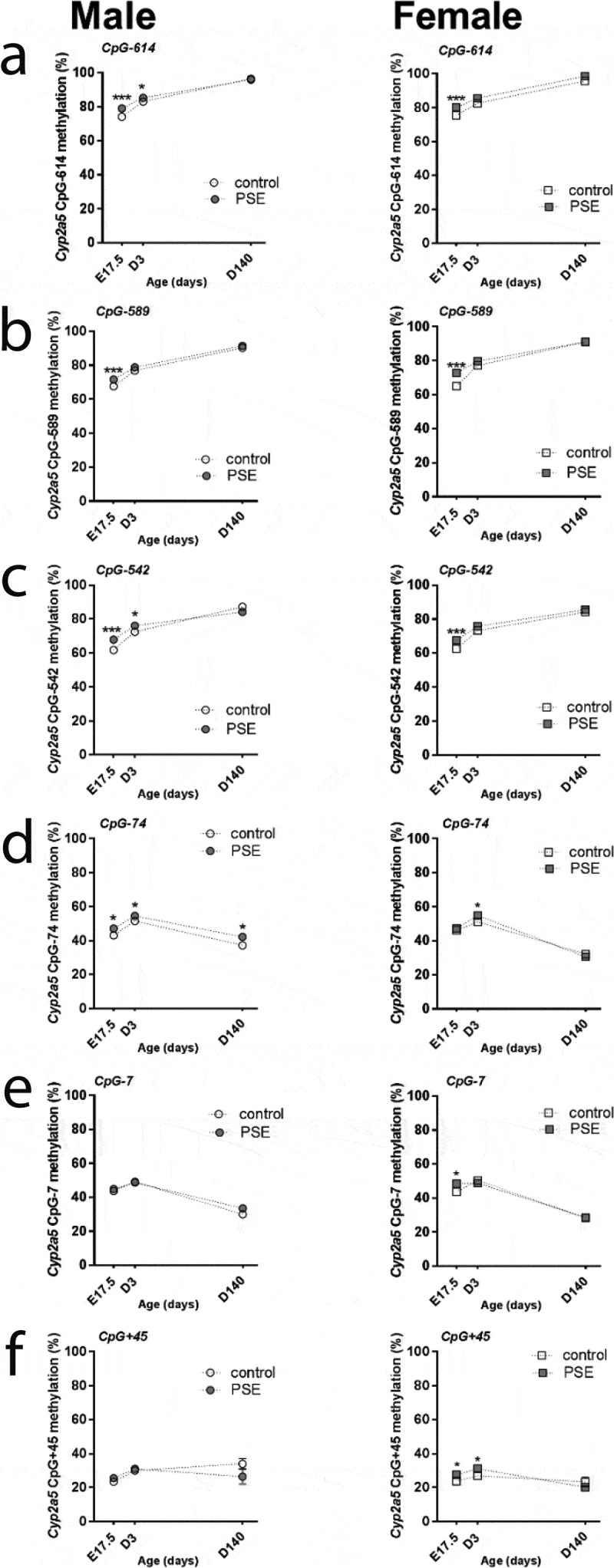
Time points comparisons of sex-dependent Cyp2a5 promoter methylation status in PSE and control groups. The ANOVA test was used to do the comparison analysis among the foetal stage (E17.5), neonatal period (D3 means three days after birth) and adulthood (D140 means 140 days later after birth). P-values<0.0001 were detected by ANOVA test in all of CpG sites over three time points. Mann–Whitney U-test was used to test the comparisons between two time points in control (open symbols) and PSE (closed symbols) groups for 6 promoter methylation CpG sites. Data are represented as mean± SEM; CpG-site annotations are relative to the ATG start codon. *p ≤ 0.05, *** p ≤ 0.001 (Mann-Whitney U-test). Circle (○) symbol(s) = male, square (□) symbol(s) = female
Prenatal smoke exposure increased the nicotine metabolism in neonatal and adult male offspring
Our final question aimed towards the understanding of whether altered Cyp2a5 mRNA levels, as observed in PSE neonatal offspring, was accompanied by an aberrant nicotine metabolism. This research question is based on the understanding that the enzyme CYP2A5 is responsible for conversion of nicotine after smoke exposure. Conversion of the in vitro nicotine metabolism to cotinine was measured by HPLC-L and MS, as shown in Figure 4(c and d).
Figure 4.
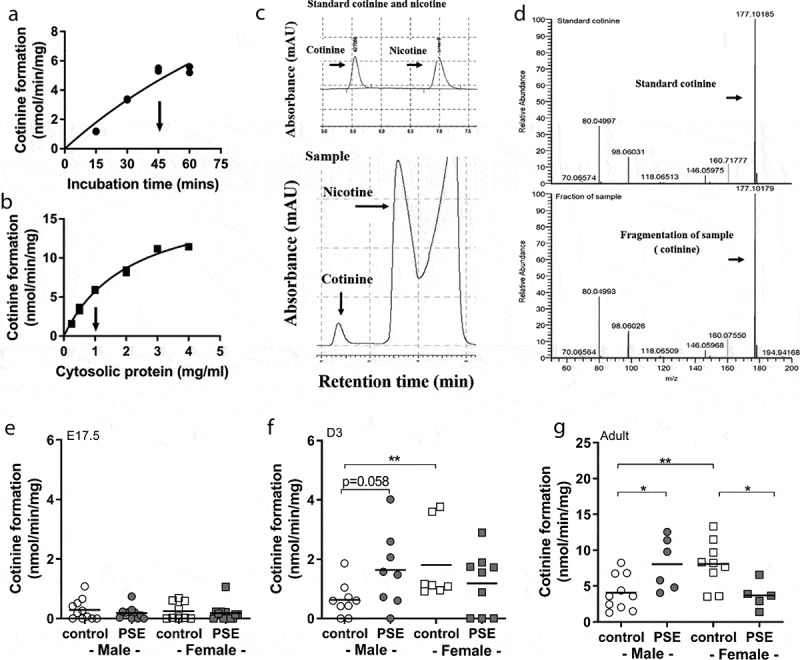
Optimization and the cotinine formation levels of in vitro nicotine metabolism in E17.5, D3 mice and adult offspring. Cotinine formation of nicotine metabolism of foetuses (E17.5), three-day-old offspring (D3) and adult offspring in PSE and control groups. The nicotine metabolism was measured by HPLC-L as described in the material and methods section. The assay was optimized with respect to incubation time (a) and cytosolic protein concentration (b). Arrows at the X-axis indicate conditions used for actual experiments. C presents the HPLC chromatogram separation of a blank sample containing the standards nicotine and cotinine and the cotinine was produced in liver microsomes of PSE male neonatal offspring. D presents the cotinine analysis by LCMS. Cotinine, corresponding with m/z 177 was detected both in standard cotinine and in the microsome sample. The levels of cotinine formation were measured in foetal (e), three-day-old offspring (f) and adult offspring (g) of both groups. Circle (○) symbol(s) = male, square (□) symbol(s) = female. Data are shown as individual values. If not stated otherwise, the difference between groups was not significant. *p ≤ 0.05, ** p ≤ 0.01 (Mann-Whitney U-test). Open symbol(s) = control group, closed symbol(s) = PSE
Interestingly, PSE-induced cotinine formation to higher levels in livers of male neonates when compared to controls (p = 0.058; Figure 4(f)). In female neonates, this PSE effect was not seen. This PSE-induced cotinine formation was even more pronounced in PSE male adult offspring (p = 0.03; Figure 4(g)) when compared to the male adult controls. In contrast, cotinine formation was decreased in PSE female offspring, when compared to female adult controls (p = 0.03; Figure 4(g)).
A comparison of basal levels of cotinine formation in male and female controls showed that female neonates and adults had a higher nicotine metabolism than their male counter parts (neonates: p = 0.007; adults: p < 0.0001; Figure 4(f and g)). In foetuses, the nicotine metabolism was not affected by PSE (Figure 4(e)), nor were there differences in control levels of cotinine formation in males compared to females.
In foetal livers, the level of cotinine formation correlated positively to Cyp2a5 expression when data of all foetuses were plotted together (r = 0.45, p = 0.003; Supplementary Figure 2(a), Table 3). This correlation was mainly driven by the control female foetuses (r = 0.78, p = 0.01; Supplementary Figure 2(c), Table 3), as no correlation was detected in PSE groups. Furthermore, a lower correlation was also seen in control when male and female foetuses were combined (r = 0.57, p = 0.01; Supplementary Figure 2(b), Table 3). In male neonates, PSE-induced cotinine formation correlated positively to Cyp2a5 expression (r = 0.81, p = 0.01; Supplementary Figure 2(e), Table 3), whereas no significant correlation was observed when data from all male neonates were combined (r = 0.53, p = ns; Supplementary Figure 2(d), Table 3). In control adult offspring, the level of the cotinine formation positively correlated with Cyp2a5 expression (r = 0.67, p = 0.002; Figure 2(g), Table 3), while no correlation was detected when data from PSE and control mice were combined, as shown in Supplementary figure 2(f).
Table 3.
Correlations between cotinine levels, Cyp2a5 mRNA expression and promoter methylation in foetal (E17.5), neonatal (D3) and adult offspring liver
| E17.5 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| correlation of / with | Cotinine formation (nmol/min/mg) | all | all male | all female | all control | all PSE | male control | male PSE | female control | female PSE | |
| Cyp2a5 (2-dCT) | r | 0.45 | 0.47 | 0.41 | 0.57 | 0.33 | 0.41 | 0.46 | 0.78 | 0.20 | |
| p-value | 0.00** | 0.03* | ns | 0.01** | ns | ns | ns | 0.01** | ns | ||
| Cyp2a5 promoter methylation [%] | CpG-614 | r | -0.15 | -0,21 | -0.14 | 0.30 | -0.07 | -0.41 | 0.19 | 0.44 | -0.27 |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-589 | r | -0.59 | 0.01 | -0.10 | 0.30 | -0.05 | 0.07 | 0.28 | 0.65 | -0.35 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-542 | r | 0.15 | 0.17 | 0.03 | 0.36 | -0.09 | 0.55 | -0.18 | 0.87 | -0.17 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | 0.00*** | ns | ||
| CpG-74 | r | 0.21 | 0.31 | 0.10 | 0.12 | 0.26 | 0.36 | 0.30 | -0.03 | 0.06 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-7 | r | 0.14 | 0.26 | 0.01 | 0.54 | -0.19 | 0.42 | 0.08 | 0.75 | -0.38 | |
| p-value | ns | ns | ns | 0.02* | ns | ns | ns | ns | ns | ||
| CpG+45 | r | 0.31 | 0.34 | 0.28 | 0.4 | 0.37 | 0.19 | 0.57 | 0.57 | 0.21 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| D3 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| correlation of / with | Cotinine formation (nmol/min/mg) | all | all male | all female | all control | all PSE | male control | male PSE | female control | female PSE | |
| Cyp2a5 (2-dCT) | r | 0.16 | 0.53 | -0.12 | 0.01 | 0.21 | 0.21 | 0.81 | 0.14 | -0,18 | |
| p-value | ns | ns | ns | ns | ns | ns | 0.01** | ns | ns | ||
| Cyp2a5 promoter methylation [%] | CpG-614 | r | -0.10 | 0,46 | -0.63 | 0.14 | -0.30 | 0.59 | 0.21 | -0.43 | -0.79 |
| p-value | ns | ns | 0.00*** | ns | ns | ns | ns | ns | 0.01** | ||
| CpG-589 | r | -0.14 | 0.48 | -0.63 | 0.01 | -0.28 | 0.69 | 0.20 | -0.61 | -0.59 | |
| p-value | ns | ns | 0.00*** | ns | ns | ns | ns | ns | ns | ||
| CpG-542 | r | -0.26 | -0.38 | -0.42 | -0.18 | -0.26 | 0.20 | -0.28 | -0.54 | -0.17 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-74 | r | -0.29 | -0.01 | -0.29 | -0.57 | -0.12 | -0.03 | -0.28 | -0.36 | 0.18 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-7 | r | 0,47 | 0,70 | 0.30 | 0.38 | 0.29 | 0.46 | 0.83 | 0.32 | 0.18 | |
| p-value | ns | 0.00*** | ns | ns | ns | ns | 0.00*** | ns | ns | ||
| CpG+45 | r | 0.19 | 0,77 | -0.52 | 0.12 | 0.21 | 0.80 | 0.86 | -0.64 | -0.44 | |
| p-value | ns | 0.00*** | ns | ns | ns | ns | 0.00*** | ns | ns | ||
| Adult |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| correlation of / with | Cotinine formation (nmol/min/mg) | all | all male | all female | all control | all PSE | male control | male PSE | female control | female PSE | |
| Cyp2a5 (2-dCT) | r | 0.23 | 0.26 | 0.21 | 0.67 | -0.67 | 0.21 | -0.37 | 0.47 | -0.41 | |
| p-value | ns | ns | ns | 0.00** | ns | ns | ns | ns | ns | ||
| Cyp2a5 promoter methylation [%] | CpG-614 | r | -0.24 | -0.42 | -0.12 | 0.02 | -0.75 | -0.50 | 0.30 | 0.42 | -0.89 |
| p-value | ns | ns | ns | ns | 0.01** | ns | ns | ns | ns | ||
| CpG-589 | r | 0.04 | 0.05 | -0.09 | 0.24 | -0.35 | -0.15 | -0.78 | 0.07 | -0.53 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-542 | r | -0.37 | -0.37 | -0.21 | -0.33 | -0.51 | -0.16 | 0.00 | 0.28 | -0.90 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG-74 | r | -0.19 | -0.18 | 0.64 | -0.28 | 0.73 | -0.17 | -0.52 | 0.48 | 0.90 | |
| p-value | ns | ns | 0.01** | ns | 0.01** | ns | ns | ns | ns | ||
| CpG-7 | r | 0.017 | 0.21 | 0.01 | -0.17 | 0.55 | 0.19 | -0.41 | -0.22 | 0.89 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG+45 | r | -0.03 | 0.23 | 0.21 | -0.15 | 0.31 | 0.65 | -0.20 | 0.03 | 0.80 | |
| p-value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
*p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001. P-values were adjusted by the Bonferroni method.
Levels of DNA methylation versus levels of cotinine formation shows that, in all cases, strong correlations were found in both male PSE neonatal and adult mice. In PSE neonatal males, levels of cotinine formation correlated strongly with CpG+45 and CpG-7 methylation (r = 0.86, p = 0.008 and r = 0.83, p ≤ 0.01; Table 3), whereas it strongly negatively correlated with CpG-614 methylation in PSE female neonates (r = −0.79, p = 0.01; Table 3). In contrast, methylation of CpG-7 (r = 0.70, p ≤ 0.01; Table 3) and CpG+45 (r = 0.77, p ≤ 0.001; Table 3) correlated with cotinine levels in all male neonatal groups, while CpG-614 (r = −0.63, p = 0.009; Table 3) and CpG-589 (r = −0.63, p = 0.009, Table 3) negatively correlated with cotinine levels in all female neonates. In adult offspring, a majority of correlations of cotinine formation levels with DNA methylation levels were detected in PSE adult offspring. Promoter methylation at CpG-614 was negatively correlated with cotinine levels in PSE adult offspring (r = −75, p = 0.01, Table 3). In contrast, for CpG-74, promoter methylation correlated positively with cotinine levels (r = −0.75, p = 0.01, Table 3), while no other correlations were observed in PSE adult offspring (Table 3).
Comparison of gene expression, methylation profiles and cotinine formation of nicotine metabolism
Comparisons of the Cyp2a5 mRNA expression, promoter methylation levels, and the nicotine metabolism across three time points in control offspring show that mRNA levels of Cyp2a5 and the level of cotinine formation were much higher in adult offspring, when compared to foetal and neonatal offspring, in both sexes (p-values<0.0001, Supplementary Figure 3(a) and (b)). In addition, basal levels of cotinine formation were higher in female neonates (p = 0.0002; Supplementary Figure 3(b)), when compared to female foetuses. This was accompanied with higher methylation levels of all measured CpG sites in both sexes, except for CpG+45 (p-values≤0.01; Supplementary Figure 3(c,d)). In adult offspring, hypermethylation was detected at three CpG sites (p-values<0.0001; CpG-614, CpG-589, and CpG-542), compared to foetuses and neonates (Supplementary Figure 3(c)). Inversely, promoter methylation was lower at two CpG sites (p-values≤0.0001; CpG-74 and CpG-7) in adult offspring, for both sexes, compared to foetuses and neonates, as shown in Supplementary Figure 3(c,d).
Discussion
In this study, we found that PSE increased the nicotine metabolism in male neonatal and adult but not in foetal offspring. This observation correlated with higher Cyp2a5 mRNA levels along with elevated promoter methylation in neonates but not in adults. In addition, while the PSE-induced nicotine metabolism increased from the foetal developmental stage to adulthood, partial to almost complete waning of the PSE-induced Cyp2a5 promoter methylation was observed over time, which was sex dependent. Finally, female neonates and adult mice had a higher hepatic nicotine metabolism than their male counterparts, and PSE had a downregulatory effect on nicotine metabolism in female adults.
Our observation that PSE increased the nicotine metabolism in neonatal and adult offspring could be relevant with respect to human smoking behaviour, as a higher nicotine metabolism is generally associated with heavier smoking, higher dependency to nicotine, and more difficulty to quit cigarette smoking [32,33]. Indeed, Kandel et al. reported aberrant smoking behaviour in children born to smoking mothers, in which PSE daughters had a higher risk to start smoking, and the smoking behaviour was more persistent than in sons in adolescence age [19]. Whether the sex-difference in smoking behaviour of PSE adolescence is based on epigenetically regulation of genes is worth to further investigate in human subjects.
In our mouse study, the PSE effect on nicotine metabolism was also sex-dependent since the effect was only seen in male offspring. This is of interest as in a study by Klein et al. in preadolescent mice, prenatal nicotine exposure induced the preference for drinking nicotine-containing water, an effect that was only seen in males, while female mice appeared to be unresponsive to treatment [25]. In addition, in a study where pregnant rats were treated with nicotine, only male hyperactive offspring exhibited a significant increase in the cortical receptor densities whereas the hyperactive females did not. Evidence suggests that only male offspring of nicotine-exposed dams are also susceptible to neurochemical effects of intrauterine nicotine exposure [34].
In the current study, for the CpG sites that were affected by PSE, methylation levels were increased, which was seen at three time points in both sexes. Promoter methylation is usually associated with repressed gene expression, but recent findings [17,22] suggest that this association could also occur inversely, which is in line with our previous studies [29,30] and studies by others [35]. Interestingly, in three development stages, PSE caused a persistent increase of methylation at CpG-74 when compared to control groups. This confirms human studies [26,36] showing that PSE-induced differentially methylated CpG sites in MYO1 G and CNTNAP2 were persistent at birth, during childhood and during adolescence in the replication cohort. Indeed, the causal link between DNA (de)methylation and gene expression and even further protein activation is complex, given that DNA methylation is only one of the known epigenetic modifications. Other modes include histone modifications, chromatin remodelling, and RNA-based mechanisms (lncRNAs/miRs). All of these modes seem to be interlinked, but their chronological order and the exact mechanism(s) that may connect these modes still need to be described to their full extent.
In this study, the PSE effect was the most striking in male neonates. PSE-induced Cyp2a5 mRNA expression was correlated with both CpG+45 methylation as well as with elevated cotinine levels. As this effect was not seen in females, it indicates that this CpG site may play a regulatory role in sex-dependent regulation of Cyp2a5 expression. Studies have reported sex-dependent methylation patterns of CpG sites in the livers of male and female mice [37,38]. It is known that sex hormones can play a major role in regulating sex-dependent DNA methylation, as described in the mouse model by Takasugi et al. [39]. In that study, male mice, treated with female-like growth hormone showed differential methylation of CpG sites of Cyp17a1, Cxcl11, and Nmt in the liver when compared to non-treated male mice, and this observation was also described in other organs [40]. In addition, Koudsi et al. demonstrated DNA methylation changes in regulating CYP2A6 expression in a pilot study of human livers. In that study, no prominent role for DNA methylation changes was observed with respect to the regulation of CYP2A6 gene expression in human livers [41]. Based on this finding, a lack of association between DNA methylation and CYP2A6 expression suggests that other gene areas, such as the proximal promotor CpG sites (1000–100 bp) in relation to the gene’s transcription start site might be relevant in regulating CYP2A6 expression, which needs to be investigated further. Moreover, in murine hepatocytes, two major regions of Cyp2a5 regulation were observed in the promoter, which are the distal (AHR and USF-1 within 2700–1500bp) and the proximal region (HNF, NF-1and C/EBP within 1000–10bp), as described by Arpiainen et al. [42]. Therefore, Arpianen and colleagues compared the distal promoter region with the proximal promoter region in the transcriptional regulation of Cyp2a5 expression, showing HNF, NF-1and C/EBP transcription factors within the proximal region are the key activators of the Cyp2a5 expression, when compared to the distal region.
Next to this, we assessed mRNA expression of the DNA methyltransferases across three developments stages. There are two different DNA methylation processes: maintenance (DNMT1) and de novo (DNMT3a and DNMT3b). Interestingly, lower Dnmt1 levels were observed in male foetuses only, supporting a study in which it has been shown that Dnmt1 enzyme can be reduced by current cigarette smoke exposure, which downregulated target genes in in vitro and ex vivo studies [43,44]. This result could be considered as the ‘current smoke’ exposure effect, which was present in the foetuses. As neonatal and adult offspring have not been exposed after they were born, this current smoke effect would not apply to these groups.
In our study, we observed that nicotine metabolism increased over time, as adult mice had a higher cotinine formation than neonates and foetuses. In addition, the PSE effect on nicotine metabolism was also observed in neonatal and adult offspring but not in foetal offspring. This result suggests that offspring in the foetal stage had a very low nicotine metabolism, which was not induced by PSE. Indeed, in a human study by Dempsey et al., it was reported that the half-life of nicotine in serum is three to four times longer in newborns than in adults [45], indicating that foetal and newborn liver might have lesser nicotine clearance than adult [46]. In addition, during pregnancy, the metabolic clearance of nicotine is relatively high [47], resulting in foetal nicotine clearance being primarily controlled by the mother’s hepatic blood flow.
With respect to Cyp2a5 methylation, this is the first study to report on methylation of Cyp2a5 over time and after PSE. We found a decline in the number of differentially methylated CpG sites over time after PSE, suggesting that PSE-induced Cyp2a5 methylation is reversible over time. One explanation could be a diverse cellular composition in all three developmental stages, which is a limitation of this study and which was also as reported in previous studies [30,48]. Indeed, in the human study, a similar result was described by Richmond and colleagues when comparing PSE newborns to adolescent offspring [26]. They found that differentially methylated CpG sites in PSE newborns have been identified in AHRR, MYO1 G, GFI1, CNTNAP2, KLF13, CYP1A1 and ATP9A genes, which are known as smoke-related genes. In that study, CpG sites in less than half of the smoking-related genes showed reversibility of the methylation pattern in PSE adolescents when compared to PSE newborns.
With respect to the functionality of the Cyp2a5 gene in foetal mice, no PSE effect was seen on nicotine metabolism, although methylation was higher in four out of six CpG sites, both in male and female PSE groups. The potential of PSE to induce foetal programming has been addressed in multiple studies, but a differential effect in males and females has only occasionally been evaluated by our research group [29,30] and others [49,50]. The present study shows that PSE-induced methylation was observed mostly in foetuses. This result could be due to the ‘current’ smoke effect, as foetuses were exposed to smoke until they were terminated. From the literature, it is known that current smoking has a profound effect on DNA methylation which for a number of genes disappears after smoking cessation [e.g., ref [51].]. Similar effects have been described by us for Igf1 and Igf1 r methylation in the same pups [30]. This ‘current’ smoke exposure effect is not present in the neonates and adults which have not been exposed to smoke after they were born. However, neonates may have been exposed to nicotine via the mothers’ milk as nicotine could have been ingested, when mothers cleaned their fur. The waning of the PSE effect over time could therefore also be explained by the ‘current’ nicotine exposure at the foetal and neonatal stages and the absence of ‘current’ nicotine exposure in adults.
We found that female neonates and female adults had a higher nicotine metabolism, which supports previous studies in humans [52,53], and which suggests that the nicotine and cotinine clearance is higher in women than in men. Indeed, other CYP enzymes may have a role in nicotine metabolism [13], and may explain the differences seen between males and females. For instance, Z. Liu et al. tested the ability of CYP2B6 to metabolize nicotine in the CYP2B6-humanized mouse model [54] showing that the nicotine-induced cotinine formation was increased in males when compared to females. In contrast, a clinical study showed that livers from female donors had a higher level of CYP2B6 mRNA, protein, and enzyme activity compared to male donors [55]. Moreover, CYP2B6 expression is known to be stimulated by oestrogen hormones [56]. However, as the CYP2B6 is a considerably lower activity enzyme in nicotine metabolism when compared to the CYP2A6 enzyme [13], we do not consider a major role for this enzyme in our model. Unfortunately, the molecular mechanism that underlies different CYP2B6 regulation in the mouse is not clear. In addition, in humans, the inherent gender difference in smoking initiation and nicotine addiction was well documented in the epidemiological studies [57,58], showing that women have greater vulnerability for heritability of smoking initiation/persistence compared to males (women 66% vs. man 49%) [59].
The nicotine metabolism is also higher in pregnant women than in non-pregnant women [47,60], suggesting a dose-dependent effect of the sex hormones oestrogen and progesterone on CYP2A6 regulation. Indeed, Higashi et al. showed that CYP2A6 mRNA was induced by oestradiol in human hepatocytes after specific binding of the oestrogen receptor to the putative oestrogen response element (ERE) on the CYP2A6 gene [61]. The sex hormone may also have a direct effect on the nicotine metabolism in rodents. In a rat experiment, ovariectomized animals showed a lower behaviour motivation effect of nicotine intake than controls [62]. Moreover, in another study, higher plasma nicotine levels were measured in intact female rats compared to male rats, and the sex difference was attenuated in ovariectomized animals [63]. Although we expected otherwise, a lower nicotine metabolism was observed in PSE female adult offspring, compared to controls. This might have resulted from PSE-induced ovarian steroidogenesis [64,65]. Indeed, studies in rats have demonstrated that in utero exposure to cigarette smoke can result in decreased oestrogen hormone [66] in female offspring, explaining a different effect of PSE in females.
There were limitations to the study. Throughout three development stages, the cellular compositions within different liver zones may be a confounding factor. Therefore, it would be interesting to assess DNA methylation patterns and nicotine metabolism in different cell compounds within a specific liver zone in future studies. In addition, foetal and neonatal offspring only provided us with limited material to do all analyses within one mouse. For the in vitro nicotine metabolism experiment, firstly, we optimized the required ratio of enzymes on higher metabolic active specimens. Selected optimized ratios were used in the actual in vitro nicotine metabolism experiment.
Conclusion: In this study, we have shown for the first time that male neonatal and adult offspring from mothers that smoked during pregnancy have a higher nicotine metabolism. In neonates, this was accompanied by a higher Cyp2a5 gene expression, which was correlated with higher DNA methylation. As studies have shown a positive relation between higher nicotine metabolism and increased nicotine dependency, this could imply that in human subjects, male offspring from smoking mothers have a higher chance of developing tobacco addiction when they start smoking.
Materials and methods
Animals & smoke exposure
C57BL/6 J mice (48 females and 48 males) were obtained from Harlan (Horst, The Netherlands) at 6 weeks of age and housed under standard conditions, with food and water provided ad libitum and at a 12-h light/dark cycle. The experimental setup was approved by the local committee on animal experimentation (DEC6589 B & C; University of Groningen, Groningen, The Netherlands) and governmental and international guidelines on animal experimentation.
Mainstream cigarette smoke was generated by a Teague10 smoking machine (Tobacco and Health Research Institute of the University of Kentucky, Lexington, KY, USA). Over a period of 7 days, 20 randomly selected primiparous female mice were adjusted to cigarette smoke by stepwise increasing the number of smoked cigarettes (3R4 cigarettes; 2.45 mg nicotine/cigarette) from two to five per smoking session. On adjustment day 5, after the end of the smoking session, all female mice were injected with pregnant mare’s serum gonadotropin (PMSG, 1.25 i.u.) to stimulate ovulation, and on day 7 with human chorionic gonadotropin (hCG, 1.25 i.u.) to induce ovulation. Subsequently, female mice were housed overnight at a 1:1 mating ratio with males. Mating was confirmed by the presence of a vaginal plug the following morning.
Throughout gestation, female mice were housed in groups and exposed to air or whole-body smoking sessions twice a day, 7 days per week. After delivery, dams and their offspring were no longer exposed to cigarette smoke. Dams were separately housed, with their offspring.
A total of 48 foetuses (24 males and 24 females) from 5 smoke-exposed dams and 4 control dams were collected at embryonic stage 17.5 (E17.5) and dams were euthanized under anaesthesia. A total of 42 pups at postnatal day 3 (D3) were randomly selected from prenatally smoke exposed (11 male, 8 female) and control (11 male, 12 female) groups for collection of the liver. Another 34 pups were randomly selected from 20 smoke-exposed (6 male, 6 female) and 19 control (10 male, 12 female) mice that were exposed to air in the Teague10 from 8 weeks of age for the following 12 weeks (adulthood) until euthanized for collection of the liver. The livers were immediately frozen in liquid nitrogen and stored at −80°C until further use.
Genetic determination of mouse sex
DNA from the tail of E17.5, D3 and adult mice was obtained by SDS denaturation, high salt extraction, and precipitation. DNA was amplified as described by Lambert et al. [67], using two primer sets which are specific for the genes Sry (forward: 5ʹ-TGGTCCC GTGGTGAGAGGC-’3, reverse: 5ʹ-TATGTGATGGCATGTGGGTTCC-’3) and IL3 (forward: 5ʹ-GGGACTCCAAGCTTCAATCA-’3, reverse: 5ʹ-TGGAGGAGGAAGAAAA GCAA-’3).
Isolation of DNA and RNA
DNA and RNA were isolated using the All Prep DNA/RNA Mini Kit (Qiagen, Cat No. 80,204), according to the manufacturer’s protocol.
mRNA expression and Pyrosequencing-based bisulphite PCR analysis
Gene expression analysis in whole liver mRNA isolates was done via qPCR using qPCR MasterMix Plus (Eurogentec, Seraing, Belgium) with commercially available primers for Cyp2a4/5 (Mm00487248_m1, TaqMan® Gene Expression Assay, Applied Biosystems, Foster City, CA, USA). Primers and probes for Dnmt1, Dnmt3a and Dnmt3b were obtained from Invitrogen (Breda, The Netherlands), which are described in details elsewhere [68]. Detection of amplification reactions was performed using the LightCycler® 480 System (Roche Diagnostics GmbH, Mannheim, Germany) with cycling conditions as follows: for 2 min at 50°C, for 10 min at 90°C, for 15 s at 40 cycles of 95°C and for 1 min at 60°C. Reactions were performed in triplicate for each sample with Gapdh (Mm99999915_m1, TaqMan® Gene Expression Assay, Applied Biosystems, Foster City, CA, USA) as a reference gene.
DNA in liver tissue of foetal, neonatal, and adult mice was isolated and prepared for bisulphite-based methylation analysis. For the assessment of promoter methylation levels of Cyp2a5, bisulphite sequencing primers were designed using PyroMark assay design software (version 2.0, Qiagen). Selection of CpG-sites was based on manual identification of CpG-dinucleotides, using ENSEMBL genome web browser (Ensembl 83: Dec 2015) and transcript location for the identification of gene promoter regions. Extracted genomic DNA was converted with sodium bisulphite (EZ DNA methylation Direct™, Zymo Research, Irvine, CA), following the manufacturer’s instructions. In short, the bisulphite conversion was carried out in the dark at 98°C for 10 minutes and 64°C for 3.5 hours, followed by desulphonation of the converted DNA. Gene amplification was done using HotStarTaq® MasterMix (Kit Qiagen, Venlo, The Netherlands). CpG-sites were identified manually in the 600bp promoter region of the mouse Cyp2a5 gene (ENSMUSG00000005547), and assessment of DNA methylation levels was performed on the PyroMarkQ24 (Qiagen) instrument. Relative levels of methylation at each CpG-site were analysed with PyroMark Q24 2.0.6 software. The amplification and sequencing primers used in this study are listed in Supplementary Table 1.
Microsomal membrane preparation
Microsomal membranes were obtained from homogenized livers of embryonic, neonatal, and adult mice for nicotine functional assay, as previously described [1,69]. Briefly, the homogenized liver tissue was centrifuged at 9000 g for 20 minutes at 4°C in phosphate buffer with EDTA. The supernatant fractions were collected and centrifuged at 100,000 g for 90 minutes at 4°C. The resultant pellets (microsomes) were collected and resuspended in phosphate buffer without EDTA. The collected supernatants (cytosolic fractions) were pooled for further use as source of aldehyde oxidase reaction in nicotine functional analysis. The content of microsomal protein was determined in each sample with an RC DC protein assay (BIO-rad kit RC DC protein Assay).
In vitro assay for nicotine metabolism
The metabolism of nicotine follows two steps, the first of which is the conversion of nicotine to nicotine- Δ1ʹ(5ʹ)-iminium ion by Cyp2a5 in the liver. Thereafter, the nicotine- Δ1ʹ(5ʹ)-iminium ion is converted to cotinine by aldehyde oxidase, which is present in cytosolic fraction. The liver microsomal protein was incubated with the pooled cytosolic fraction and nicotine-containing solution. To ensure that the nicotine conversion was linear with time, and aldehyde oxidase availability was not rate-limiting, 45 minutes with incubation time (optimization: 15, 30, 45, and 60 min) and 1 mg/ml with concentration of cytosolic protein (optimization: 0.25, 0.5, 1, 2, 3, 4, 6, 8 and 10 mg/ml) [69] were used, as shown in Figure 4(a,b). Briefly, a normalized 0.5 mg/ml liver microsomal protein and 80 µl cytosolic fraction with a protein content of 1.0 mg/ml were added to a freshly prepared nicotine-containing solution 80 µl, which is a mix of 4.4 µl S(-)-nicotine (≥99%, N3876, Sigma-Aldrich, St.Louis, MO), 1.5 ml demineralized water and 28.5 ml of 100 mM sodium phosphate buffer pH 7.4. The mixture was supplemented with phosphate buffer (pH 7.4) to a total volume of 220 µl. After preheating this mixture at 37°C for 5 minutes, 180 µl of freshly prepared NADPH in phosphate buffer containing 12.3 mM magnesium chloride (Sigma-Aldrich, St.Louis, MO), 55.8 mM potassium chloride, 1.17 mM D-Glucose-6-phosphate (≥98%, G7250, Sigma-Aldrich, St.Louis, MO) and 5.2 µl glucose-6-phosphate dehydrogenase (Roche, Germany; Grade II from yeast) were added to the mixture for supplying the energy to recycle the CYP’s active site after oxidation. This mixture (400 µl in total) was incubated at 37°C for 45 minutes in an incubator for the actual biotransformation.
Analysis for HPLC and LCMS
For the analysis of the biotransformation products of nicotine metabolism, the samples were prepared as previously described by E.S. Messina et al. [1] and H. Raunio et al. [2]. Briefly, the samples were alkalized by adding 20 µl of 10 M sodium hydroxide and mixing for 2–3 seconds on a vortex, to stop the reaction after the biotransformation assay, and to deprotonate the analytes. From this, the uncharged analytes were extracted in a two-step extraction. Each extraction step consisted of adding 1 ml of dichloromethane, mixing thoroughly for 60 seconds on a vortex, spinning for 3 minutes at 10,000 g to separate the layers, after which the organic (lower) phase was collected. 20 µl of 2.5 M hydrochloric acid was added to the combined dichloromethane fractions, as suggested by A.M. Massadeh et al. [70]. Before evaporating the solvent under a nitrogen atmosphere overnight at room temperature until dryness. Dried samples were reconstituted in 100 µl of HPLC buffer, which is a mix of 15% acetonitrile and 85% of 20 mM phosphate buffer, pH 3.0, containing 1,0 g/L heptane sulphonate sodium salt acid. The sample was analysed by reverse phase HPLC with UV detection (245 nm), as shown in Figure 4(c). The separation was performed on HPLC-L, a Hitachi system consisting of a L2130 pump (set at isocratic elution of 0,6 ml/min) and a L2300 column oven set at 25°C. To confirm the identity of the peak in the chromatogram a fragment of the cotinine fraction from HPLC system was sent to Interfaculty Mass Spectrometry Centre (IMSC) at the University of Groningen. The sample was dried again under a nitrogen atmosphere and reconstituted in eluent (water and acetonitrile both containing formic acid) for the liquid chromatography separation. The sample was injected into LCMS system, which was measured in positive mode from m/z 100–1000 and parallel reaction monitoring was included in the analysis for targeted analysis of the compounds with m/z 177. We identified that cotinine of the nicotine metabolism experiment displayed the same retention time as the cotinine standard. In the LCMS analysis, both the cotinine standard and the cotinine of the metabolism experiment were detected at an m/z of 177.10 (Figure 4(d)).
Calculations and statistical methods
For statistical evaluation of the different groups, two-tailed Mann–Whitney U-test and Spearman’s correlation test were used (GraphPad Prism 7.02 Software, San Diageo, CA and SPSS Statistics 23, IBM, The Netherlands). Comparisons of the methylation data among three different time points were conducted using one-way Analysis of Variance (ANOVA). P values were adjusted by the Bonferroni method. P ≤ 0.05 was considered significant. Relative gene expression (2−ΔCt method) as well as mean percent methylation and standard error of the mean (SEM) were calculated in Microsoft® Office Excel 2003.
Supplementary Material
Funding Statement
This work was supported by grants from the Lung Foundation Netherlands (LF3.2.11.013 to M.N. Hylkema) and a Fellowship from the Mongolian State Training Fund and UMCG (to K. Lkhagvadorj).
Author’s contributions
K.L, K.M and M.H designed the study, and K.M, K.L, L.V, W.K and M.R performed experiments. H.D was involved in the in vitro nicotine metabolism experiment, and I.G contributed to the interpretation of the in vitro nicotine metabolism result. K.L drafted the manuscript, which is finalized by T.P and M.H. All authors were involved in writing and had final approval of the submitted version.
Disclosure statement
The authors declare that they have no competing interests.
Ethical approval and consent to participate
The experimental setup was approved by the local committee on animal experimentation (DEC6589 B & C; University of Groningen, Groningen, The Netherlands) and governmental and international guidelines on animal experimentation.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Messina ES, Tyndale RF, Sellers EM.. A major role for CYP2A6 in nicotine C-oxidation by human. J Pharmacol Exp Ther [Internet]. 1997;282(3):1608–1614. Available from: http://jpet.aspetjournals.org/content/282/3/1608.long#page [PubMed] [Google Scholar]
- [2].Raunio H, Pokela N, Puhakainen K, et al. Nicotine metabolism and urinary elimination in mouse: in vitro and in vivo. Xenobiotica. 2008;38(1):34–47. [DOI] [PubMed] [Google Scholar]
- [3].Siu ECK, Tyndale RF, Metabolism D, et al. Characterization and comparison of nicotine and cotinine metabolism in vitro and in vivo in DBA/2 and C57BL/6 mice. Nicotine Tob Res [Internet]. 2009;71(3):826–834. Available from: http://molpharm.aspetjournals.org/cgi/doi/10.1124/mol.106.032086 [DOI] [PubMed] [Google Scholar]
- [4].Cho MH, Castaldi PJ, Wan ES, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet. 2012;21(4):947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Furberg H, Kim Y, Dackor J, et al. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thorgeirsson TE, Gudbjartsson DF, Surakka I, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42(5):448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kubota T, Nakajima-Taniguchi C, Fukuda T, et al. CYP2A6 polymorphisms are associated with nicotine dependence and influence withdrawal symptoms in smoking cessation. Pharmacogenomics J. 2006;6(2):115–119. [DOI] [PubMed] [Google Scholar]
- [8].Lee NP, Fry SJ.. Systematic review of the evidence relating FEV1 decline to giving up smoking. BMC Med [Internet]. 2010;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Amos CI, Spitz MR, Cinciripini P. Chipping away at the genetics of smoking behavior. Nat Genet [Internet]. 2010;42(5):366–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dempsey D, Tutka P, Jacob P, et al. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther. 2004;76(1):64–72. [DOI] [PubMed] [Google Scholar]
- [11].Nakajima M, Yoshihiko T, Noriaki K, et al. ROLE OF HUMAN CYTOCHROME P450A6 IN C-OXIDATION OF NICOTINE. Drug Metab Dispos. 1996;24(11):1212–1217. [PubMed] [Google Scholar]
- [12].Siu ECK, Tyndale RF. Characterization and comparison of nicotine and cotinine metabolism in vitro and in vivo in DBA/2 and C57BL/6 mice. Mol Pharmacol [Internet]. 2006;71(3):826–834. Available from: http://molpharm.aspetjournals.org/cgi/doi/10.1124/mol.106.032086 [DOI] [PubMed] [Google Scholar]
- [13].Zhou X, Zhuo X, Xie F, et al. Role of CYP2A5 in the clearance of nicotine and cotinine: insights from studies on a Cyp2a5-null mouse model. J Pharmacol Exp Ther. 2010;3332:578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Murphy SE, Raulinaitis V, Brown KM. Nicotine 5′-oxidation and methyl oxidation by P450 2A enzymes. Drug Metab Dispos. 2005;33(8):1166–1173. [DOI] [PubMed] [Google Scholar]
- [15].Blacquie`re MJ, Timens W, Berg AVD, et al. Maternal smoking during pregnancy decreases Wnt signalling in neonatal mice. Thorax. 2010;65(6):553. [DOI] [PubMed] [Google Scholar]
- [16].Stevens DR, Malek AM, Laggis C, et al. In utero exposure to tobacco smoke, subsequent cardiometabolic risks, and metabolic syndrome among U.S. adolescents. Ann Epidemiol. 2018;28(9):619–624.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wiklund P, Karhunen V, Richmond RC, et al. DNA methylation links prenatal smoking exposure to later life health outcomes in offspring. Clin Epigenetics. 2019;11(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kandel DB, Udry JR. Prenatal effects of maternal smoking on daughters’ smoking: nicotine or testosterone exposure? Am J Public Health. 1999;89(9):1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kandel DB, Wu P, Davies M. Maternal smoking during pregnancy and smoking by adolescent daughters. Am J Public Health. 1994;84(9):1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Savran O, Ulrik CS. Early life insults as determinants of chronic obstructive pulmonary disease in adult life. Int J COPD. 2018;13:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Blacquière MJ, Timens W, Melgert BN, et al. Maternal smoking during pregnancy induces airway remodelling in mice offspring. Eur Respir J. 2009;33:1133–1140. [DOI] [PubMed] [Google Scholar]
- [22].Krauss-Etschmann S, Meyer KF, Dehmel S, et al. Inter-and transgenerational epigenetic inheritance: evidence in asthma and COPD? Clin Epigenetics [Internet]. 2015;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ferrini M, Carvalho S, Cho YH, et al. Prenatal tobacco smoke exposure predisposes offspring mice to exacerbated allergic airway inflammation associated with altered innate effector function. Part Fibre Toxicol. 2017;14(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dehmel S, Nathan P, Bartel S, et al. Intrauterine smoke exposure deregulates lung function, pulmonary transcriptomes, and in particular insulin-like growth factor (IGF)-1 in a sex-specific manner/631/136/3194/631/443/1784/692/699/1785/31/38/61/38/90/38/39/59/64/64/60 article. Sci Rep. 2018;8(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Klein LC, Stine MM, Pfaff DW, et al. Maternal nicotine exposure increases nicotine preference in periadolescent male but not female C57B1/6J mice. Nicotine Tob Res. 2003;5(1):117–124. [DOI] [PubMed] [Google Scholar]
- [26].Richmond RC, Simpkin AJ, Woodward G, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet. 2015;24(8):2201–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Maccani JZJ, Koestler DC, Houseman EA, et al. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics. 2013;5(6):619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Breton CV, Siegmund KD, Joubert BR, et al. Prenatal tobacco smoke exposure is associated with childhood DNA CpG methylation. PLoS One. 2014;9(6):e99716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Meyer KF, Krauss-Etschmann S, Kooistra W, et al. Prenatal exposure to tobacco smoke sex dependently influences methylation and mRNA levels of the Igf axis in lungs of mouse offspring. Am J Physiol-Lung Cell Mol Physiol [Internet]. 2017;312(4):L542–55. Available from: http://ajplung.physiology.org/lookup/doi/10.1152/ajplung.00271.2016 [DOI] [PubMed] [Google Scholar]
- [30].Meyer KF, Verkaik-Schakel RN, Timens W, et al. The fetal programming effect of prenatal smoking on Igf1r and Igf1 methylation is organ- and sex-specific. Epigenetics [Internet]. 2017;12(12):1076–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Eyring KR, Pedersen BS, Yang IV, et al. In utero cigarette smoke affects allergic airway disease but does not alter the lung methylome. PLoS One. 2015;10(12):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Benowitz NL. Nicotine Addiction. N Engl J Med. 2010;362(24):2295–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ho MK, Mwenifumbo JC, Al Koudsi N, et al. Association of nicotine metabolite ratio and CYP2A6 genotype with smoking cessation treatment in African-American light smokers. Clin Pharmacol Ther. 2009;85(6):635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tizabi Y, Popke EJ, Rahman MA, et al. Hyperactivity induced by prenatal nicotine exposure is associated with an increase in cortical nicotinic receptors. Pharmacol Biochem Behav. 1997;58(1):141–146. [DOI] [PubMed] [Google Scholar]
- [35].van Otterdijk SD, Binder AM, Michels KB. Locus-specific DNA methylation in the placenta is associated with levels of pro-inflammatory proteins in cord blood and they are both independently affected by maternal smoking during pregnancy. Epigenetics [Internet]. 2017;12(10):875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee KWK, Richmond R, Hu P, et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ Health Perspect. 2015;123(2):193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yokomori N, Moore R, Negishi M. Sexually dimorphic DNA demethylation in the promoter of the Slp (sex-limited protein) gene in mouse liver. Proc Natl Acad Sci U S A [Internet]. 1995;92(5):1302–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yokomori N, Kobayashi R, Moore R, et al. A DNA methylation site in the male-specific P450 (Cyp 2d-9) promoter and binding of the heteromeric transcription factor GABP. Mol Cell Biol [Internet]. 1995;15(10):5355–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Takasugi M, Hayakawa K, Arai D, et al. Age- and sex-dependent DNA hypomethylation controlled by growth hormone in mouse liver. Mech Ageing Dev [Internet]. 2013;134(7–8):331–337. [DOI] [PubMed] [Google Scholar]
- [40].Nugent BM, Schwarz JM, Mccarthy MM. Hormonally mediated epigenetic changes to steroid receptors in the developing brain: implications for sexual differentiation. Horm Behav [Internet]. 2011;59(3):338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Al Koudsi N, Hoffmann EB, Assadzadeh A, et al. Hepatic CYP2A6 levels and nicotine metabolism: impact of genetic, physiological, environmental, and epigenetic factors. Eur J Clin Pharmacol. 2010;66(3):239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].The ROF, Cytochrome H . Transcriptional regulation of the hepatic cytochrome P450 2a5 gene [Internet]. 2007. 1–130 p. Available from: http://herkules.oulu.fi/isbn9789514285653/isbn9789514285653.pdf%5Cnpapers3://publication/uuid/04328BDE-78AD-421C-BB7F-E84F9DAEA6E7
- [43].Liu F, Killian JK, Yang M, et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene. 2010;29(25):3650–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sundar IK, Rahman I. Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: implications for COPD and lung cancer. Am J Physiol Lung Cell Mol Physiol. 2016;311(6):L1245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dempsey D, Jacob P, Benowitz NL. Nicotine metabolism and elimination kinetics in newborns. Clin Pharmacol Ther. 2000;67(5):458–465. [DOI] [PubMed] [Google Scholar]
- [46].Gow PJ, Ghabrial H, Smallwood RA, et al. Neonatal hepatic drug elimination. Toxicol Pharmacol. 2001;88(1):3–15. [DOI] [PubMed] [Google Scholar]
- [47].Dempsey D, Jacob P, Benowitz N. Accelerated metabolism of nicotine and cotinine in pregnant smokers. J Pharmacol Exp Ther [Internet]. 2002;301(2):594–598. [DOI] [PubMed] [Google Scholar]
- [48].Novakovic B, Ryan J, Pereira N, et al. Postnatal stability and tissue- and time-specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics. 2013;9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Massart R, Nemoda Z, Suderman MJ, et al. Early life adversity alters normal sex-dependent developmental dynamics of DNA methylation. Dev Psychopathol. 2016;28(4):1259–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chhabra D, Sharma S, Kho AT, et al. Fetal lung and placental methylation is associated with in utero nicotine exposure. Epigenetics. 2014;9(11):1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gao X, Jia M, Zhang Y, et al. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics [Internet]. 2015;7(1). DOI: 10.1186/s13148-015-0148-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Benowitz NL, Lessov-Schlaggar CN, Swan GE, et al. Female sex and oral contraceptive use accelerate nicotine metabolism. Clin Pharmacol Ther. 2006;79(5):480–488. [DOI] [PubMed] [Google Scholar]
- [53].Berlin I, Gasior MJ, Moolchan ET. Sex-based and hormonal contraception effects on the metabolism of nicotine among adolescent tobacco-dependent smokers. Nicotine Tob Res. 2007;9(4):493–498. [DOI] [PubMed] [Google Scholar]
- [54].Liu Z, Li L, Wu H, et al. Characterization of CYP2B6 in a CYP2B6-humanized mouse model: inducibility in the liver by phenobarbital and dexamethasone and role in nicotine metabolism in vivos. Drug Metab Dispos. 2015;43(2):208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lamba V, Lamba J, Yasuda K, et al. Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (Constitutive Androstane Receptor) expression. J Pharmacol Exp Ther. 2003;307(3):906–922. [DOI] [PubMed] [Google Scholar]
- [56].Dickmann LJ, Isoherranen N. Quantitative prediction of CYP2B6 induction by estradiol during pregnancy: potential explanation for increased methadone clearance during pregnancy. Drug Metab Dispos. 2013;41(2):270–274. [DOI] [PubMed] [Google Scholar]
- [57].Pogun S, Yararbas G, Nesil T, et al. Sex differences in nicotine preference. J Neurosci Res. 2017;95(1–2):148–162. [DOI] [PubMed] [Google Scholar]
- [58].Shiffman S, Paton S. Individual differences in smoking: gender and nicotine addiction. Nicotine Tob Res. 1999;1(1):153–157. [DOI] [PubMed] [Google Scholar]
- [59].Hamdani N, Ades J, Gorwood P. [Heritability and candidate genes in tobacco use]. Encephale. 2006;32:966–975. [DOI] [PubMed] [Google Scholar]
- [60].Arger CA, Taghavi T, Heil SH, et al. Pregnancy-Induced increases in the nicotine metabolite ratio: examining changes during antepartum and postpartum. Nicotine Tob Res. 2018;12(12)1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Higashi E, Fukami T, Itoh M, et al. Human CYP2A6 is induced by estrogen via estrogen receptor. Drug Metab Dispos. 2007;35(10):1935–1941. [DOI] [PubMed] [Google Scholar]
- [62].Torres OV, Natividad LA, Tejeda HA, et al. Female rats display dose-dependent differences to the rewarding and aversive effects of nicotine in an age-, hormone-, and sex-dependent manner. Psychopharmacology (Berl). 2009;206(2):303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Harrod SB, Booze RM, Mactutus CF. Sex differences in nicotine levels following repeated intravenous injection in rats are attenuated by gonadectomy. Pharmacol Biochem Behav. 2007;86(1):32–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Weinberg CR, Wilcox AJ, Baird DD. Reduced fecundability in women with prenatal exposure to cigarette smoking. Am J Epidemiol. 1989;129(5):1072–1078. [DOI] [PubMed] [Google Scholar]
- [65].Jensen TK, Henriksen TB, Hjollund NHI, et al. Adult and prenatal exposures to tobacco smoke as risk indicators of fertility among 430 Danish couples. Am J Epidemiol. 1998;148(10):992–997. [DOI] [PubMed] [Google Scholar]
- [66].Holloway AC, Kellenberger LD, Petrik JJ. Fetal and neonatal exposure to nicotine disrupts ovarian function and fertility in adult female rats. Endocrine. 2006;30(2):213–216. [DOI] [PubMed] [Google Scholar]
- [67].Lambert JF, Benoit BO, Colvin GA, et al. Quick sex determination of mouse fetuses. J Neurosci Methods. 2000;95(2):127–132. [DOI] [PubMed] [Google Scholar]
- [68].EME VS, Bloks VW, Huijkman NCA, et al. The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am J Epidemiol. 2010;298(2):275–282. [DOI] [PubMed] [Google Scholar]
- [69].Siu ECK, Wildenauer DB, Tyndale RF. Nicotine self-administration in mice is associated with rates of nicotine inactivation by CYP2A5. Psychopharmacology (Berl). 2006;184(3–4):401–408. [DOI] [PubMed] [Google Scholar]
- [70].Massadeh AM, Gharaibeh AA, Omari KW. A single-step extraction method for the determination of nicotine and cotinine in Jordanian smokers ’ blood and urine samples by RP-HPLC and GC – MS. J Chromatogr Sci. 2009;47(February):170–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.