

ABSTRACT
Interactions between the cytoplasmic domains of viral transmembrane proteins and host machinery often determine the outcome of viral infection. The M2 protein of influenza A has been identified as a key player in autophagy-mediated viral replication. Here, we describe the engineering and validation of an antibody specific for the cytoplasmic domain of the M2 protein. Through phage and yeast display selection techniques, we obtained an antibody that recognizes: 1) the M2 cytoplasmic domain purified from bacterial inclusion bodies and refolded, 2) full-length M2 recombinant protein expressed in mammalian cells, and 3) native M2 protein in influenza A infected cells. This antibody can serve as a molecular tool to enhance our knowledge of protein–protein interactions between influenza A virus and the host cell machinery. We anticipate the methods described herein will further the development of antibodies specific to the cytoplasmic domains of transmembrane proteins.
KEYWORDS: Influenza A, M2 cytoplasmic domain, antibodies, transmembrane proteins, phage display, yeast display, antibody formats
Introduction
Although lipid bilayers provide the structure of cell membranes, proteins perform most of the specific functions associated with these membranes. Membrane proteins, which play key roles in molecular transport and signal transduction, comprise 20–30% of all proteins in living organisms and more than half of all drug targets. Transmembrane proteins generally extend through the lipid bilayer (often more than once) and usually consist of ectodomain(s), transmembrane domain(s), and cytoplasmic or endodomain(s).1 Large cytoplasmic domains are a common feature of many transmembrane proteins; often these domains have key functions in a variety of signal transduction pathways, including apoptosis, gene expression, and cellular differentiation in mammalian cells.2
Transmembrane proteins of enveloped viruses play critical roles in viral entry, replication, and packaging through interaction with host cell surface proteins and, following virus internalization, with cellular cytosolic components.2 The cytosolic components of viral transmembrane proteins can be very large in size (envelope glycoproteins of lentiviruses can exceed 150 kD),3 or very small, as is the case with Influenza virus M2 protein. Influenza virus entry into host cells is triggered by low pH, which causes a conformational change in the hemagglutinin (HA) envelope protein, initiating viral entry.4 M2 forms a pH-gated proton channel and facilitates HA-mediated fusion during viral entry.5 M2 is a 97-amino acid protein of which approximately half (aa 47–97) comprises the cytoplasmic domain,6 which is highly conserved among virus strains, even though it is not required for viral entry.7 Though small in size, the M2 cytoplasmic domain interacts directly with mammalian host cell proteins (e.g., in the autophagy pathway) and plays an important role in regulating viral replication.8
Many commercial antibodies are raised against specific peptides derived from cytoplasmic domains, generating antibodies that can recognize post-translational modifications (e.g., phosphorylation) in cytoplasmic domains involved in signal transduction. However, many such antibodies are polyclonal, which suffer from batch-to-batch variation and poly-reactivity.9,10 Although this is somewhat mitigated by the use of monoclonal antibodies, even these can suffer from off-target binding.11 Recombinant antibodies (including both monoclonal antibodies and polyclonal sera) can solve many of the problems of batch-to-batch discrepancy, provided they are carefully validated and bind the expected target.12 In vitro display technologies have been used over the past 30 years to screen recombinant antibodies against a variety of targets.13 In a previous study, we used phage and yeast display technologies to generate monoclonal antibodies against phosphorylated peptides derived from the cytoplasmic domain of IgE receptor proteins;14 these antibodies also recognized the in vivo phosphorylated proteins. The use of in vitro technologies on native or near-native cytoplasmic domains of transmembrane proteins has not been widely explored, and there are no published reports of antibodies specific for the cytoplasmic domain of influenza A (IVA) M2. In this study, we describe phage and yeast display selection and screening strategies, followed by mammalian cell-based verification, to develop antibodies specific for the IVA M2 cytoplasmic domain. We believe analogous methods can be used to develop antibodies against the cytoplasmic domains of other viruses, as well as medically important human transmembrane proteins.
Results
Cloning and purification of M2-cytoplasmic domain
A codon-optimized gene fragment encoding for the cytoplasmic domain of influenza A/PR/8/34 M2 protein (PR8-M2, Genebank accession # AF389121.1)15 was cloned into a pET-based protein expression vector and produced in E. coli (BL21 DE3). As the M2 cytodomain fragment was insoluble, it was purified from inclusion bodies in the presence of guanidium chloride, then re-folded and stabilized prior to antibody selection. The protocol for the selection of antibodies recognizing the recombinant peptide is summarized in (Figure 1(a,b)).
Figure 1.
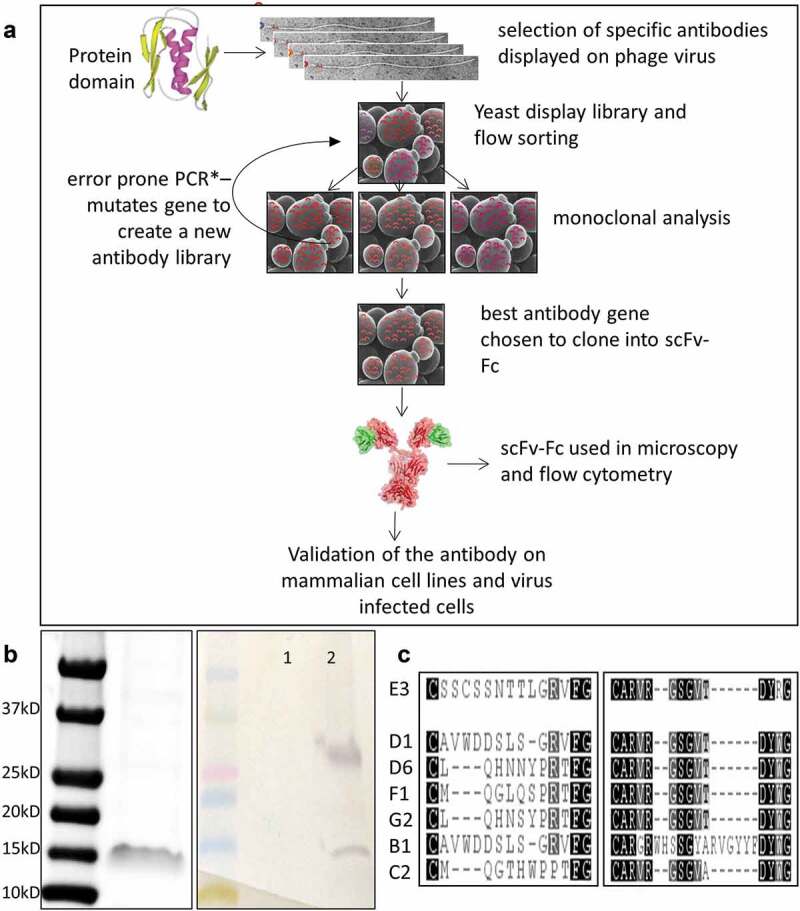
Selection of anti-M2cyto antibodies using phage and yeast display techniques. Panel A depicts a schematic diagram of the phage and yeast display protocols used in the antibody selection process. Panel B shows the size of the M2 cytodomain used as the antigen in the antibody selection process which corresponds to dimeric form at ~15kD. A Western blot shows unbiotinylated (1) and the biotinylated M2 cytodomain (2) used as the antigen in our selection process. The Western blot was probed with streptavidin alkaline phosphatase (AP) to detect the biotinylated antigen and shows bands corresponding in size to dimeric and tetrameric forms of M2 cytodomain in line 2. Panel C shows CDR3 amino acid sequences from the light-chain (left) and heavy-chain (right) of multiple scFv antibodies selected via this process. These antibody encoding sequences were cloned into yeast scFv-Fc expression vectors, and the secreted scFv-Fcs antibodies were used in subsequent characterization assays
Phage display and yeast display based antibody selection and screening
The purified M2 cytodomain was biotinylated and used as a target for phage display-based single-chain variable fragment (scFv) antibody selection.16 We also used CR1, an unlabeled unrelated protein, as a subtraction agent to eliminate any antibodies recognizing the expression and purification tags on the target. After two rounds of phage selection, scFv genes from 107 colonies were cloned into a yeast display vector,17 and the yeast display library (105 diversity) was sorted for binding. After three rounds of sorting with decreasing amounts of antigen, error-prone polymerase chain reaction (EP PCR) was performed on the binding scFv gene population to improve affinity. After two rounds of EP PCR and nine total rounds of sorting, displayed scFv clones were able to recognize the biotinylated M2 cytodomain at concentrations of 1–2 nM. Sequencing of 96 clones from the final sort population revealed a predominant variable heavy (VH) domain and variable light (VL) domain grouped into six distinct clusters. Single amino acid variations were also observed, indicating the effects of EP PCR. Seven scFv clones with differences in light-chain complementarity-determining region 3 (CDR3), and one scFv clone with a distinct heavy-chain CDR3, were selected to represent the different sequence clusters (Figure 1c). The selected clones were isolated, and plasmid DNA was used to prepare yeast secretion vectors. ScFvs C2 and E3 yielded the highest signals for binding to the M2 cytoplasmic domain and therefore were carried forward for further analysis.
Recognition of M2 cytodomain in mammalian cell line
In order to validate antibody selectivity in a native environment, the antibodies were tested against HEK cells expressing full-length M2 protein (HEK M2 cells, provided by Dr. M. Tompkin, University of Georgia15). We chose the scFv-Fc format for these assays to take advantage of the bivalent binding, longer shelf life, and availability of Fc–specific secondary antibodies. Figure 2a provides fluorescent microscopy images for anti-M2cyto C2. Fluorescent signals are visible lining the internal cell wall of HEK M2 cells after fixation and permeabilization. No signal is visible in HEK cells that do not express M2, or when the cells were not permeabilized. The location of M2cyto antibody staining was also investigated with an intrinsically fluorescent construct of Z318,19 (single-chain green fluorescent protein (scGFP-Z3)), which recognizes the ectodomain of this transmembrane protein. Co-staining with two different fluorescent dyes allows visualization of the M2 cytodomain antibodies in the cytoplasm of HEK M2 cells. Figure 2b provides population-level data for the recognition patterns of scFv-Fc Z3, scFv-Fc C2, scFv-Fc E3, and a nonspecific scFv-Fc against mycolactone (ML).20 The Z3 antibody was derived from a previously characterized M2 antibody recognizing the M2 ectodomain,21 and was used as a positive control to develop assay protocols. Since permeabilization of the cells should be required for access to the M2 cytoplasmic domain, cytoplasmic domain antibodies should not bind to cells that have been fixed and not permeabilized. On the other hand, permeabilization should not be required for binding of the anti-M2ecto scFv Z3 antibody. The data in Figure 2b show that anti-M2ecto Z3 recognizes both fixed and permeabilized HEK M2 and fixed HEK M2 cells without permeabilization as expected, whereas the mean fluorescence intensity (MFI) values for HEK cells not expressing M2 is 2–3 times lower. The anti-M2cyto C2 antibody only recognizes fixed and permeabilized HEK M2 cells as expected, while anti-M2cyto E3 was found to nonspecifically recognize HEK cells, as well as HEK M2 cells. The data also confirm that the nonspecific and secondary antibodies do not recognize the M2 protein expressed in mammalian cells.
Figure 2.
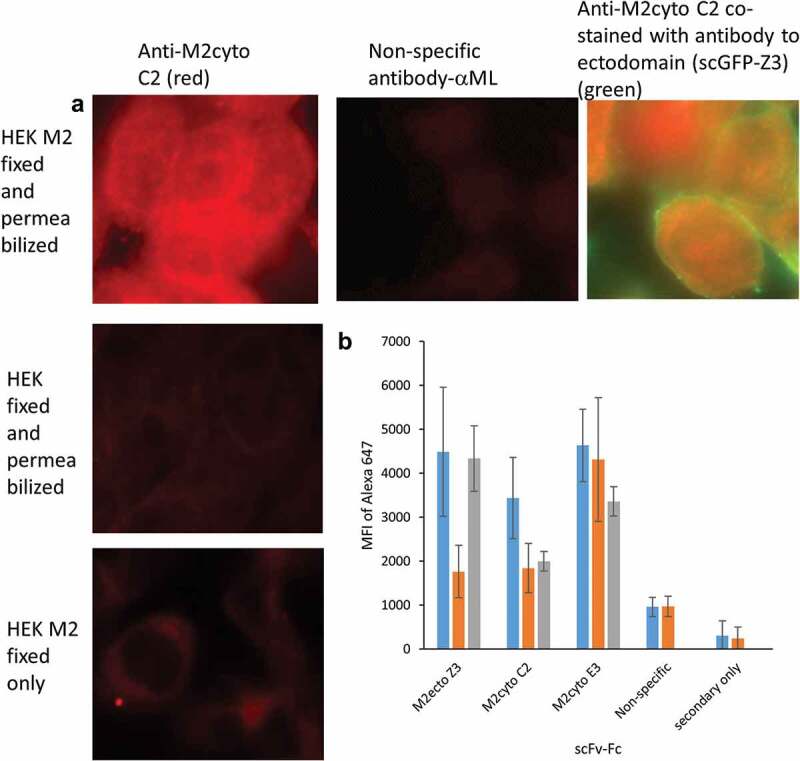
Recognition of full-length M2 protein expressed in mammalian HEK cells. HEK M2 cells were induced to produce M2 protein, then fixed and permeabilized. Panel A shows fluorescence microscopy images from M2 detection performed in HEK M2 cells using the scFv-Fc format of the anti-M2cyto C2 antibody, indicating cytoplasmic staining. Nonspecific scFv-Fc antibody, HEK (untransfected) cells, and HEK M2 cells without permeabilization were used as controls. Panel A also shows co-stained HEK M2 cells with anti-M2cyto C2 (red) and M2 extracellular antibody; scGFP-Z3 (green). Panel B depicts population level data assessed by flow cytometry using 10,000 cells per sample. The mean fluorescence intensity (MFI) of the secondary antibody is given for fixed and permeabilized HEK M2 cells (blue), fixed and permeabilized HEK cells (orange), and HEK M2 cells that have been fixed but not permeabilized (gray)
Figure 3 provides further evidence for anti-M2cyto C2 recognition of the cytoplasmic M2 domain. Biochemical analysis was performed using a styrene-maleic acid (SMA) copolymer, which inserts into the lipid bilayer and solubilizes the membrane into small discs encircled by SMA, thereby creating SMA lipid particles (SMALPs).22 In principle, both the ectodomain and cytodomain of the M2 protein on SMALPs should be accessible to antibodies. Membrane vesicles were prepared from both HEK cells and HEK M2 cells; Western blot analysis using a commercial IVA M2 monoclonal antibody (m14C2 IgG) showed that M2-containing vesicles were obtained exclusively from HEK M2 cells. The m14C2 IgG is sold as an antibody capable of immunoprecipitating (IP) M2 from cell lysate; specific IP of M2 from HEK M2 cell lysates were further verified in-house prior to using m14C2 IgG to IP M2-SMALPs. In the yeast-based SMALP experiments, SMALPs from HEK M2 cells were allowed to bind to yeast displaying M2cyto C2, M2ecto Z3, and a nonspecific scFv YP223. The bound SMALPs were detected using an intrinsically fluorescent construct of anti-M2ecto 14C2 (scGFP-14C2); another antibody that recognizes the M2 ectodomain,15 engineered to use GFP as a linker between the VH and VL regions.18,19 Figure 3 shows that yeast displaying scFv C2 bind to M2 SMALPs, while allowing simultaneous binding of scGFP-14C2, which recognizes the external domain. In contrast, there is no binding when scFv Z3 is displayed on yeast and stained with scGFP-14C2, as the two antibodies compete for binding to the small ectodomain. The data also show that the nonspecific antibody (YP2) does not recognize the M2 SMALPs and is not stained by scGFP-14C2. The scFv sequence for the anti-M2cyto C2 antibody is given in Table 1.
Figure 3.
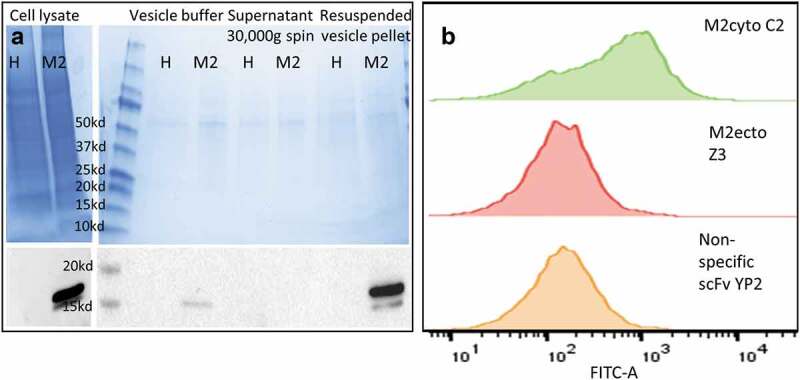
Preparation of membrane vesicles and recognition of transmembrane protein using SMALP-stabilized plasma membrane vesicle assay. Panel A shows Coomassie staining (upper-left) and Western blot analysis (lower-left) of membrane vesicles used to prepare SMALPs. ‘H’ denotes HEK cell line and ‘M2ʹ denotes HEK M2 cell line. For Western blot analysis, the nitrocellulose membranes were probed with monoclonal 14C2 IgG, followed by anti-mouse-HRP. Panel B shows recognition of SMALP-stabilized M2 vesicles by yeast displayed scFv antibodies using flow cytometry. Histograms show the binding patterns for three different scFv antibodies. The yeast bound vesicles were detected using scGFP-14C2, which binds to the ectodomain of M2. The yeast displayed anti-M2cyto C2 antibody recognizes the transmembrane M2 protein in the plasma membrane vesicles (green). Panel B also shows data obtained with the anti-M2ecto Z3 antibody (red), which recognizes the M2 ectodomain, and the nonspecific scFv YP2 antibody (yellow). The mean fluorescence intensity (MFI) of binding for C2, Z3, and YP2 scFvs was 999, 539, and 202 respectively
Table 1.
Amino acid sequence for the anti-M2 cyto C2 scFv antibody with linker and SV5 & HIS tags. Grey letters indicate vector sequences
| VL | DIVMTQTPLSLPVTLGQPASISCRSSQSLVSSDGNTYLNWFQQRPGQSPRRLIYKVSNRDSGVPDRFSGSGSGTDFSLKISRVEAEDVGVYYCMQGTHWPPTFGQGTKVEIK |
| Linker | SGGSTITSYNVYYTKLSSSGT |
| VH | QVQLVESGPGLVKPSETLSLTCTVSGYSISSGYYWGWIRQPPGKGLEWIGSIYHSGSTYYNPSLKSRVTISVDTSKNQFSLKLSSVSAADTAVYYCARVRGSGVADYWGQGTLVTVSSASGKPIPNPLLGLDSTHHHHHH |
Recognition of the M2 cytodomain in IVA-infected cells
Finally, we tested the ability of the anti-M2cyto C2 antibody to recognize native M2 protein produced during IVA infection of MDCK cells with the H3N2 (A/PCh/1/73) and H1N1 (A/PR/8/34) strains of influenza. We observed efficient and specific recognition of native M2 protein by the scFv-Fc C2 via fluorescent microscopy and flow cytometry (Figure 4(a,b)). We detected a clear increase in fluorescence intensity in IVA-infected cells compared to mock-infected cells (Figure 4b). A population-level analysis of IVA-infected cells labeled with four different antibodies revealed that the newly generated C2 antibody is comparable as an analytical tool in IVA infection studies to commercial antibodies specific for the M2 ectodomain (m14C2), nucleoprotein (NP), or in-house scFv-Fc Z3 antibody specific for the M2 ectodomain (Figure 4c).
Figure 4.
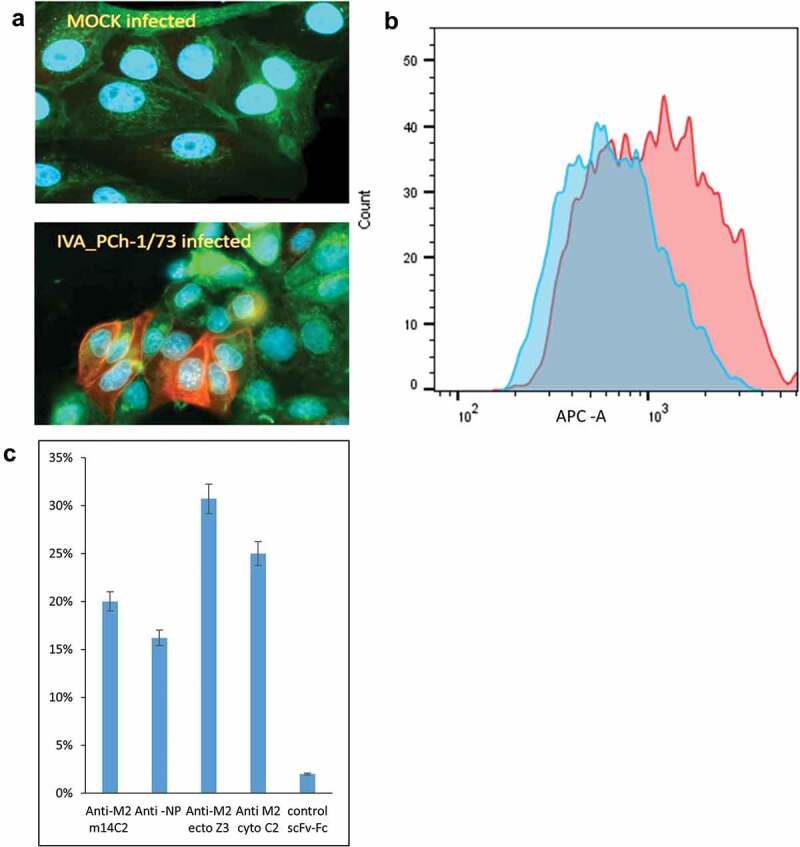
Recognition of M2 protein in MDCK cells infected with Influenza A (IVA) virus
MDCK cells infected with IVA and mock-infected cells were fixed, permeabilized, and stained with scFv-Fc C2 antibody and anti-rabbit Alexa 647 secondary antibody. Panel A shows fluorescence microscopy images for the specific recognition of native M2 by the anti-M2cyto C2 antibody. Panels B and C show population level data obtained using flow cytometry. Panel B depicts the positive shift in fluorescence for anti-M2cyto C2-stained IVA-infected cells (red) compared to mock-infected cells (blue). In panel C, IVA-infected cells were assessed by flow cytometry using four different antibodies. The data show percentage infected cells labeled by IVA-specific antibodies (m14C2, NP, Z3, and C2) and a nonspecific control scFv-Fc (anti-ML). The anti-M2 m14C2 and anti-NP IgGs are commercial antibodies. The anti-M2ecto Z3, anti-M2cyto C2, and the nonspecific control were all expressed as scFv-Fc antibodies in our yeast secretory system. Shown is a representative of five independent infectionexperiments.
Discussion
The cytoplasmic domains of viral transmembrane proteins play a critical role in viral replication through their interaction with various host cell proteins. Developing reagents that specifically recognize viral cytoplasmic domains is an opportunity to discover new drug targets.13 Here, we describe the validation of an antibody selected against the cytoplasmic domain of the M2 protein of IVA using a combination of phage and yeast display, bacterial- and mammalian cell-expressed target proteins, and various antibody formats (scFv, scFv-Fc, IgG, and scGFP). The use of both phage and yeast display together17 has been previously shown to result in the generation of many more antibodies than phage display alone, exploiting access to highly diverse antibody libraries displayed on phage with the subsequent precision afforded by yeast display-based sorting. The binding of the antibody (anti-M2cyto C2) to the cytoplasmic domain of IVA M2 was confirmed in three different ways: 1) M2 expressed in mammalian cells was only recognized by the C2 antibody when cells were permeabilized (enabling antibody access to the cytoplasm), and the C2 antibody showed a combined membrane and intracellular distribution by microscopy, 2) SMALPs containing M2 could be simultaneously recognized by the C2 antibody displayed on yeast (which binds the M2 cytodomain) and by the fluorescent scGFP-14C2 (which binds the M2 ectodomain), and 3) IVA-infected (but not mock-infected) cells were recognized by the C2 antibody. The anti-M2cyto C2 antibody as well as the in vitro selection techniques described herein, including the novel use of SMALPs to isolate correctly folded cytoplasmic domains for antibody studies, provide key tools to facilitate better understanding of Influenza host-pathogen protein interactions, and provide a roadmap for generating cytoplasmic domain-specific antibodies for other transmembrane proteins, including both host and viral proteins.
Host-directed therapy is a novel approach proposed to counter the effect of anti-microbial resistance in bacteria and viruses,24 in which host proteins interacting with the pathogen are targeted, rather than the pathogens themselves. Understanding how host proteins interact with viral proteins to regulate host–pathogen interactions is essential for this approach. Cellular pathways such as autophagy and apoptosis are often involved in adaptive immunity.25 Accumulation of auto-phagosomes during influenza infection has been established.26 However, the underlying mechanisms and the host and viral proteins involved in the regulation of autophagy are still under investigation. The cytoplasmic domain of IVA M2 has been postulated as a key regulator of autophagy in IVA-infected cells.27 The anti-M2cyto antibody C2 reported here would be an ideal reagent to study M2 interactions with various host proteins in humans and other animals. Our results also show that our antibody recognizes two different strains of IVA (PR8 and Port Chalmers), and therefore may be useful in studies involving different influenza strains. In our experiments, the stably transfected M2 cell line encoded sequence derived from the PR8 strain of IVA. In order to verify cross-strain specificity of our antibody, we used the Port Chalmers strain of IVA in the viral infection studies presented in Figure 4.
In this report, we also demonstrate the general utility of intrinsically fluorescent scGFP constructs18,19 based on scFvs, with an exceptionally well-folded fluorescent protein28 acting as the linker between the VL and VH regions. This construct combines the specificity of antibody recognition with the efficiency of fluorescence-based detection in a one-step format. Beyond the general utility of the scGFP format, scGFP-Z3 and scGFP-14C2 are particularly useful for the identification and staining of M2 protein in various assays where a direct fluorescence readout is required. Furthermore, given the ability of scGFP constructs to be functionally expressed in the cytoplasm, particularly if the GFP is extremely well-folded,14,16,17,28 they may also be useful in studying the role of the M2 cytoplasmic domain by binding intracellularly and potentially inhibiting protein–protein interactions.
Materials and methods
Antigen production
The IVA M2 cytoplasmic domain gene was synthesized by Genscript Inc. The gene fragment included BssH II and Not I restriction enzyme sites, used for cloning and a TEV tag,29 and the last two amino acids (RL) of the M2 transmembrane domain. The nucleotide and amino acid sequences of the M2cyto gene used are:
GCGCGCGTCGACTGTTTTTCAAGTGTATATACAGGCGCTTTAAGTACGGGCTCAAGGGTGGCCCATCTACTGAGGGAGTACCCAAATCCATGCGCGAGGAGTATCGAAAAGAACAACAGTCCGCAGTCGACGCGGACGATGGGCACTTTGTGAGCATTGAGCTTGAAGCTAGCGAGAATCTGTATTTTCAGGGAAGCCCGCGGCCGC
(ARRLFFKCIYRRFKYGLKGGPSTEGVPKSMREEYRKEQQSAVDADDGHFVSIELEASENLYFQGSPRP)
BssH II and Not I sites were used to clone the M2cyto gene into the pET-CK3 protein expression vector30 (pET-C6His28 based on pET-28). The pET-CK3-M2cyto vector was transformed into BL21 competent E. coli and protein expression was performed using auto-induction media at 30 °C overnight. The cell pellet was collected by centrifugation at 10,000 g for 20 minutes. Cell lysis was performed using the Emulsiflex C5 homogenizer (Avestin Inc.) in TNG buffer (30 mM Tris pH 8.0, 150 mM NaCl, and 10% glycerol). Soluble and insoluble fractions were separated by centrifugation at 35,000 g for 30 minutes. Ni-NTA agarose-based purification of the His-tagged protein was performed according to manufacturer’s instructions. The wash buffers contained 20 mM imidazole and the elution buffer contained 250 mM imidazole. Protein purification from the soluble fraction yielded very little M2 cytodomain protein. However, Coomassie staining of the boiled insoluble fraction revealed that the majority of the M2 cytodomain protein resided in this fraction. Therefore, the cell pellet was dissolved in TNG buffer with 6 M guanidium chloride to allow denaturation of the insoluble protein. Ni-NTA agarose-based purification was repeated in buffers containing guanidium chloride and the protein was re-folded by dialysis in 1x phosphate-buffered saline (PBS) with 10% glycerol overnight in the presence of 1 mM dithiothreitol. The presence of 10% glycerol was found to be crucial for the protein stability of this subdomain. Quality of the purified and re-folded M2 cytodomain was evaluated using Coomassie staining, and quantification of the protein was determined by absorbance at 280 nm. A portion of the purified protein was biotinylated using the EZ-Link Sulfo-NHS-LC-LC-biotinylation kit (ThermoFisher, cat# 21,338) for 1 h on ice, and unconjugated biotin was subsequently removed by dialysis in 1xPBS with 10% glycerol for 2 h. Protein loss was observed during biotinylation and dialysis, thus shorter times were required for both. Biotinylation of the cytodomain was evaluated via Western blots probed with streptavidin conjugated to alkaline phosphatase. Western blots showed two bands: the dimeric M2 cytodomain at 15 kD, and a second band at ~30 kD, which was confirmed to be an M2 tetramer by mass spectrometry.
Phage-based scFv antibody selection
The phage antibody library described by Sblattero et al. was used.16 The 180 L of phage library was blocked with 20 L of 5% bovine serum albumin (BSA) in 1xPBS for 30 minutes with rotation, followed by addition of a nonspecific protein (CR1) that contained the same expression and purification tags. CR1 was added to subtract binders to any tags on the M2cyto antigen. Selection using the phage library without CR1 subtraction was also performed. Biotinylated M2 cytodomain (bio-M2cyto) was used at a concentration of 5 M for phage selection, performed as previously described in Velappan et al.14 Acid-based phage elution and phage propagation for the second round of selection was also performed as described earlier.14,16 Plasmid DNA was prepared from the second round phage selection output and the scFv genes were amplified using specific primers containing regions of DNA overlapping with the yeast display vector pDNL6. Yeast vector preparation and cloning of the second round phage selection output into pDNL6 by homologous recombination in yeast were performed as described earlier.14
Yeast library sorting
The yeast library was evaluated for antigen recognition at concentrations of 1000 nM and 200 nM bio-M2cyto for the first round of sorting. Biotinylated lysozyme at 1000 nM and streptavidin only were used as negative control antigens. Yeast staining was performed as previously described in Velappan et al.14 A competitive assay with bio-M2cyto and M2cyto at a 5x excess amount was also used to evaluate the library for M2cyto-specific binders and to exclude biotin binders. Three rounds of sorting were conducted by progressively lowering the specific antigen concentration in a stepwise manner from 1000 nM to 100 nM. After each round of sorting, the yeast library was grown, induced, and stained at 1000 nM, 500 nM, 100 nM, and 50 nM of bio-M2cyto along with negative control antigens as described earlier.14
After three rounds of sorting, plasmid DNA was prepared and used as a template for EP PCR, performed with a differential ratio of deoxynucleoside triphosphates (1 mM T/C and 200 µM A/G) in the presence of dimethyl sulfoxide (10%), MnCl2 (0.2 mM), and MgSO4 (2 mM), using Taq polymerase (New England Biolabs, cat# M0273L).14 The amplification product was re-cloned into pDNL6 by homologous recombination and sorting was continued with decreasing concentrations of bio-M2cyto. This recombined library (R2) was also subjected to three rounds of sorting with decreasing concentrations of antigen down to 2 nM. Plasmid DNA from R2 S3 was transformed into bacterial cells (Omnimax T1) and 96 colonies were analyzed by DNA sequencing. Eight different unique scFv clones were identified using the Geneious® sequence analysis software. These scFvs were sub-cloned into a yeast scFv-Fc expression vector; pDNL9-Rabbit Fc; and expressed in YVH10 cells as previously described.14 Culture supernatant was used as the binding agent in subsequent assays.
Recognition of M2 cytodomain in mammalian cells
The HEK 293 M2 cell line and the corresponding HEK 293 untransfected control cell line were kindly supplied by Dr. Mark Tompkins (University of Georgia). Cell propagation and M2 induction were performed as described in Gabbard et al.15 For microscopy experiments, the cells were seeded onto 8-chambered borosilicate glass slides (Nunc LabTek, cat# 155411). The cells were fixed using 4% paraformaldehyde (PFA) in 1xPBS for 15 minutes and washed twice with 1xPBS. The cells were then permeabilized using 0.3% Triton-X100 in 1xPBS for 10 minutes. The permeabilization solution was washed away with 1xPBS and the cells were blocked using 2% BSA in 1xPBS for at least 30 minutes (up to 24 h). A total of 70 μL of scFv-Fc containing yeast culture supernatant and 30 µL of 2% BSA were added after discarding the blocking solution. The antigen-antibody binding reaction was allowed to proceed for 2 h, then unbound antibody was removed by washing three times with PBS/T (1xPBS with 0.05% Tween-20) and another three times with 1xPBS. Bound antibody was detected using anti-rabbit Alexa 647 secondary antibody (ThermoFisher, cat# A27040). The cells used for flow cytometry assays were similarly propagated and induced. The live cells were lifted from culture flasks in 1xPBS and washed by centrifugation at 8000 g for 5 minutes. The scFv-Fc Z3, used as a control in these experiments, was cloned and expressed in the same vector system14 as the M2cyto antibodies. The scGFP Z3 was prepared by cloning GFP protein in between the VL and VH domains. The subsequent construct was expressed in BL21 E.coli cells and purified using Ni-NTA agarose.
SMALP based analysis
HEK 293 M2 cells were cultured and induced for M2 production (as above) in 6-well tissue culture plates. Each well was rinsed twice with 1 mL of 30% PBS at ambient temperature for 1 minute, then 1 mL of vesiculation buffer (200 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, 0.75 mM CaCl2, buffered at pH 8.5 by 100 mM bicine) was added to each well and the plate incubated for 13 h at 37 °C. After incubation, the supernatant was harvested, leaving the cells on the plate. The supernatant was briefly centrifuged at 1000 g for 5 min to remove any remaining cells prior to vesicle isolation with high speed centrifugation.The supernatant was centrifuged at 30,000 g for 30 minutes at 4 °C, the resulting vesicle pellet was resuspended in 1 mL 1xPBS, and the protein concentration was measured by absorbance at 280 nm. SMA powder, kindly provided by Tim Dafforn (University of Birmingham), was dissolved in freshly prepared SMA solubilization buffer (50 mM Tris pH 8.0, 500 mM NaCl and 10% glycerol in H2O) to make a 5% solution and warmed to 42 °C. 1 mL of the warm SMA was added to 1 mL of prepared membrane fraction to achieve a 1:1 (vol/vol) ratio. This solution was allowed to incubate at ambient temperature with gentle agitation for 2 h. Protein gel electrophoresis followed by Coomassie staining and Western blot analyses was used to characterize vesicles from HEK and HEK M2 cells. Western blot detection was performed using m14C2 IgG (ThermoFisher, cat# MA1-082) and anti-mouse HRP (ThermoFisher cat# 62–6520). Chemiluminescence detection was performed using SuperSignal West Dura substrate (ThermoFisher cat #37071). Purification of M2 SMALPs was performed using the anti-M2 mouse monoclonal 14C2 IgG (m14C2) covalently coupled to magnetic Protein-G beads (ThermoFisher, cat# 88848). A total of 50 μL of magnetic Protein-G beads was washed in 1xPBS and blocked with 2% BSA for 30 minutes. In total, 10 μL of m14C2 (at 1 mg/mL) was added and incubated for 30 minutes to coat the magnetic beads, washed and conjugated. Prepared SMALPs were added to this mixture and incubated for 1–2 h. The beads were washed three times in PBS/T and three times in 1xPBS. Purified M2 SMALPs were eluted in 200 μL of 0.1 M glycine buffer with an incubation period of 15 minutes on a rotator. The solution was neutralized using 100 μL of 1.5 M Tris (pH 8.0). Traditional SDS polyacrylamide denaturing Western blot analysis with SMALP particles was attempted, but this was unsuccessful, as an M2-specific band at the expected molecular weight was not observed in the sample used as input to IP (in fact, no visible band was observed between 10–250KD), likely representing interference of the PAGE by the SMALPs. However, we verified the ability of m14C2 IgG to immunoprecipitate M2 using HEK M2 cell lysates as specified by the manufacturer; HEK cell lysate was used as control. The immunoprecipitated M2 was detected by Western blotting as described above. We used a yeast-based analysis technique for SMALP binding assay. Here, SMALPs were incubated with yeast displaying scFvs for 1 h and washed as previously described.14 Bound SMALPs were detected using 1 μg of scGFP-14C2 after incubation for 1 h followed by washing of the yeast. The scGFP 14C2 was prepared by cloning GFP protein in between the VL and VH domains. The subsequent construct was expressed in BL21 E.coli cells and purified using Ni-NTA agarose. The yeast scFv-M2 SMALPs-scGFP 14C2 were analyzed using a BD FACSAria II flow cytometer.
Cell culture, viruses, and infection
Madin-Darby canine kidney (MDCK) cells (ATCC, cat# CCL-34) were cultured in DMEM-5%: Dulbecco’s Modified Eagle Medium (DMEM) containing high glucose, sodium pyruvate, and GlutaMAX (Gibco, cat# 10,569,044), supplemented with 5% fetal bovine serum (Gibco, cat# 26140079). In-house stocks of IVA strains A/Puerto Rico/8/1934 (H1N1) and A/Port Chalmers/1/1973 (H3N2) were amplified in MDCK cells infected with a multiplicity of infection (MOI) of 0.01 at 37 °C and collected 48 h post-infection. Virus titers were calculated by the Reed-Muench method as log10 infective dose per mL.31 Prior to virus infection for experiments, 106 MDCK cells in DMEM-5% were seeded into 6-well tissue culture plates and incubated at 37 °C with 5% CO2 until 90% confluent (24–48 h). Viral stocks were diluted in serum-free DMEM to achieve an MOI of 1.0 relative to the number of target cells, then 1 μg/mL TPCK Trypsin (Worthington Biochemical Corporation, Lakewood, NJ) was added to the viral solution to activate HA cleavage, which is essential for viral particle docking and endocytosis by host cells. After 1 h of incubation at 30 °C with 5% CO2, the virus inoculum was removed and replaced with fresh DMEM-5%, then cells were incubated for 24 h at 37 °C prior to analysis. Mock-infected cells were incubated in serum-free DMEM containing 1 μg/mL TPCK Trypsin for 1 h at 30 °C with 5% CO2, followed by incubation in DMEM-5% for 24 h at 37 C.
For immunohistochemistry (IHC) analysis, infected MDCK cells were washed twice with cold 1xPBS, fixed with 4% PFA in 1xPBS for 1 h at ambient temperature, and washed twice with cold 1xPBS. Cells were permeabilized with 0.3% Triton-X100 in 1xPBS for 10 minutes. For flow cytometry analysis, infected MDCK cells were lifted into suspension with 0.25% Trypsin-EDTA solution (Gibco, cat# 25200056), the Trypsin was washed away with 1xPBS by centrifugation at 500 g for 2 minutes, and the cell pellets were resuspended and fixed in 4% PFA in 1xPBS for 1 h at ambient temperature, then permeabilized as described above. Cells for IHC and flow cytometry were incubated in blocking solution (2% BSA in 1xPBS) for 1 h prior to staining with primary and secondary antibodies. In total, 100 μL of scFv-Fc, 100 µL 2% BSA, and 300 μL of PBS were used as primary antibody. The antigen-antibody binding reaction was allowed to proceed for 2 h, then unbound antibody was removed by washing three times with PBS/T (1xPBS with 0.05% Tween-20) and another three times with 1xPBS. Bound antibody was detected using anti-rabbit Alexa 647 secondary antibody (ThermoFisher, cat# A27040) at 1:1000 dilution. Unbound reagents were removed by washing three times with PBS/T (1xPBS with 0.05% Tween-20) and another three times with 1xPBS. The antibody-bound cells were analyzed using a BD FACSAria II flow cytometer. The statistical analysis was performed by the DIVA software.
Funding Statement
This project was funded by LDRD – DR grant (20160054DR (Waldo) Countering pathogen interference with human defenses) from Los Alamos National Laboratory.
Abbreviations
| CDR3 | complementarity-determining region 3 |
| EP PCR | error-prone polymerase chain reaction |
| IP | immunoprecipitation |
| IVA | influenza A |
| MFI | mean fluorescence intensity |
| ML | mycolactone |
| NP | nucleoprotein |
| scFv | single-chain variable fragment |
| scGFP | Single-chain green fluorescent protein |
| SMA | styrene-maleic acid |
| SMALP | SMA lipid particle |
| VH | variable heavy domain |
| VL | variable light domain |
Author contributions
NV, SMV, SHA, AML performed the experimental work. NV, SMV, GSW, and ARMB wrote initial drafts of the paper. All authors provided important revisions.
Disclosure statement
No potential conflicts of interest were disclosed.
References
- 1.Alberts B, Johnson A, Lewis J, Alberts B, et al. MeMolecular biology of the cell. 4th. Garland Science; https://www.ncbi.nlm.nih.gov/books/NBK26878/ [Google Scholar]
- 2.Wegener KL, Campbell ID.. Transmembrane and cytoplasmic domains in integrin activation and protein-protein interactions (Review). Mol Membr Biol. 2008;25(5):376–10. doi: 10.1080/09687680802269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Postler TS, Desrosiers RC.. The Tale of the Long Tail: the Cytoplasmic Domain of HIV-1 gp41. J Virol. 2013;87(1):2–15. doi: 10.1128/JVI.02053-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43(3):189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451(7178):591–95. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cady SD, Luo W, Hu F, Hong M. Structure and function of the Influenza A M2 proton channel. Biochemistry. 2009;48(31):7356–64. doi: 10.1021/bi9008837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCown MF, Pekosz A. Distinct domains of the Influenza A virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J Virol. 2006;80(16):8178–89. doi: 10.1128/JVI.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beale R, Wise H, Stuart A, Ravenhill BJ, Digard P, Randow F. A LC3-Interacting Motif in the Influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014;15(2):239–47. doi: 10.1016/j.chom.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradbury A, Plückthun A. Reproducibility: standardize antibodies used in research. Nature. 2015;518(7537):27–29. doi: 10.1038/518027a. [DOI] [PubMed] [Google Scholar]
- 10.Bradbury ARM, Plückthun A. Getting to reproducible antibodies: the rationale for sequenced recombinant characterized reagents. Protein Eng Des Sel. 2015;28(10):303–05. doi: 10.1093/protein/gzv051. [DOI] [PubMed] [Google Scholar]
- 11.Bradbury ARM, Trinklein ND, Thie H, Wilkinson IC, Tandon AK, Anderson S, Bladen CL, Jones B, Aldred SF, Bestagno M, et al. When monoclonal antibodies are not monospecific: hybridomas frequently express additional functional variable regions. mAbs. 2018;10(4):539–46. doi: 10.1080/19420862.2018.1445456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradbury AM, Plückthun A. Antibodies: validate recombinants once. Nature. 2015;520(7547):295–295. doi: 10.1038/520295b. [DOI] [PubMed] [Google Scholar]
- 13.Bradbury ARM, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Velappan N, Mahajan A, Naranjo L, Velappan P, Andrews N, Tiee N, Chakraborti S, Hemez C, Gaiotto T, Wilson B, et al. Selection and characterization of FcεRI phospho-ITAM specific antibodies. mAbs. 2019;11(7):1206–18. doi: 10.1080/19420862.2019.1632113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabbard J, Velappan N, Di Niro R, Schmidt J, Jones CA, Tompkins SM, Bradbury ARM. A humanized anti-M2 scFv shows protective in vitro activity against influenza. Protein Eng Des Sel. 2008;22(3):189–98. doi: 10.1093/protein/gzn070. [DOI] [PubMed] [Google Scholar]
- 16.Sblattero D, Bradbury A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 2000;18(1):75–80. doi: 10.1038/71958. [DOI] [PubMed] [Google Scholar]
- 17.Ferrara F, Naranjo LA, Kumar S, Gaiotto T, Mukundan H, Swanson B, Bradbury ARM. Lenz LL. Using phage and yeast display to select hundreds of monoclonal antibodies: application to antigen 85, a tuberculosis biomarker. PLoS ONE. 2012;7(11):e49535. doi: 10.1371/journal.pone.0049535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markiv A, Beatson R, Burchell J, Durvasula RV, Kang AS. Expression of recombinant multi-coloured fluorescent antibodies in gor−/trxB− E. coli cytoplasm. BMC Biotechnol. 2011;11(1):117. doi: 10.1186/1472-6750-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markiv A, Anani B, Durvasula RV, Kang AS. Module based antibody engineering: A novel synthetic REDantibody. J Immunol Methods. 2011;364(1–2):40–49. doi: 10.1016/j.jim.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naranjo L, Ferrara F, Blanchard N, Demangel C, D’Angelo S, Erasmus MF, Teixera AA, Bradbury ARM. Recombinant Antibodies against Mycolactone. Toxins. 2019;11(6):346. doi: 10.3390/toxins11060346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang R, Song A, Levin J, DENNIS D, ZHANG N, YOSHIDA H, KORIAZOVA L, MADURA L, SHAPIRO L, MATSUMOTO A, et al. Therapeutic potential of a fully human monoclonal antibody against influenza A virus M2 protein. Antiviral Res. 2008;80(2):168–77. doi: 10.1016/j.antiviral.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Lee SC, Knowles TJ, Postis VLG, Jamshad M, Parslow RA, Lin Y-P, Goldman A, Sridhar P, Overduin M, Muench SP, et al. A method for detergent-free isolation of membrane proteins in their local lipid environment. Nat Protoc. 2016;11(7):1149–62. doi: 10.1038/nprot.2016.070. [DOI] [PubMed] [Google Scholar]
- 23.Lillo AM, Ayriss JE, Shou Y, Graves SW, Bradbury ARM. Development of Phage-Based Single Chain Fv Antibody Reagents for Detection of Yersinia pestis. PLOS ONE. 2011;6(12):e27756. doi: 10.1371/journal.pone. 0027756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov. 2018;17(1):35–56. doi: 10.1038/nrd.2017.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, et al. Matrix protein 2 of Influenza a virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6(4):367–80. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dumit VI, Dengjel J. Autophagosomal protein dynamics and influenza virus infection. Front Immunol. 2012:3. doi: 10.3389/fimmu.2012.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.J-D P, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24(1):79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 29.Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1′ specificity of tobacco etch virus protease. Biochem Biophys Res Commun. 2002;294(5):949–55. doi: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- 30.Kiss C, Fisher H, Pesavento E, Dai M, Valero R, Ovecka M, Nolan R, Phipps ML, Velappan N, Chasteen L, et al. Antibody binding loop insertions as diversity elements. Nucleic Acids Res. 2006;34(19):e132–e132. doi: 10.1093/nar/gkl681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reed LJ, Muench H. A Simple method of estimating fifty per cent endpoints 12. Am J Epidemiol. 1938;27(3):493–97. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]