

Abstract
Hydrophobic residues provide much of the thermodynamic driving force for the folding, self-assembly, and consequent hydrogelation of amphiphilic β-hairpin peptides. We investigate how the identity of hydrophobic side chains displayed from the hydrophobic face of these amphiphilic peptides influences their behavior to expound on the design criteria important to gel formation. Six peptides were designed that globally incorporate valine, aminobutyric acid, norvaline, norleucine, phenylalanine, or isoleucine on the hydrophobic face of the hairpin to study how systematic changes in hydrophobic content, β-sheet propensity, and aromaticity affect gelation. Circular dichroism (CD) spectroscopy indicates that hydrophobic content, rather than β-sheet propensity, dictates the temperature- and pH-dependent folding and assembly behavior of these peptides. Transmission electron microscopy (TEM) and small-angle neutron scattering (SANS) show that the local morphology of the fibrils formed via self-assembly is little affected by amino acid type. However, residue type does influence the propensity of peptide fibrils to undergo higher order assembly events. Oscillatory rheology shows that the mechanical rigidity of the peptide gels is highly influenced by residue type, but there is no apparent correlation between rigidity and residue hydrophobicity nor β-sheet propensity. Lastly, the large planar aromatic side chain of phenylalanine supports hairpin folding and assembly, affording a gel characterized by a rate of formation and storage modulus similar to the parent valine-containing peptide.
Graphical Abstract
1. INTRODUCTION
Peptides represent unique building blocks for the preparation of self-assembled materials. Peptide helices,1–3 β-strands,4–19 and sheet motifs20–23 have been employed to construct novel assembled architectures with applications in biomedicine.24–26 We have been developing self-assembling β-hairpin peptides that undergo environmentally triggered gelation.27–32 Our first de novo designed sequence named MAX1 is a small, 20-residue peptide that contains two strands of alternating lysine (Lys) and valine (Val) residues connected by a four-residue type II′ turn sequence (-VDPPT-) (Figure 1).31 When the peptide is initially dissolved in an aqueous buffer of low ionic strength at neutral or acidic pH, the positively charged Lys side chains keep the peptide soluble and in its monomeric unfolded state due to inter-residue electrostatic repulsion.
Figure 1.
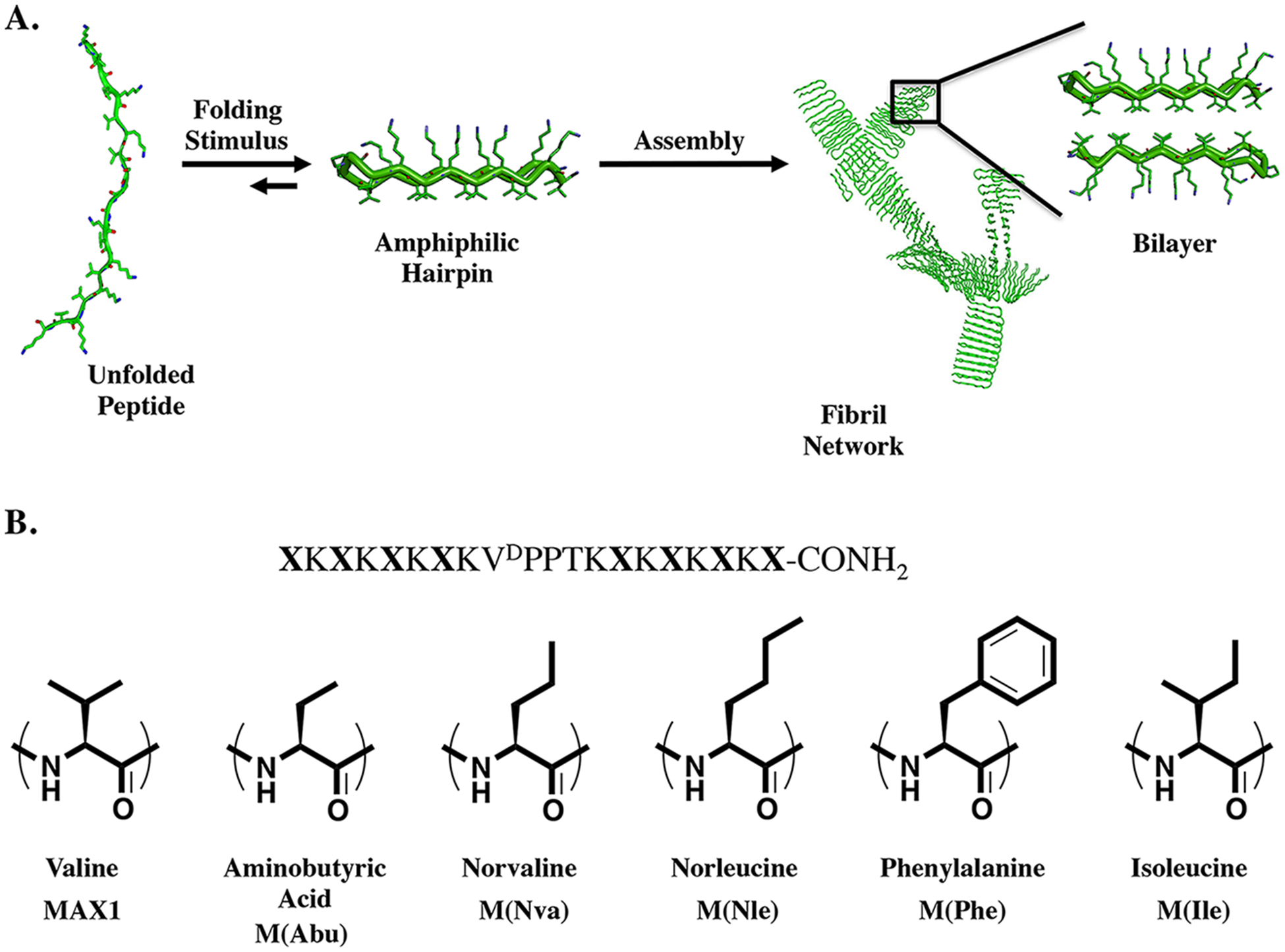
(A) Folding and self-assembly mechanism leading to fibril formation and ultimate gelation. Hydrophobic interactions drive bilayer formation, a critical feature of fibrillization. (B) Peptide sequence of studied peptides and the structures of residues occupying the hydrophobic face positions (X) of the amphiphilic hairpin.
However, the peptide can be triggered to fold into an amphiphilic β-hairpin conformation that rapidly self-assembles into a fibrillar network that constitutes the formation of a hydrogel. Thus, by triggering the initial peptide folding, one can trigger gelation, affording temporal control over material formation. Folding and assembly can be triggered by increasing the solution pH to deprotonate some of the lysines31 or by increasing the solution’s ionic strength,28 which screens the lysine-borne charge. In addition to these environmental triggers, increasing the temperature of the solution can drive folding and assembly via the hydrophobic effect.29 These three triggers are interdependent and can be used in concert to tune the solution conditions that support gelation. For example, a low ionic strength solution of MAX1 at pH 9 and 20 °C can be triggered to gel by simply raising the temperature. At pH 9.0, although most of the Lys side chains are protonated, a few are neutral, priming the peptide toward folding. Raising the temperature provides the needed energy to surmount the activation barrier en route to the folded and assembled state. In this mechanism, the ability of water to solubilize hydrophobic side chains, such as valine, is temperature dependent.33 At 20°C, the valine side chains are solubilized by ordered water. However, increasing the temperature drives the entropic release of the ordered water, which desolvates the valine side chains. In response, MAX1 folds and self-assembles to bury its hydrophobic isopropyl chains within the fibril where they are shielded from water. Thus, the free energy gained in shielding the hydrophobic portion of MAX1 from water outweighs the electrostatic repulsions that must be overcome for the peptide to fold. Transmission electron microscopy (TEM), atomic force microscopy (AFM), small-angle neutron scattering (SANS), and rheological experiments indicate that self-assembly results in the formation of a fibrillar network where each fibril is composed of a bilayer of hairpins that have intermolecularly hydrogen bonded along the long axis of a given fibril. The formation of the bilayer results from association of the hydrophobic valine-rich faces of the hairpin amphiphiles (Figure 1A). During assembly, noncovalent branch points are formed in the fibril network, which serve as physical cross-links that help define the mechanical properties of the gel.
This folding and assembly mechanism predicts that the formation of hydrophobic interactions, such as those responsible for bilayer formation, is important in driving the gelation of the peptide. In the folded and assembled state, the facially amphiphilic peptide packs its valine side chains into the interior of the fibril, providing a large part of the thermodynamic driving force for gelation. Valine was chosen in the initial design of MAX1 due to its hydrophobic content and experimentally measured propensity to adopt dihedral angles consistent with β-sheet secondary structure.34 With respect to residue placement, valine was incorporated uniformly on the peptide’s hydrophobic face (with the exception of one isostructural threonine residue proximal to the β-turn). When folded, this provides a smooth hydrophobic surface, void of topographical features, that can nonspecifically pack against like surfaces provided by other hairpins during self-assembly. Thus, the peptide has the opportunity to facially assemble not only in an ordered fashion where hairpins in the bilayer are in register, as is evident along the long axis of a given fibril, but also in an irregular fashion. Here, the hydrophobic face of one hairpin slips and rotates relative to its partner in the bilayer. This creates a nucleation point for the growth of a new fibril and constitutes the formation of a branch point in the network (Figure 1A). This nonspecific mode of packing is in contrast to the specific hydrophobic interactions that are formed during the folding of native proteins, such as the “knobs into holes” and “ridges into grooves” types of interactions.35 When proteins fold, the formation of these specific hydrophobic interactions helps to specify the formation of a well-defined native state conformation. Interestingly, the nonspecific hydrophobic interactions designed for in MAX1 are reminiscent of those observed in early de novo designed coiled coils that fold into a broad distribution of molten globule states.36
Although valine performs well in the context of MAX1, other hydrophobic residues, and even aromatic residues, should be capable of supporting self-assembly with the potential to modulate material behavior. For example, we recently reported on the design of β-hairpin peptides that incorporate alanine and naphthylalanine residues to impart specific packing interactions into the hydrophobic core of the bilayer.37 As expected, the specific packing of these side chains strongly hinders the fibril branching observed for the MAX1 networks. Aside from this limited study, little is known concerning the overall design plasticity of the hydrophobic face of the molecule. For example, although it seems reasonable that an amino acid must be hydrophobic to occupy a position on the hairpin’s apolar face, its unknown whether or not a high β-sheet propensity is mandatory. Herein, we investigate the effect of varying the identity of the residues occupying the hydrophobic face of MAX1 on its folding, self-assembly, fibril morphology, and the bulk rheological properties of its gels.
2. EXPERIMENTAL SECTION
2.1. General Methods and Materials.
Acetic acid (ammonium salt), anisole, boric acid, deuterated water, diisopropylethylamine (DIEA), 1,2-ethanedithiol, monohydrate of 2-morpholinoethanesulfonic acid (MES), piperidine, potassium fluoride, thioanisole, and trifluoroacetic acid (TFA) were purchased from Acros. Bis-tris propane (BTP) and boric acid were purchased from Sigma-Aldrich. Sodium chloride was purchased from Fisher Scientific. O-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole (HOBT) (anhydrous) were purchased from Senn Chemicals. Appropriately side-chain-protected Fmoc amino acids were purchased from Novabiochem. PL-Rink Resin was purchased from Polymer Laboratories. Sodium deuteroxide (6 M) was carefully prepared by adding small amounts of solid sodium metal to deuterated water chilled in an ice bath and then filtering away any particulate remaining in the liquid. Hydrogel formulations are represented in weight percent, as opposed to molarity, since each of the peptide’s molecular weights differ slightly. In general, a 1 wt % gel corresponds to a 2.75–3.18 mM peptide concentration range.
2.2. Peptide Synthesis.
All the peptides were prepared chemically by Fmoc-based solid phase peptide synthesis and purified extensively to near homogeneity (>98% purity).38 RP-HPLC chromatograms and mass spectroscopy data are provided in Figures S11–S16. The crude peptides (M(Abu), M(Nva), M(Nle), M(Phe), M(Ile)) were purified by RP-HPLC (preparative Vydac C18 peptide/protein column for M(Abu) and M(Nva); preparative Vydac C4 peptide/protein column for M(Nle), M(Phe), and M(Ile), starting with Std A followed by increasing concentrations of Std B or Std C (standards defined below), with a flow rate of 8 mL/min. M(Abu), M(Nva), M(Nle), and M(Phe) were purified at 20 °C using the Std A/Std B gradient, while M(Ile) was purified at 40 °C using the Std A/Std C gradient. Solvent A is 0.1% TFA in water; solvent B is 90% acetonitrile, 10% water, and 0.1% TFA; and solvent C is 60% acetonitrile, 30% isopropanol, 10% water, and 0.1% TFA. M(Abu) was purified using an isocratic gradient at 0% for 2 min and then linear gradients from 0% to 14% for 8 min and 14% to 100% for 166 min, with desired product elution at 32 min. M(Nva) was purified using an isocratic gradient at 0% for 2 min and then linear gradients from 0% to 22% over 3 min and 22% to 100% for 370 min, with desired product elution at 30 min. M(Nle) was purified using an isocratic gradient at 0% for 2 min and then linear gradients from 0% to 28% over 8 min and 28% to 100% for 144 min, with desired product elution at 26 min. M(Phe) was purified using an isocratic gradient at 0% for 2 min and then linear gradients from 0% to 30% over 3 min and 30% to 100% over 345 min, with desired product elution at 24 min. M(Ile) was purified using an isocratic gradient at 0% for 2 min and then linear gradients from 0% to 25% over 3 min and 25% to 100% over 150 min with desired product elution at 36 min. MAX1, ESI (M + 2H)2+ calcd 1115.8, obsd 1116.0; M(Abu), ESI (M + 2H)2+ calcd 1059.7, obsd 1059.7; M(Nva), ESI (M + 2H)2+ calcd 1115.8, obsd 1115.8; M(Nle), ESI (M + 2H)2+ calcd 1171.9, obsd 1171.9; M(Phe), ESI (M + 2H)2+ calcd 1307.8, obsd 1307.7; M(Ile), ESI (M + 2H)2+ calcd 1171.9, obsd 1171.8.
2.3. Circular Dichroism Spectroscopy.
CD spectra were collected on either an Aviv model 215 or a Jasco model J-810 spectropolarimeter. All wavelength and temperature-dependent spectra for all peptides were obtained in a 1 mm quartz cell. Wavelength scans were collected by scanning in 2 nm step intervals with a 3 s averaging time. Temperature-dependent experiments were performed starting at 5 °C and increasing the temperature in 5 °C steps through the thermal transition for each peptide using a 10 min equilibration time at each temperature. Data were averaged for 60 s at each temperature interval. All peptide sample solutions (pH 9, 125 mM borate and 10 mM KF or NaCl) were prepared by dissolving the peptide lyophilizate in water to half the final volume and chilling in an ice bath, followed by an equal volume addition of ice-cold, stock buffer solution (pH 9, 250 mM borate and 20 mM KF or NaCl) affording a final sample composed of 150 μM peptide, 125 mM borate, 10 mM KF or NaCl, pH 9.0. The pH was determined before and after buffer stock solution addition and after each CD experiment. The same approach was used for the preparation of pH-dependent (50 mM buffer, 10 mM NaCl) samples. The buffers used for the pH study were as follows: ammonium salt of acetic acid (pH 5); MES (pH 6); BTP (pH’s 7 and 8); boric acid (pH’s 9 and 9.8). Determination of concentration was achieved by monitoring absorbance at 220 nm (ε = 15 750 cm−1 M−1) for M(Abu), M(Nva), M(Nle), and M(Ile), and 257 nm (ε = 191 cm−1 M−1) for M(Phe). ε220 was used from the parent peptide (MAX1), which was determined by amino acid analysis, and ε257 was taken from literature.39 Mean residue ellipticity [θ] was calculated using the equation [θ] = (θobs/10lc)/r, where θobs is the observed ellipticity in millidegrees, l is the length of the cell (cm), c is the concentration (M), and r is the number of residues.
2.4. Oscillatory Rheology.
Dynamic frequency, strain, and time sweep rheology experiments were performed on either an Anton Paar-Physica modular compact rheometer or an Ares AR 2000 rheometer, equipped with a steel 25 mm parallel plate geometry tool. Samples were prepared by dissolving the peptide lyophilizate in ice chilled water to make a 2 wt % solution. This solution was diluted 1:1 with a 250 mM borate, 20 mM NaCl, pH 9 buffer solution, resulting in a 1 wt % peptide solution. Time-sweep experiments, monitoring the storage modulus (G′) and loss modulus (G″), were performed by adding the respective peptides solution to a cooled (5 °C) rheometer, and the tool was lowered to a height of 0.5 mm. The samples were protected from dehydration by the application of a layer of S6 viscosity reference standard oil (Cannon Instrument Company) around the edge of the tool during the experiment. A linear temperature ramp was then applied from 5 to 50 °C over 100 s for MAX1, M(Nva), M(Nle), M(Phe), and M(Ile) and held at 50 °C (well above the Tg for each of the peptides) for 60 min at constant strain (0.2%) and frequency (6 rad/s). For M(Abu), the temperature was ramped to 80 °C, a temperature above its Tg. Dynamic frequency and strain sweep experiments were performed to ensure that the time-sweep data were collected in the linear regime of strain and frequency. Briefly, immediately following the dynamic time sweep experiments, the temperature and strain (0.2%) were held constant, and the frequency varied from 0.1 to 100 rad/s. A dynamic strain sweep was subsequently performed in which the temperature and frequency (6 rad/s) were held constant and the strain varied from 0.1 to 1000% strain. Dynamic frequency and strain sweep data are provided in the Supporting Information.
2.5. Transmission Electron Microscopy.
Fibrils were isolated by diluting preformed hydrogels. The following methodology is representative for all peptide samples. A copper TEM grid, with a 20–30 nm film of pure carbon deposited on one side, was held at the edge of self-clamping tweezers. A small drop of the fibril sample was placed on the grid to form a bead. A drop of staining solution (2 wt % uranyl acetate in deionized water) was immediately added to prevent drying. After setting for 1–2 min, the stain was blotted off by carefully contacting with a piece of filter paper. Images were taken in bright-field mode with a JEOL 2000 transmission electron microscope at 200 kV accelerating voltage. Average fibril diameters were measured via ImageJ software by taking 15 independent measurements from distinct fibrils in the field of view.
2.6. Small-Angle Neutron Scattering.
Small-angle neutron scattering experiments were performed on 1 wt % peptide hydrogels in D2O with 50 mM BTP, 10 mM NaCl at pH 9.0. Data were collected at the National Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD, using the 30 m instrument installed on neutron guide 3 (NG-3). The neutron beam was monochromated to a mean wavelength of 6 Å with a mechanical velocity selector. The band-pass resolution of the selector for wavelength spread (Δλ/λ) was 0.15 (fwhm). The scattered neutrons were detected at three different sample-to-detector distances: 1.3, 4.0, and 13 m. These configurations allowed values of the scattering wave vector q in the range 0.004 < q (Å−1) < 0.500. Here, q is the scattering wave vector defined as q = (4/λ) sin(θ/2), where λ is the neutron wavelength (6 Å) and θ is the scattering angle. The spatial resolution of the 64 cm × 64 cm 2-D position sensitive proportional detector was 0.5 × 0.5 cm2 (fwhm). The resulting data were corrected for background electronic noise and radiation, detector inhomogeneity, and empty cell scattering. Intensities were normalized to an absolute scale relative to main-beam transmission measurements through the sample. The uncertainties of the I(q) versus q individual data points were calculated statistically from the number of averaged detector counts and are within the limits of the data point symbols. Cylinder form factor fits and the modified Guinier analyses were performed as previously described for MAX1.40
3. RESULTS AND DISCUSSION
3.1. Peptide Design.
A family of six peptides was studied, which included MAX1 (Figure 1B). The family was constructed by globally replacing the eight valines of MAX1 with aliphatic and aromatic residues. The peptides M(Abu), M(Nva), and M(Nle) contain aminobutyric, norvaline, and norleucine residues, respectively. The nomenclature represents the MAX1 peptide in which the eight valines have been globally replaced by the residue side chain indicated in the parentheses. This subfamily of three peptides replaces the β-branched, high sheet-forming propensity, isopropyl side chains of MAX1 with linear aliphatic groups whose length incrementally increases by one methylene. These peptides investigate if β-branched residues are requisite for β-sheet formation and provide a nicely controlled series where the side-chain length and hydrophobic character of the residue are varied incrementally. The remaining two peptides contain phenylalanine (M(Phe)) and isoleucine (M(Ile)). The M(Phe) peptide investigates the ability of aromatic side chains to support folding and assembly. In addition to being hydrophobic, Phe side chains can engage in potentially stabilizing inter-residue electronic interactions. For example, edge-to-face interactions and offset stacked interactions are observed in naturally occurring proteins and are thought to help stabilize and provide specificity to the folded state of the protein.41 Lastly, isoleucine, a β-branched residue, was incorporated affording M(Ile). Isoleucine is slightly more hydrophobic than valine and is regarded as having a higher propensity to form β-sheet structure.34 However, measured β-sheet propensity values for the naturally occurring amino acids are known to be context-dependent,42 so one must be careful in ascribing observed differences in peptide behavior alone to β-sheet propensity.
3.2. Peptide Folding and Assembly.
The propensity of each peptide to fold and assemble into β-sheet rich fibrils was assessed by circular dichroism (CD) spectroscopy by following the transition from random coil to β-sheet in response to an environmental trigger. Since each of the peptides varies with respect to the identity of its hydrophobic residues, modulating the solution temperature is a convenient way to trigger folding and assembly. Here, increasing the solution temperature drives the hydrophobic effect, which induces folding and assembly. This is exemplified in Figure 2 for M(Nva). At 5 °C, the peptide displays a spectrum having a negative value of mean residue ellipticity [θ] at 198 nm, which indicates the peptide is unfolded. However, raising the temperature to 80 °C affords a spectrum having a maximum at around 200 nm and a new minimum at around 218 nm. These bands are consistent with π−π* and n−π* electronic transitions for a peptide adopting β-sheet structure.43 In these experiments, 150 μM solutions of peptide are employed, which is convenient for CD spectroscopy. This concentration is not large enough to gel the sample volume but sufficient to support folding and assembly of the peptide into soluble fibrils. At higher peptide concentrations, such as those used in the rheology and SANS experiments (vide supra), a sufficient number of fibrils are formed that can percolate the solution to form a self-supporting gel. At any rate, the folding behavior of the nonbranched M(Nva) indicates that the inclusion of β-branched residues is not mandatory to support hairpin folding and assembly.
Figure 2.

Temperature-dependent CD measuring mean residue ellipticity as a function of wavelength for M(Nva); 150 μM peptide solution (pH 9, 125 mM borate, 10 mM NaCl).
Temperature-dependent CD was also used to assess the influence of hydrophobic content on folding and assembly. Since the hydrophobic effect is temperature-dependent,44 one might expect the more hydrophobic peptides to require less thermal energy to fold. This is borne out in Figure 3 where the evolution of β-sheet structure is measured as a function of temperature. Here the fraction of β-sheet (fβ) is plotted as a function of temperature as measured by CD. Panel A shows data for the linear aliphatic series M(Abu), M(Nva), and M(Nle). At low temperatures, both M(Abu) and M(Nva) are unfolded. As the temperature is raised, both fold and assemble into β-sheet rich fibrils in a temperature-dependent manner. At 5 °C, the lowest temperature investigated, the most hydrophobic peptide (M(Nle)) is largely already folded. The midpoint of the thermal transition (Tg) is the temperature at which half the peptide population has folded and provides a convenient parameter to compare the peptides. The Tg values for M(Abu), M(Nva), and M(Nle) are 70, 30, and <5 °C, respectively, indicating that as the residue side chain length increases, and thus its hydrophobicity, the temperature necessary to induce folding and assembly decreases.
Figure 3.
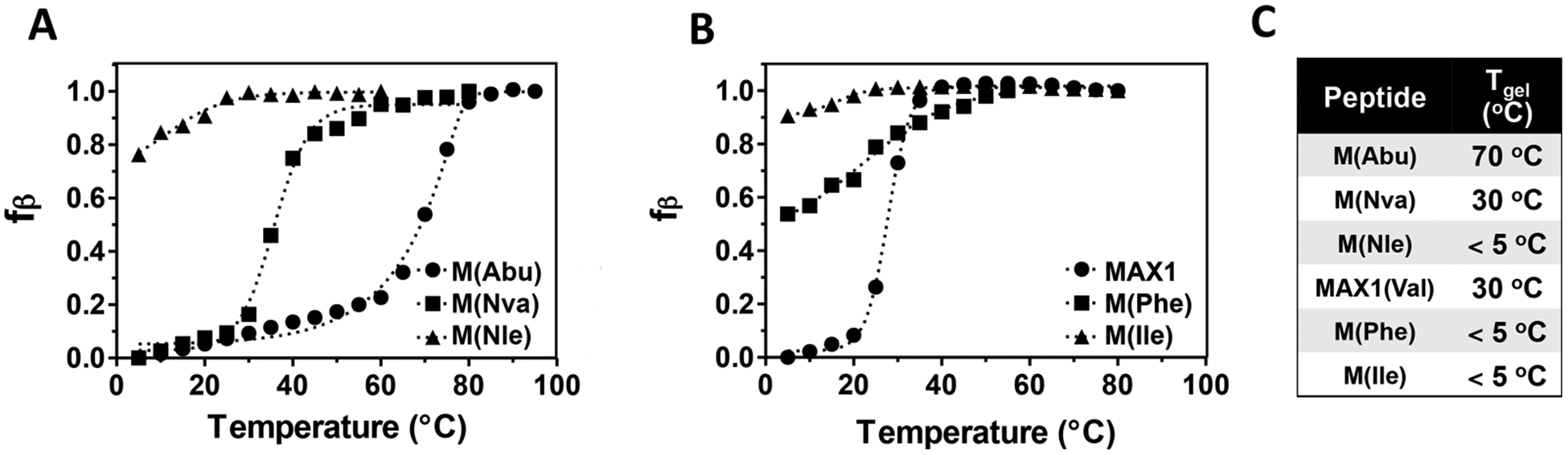
Temperature-dependent folding and assembly of peptides M(Abu), M(Nva), and M(Nle) (A) and MAX1, M(Phe), and M(Ile) (B). Fraction of β-sheet (fβ) was derived from CD data, which measured the increase in negative ellipticity at 218 nm as a function of temperature for 150 μM solutions of peptide (pH 9, 125 mM borate, 10 mM NaCl). (C) Midpoint of thermal transition temperatures.
Panel B shows similar data for MAX1, M(Phe), and M(Ile). The Tg for MAX1 is 30 °C. In comparison, the Tg for M(Ile) is less than 5 °C, indicating that the side chain of Ile facilitates folding and assembly more so than that of valine. This could be due to an increase in side chain hydrophobicity or inherent β-sheet propensity, as discussed earlier, or a combination of both. This question can be addressed by comparing the data for MAX1 and M(Nva). Both peptides contain isomeric side chains of the same number of carbons but differ in their arrangement. The β-branch valines of MAX1 have high β-sheet propensity whereas the n-propyl-containing norvaline residues should have a lower propensity to form sheet structure. Although their β-sheet propensities differ, these residues have similar calculated log P values (0.19 versus 0.20 for Val and Nva, respectively).45 Interestingly, MAX1 and M(Nva) have very similar temperature-dependent folding behavior with identical values of Tg (Figure 3C). The same trend is observed when comparing M(Nle) and M(Ile), which also have similar temperature-dependent folding behavior. Nle and Ile have different β-sheet propensities (based on the fact that Ile is β-branched and Nle is not) but similar log P values (0.60 versus 0.62 for Ile and Nle, respectively). This analysis suggests that residue hydrophobic content, as opposed to β-sheet propensity, plays the dominant role in defining the temperature-dependent folding in this family of peptides. Lastly, M(Phe) is characterized by a Tg of about 5 °C. Phenylalanine, although being aromatic is also hydrophobic, and thus M(Phe) requires little thermal energy to fold and assemble. Taken together, the CD data indicate that the side-chain identity of residues occupying the hydrophobic face of the hairpin amphiphile has a pronounced effect on the ability of these peptides to fold and assemble.
Solution pH can also be used to trigger folding and assembly. Figure 4 shows the fraction of β-sheet for the family of peptides as a function of pH, as determined by CD. In this experiment, the solution temperature is held constant at 60 °C, a common temperature where the entire class of peptides demonstrate sensitivity to changes in pH. As the solution pH is varied from acidic to basic, a population of the lysine side chains becomes deprotonated, enabling folding and assembly. Because of the high electropositive charge density present in the folded and assembled state, the intrinsic pKa of the Lys side chains is perturbed, and the magnitude of this perturbation is context dependent. For example, M(Abu), the least hydrophobic peptide, demands a high solution pH (>9) to begin its folding transition (Figure 4A). As the side-chain hydrophobicity progressively increases, as in M(Nva) and M(Nle), folding and assembly can occur at progressively lower values of pH. This behavior suggests that the driving force for folding/assembly imparted by the formation of hydrophobic contacts is large enough to significantly alter the intrinsic pKa of the Lys residues; as the hydrophobic content of the peptide increases, the Lys pKa decreases. This trend is also observed when comparing MAX1 and M(Ile) (Figure 4B). The Ile-containing peptide can fold and assemble at pH values as low as 7.0. Of note, the apparent decrease in fβ at pH 9.8 for MAX1 and M(Nle) is due to peptide precipitation and concomitant loss of CD signal. The data in panels A and B also suggest that the observed pH dependency for this family of peptides is largely driven by hydrophobicity and not differences in residue β-sheet propensity. For example, the pH-dependent behavior of M(Nle) and M(Ile) (panels A and B, respectively) are extremely similar despite having different propensities to form sheet structure. Interestingly, the phenyalanine-containing peptide, M(Phe), displays an almost linear dependency on pH that is distinct from all the other peptides, suggesting that Phe-Phe electronic interactions, in addition to hydrophobic interactions, may be playing a role in its behavior. Taken together, the data indicate that the temperature- and pH-dependent folding/assembly behavior of these amphiphilic hairpins is dependent on their hydrophobic content, which can be easily modulated by altering the residues on their hydrophobic face. The data also show that simple design principles can be used to generate peptides that fold, assemble, and gel under very specific solution conditions.
Figure 4.
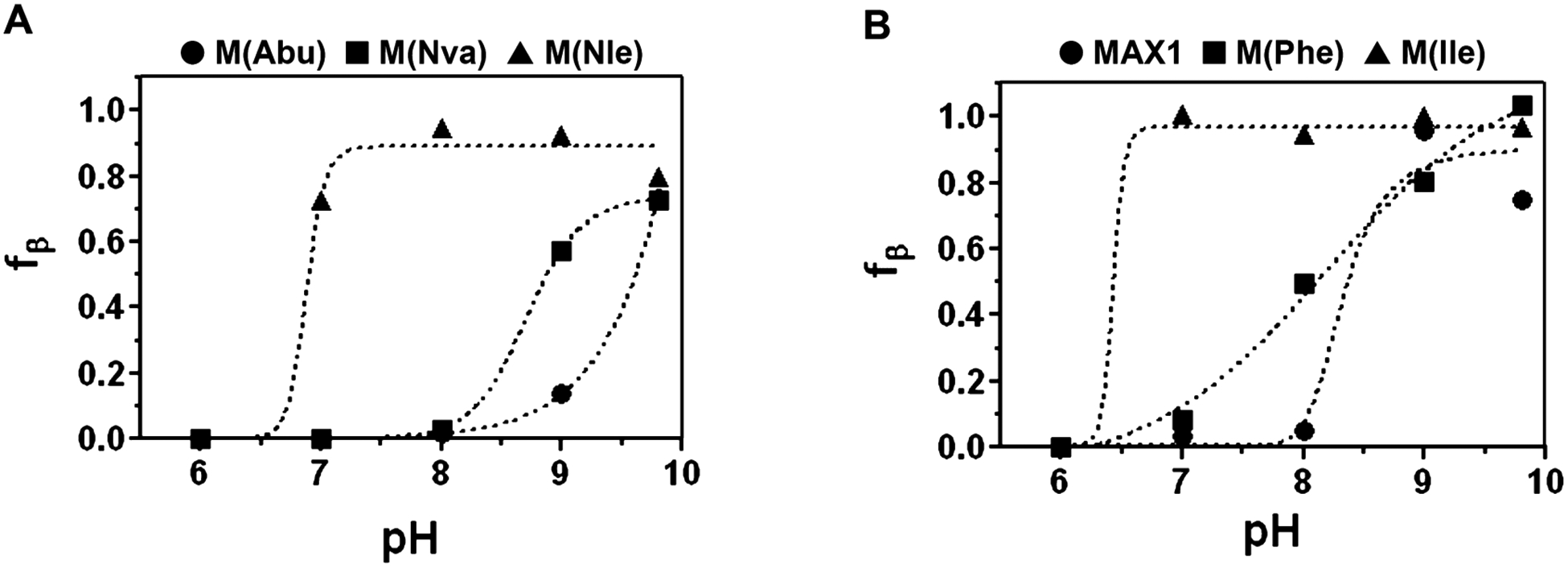
pH-dependent folding and assembly of peptides M(Abu), M(Nva), and M(Nle) (A) and MAX1, M(Phe), and M(Ile) (B). Fraction of β-sheet (fβ) was derived from CD data, which measured the increase in negative ellipticity at 218 nm as a function of solution pH for 150 μM solutions of peptide (50 mM buffer, 10 mM NaCl, 60 °C).
3.3. Rheological Properties of Peptide Hydrogels.
As opposed to the micromolar solutions of peptide used in the CD experiments outlined above, 1 wt % (i.e., millimolar) solutions of peptide will form self-supporting hydrogels after folding and assembly is triggered. The evolution of the fibril network constituting the formation of the gel can be followed in time-sweep rheological experiments that monitor the storage modulus (G′), which is a measure of the gel’s mechanical rigidity, as a function of time. Here, 1 wt % solutions of peptide (pH 9) are deposited on the rheometer at 5 °C. Folding and assembly is initiated by ramping the temperature above the Tg, which was previously determined by CD spectroscopy. For M(Abu), the temperature was ramped to 80 °C, and for the remaining family members, a temperature of 50 °C was employed. Figure 5A shows data for M(Abu). The initial lag time corresponds to the time necessary to ramp the temperature from 5 to 80 °C. After which time, the peptide folds and assembles quickly forming a hydrogel within seconds that further rigidifies with time. The resultant gel is characterized by a final storage modulus of ~1090 Pa. Panel B shows time-sweep data for the remaining peptides. It is clear from the data that the identity of the amino acid side chains occupying the hydrophobic face of the hairpin influences the mechanical properties of the hydrogel. For example, the linear propyl group of norvaline results in the stiffest gel (3300 Pa) whereas norleucine, the weakest (680 Pa). Close inspection of the data also indicates that material stiffness does not correlate with peptide hydrophobic content. For example, in the linear side-chain series, M(Nva) and M(Nle), the most hydrophobic peptide affords the weakest gel network. This is also the case for MAX1 versus M(Ile), with M(Ile) forming the weaker gel. Material stiffness also does not necessarily correlate with the β-sheet propensities of the residues comprising the peptides (compare M(Nva) to MAX1). Dynamic frequency- and strain-sweep experiments Figures S1–S6 were performed for each peptide gel to ensure that the time-sweep data were collected in the linear viscoelastic regime.
Figure 5.
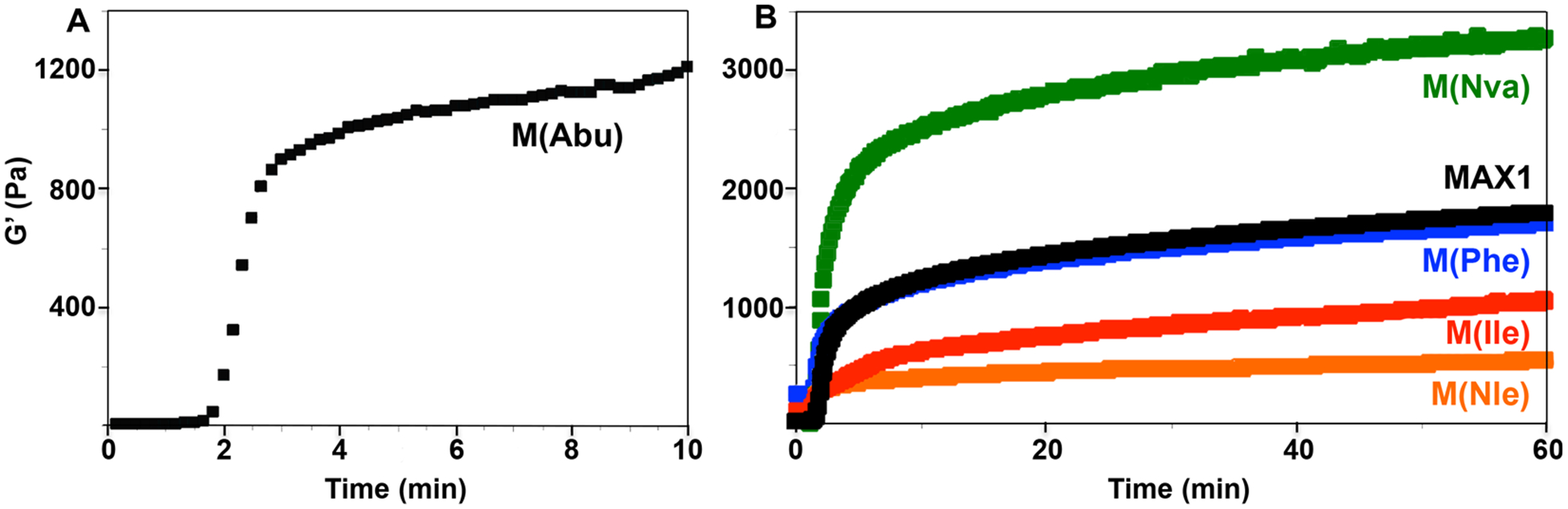
Time-sweep oscillatory rheology (frequency = 6 rad/s and 0.2% strain) measuring the storage modulus as a function of time after gelation is initiated for (A) M(Abu) and (B) M(Nva), M(Nle), M(Phe), M(Ile), and MAX1.
The fact that material stiffness does not correlate with general descriptors such as hydrophobic content nor amino acid secondary structure propensities is not surprising. Rather, material stiffness for semiflexible networks should be dependent on both the cross-link density of the network and the stiffness of the individual fibrils comprising the network.46 Both of these factors should, in part, depend on the exact structure of the side chains and their specific spatial presentation from the hydrophobic face of the hairpin. For example, side-chain packing interactions within the fibril bilayer that lead to stiffer fibrils would, in turn, lead to stiffer gels. Likewise, side chains capable of forming smooth featureless hydrophobic surfaces when displayed from the hydrophobic face of a hairpin may promote the formation of branch points in the network, increasing network stiffness.
3.4. Local Fibril Morphology.
Transmission electron microscopy (TEM) was employed to gain insight into how side chain type might be influencing local fibril morphology. For these studies M(Abu) was excluded due to the high temperature necessary to induce fibrillization. As a reference, Figure 6A shows a representative micrograph of MAX1 fibrils formed from its self-assembly. It is clear that MAX1 assembles with high fidelity, forming single fibrils that are extremely homogeneous in morphology. The measured width of the fibrils (3.5 nm, Figure 6F) is similar to the length of a folded hairpin in the self-assembled state (~3 nm). Earlier AFM measurements confirmed a fibril height of 2.5 nm.47 Thus, the fibrils imaged in panel A consist of a bilayer of hairpins that have assembled laterally along the fibril’s long axis, consistent with the model shown in Figure 1A. The fibrils formed by M(Nva), shown in panel B, show similar morphology at the level of single fibrils with an average width of 3.3 nm. M(Nle), M(Phe), and M(Ile) also assemble into fibrils having similar local morphology with average fibril widths of 3.2, 3.4, and 3.3 nm, respectively (panel F). Thus, TEM suggests that fibril formation is tolerant of residue identity for this series of peptides. The micrographs of M(Nva), M(Nle), and M(Phe) suggest that fibrils formed by these peptides may have the propensity to undergo higher order assembly processes forming laminates, ropes and braids, respectively. However, one must be conservative in making such assertions based on TEM data alone. Although TEM accurately reports on the local structure of the fibrils as they are presented on a two-dimensional surface, the micrographs in Figure 6 cannot be used to make inferences regarding higher-order structures that may be present in the three-dimensional network of the gel. In order to investigate fibril morphology in the gel state, small-angle neutron scattering (SANS) of intact gels was employed.
Figure 6.
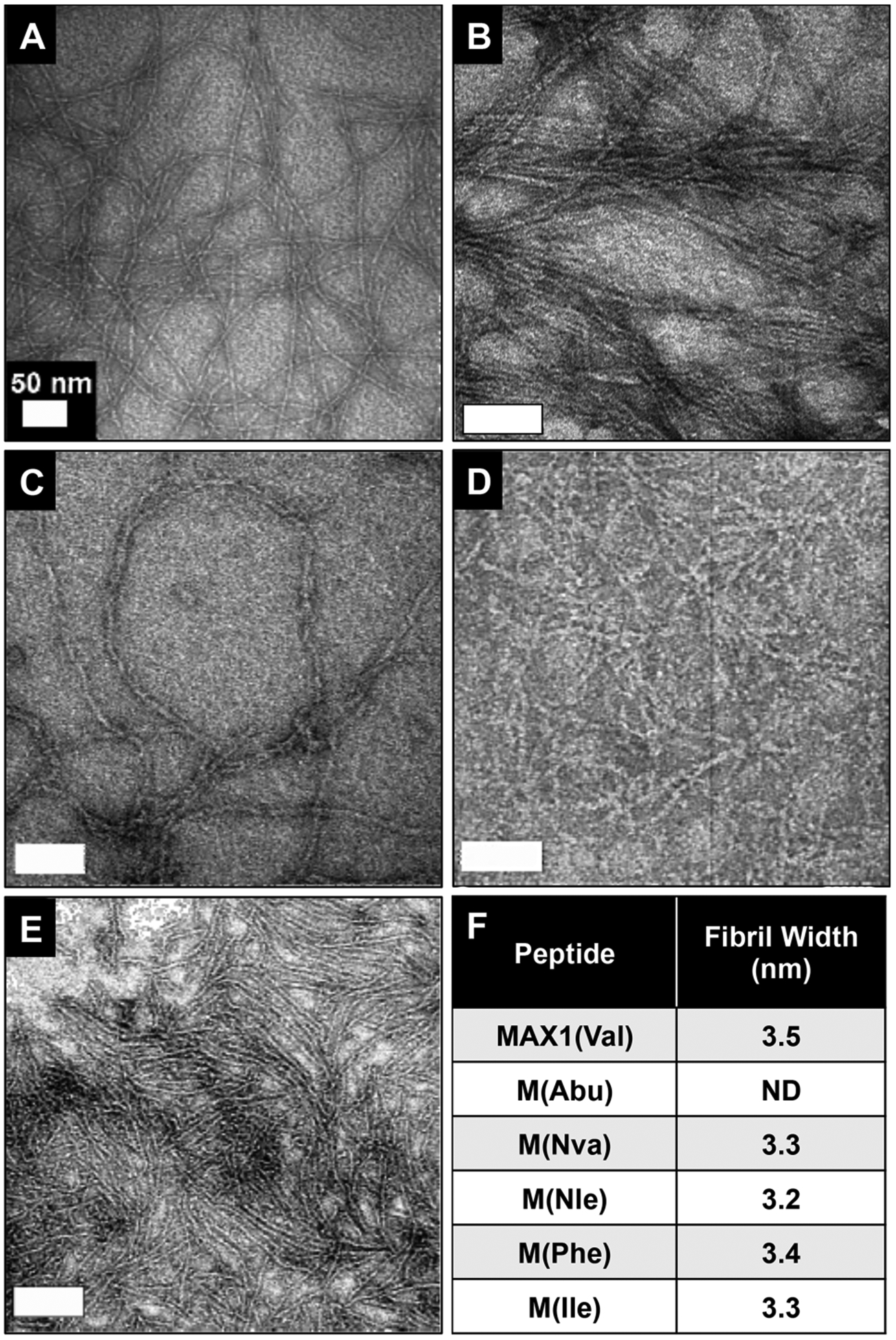
TEM of 1 wt % (A) MAX1, (B) M(Nva), (C) M(Nle), (D) M(Phe), and (E) M(Ile) fibrils prepared in 125 mM borate buffer, 10 mM NaCl at pH 9 and stained with uranyl acetate. (F) Average fibril widths calculated from TEM via direct multiple measurements, n = 15; ND (not determined).
Figure 7A shows the scattering curve for a M(Nva) gel fit to a cylinder form factor.40 There is good agreement between the fit and the data indicating that the fibrils can be approximated by cylindrical geometries. A plot of log I(q) versus log q using data from the low- to mid-q regime (0.01–0.08 Å−1) provides a slope of −1, suggesting that the self-assembled fibrils are rodlike objects (Figure S7 inset). The excellent fit in Figure 7A provides a cylindrical diameter of 3.4 nm, which is further supported by the modified Guinier analysis48 shown in Figure 7B. The Guinier analysis provides a cross-sectional radius of gyration of a rod, Rc, which can be converted to a radius R = Rc√2 and diameter of 2.9 nm. The diameters measured directly by TEM and through SANS analysis are internally consistent and support the fibril model shown in Figure 1. Concerning the possible higher-order formation of laminates suggested by TEM (Figure 6B), the SANS data shown in Figure 7A are featureless in the mid-q regime (~0.08 Å−1), indicating that laminates most likely do not exist in the gel state. Thus, the laminates observed in Figure 6B are perhaps formed as a result of drying or surface effects experienced during sample preparation.
Figure 7.
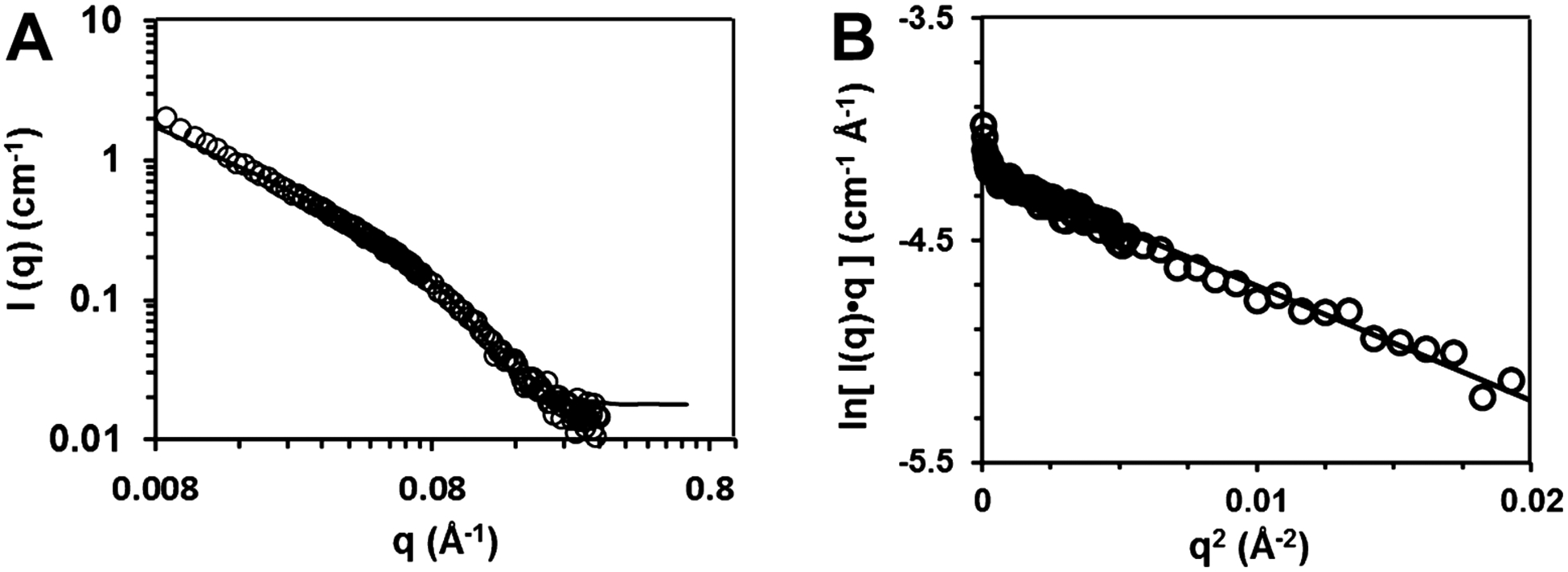
SANS data for 1 wt % M(Nva) hydrogels in D2O with 50 mM BTP, 10 mM NaCl at pH 9. (A) Plot of log I (intensity) vs log q (scattering vector) fit using a rigid-cylindrical form factor (solid line) to extract values of cylindrical diameter. (B) Plot of ln[I(q)q] vs q2 of data from the low-q regime. The slope of a linear fit to the data (solid line) allowed for calculation of fibril cross-sectional diameter using a modified Guinier analysis.
Plots of log I(q) versus log q using data from the low-q regime (0.01–0.08 Å−1) for M(Nle), M(Phe), and M(Ile) gels all provided slopes near −1, indicating that fibrils formed by these peptides behave as rodlike objects (Figures S8–S10 insets). Interestingly, M(Nle) and M(Phe) gels provided plots of I(q) versus q that have distinct features in the mid-q regime (~0.08 Å−1), suggesting that the higher-order assemblies observed by TEM likely exist in the gel state (Figures S8 and S9). The scattering curve for the M(Ile) gel was featureless in the mid-q regime, indicating that its fibrils do not undergo higher-order assembly processes in the gel state (Figure S10A). The scattering curve of the M(Ile) gel was further analyzed in a similar manner as the M(Nva) gel, providing a cylindrical diameter of 3.8 nm, which was further supported by a modified Guinier analysis that provided a diameter of 3.4 nm (Figure S10B). Again, the diameters derived from TEM and SANS are similar, providing further confidence in the proposed mechanism of fibril formation. Taken together, the TEM and SANS data show that while all the peptides can form fibrils of similar local morphology, the identity of the amino acids on the hairpin’s hydrophobic face can influence the potential to form higher-order assemblies. For example, in the self-assemble fibrillar state of a given peptide, if portions of its hydrophobic side chains are not well-packed and thus not fully buried within the interior of a single fibril, these exposed surfaces could facilitate the formation of interfibril hydrophobic contacts, resulting in higher-order assemblies.
4. CONCLUSION
The influence of amino acid type displayed from the hydrophobic face of amphiphilic self-assembling β-hairpin peptides on their folding, self-assembly, local fibril morphology, and hydrogel rheological properties was examined. We find that hydrophobic content, and not necessarily β-sheet propensity, influences the temperature- and pH-dependent folding and assembly behavior of this class of amphiphiles. Peptides containing residues of greater hydrophobic content displayed from their hydrophobic faces have a greater propensity to fold and self-assemble. More hydrophobic peptides fold and assemble at lower temperatures and lower values of pH. The local morphology of the fibrils formed by this peptide class is influenced little by amino acid type. As was shown, when the hydrophobic residue was valine, aminobutyric acid, norvaline, norleucine, phenyalanine, or isoleucine, fibrils of well-defined local morphology were realized. However, residue type does dictate the potential of these fibrils to undergo higher-order assembly into ropes (Nle) and braids (Phe). Lastly, we find that amino acid type strongly influences the rheological properties of the gel. However, there is no direct correlation between amino acid hydrophobicity nor β-sheet propensity on the mechanical rigidity of the gels formed from this class of peptides. Thus, the hydrophobic face of these self-assembling hairpin peptides can be modulated by design in a straightforward manner to facilitate gelation at desired solution conditions, but as of yet, it is impossible to predict how changes of amino acid composition effects material stiffness nor higher-order assembly processes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by funding from the Intramural Research Program of the National Cancer Institute of the National Institutes of Health and also by the cooperative agreement 70NANB12H239 from NIST, U.S. Department of Commerce, and the UD-NIST Center for Neutron Science. The statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the view of NIST or the Department of Commerce.
Footnotes
The authors declare no competing financial interest.
Supporting Information
Figures S1–S16. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Fletcher JM; Harniman RL; Barnes FRH; Boyle AL; Collins A; Mantell J; Sharp TH; Antognozzi M; Booth PJ; Linden N; Miles MJ; Sessions RB; Verkade P; Woolfson DN Science 2013, 340 (6132), 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).O’Leary LER; Fallas JA; Bakota EL; Kang MK; Hartgerink JD Nat. Chem 2011, 3 (10), 821–828. [DOI] [PubMed] [Google Scholar]
- (3).Stahl PJ; Yu SM Soft Matter 2012, 8 (40), 10409–10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Amit M; Cheng G; Hamley IW; Ashkenasy N Soft Matter 2012, 8 (33), 8690–8696. [Google Scholar]
- (5).Bakota EL; Sensoy O; Ozgur B; Sayar M; Hartgerink JD Biomacromolecules 2013, 14 (5), 1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Davies RPW; Aggeli AJ Pept. Sci 2011, 17 (2), 107–114. [DOI] [PubMed] [Google Scholar]
- (7).Feng Y; Taraban M; Yu YB Soft Matter 2012, 8 (46), 11723–11731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Geisler IM; Schneider JP Adv. Funct. Mater 2012, 22 (3), 529–537. [Google Scholar]
- (9).Jiang T; Xu CF; Liu Y; Liu Z; Wall JS; Zuo XB; Lian TQ; Salaita K; Ni CY; Pochan D; Conticello VP J. Am. Chem. Soc 2014, 136 (11), 4300–4308. [DOI] [PubMed] [Google Scholar]
- (10).Khoe U; Yang YL; Zhang SG Langmuir 2009, 25 (7), 4111–4114. [DOI] [PubMed] [Google Scholar]
- (11).Lin YA; Ou YC; Cheetham AG; Cui HG Biomacromolecules 2014, 15 (4), 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lowik D; Garcia-Hartjes J; Meijer JT; van Hest JCM Langmuir 2005, 21 (2), 524–526. [DOI] [PubMed] [Google Scholar]
- (13).Lu K; Jacob J; Thiyagarajan P; Conticello VP; Lynn DG J. Am. Chem. Soc 2003, 125 (21), 6391–6393. [DOI] [PubMed] [Google Scholar]
- (14).Matsumura S; Uemura S; Mihara H Chem.-Eur. J 2004, 10 (11), 2789–2794. [DOI] [PubMed] [Google Scholar]
- (15).Roberts D; Rochas C; Saiani A; Miller AF Langmuir 2012, 28 (46), 16196–16206. [DOI] [PubMed] [Google Scholar]
- (16).Rudra JS; Mishra S; Chong AS; Mitchell RA; Nardin EH; Nussenzweig V; Collier JH Biomaterials 2012, 33 (27), 6476–6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sakai H; Watanabe K; Asanomi Y; Kobayashi Y; Chuman Y; Shi LH; Masuda T; Wyttenbach T; Bowers MT; Uosaki K; Sakaguchi K Adv. Funct. Mater 2013, 23 (39), 4881–4887. [Google Scholar]
- (18).Segman S; Lee MR; Vaiser V; Gellman SH; Rapaport H Angew. Chem., Int. Ed 2010, 49 (4), 716–719. [DOI] [PubMed] [Google Scholar]
- (19).Swanekamp RJ; DiMaio JTM; Bowerman CJ; Nilsson BL J. Am. Chem. Soc 2012, 134 (12), 5556–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Colfer S; Kelly JW; Powers ET Langmuir 2003, 19 (4), 1312–1318. [Google Scholar]
- (21).Rughani RV; Salick DA; Lamm MS; Yucel T; Pochan DJ; Schneider JP Biomacromolecules 2009, 10 (5), 1295–1304. [DOI] [PubMed] [Google Scholar]
- (22).Valery C; Paternostre M; Robert B; Gulik-Krzywicki T; Narayanan T; Dedieu JC; Keller G; Torres ML; Cherif-Cheikh R; Calvo P; Artzner F Proc. Natl. Acad. Sci. U. S. A 2003, 100 (18), 10258–10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang C; Huang LX; Wang LJ; Hong YK; Sha YL Biopolymers 2007, 86 (1), 23–31. [DOI] [PubMed] [Google Scholar]
- (24).Branco MC; Schneider JP Acta Biomater. 2009, 5 (3), 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Branco MC; Sigano DM; Schneider JP Curr. Opin. Chem. Biol 2011, 15 (3), 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Veiga AS; Schneider JP Biopolymers 2013, 100 (6), 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Knerr PJ; Branco MC; Nagarkar R; Pochan DJ; Schneider JP J. Mater. Chem 2012, 22 (4), 1352–1357. [Google Scholar]
- (28).Ozbas B; Kretsinger J; Rajagopal K; Schneider JP; Pochan DJ Macromolecules 2004, 37 (19), 7331–7337. [Google Scholar]
- (29).Pochan DJ; Schneider JP; Kretsinger J; Ozbas B; Rajagopal K; Haines LJ Am. Chem. Soc 2003, 125 (39), 11802–11803. [DOI] [PubMed] [Google Scholar]
- (30).Rughani RV; Branco MC; Pochan D; Schneider JP Macromolecules 2010, 43 (19), 7924–7930. [Google Scholar]
- (31).Schneider JP; Pochan DJ; Ozbas B; Rajagopal K; Pakstis L; Kretsinger JJ Am. Chem. Soc 2002, 124 (50), 15030–15037. [DOI] [PubMed] [Google Scholar]
- (32).Sinthuvanich C; Haines-Butterick LA; Nagy KJ; Schneider JP Biomaterials 2012, 33 (30), 7478–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Privalov PL Crit. Rev. Biochem. Mol. Biol 1990, 25 (4), 281–305. [DOI] [PubMed] [Google Scholar]
- (34).Minor DL; Kim PS Nature 1994, 367 (6464), 660–663. [DOI] [PubMed] [Google Scholar]
- (35).Lupas AN; Gruber M Fibrous Proteins 2005, 70, 37−+. [Google Scholar]
- (36).Hill RB; Raleigh DP; Lombardi A; Degrado WF Acc. Chem. Res 2000, 33 (11), 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sathaye SZH; Sonmez C; Schneider JP; MacDermaid CM; Von Bargen CD; Saven JG; Pochan DJ Biomacromolecules 2014, 15 (11), 3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nagarkar RP; Schneider JP; Gazit E; Nussinov R Methods Mol. Biol 2008, 474, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Fasman GD Handbook of Biochemistry and Molecular Biology; CRC Press: Boca Raton, FL, 1976; Vol. 1. [Google Scholar]
- (40).Ozbas B; Rajagopal K; Schneider JP; Pochan DJ Phys. Rev. Lett 2004, 93 (26), 268106. [DOI] [PubMed] [Google Scholar]
- (41).Singh J; Thornton JM FEBS Lett. 1985, 191 (1), 1–6. [Google Scholar]
- (42).Minor DL; Kim PS Nature 1994, 371 (6494), 264–267. [DOI] [PubMed] [Google Scholar]
- (43).Manning MC; Illangasekare M; Woody RW Biophys. Chem 1988, 31 (1–2), 77–86. [DOI] [PubMed] [Google Scholar]
- (44).Privalov PL; Gill SJ Adv. Protein Chem 1988, 39, 191–234. [DOI] [PubMed] [Google Scholar]
- (45).Log P values were calculated for Ac-Val-OMe, A.-N.-O., Ac-Ile-OMe, and Ac-Nle-OMe using Spartan software.
- (46).Mackintosh FC; Kas J; Janmey PA Phys. Rev. Lett 1995, 75 (24), 4425–4428. [DOI] [PubMed] [Google Scholar]
- (47).Nagy KJ; Giano MC; Jin A; Pochan DJ; Schneider JP J. Am. Chem. Soc 2011, 133 (38), 14975–14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Burkoth TS; Benzinger TLS; Urban V; Morgan DM; Gregory DM; Thiyagarajan P; Botto RE; Meredith SC; Lynn DG J. Am. Chem. Soc 2000, 122 (33), 7883–7889. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.