

Abstract
While low-temperature (−78 °C) reaction of the lithium dithiolene radical (1•) with boron bromide gives the dibromoboron dithiolene radical (2•), the parallel reaction of 1• with (C6H11)2BCl (0 °C) affords the dicyclohexylboron dithiolene radical (3•). Radicals 2• and 3• are characterized by single crystal X-ray diffraction, UV-vis, and EPR spectroscopy. The nature of these radicals was also probed computationally. Under mild conditions, 3• undergoes unexpected thiourea-mediated B–C bond activation, giving zwitterion 4, which may be regarded as an anionic dithiolene-modified-carbene complex of the sulfenyl cation (RS+, R = cyclohexyl).
Keywords: B-C activation, boron, carbenes, dithiolene, radicals
Graphical Abstract
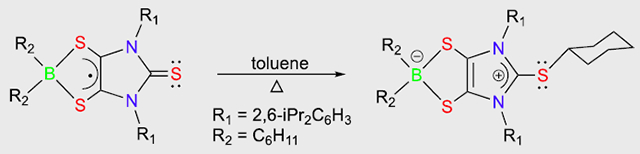
The dicyclohexylboron dithiolene radical undergoes unexpected thiourea-mediated B–C bond activation, giving a zwitterionic product, which may be regarded as an anionic dithiolene-modified-carbene complex of the sulfenyl cation (RS+, R = cyclohexyl).
The unique optical, conductive, magnetic, and catalytic properties of transition metal-dithiolene complexes have long fascinated chemists.[1] The “non-innocent” character[1c] of dithiolene ligands (Figure 1) has been shown to play a pivotal role in such disparate fields as materials science[1d,1j] and biological systems.[1b,1k] Indeed, such “non-innocent” character of transition metal-dithiolenes were recently probed by sulfur K-edge X-ray absorption spectroscopy (XAS).[1h]
Figure 1.
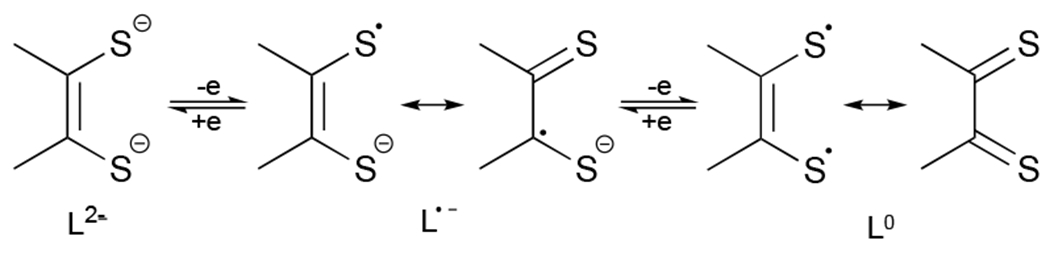
Redox non-innocence of a dithiolene ligand.[2]
The presence of radical anionic ligands (L•−) in transition metal-dithiolene complexes was notably predicted by Gray more than five decades ago.[3] Subsequently, the radical character of ligands in transition metal-dithiolenes has been extensively studied.[1h,4] Notably, the literature does not reveal reports of p-block element-containing dithiolene radicals. This laboratory recently reported the first structurally characterized anionic dithiolene radical (1•)[5] (Scheme 1) via tri-sulfurization of the corresponding anionic N-heterocylic dicarbene (NHDC).[6] Significantly, 1• provides a unique platform from which a variety of dithiolene-based p-block species may be conveniently accessed.
Scheme 1.
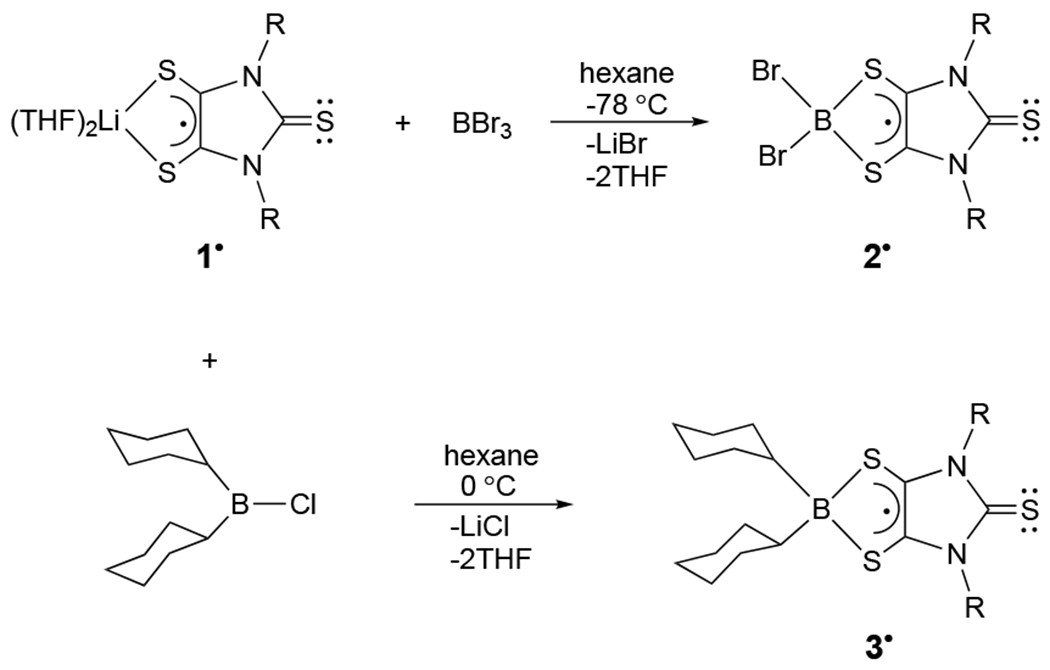
Synthesis of boron dithiolene radicals 2• and 3• (R = 2,6-diisopropylphenyl).
Radicals involving boron atoms[7] are intriguing not only for their interesting structures and bonding motifs, but also due to their potentially broad applications in chemical sensors, polymerization initiators, reagents for organic synthesis and building blocks for magnetic systems.[8] Compared to anionic boron radicals (BR3•−), neutral radicals (BR2•) are more reactive due to the enhanced electron deficiency of the two-coordinate boron atom (which are usually complexed by Lewis bases). The reported neutral three-coordinate L:BR2• species usually employ amines, phosphines, N-heteroaryls, and carbenes as the Lewis base ligands (L:).[7b] In addition, neutral β-diiminate-based three-coordinate diazaborocyclic radical (Figure 2a) has been reported, wherein the unpaired electron is delocalized over the six-membered C3N2B ring.[9] Bidentate heterocyclic ligands are effective for achieving boron-containing radicals. Indeed, five-membered diazaborocyclic (Figure 2b and 2c)[10] and dioxoborocyclic (Figure 2d)[11] radicals, containing four-coordinate boron, have been synthesized. Interestingly, synthesis of dioxoborocycle (Figure 2d) and its 4,5-pyrenedione-derived analogues involve H2 activation with polyaromatic diones and B(C6F5)3 as frustrated Lewis pairs.[11] Notably, the dithioborocyclic radicals have not been reported. Herein, we report the synthesis,[12] structures,[12] EPR,[12] UV-vis,[12] and computations[12] of the first boron dithiolene radicals (2• and 3•, Scheme 1). Surprisingly, 3• readily undergoes thiourea-mediated boron-carbon bond activation under mild conditions, giving diamagnetic zwitterion 4 (Scheme 2), which may be regarded as an anionic dithiolene-modified-carbene complex of the sulfenyl cation (RS+, R = cyclohexyl). Activation of boron-carbon bonds with high bond dissociation energy (BDEB-C = ca. 360 KJ mol−1 vs. BDEC-C = ca. 350 KJ mol−1) under mild conditions has been well explored.[13] Transition-metal species are well known for their utility in boron-carbon bond activation [i.e., palladium(0) species in Suzuki coupling[14] between organoboranes and organic halides].[15] In contrast, main group species-mediated B–C bond activation has only been scarcely documented.[13,16] The synthesis of 4 represents the first example of thiourea-mediated B–C bond activation.
Figure 2.
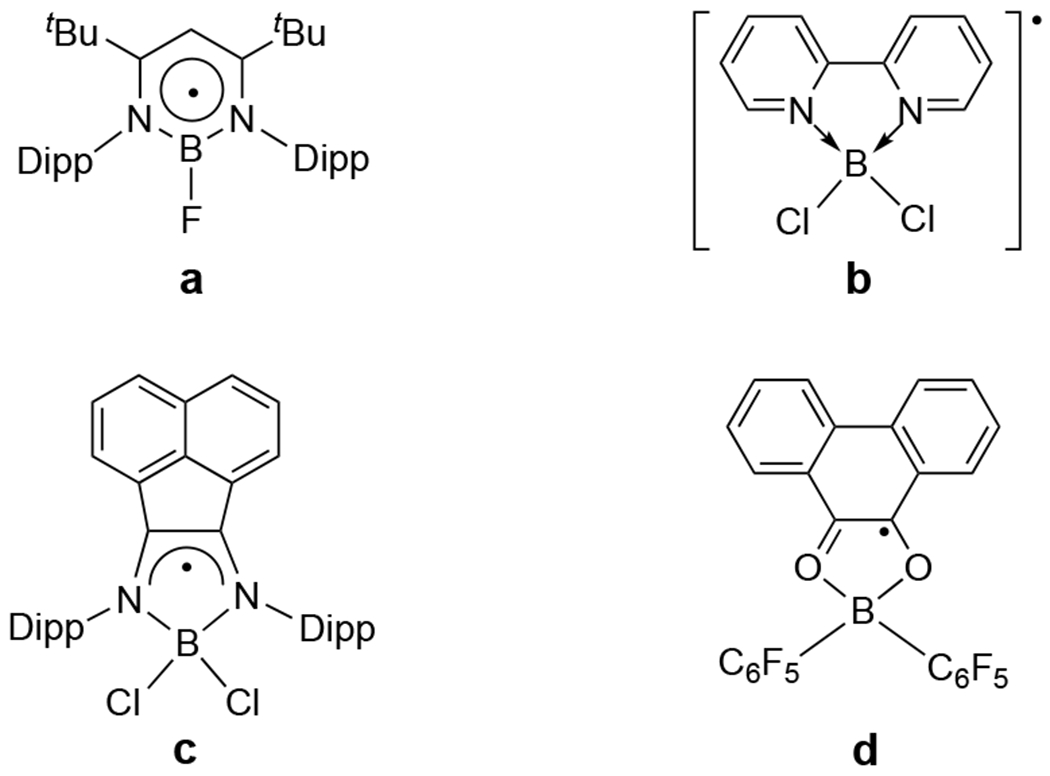
Representative neutral diazaborocyclic (a–c) and dioxoborocyclic (d) radicals (Dipp = 2,6-diisopropylphenyl).
Scheme 2.
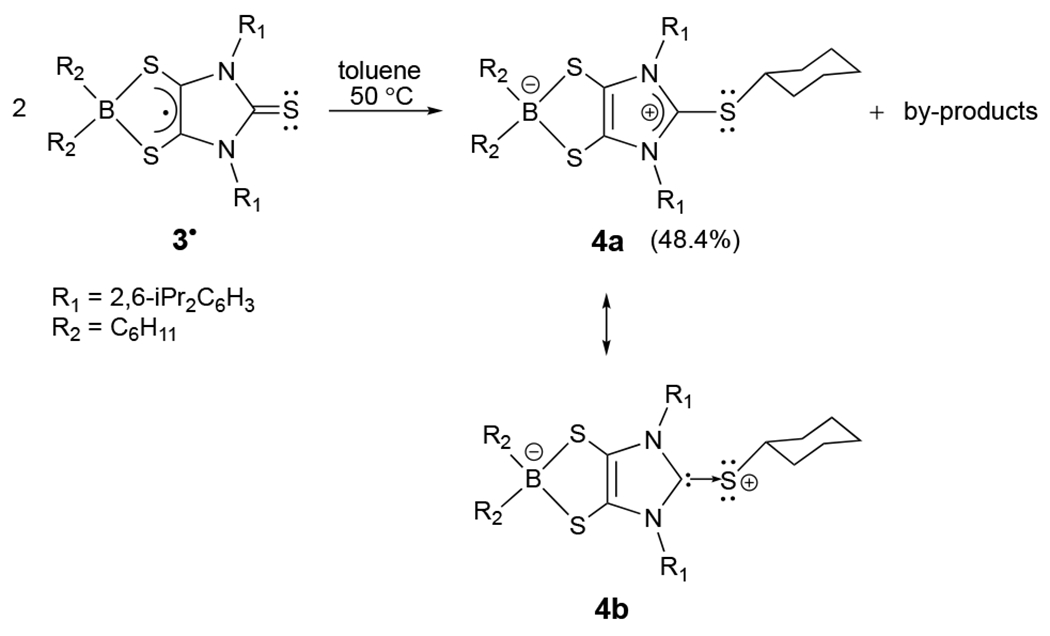
Synthesis of zwitterion (4) via thiourea-mediated B–C bond activation. Compound 4 has two major resonance forms: 4a and 4b (anionic boron-dithiolene-modified-carbene complex of sulfenyl cation).
Reaction of the purple lithium dithiolene radical (1•) with excess BBr3 in hexane (−78 °C) gives 2• as dark blue crystals (via recrystallization from a toluene solution) (Scheme 1). The parallel reaction of 1• with (C6H11)2BCl (0 °C) affords the dark blue radical 3• (Scheme 1). Two strong UV-vis absorption peaks for 2• (606 and 654 nm) and 3• (596 and 630 nm) were observed.[12] The paramagnetic properties of radicals 2• and 3• are characterized by room temperature EPR spectroscopy in toluene. The EPR spectrum of 2• (Figure 3A) displays an S = 1/2 septet due to hyperfine coupling with two equivalent bromine atoms. Spectral simulations indicate gav = 2.008 and hyperfine coupling constants of Aav = 28.95 MHz for 79Br (I = 3/2, gn=1.404) and Aav = 31.20 MHz for 81Br (I = 3/2, gn = 1.514). Molecular orbital calculations indicate that the SOMOs of 2• (Figure 3C) and [3-Ph]• (Figure 3D) are predominantly ligand-based, involving C–C π-bonding and C–S π-antibonding character. While computations indicate that the majority of the spin density of 2• (0.71) and [3-Ph]• (0.77), similar to that for 1• (0.88),[5] resides on the C2S2 unit of the dithiolene ligand, the hyperfine values suggests significant spin density resides on the two equivalent bromine atoms in 2•. For instance, after correcting for the difference in gn values, the bromine atoms have two-three-fold higher spin density than the two equivalent nitrogen atoms in 2• and its precursor 1•.[5] The large linewidths (0.6 mT) (Figure 3A) reflect the presence of two I = 3/2 bromine isotopes with different gn values and, most likely, the presence of unresolved 10B (I = 3), 11B (I = 3/2) and 14N (I = 1) hyperfine interactions. Although 3• exhibits a complex EPR spectrum with partially resolved hyperfine splitting (Figure 3B), it is well simulated with gav = 2.015, linewidth = 0.137 mT, and hyperfine coupling constants of Aav = 5.64 MHz for 11B (I = 3/2, gn = 1.404), Aav = 1.97 MHz for 10B (I = 3, gn = 0.600), and Aav = 2.8 MHz for two equivalent 14N (I = 1, gn = 0.404). After correction for the differences in gn values, the hyperfine interactions indicate approximately two-fold greater spin density on the two equivalent nitrogen atoms than the boron atom.
Figure 3.

Room-temperature X-band EPR spectra of 2• (A) and 3• (B) in toluene. Spectra (black lines) were recorded at 9.581 GHz with a modulation amplitude of 0.02 mT and a microwave power of 0.1 mW for 2• and 1.0 mW for 3•. Spectral simulations (red lines) used the following parameters: (A) Linewidth, 0.6 mT; gav = 2.008; 79Br (I = 3/2) Aav = 28.95 MHz; 81Br (I = 3/2) Aav = 31.20 MHz. (B) Linewidth, 0.137 mT, gav = 2.015; 10B (I = 3) Aav = 1.97 MHz; 11B (I = 3/2) Aav = 5.64 MHz; 14N (I = 1) Aav = 2.80 MHz. (C) SOMO of 2• and (D) SOMO of the simplified model [3-Ph]•.
The solid-state structures of both 2• and 3• (Figure 4)[12] feature a bent five-membered BS2C2 ring. However, the BS2C2 rings of the 2• and [3-Ph]• models in the gas phase are computed as planar.[12] The bent angle (η) between the BS2 plane and the S2C2 plane [13.5° (2•), 16.2° (3•)] in the solid-state may be a consequence of a combination of packing effects and the steric repulsion between the substituents of boron and the dithiolene ligand.[17] The experimental B–S bond distances of 2• [1.927 (4) Å] and 3• [2.027(4) Å, av] are comparable to the calculated values of 2• [1.985 Å] and [3-Ph]• [2.058 Å].[12] The boron atoms in 2• and in 3• are four-coordinate and adopt a distorted tetrahedral geometry. As those in precursor 1• [dC–C = 1.417(3) Å, dC–S = 1.677(3) Å, av],[5] the C2S2 units in both 2• [dC–C = 1.394(7) Å, dC–S = 1.682(3) Å] and 3• [dC–C = 1.388(4) Å, dC–S = 1.687(3) Å, av] exhibit elongated carbon–carbon bonds and shortened carbon–sulfur bonds compared to those in dithiolates [i.e., (NMe4)2(C3S5),[18] dC–C = 1.371(8) Å, dC–S = 1.724(6) Å, av].[1b] This is consistent with the presence of a SOMO in 2• (Figure 3C) and in 3• (Figure 3D), which has both C–C π-bonding and C–S π-antibonding character.[12]
Figure 4.
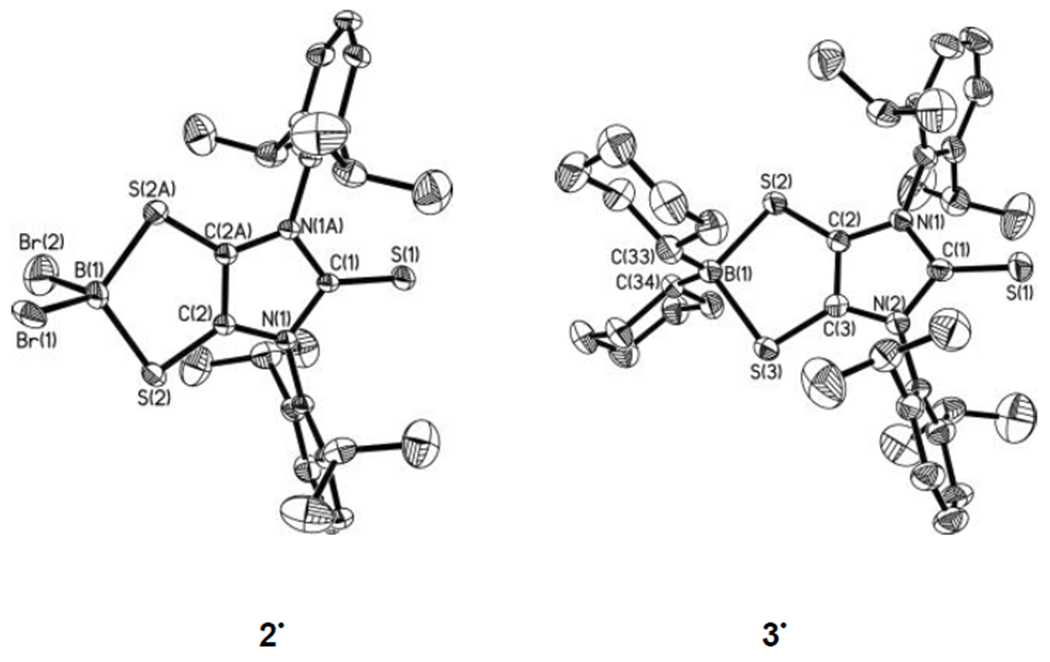
Molecular structures of 2• and 3• (thermal ellipsoids represent 30% probability; hydrogen atoms on carbons omitted for clarity). Selected bond distances (Å) and angles (deg) for 2•: S(1)–C(1), 1.623(5); C(2)–C(2A), 1.394(7); S(2)–C(2), 1.682(3); S(2)–B(1), 1.927(4); B(1)–Br(1), 1.982(8) (av); B(1)–Br(2), 1.984(10) (av); B(1)–S(2)–C(2), 92.4(2); S(2)–C(2)–C(2A), 121.69(12); S(2)–B(1)–S(2A), 110.3(3); Br(1)–B(1)–Br(2), 107.4(4) (av). For 3•: S(1)–C(1), 1.633(4); C(2)–C(3), 1.388(4); S(2)–C(2), 1.680(3); S(3)–C(3), 1.694(3); S(2)–B(1), 2.026(4); S(3)–B(1), 2.028(4); B(1)–C(33), 1.607(6); B(1)–S(2)–C(2), 94.94(16); S(2)–C(2)–C(3), 122.9(3); S(2)–B(1)–S(3), 103.27(18); C(33)–B(1)–C(34), 118.8(3).
In contrast to 2•, radical 3• readily undergoes thiourea-mediated boron-carbon bond activation, resulting in diamagnetic zwitterion 4 (in 48.4% yield; Scheme 2).[12] While the mechanism remains unclear, the formation of 4 appears to involve thiourea-mediated intermolecular cyclohexyl radical transfer. Notably, the spin density[12] residing on S(1) (0.228) in 3• (Figure 4) is comparable to that of S(2) (0.234) and S(3) (0.234). Furthermore, reactions of thiourea with carbon-centered radicals have previously been reported.[19] While compound 4 was routinely isolated as pale yellow-green crystals (Scheme 2), repeated attempts to isolate and characterize other by-products from the thick residue were unsuccessful. Interestingly, Stephan recently reported that phenanthrenedione- (Figure 2d) and pyrenedione-derived borocyclic radicals exhibit unique reactivity with nucleophiles [i.e., phosphines, carbenes, amines, and pyridines].[20]
X-ray structural analysis[12] (Figure 5) shows that, in contrast to the bent boron dithiolene ring in 3• [dC–C = 1.388(4) Å, dC–S = 1.687(3) Å, av], the five-membered BS2C2 ring in 4 is almost planar, containing a shortened carbon–carbon bond and concomitantly elongated carbon–sulfur bonds [dC–C = 1.365(2) Å, dC–S = 1.7099(18) Å, av]. The C(2)–C(3) bond in 4 [1.365(2) Å] compares well to the reported imidazole C=C double bonds.[21] The four-coordinate boron in 4 exhibits a 11B NMR resonance (18.9 ppm), which is comparable to those values reported for the zwitterions containing dioxoborocyclic moieties (10.2 to 11.1 ppm).[20] While comparing well to those [1.732(4)–1.737(2) Å] in carbene-stabilized sulfenyl cations,[22] the C(1)–S(1) bond distance in 4 [1.7256(18) Å] is ca. 0.09 Å longer than that for 3• [1.633(9) Å], but ca. 0.11 Å shorter than the C(40)–S(1) single bond distance in 4 [1.834(4) Å]. Our computations[12] indicate that the C(1)–S(1) bond of 4 is polarized predominantly toward the C(1) atom (55.7%). The C(1) and S(1) atoms bear +0.21 and +0.23 positive charges, respectively. The Wiberg bond index (1.12) of the C(1)–S(1) bond in 4 is only marginally less than that (1.17) of the carbene-stabilized parent sulfenyl cation (HS+),[22] thereby suggesting modest electron back donation from the S(1) atom to the empty p orbital of the C(1) atom. Consequently, two resonance structures (4a and 4b in Scheme 2) may be proposed for 4. Resonance form 4b represents an anionic boron-dithiolene-modified-carbene complex of the sulfenyl cation (RS+, R = cyclohexyl). In both the solid state and the gas phase, the C(1)–S(1) bond of 4 is slightly puckered out of the imidazole plane [the S(1)-C(1)-N(1)-C(2) torsion angle = −168.7° (solid state), −162.5° (gas phase)], which may be ascribed to the steric repulsion between two flanking 2,6-diisopropylphenyls and the sulfur-bonded cyclohexyl group.
Figure 5.
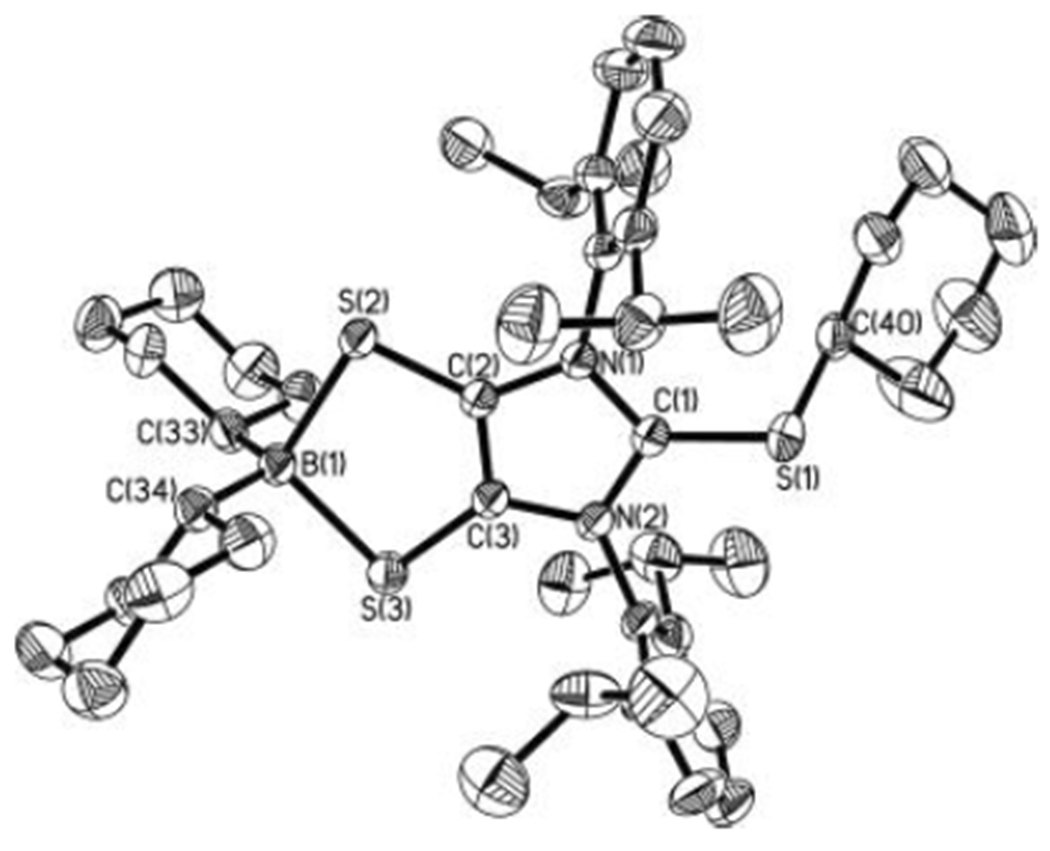
Molecular structure of 4 (thermal ellipsoids represent 30% probability; hydrogen atoms on carbons omitted for clarity). Selected bond distances (Å) and angles (deg): S(1)–C(1), 1.7256(18); S(1)–C(40), 1.834(4); C(2)–C(3), 1.365(2); S(2)–C(2), 1.7078(18); S(3)–C(3), 1.7120(18); S(2)–B(1), 2.027(2); S(3)–B(1), 2.031(2); B(1)–C(33), 1.611(3); B(1)–S(2)–C(2), 94.96(8); S(2)–C(2)–C(3), 123.01(14); S(2)–B(1)–S(3), 104.64(9); C(33)–B(1)–C(34), 117.05(16).
The first boron-dithiolene radicals 2• and 3• have been synthesized by combining 1• with the corresponding borane agents at low temperatures. Under mild conditions, radical 3• is further converted into zwitterion 4 via thiourea-mediated B–C bond activation. Notably, 4 may be regarded as an anionic dithiolene-modified-carbene complex. The unique redox-active character of dithiolenes and diverse utility of carbenes have both been well documented. This study demonstrates the possibility of developing novel chemistry at the dithiolene-carbene interface. The utility of zwitterionic 4 is being explored in this laboratory.
Supplementary Material
Acknowledgments
We are grateful to the National Science Foundation for support: CHE-1565676 (G.H.R., Y.W.) and CHE-1661604 (H.F.S.). EPR studies were supported by a grant from the National Institutes of Health R37-GM62524 (M.K.J.).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org.
References
- [1].a) McCleverty JA, Prog. Inorg. Chem 1968, 10, 49–221; [Google Scholar]; b) Stiefel EI, Ed., Dithiolene Chemistry: Synthesis, Properties, and Applications, John Wiley & Sons, Hoboken, NJ, 2004; [Google Scholar]; c) Eisenberg R, Gray HB, Inorg. Chem 2011, 50, 9741–9751; [DOI] [PubMed] [Google Scholar]; d) Kato R, Chem. Rev 2004, 104, 5319–5346; [DOI] [PubMed] [Google Scholar]; e) Hine FJ, Taylor AJ, Garner CD, Coord. Chem. Rev 2010, 254, 1570–1579; [Google Scholar]; f) Rabaca S, Almeida M, Coord. Chem. Rev 2010, 254, 1493–1508; [Google Scholar]; g) Garreau-de Bonneval B, Ching KIMC, Alary F, Bui TT, Valade L, Coord. Chem. Rev 2010, 254, 1457–1467; [Google Scholar]; h) Sproules S, Wieghardt K, Coord. Chem. Rev 2011, 255, 837–860; [Google Scholar]; i) Sproules S, Prog. Inorg. Chem 2014, 58, 1–144; [Google Scholar]; j) Kobayashi A, Fujiwara E, Kobayashi H, Chem. Rev 2004, 104, 5243–5264; [DOI] [PubMed] [Google Scholar]; k) Holm RH, Kennepohl P, Solomon EI, Chem. Rev 1996, 96, 2239–2314. [DOI] [PubMed] [Google Scholar]
- [2].Lim BS, Fomitchev DV, Holm RH, Inorg. Chem 2001, 40, 4257–4262. [DOI] [PubMed] [Google Scholar]
- [3].a) Gray HB, Billig E, J. Am. Chem. Soc 1963, 85, 2019–2020; [Google Scholar]; b) Stiefel EI, Waters JH, Billig E, Gray HB, J. Am. Chem. Soc 1965, 87, 3016–3017. [Google Scholar]
- [4].Filatre-Furcate A, Bellec N, Jeannin O, Auban-Senzier P, Fourmigue M, Vacher A, Lorcy D, Inorg. Chem 2014, 53, 8681–8690. [DOI] [PubMed] [Google Scholar]
- [5].Wang Y, Hickox HP, Xie YM, Wei PR, Blair SA, Johnson MK, Schaefer HF III, Robinson GH, J. Am. Chem. Soc 2017, 139, 6859–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang Y, Xie Y, Abraham MY, Wei P, Schaefer HF III, Schleyer P. v. R., Robinson GH, J. Am. Chem. Soc 2010, 132, 14370–14372. [DOI] [PubMed] [Google Scholar]
- [7].a) Power PP, Chem. Rev 2003, 103, 789–809; [DOI] [PubMed] [Google Scholar]; b) Su YT, Kinjo R, Coord. Chem. Rev 2017, 352, 346–378. [Google Scholar]
- [8].a) Kaim W, Hosmane NS, Zalis S, Maguire JA, Lipscomb WN, Angew. Chem. Int. Ed 2009, 48, 5082–5091; [DOI] [PubMed] [Google Scholar]; b) Renaud P, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, 2012. [Google Scholar]
- [9].Aramaki Y, Orniya H, Yamashita M, Nakabayashi K, Ohkoshi S, Nozaki K, J. Am. Chem. Soc 2012, 134, 19989–19992. [DOI] [PubMed] [Google Scholar]
- [10].a) Mansell SM, Adams CJ, Bramham G, Haddow MF, Kaim W, Norman NC, McGrady JE, Russell CA, Udeen SJ, Chem. Commun 2010, 46, 5070–5072; [DOI] [PubMed] [Google Scholar]; b) Wood TK, Piers WE, Keay BA, Parvez M, Chem. Commun 2009, 5147–5149; [DOI] [PubMed] [Google Scholar]; c) Hinchliffe A, Mair FS, McInnes EJL, Pritchard RG, Warren JE, Dalton Trans. 2008, 222–233; [DOI] [PubMed] [Google Scholar]; d) Fedushkin IL, Markina OV, Lukoyanov AN, Morozov AG, Baranov EV, Maslov MO, Ketkov SY, Dalton Trans. 2013, 42, 7952–7961. [DOI] [PubMed] [Google Scholar]
- [11].Longobardi LE, Liu L, Grimme S, Stephan DW, J. Am. Chem. Soc 2016, 138, 2500–2503. [DOI] [PubMed] [Google Scholar]
- [12].See the supporting information for synthetic, spectral, computational, and crystallographic details. CCDC 1831276 (2•), 1831277 (3•), and 1831278 (4) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
- [13].Kelch H, Kachel S, Celik MA, Schafer M, Wennemann B, Radacki K, Petrov AR, Tamm M, Braunschweig H, Chem. - Eur. J 2016, 22, 13815–13818. [DOI] [PubMed] [Google Scholar]
- [14].Miyaura N, Suzuki A, Chem. Rev 1995, 95, 2457–2483. [Google Scholar]
- [15].a) Yu Y, Brennessel WW, Holland PL, Organometallics 2007, 26, 3217–3226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pettinari R, Pettinari C, Marchetti F, Monari M, Mosconi E, De Angelis F, Organometallics 2013, 32, 3895–3902. [Google Scholar]
- [16].a) Dasgupta R, Panda A, Pal S, Muhasina PV, De S, Parameswaran P, Khan S, Dalton Trans. 2017, 46, 15190–15194; [DOI] [PubMed] [Google Scholar]; b) Kowalewski M, Krumm B, Mayer P, Schulz A, Villinger A, Eur. J. Inorg. Chem 2007, 5319–5322. [Google Scholar]
- [17].Steed JW, in Frontiers in Crystal Engineering, 1st ed (Eds.: Tiekink ERT, Vittal J), John Wiley & Sons, Ltd, Chichester, UK, 2006, pp. 67–90. [Google Scholar]
- [18].Breitzer JG, Smirnov AI, Szczepura LF, Wilson SR, Rauchfuss TB, Inorg. Chem 2001, 40, 1421–1429. [DOI] [PubMed] [Google Scholar]
- [19].Ricci P, Khotavivattana T, Pfeifer L, Medebielle M, Morphy JR, Gouverneur V, Chem. Sci 2017, 8, 1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Longobardi LE, Zatsepin P, Korol R, Liu L, Grimme S, Stephan DW, J. Am. Chem. Soc 2017, 139, 426–435. [DOI] [PubMed] [Google Scholar]
- [21].Arduengo AJ III, Krafczyk R, Schmutzler R, Craig HA, Goerlich JR, Marshall WJ, Unverzagt M, Tetrahedron 1999, 55, 14523–14534. [Google Scholar]
- [22].Liu L, Zhu D, Cao LL, Stephan DW, Dalton Trans. 2017, 46, 3095–3099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.