

Abstract
Injection into the heart tissue is a direct route for optimally placing mesenchymal stem cells (MSC) to regulate local inflammation following a heart attack. The retention of MSCs at the injection site is severely limited by the fluid flows that rapidly wash cells away and minimize their capacity to modulate cardiac inflammation. To prevent this loss of MSCs and their function, antibody coatings were designed for the surface of MSCs to enhance their adhesion to the inflamed tissue. MSCs were biotinylated, and biotinylated antibodies against intercellular cell adhesion molecules were conjugated to the cell surface through an intermediate layer of streptavidin. MSC surfaces were modified with ~7,000 biotin/μm2 and ~23 antibodies/μm2. The heart tissue injection of antibody-coated MSCs offered a 3-fold increase of cell retention in an infarcted heart over the injection of uncoated MSCs. We supported the mechanism of adhesion through analysis of MSC adhesion to inflamed endothelial cells and also surfaces of purified adhesion molecules on glass under microfluidic shear flow.
Keywords: Cell surface coating, endothelium targeting, ICAM1 antibody, intramyocardial delivery, targeted cell delivery
Graphical Abstract
INTRODUCTION
A myocardial infarction (MI or heart attack) is a severe cardiovascular event where the occlusion of the blood supply damages local tissues. Without access to oxygen and essential nutrients, cells within the infarct zone become necrotic and release waves of cytokines driving local inflammation to recruit additional inflammatory cells1, 2 and altering the phenotype of local immune cells3. The spike in inflammation drives the spread of damage to tissue outside the initial infarct site2, and this wave of inflammation-driven damage continues to spread out into healthy heart tissue for weeks following the initial occlusion4. As a result, MI-associated damage is dramatically worsened by unchecked inflammation following the initial injury.
Mesenchymal stem cells (MSCs) are potent modulators of inflammation and have been effectively utilized in vivo to promote immunomodulatory effects of immunocytes5–8. Additionally, MSCs that are prestimulated release immunosuppressive factors9, and these factors drive pro-inflammatory monocytes towards an anti-inflammatory state10, 11. Beyond their immunomodulatory effects, MSCs are effective at driving the development of new blood vessels12, 13 and differentiation14, 15 to further promote repair to damaged tissues. Through the cooperative action of these mechanisms, the introduction of MSCs into the heart has been effective in promoting cardiovascular function and limiting the expansion of tissue injury following MI16, 17.
Direct MSC injection into the healthy tissue surrounding the infarct concentrates these therapeutic cells at the site of injury to promote a localized therapeutic effect18. Unfortunately, the injected cells are rapidly cleared from the injection site, limiting their opportunity to modulate the infarct environment. For most injected cell therapies, the vast majority of the injected cells are rapidly lost from the injection site19–22 owing to the injected cells and buffer swelling the tissue. The elastic recovery of the heart drives fluid escape from the tissue, and the injected cells are swept away22. This pressure driven flow is further exacerbated by heart muscle contractions and other tissue dynamics in a pumping heart23–26. In MSC therapies for MI, these factors combine to yield as little as 0.44% of the injected MSCs remaining in the heart 4 days after injection27. Since a larger MSC dose carries a greater risk of mortality due to micro-vessel obstruction28, 29, the maximum number of MSCs present in the heart post-MI is fundamentally limited by the retention of these cells in the peri-infarct tissue. The loss of cells from the infarct site is a critical challenge for the future of MSC-based cardiovascular therapies22, 30.
We seek to increase the number of MSCs retained in the peri-infarct tissue through engineering of the MSC peripheral membrane using artificial ligands on the MSC plasma membrane. The inflammatory response to ischemia drives the secretion of proinflammatory factors which alter the endothelium to recruit leukocytes and other immune cells. The inflamed endothelium overexpresses cell adhesion molecules, making these endothelium sheets important targets31–33.
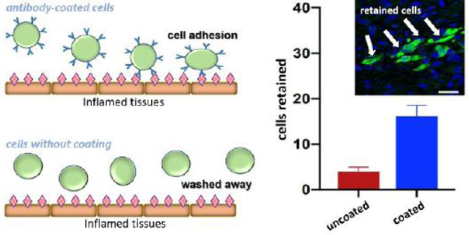
Here, we developed a cell surface coating to present antibodies to adhere to intercellular cell adhesion molecules (ICAM1) on the endothelium of infarcted hearts (Figure 1a). Through injection into the heart tissue, we confirmed that the delivery of antibody-coated MSCs provides 3-fold higher retention in infarcted heart tissues when compared to uncoated MSC injection. The adhesion mechanism is supported through the increased cell adhesion of coated MSCs to activated endothelial cells and ICAM1-coated glass under physiological shears. In all, this straightforward coating design significantly enhances the adhesion of MSCs to an infarcted heart through specific targeting of overexpressed endothelial ICAM in response to local inflammation.
Figure 1. Scheme of intramyocardial delivery of antibody-coated MSCs.
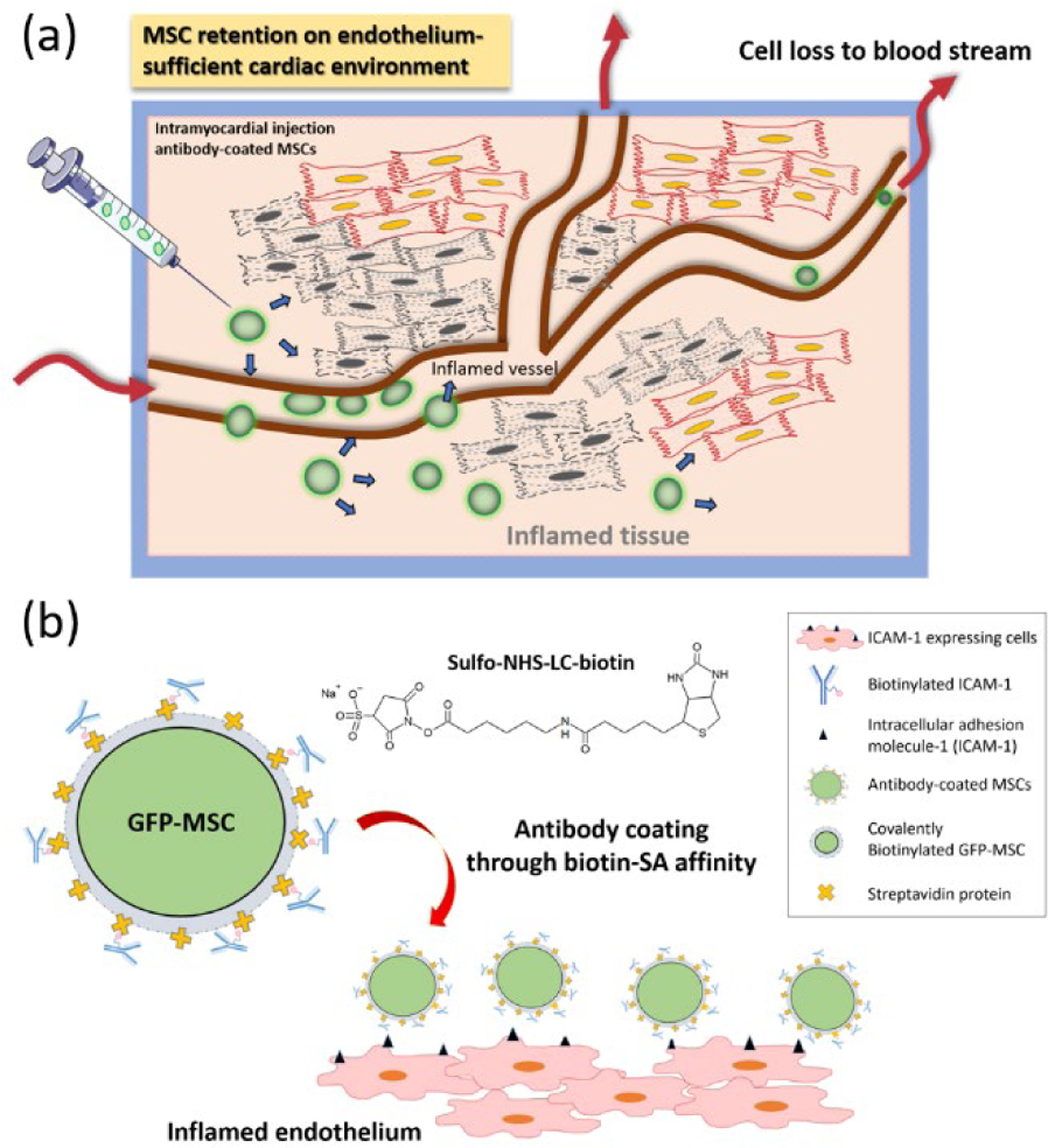
(a) Direct intramyocardial injection of antibody-coated MSCs and subsequent interaction of MSCs with inflamed endothelium. (b) Incorporation of inflammation-targeted antibody on MSCs through biotin-streptavidin affinity.
EXPERIMENTAL SECTION
MSC isolation and culture
Briefly, 6–8 weeks old C57BL/6-Tg (CAGEGFP) mice (Jackson Laboratory, BarHarbor, ME) were sacrificed, femurs, tibias and hip bones were collected and then flushed with PBS supplemented with 10% FBS.
Mouse MSCs isolated from C57BL/6 or C57BL/6-Tg (CAG-EGFP)131Osb/LeySopJ mice (Jackson Laboratory, BarHarbor, ME). Mice were sacrificed, femurs, tibias and hip bones were collected and then flushed with PBS supplemented with 10% FBS. MSCs used in this study were of a passage less than 10. MSCs were cultured in pre-warmed mouse MesenCult™ basal medium, supplemented with MesenCult™ 10X Supplement (STEMCELL Technologies), 10% USDA Approved Origin FBS (VWR), 1% 200mM L-glutamine (Gibco) and 1% streptomycin/penicillin (Gibco). MesenPure™ (STEMCELL Technologies) was added in MSC medium at 1:1000 dilution right before cell seeding. MSCs were cultured under hypoxic conditions (less than 5% O2 supply) for MSC cultivation was needed by introducing ultrapure nitrogen into a 37°C humidified incubator, with 5% CO2. Cells were seeded into T175 vented tissue culture flask (VWR) until over 90% confluency. For cell collection, medium supernatant was removed, and cells were rinsed with sterile PBS (Corning) and treated with 6 mL of 0.25% trypsin/EDTA (Gibco) for 5 min at 37°C. 6 mL of Serum-containing medium was added to neutralize the trypsin. Cells were centrifuged at 300 g for 5 min and the supernatant was removed by pipetting. The cell pellet was washed once and resuspended in ice-cold DPBS before use.
Preparation of anti-ICAM1 coated MSCs
All incubation steps for cell membrane antibody incorporation were processed on ice. MSC pellet was rinsed twice with 2mL of cold PBS in a 15 mL centrifuge tube. 0.1 mM or 1.0 mM sulfo-NHS-LC-biotin (Thermo scientific) was prepared in PBS immediately before cell incubation, 2 mL of biotin solution per 106 cells is added for 1 h incubation. Cells were then rinsed twice with PBS, and 2 mL of 25 μg/mL streptavidin (Thermo scientific) in PBS was added for every million cells for 1 h. After two more PBS rinses, the streptavidin coated cells were then incubated for 1h in 30, 50, or 100 μg/mL biotinylated monoclonal mouse antibody against human ICAM1 ((Invitrogen, MA119527) or 100 μg/mL biotinylated monoclonal rat antibody against mouse ICAM1 (clone YN1/1.7.4, Invitrogen) in PBS. Coated cells were ready for use after two more PBS rinses.
MTT viability assay
Metabolic activity detection depending on mitochondrial function was evaluated by using MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) reagents (M6494, Invitrogen). A 12mM MTT solution was prepared in PBS (5 mg/mL). 50,000 cells were diluted in 1mL MSC culture medium. 200 μL of cell solution was pipetted into 96 well plate and then incubated with 20 μL of MTT stock for 4 h at 37 °C. Cell supernatant was gently removed after centrifuge (1 rpm, 5 minutes) and 200 μL of DMSO was added to dissolve the reacted product. A plate reader (Biotek, Winooski, VT) was used to examine the sample absorbance at 570 nm. The percentage of cell viability was determined by normalizing the sample absorbance with the positive control.
Myocardial infarction surgery and intramyocardial cell transplantation
Myocardial infarction was induced by permanent left anterior descending artery (LAD) ligation in 8–10 week female C57BL/6 mice under 1–3% isoflurane anesthesia using a small animal vaporizer system. Immediately after ligation, the animals were randomized to receive 25 μL of uncoated MSCs (n=4) or anti-ICAM1 modified MSCs (n=3; 125,000 cells per 25 μL injection per mouse) via intramyocardial injection to the peri-infarct region. Pain medications were administrated after surgery for 24–48 hours.
Immunohistochemistry analysis
The hearts were harvested 3 days after MI and MSC injection. Immediately after cervical dislocation, the hearts were perfused with cold PBS with 4% paraformaldehyde and fixed for 24 hours prior to embedding in a paraffin block. 4-μm thick heart tissue sections were used for immunofluorescent analysis of heart for evidence of the GFP+ MSCs. Briefly, heart tissue sections were exposed to pH 6.0 antigen retrieval buffer (Vector Laboratories, Burlingame CA) following the company protocol. Slides were blocked with normal goat serum for 16 hours at 4°C and stained with primary rabbit anti-GFP antibodies (Abcam, Cambridge, United Kingdom) following by incubation with Alexa 488 secondary goat anti-rabbit antibodies (Abcam, Cambridge, United Kingdom).
Flow Cytometry
Single-cell suspensions were prepared from digested cardiac tissue as previously described34. Four hearts in the coated MSC treatment and 5 hearts in the uncoated cell treatment were used. Each heart was analyzed as one sample as the whole heart is digested. Briefly, hearts collected 3 days after MI were minced then digested using a collagenase B (Roche, Indianapolis, IN) and dispase II (Roche, Indianapolis, IN) solution for 30 minutes at 37°C with mixing every 5 minutes. The heart cell suspensions were strained using 40 μm strainers (VWR, cat#10199–654) to remove tissue debris and undigested tissue. Cells were washed with flow buffer by centrifugation (400×g for 10 min at 4 °C) and then suspended in Flow Buffer. Cells were stained with eFluor 660 conjugated anti-GFP antibody (eBioscience, Thermo Fisher Scientific, Waltham MA) and analyzed using an LSR II (Becton Dickinson) in the University of Kentucky Flow Cytometry Core. We utilized FlowJo v10 (FlowJo, FlowJo Ashland OR) software to generate dot plots and analyze the data.
Macrophage cytokine secretion assay
Primary BMDMs (Bone marrow-derived macrophage) were isolated and differentiated using previously published protocols35. Primary macrophages were derived from bone marrow cells and cultured for 7 days in DMEM (with 10% L929 cell-conditioned medium, and 10% fetal bovine serum). For co-culturing, 5×105 BMDMs were seeded into a twelve-well plate. The next day, 20,000 unmodified MSCs or anti-ICAM1 coated MSCs were placed into the twelve-well plate with the BMDMs. Co-culture were incubated for 3 hours with or without 100 ng/mL lipopolysaccharide (LPS-B5, Invitrogen) and IFN-γ (20 ng/ml).
Conditioned media were obtained from BMDMs alone, co-cultured with BM-MSCs, or anti-ICAM1-coated BM-MSCs as described above. The secreted TNF-α and IL-10 in the conditioned media were evaluated using enzyme-linked immunosorbent assay ELISA kits (BD OptEIA™) according to the manufacturer instructions.
Endothelial cell culture, activation and shear assays
Human Umbilical Vein Endothelial Cells (HUVEC) #00191027) was purchased from Lonza. HUVEC culture medium was prepared by adding 1% streptomycin/penicillin and EGMTM-2 SingleQuots™ Supplements (CC-4176) into EGMTM-2 Basal Medium (CC-3156). The EGMTM-2 SingleQuots™ Supplements include 2% FBS, 0.2 mL hydrocortisone, 2 mL hFGF-B, 0.5 mL VEGF, 0.5 mL IGF-1, 0.5 mL ascorbic acid, 0.5 mL hEGF, 0.5 mL GA-1000, and 0.5 mL heparin. Cells were seeded in T-75 tissue culture flask (VWR) and cultured in a 37°C incubator with 5% CO2. Cells were rinsed with sterile PBS and detached by 5mL of TrypLE treatment for 5 min at 37°C. The trypsin was neutralized with culture medium and centrifuged at 300 g for 5 min. The supernatant was removed, and cells were resuspended in pre-warmed medium or PBS before use or next passage.
To create a HUVEC monolayer on glass slides, attachment factor (Gibco) was used to enhance HUVEC attachment to the glass. Microscope slides (25mm × 75mm, VWR) were placed in a 50 mL conical tube and presterilized overnight in 70 vol% ethanol and dried before use. Each sterilized slide was loaded into a well (3×8cm) in a 4-well rectangular dishes (Thomas scientific) and rinsed at least twice in PBS. Each slide was incubated with 4 mL 1X attachment factor at 37°C for at least 3 h. Excess solution removed and 1×106 of HUVEC in 4 mL EGM™-2 medium was seeded directly onto each slide. Each HUVEC-seeded slide was used for MSC adhesion studies after reaching 95% confluency.
HUVECs were activated through the addition of 4 mL of 10 ng/mL recombinant human TNF-α protein (R&D systems) in growth factor and serum-suppled EGM-2 medium directly onto a HUVEC-adhered microscope slide in a 4 well dish at 37°C incubator for 5 or 20 h.
Adherent HUVEC slides were loaded into a Whatman Chip Clip and rinsed with PBS. ICAM1 expression on adherent HUVECs was qualitatively labeled with 400 μL of 25 μg/mL biotinylated monoclonal antibody against human ICAM1 in PBS for 45 min, rinsed twice with PBS and stained with 400 μL of 2 μg/mL Alexa647-labeled secondary antibodies against mouse IgG (Invitrogen) for 45 min. Cells were rinsed in PBS and imaged in the far-red channel of a Nikon Ti-U Epifluorescent microscope.
In cell-cell adhesion studies, HUVEC cells were stained with calcein red (Invitrogen) for tracking. HUVECs were rinsed with serum-free (SF) EGM-2 medium twice in a 4-well rectangular dish. Calcein-red orange powder was dissolved in 63 μL of pure DMSO and stored in −20°C before use. HUVEC slides were incubated with in a 1:1000 dilution of the calcein-red orange stock in SF-EGM-2 (4mL) for 3 min and rinsed twice with SF-EGM-2. Cell slides were blocked with 4 mL of 5% BSA in SF-EGM-2 at least for 1 hour. For the in vitro shear assays of GFP-MSCs on calcein red stained HUVECs, one milliliter of 0.5×106 MSCs/mL in SF-EGM-2 was pipetted onto a slide of 95% confluent HUVEC cells blocked with BSA. The coculture was incubated for 5min at 37°C. Cells were sheared using a rectangular parallel plate flow chamber from Glycotech (#31–010) with a 0.01-inch-thick gasket. A vacuum pump was used to seal the device to a cell-coated slide immersed in PBS. The flow rate of fluid through the chamber was controlled with a syringe pump (NEWERA, #4000-US) and two 60 mL syringes withdrawing fluid to pull PBS from a reservoir through the chamber at the indicated flow rate for 15 s. The initial number of attached MSCs was counted before shear and used to calculate fractional retention. The fluorescence images of attached GFP-positive MSCs under different shears and the number of retained cells were counted with ImageJ analysis. Shear stress acting on the cell was calculated as the wall shear stress under Newtonian flow in a rectangular cross section based on the following equation (Q= flow rate, μ= fluid viscosity, H= gap height, W= gap width, τw= shear stress at wall).
Quantitation of cell surface group density
Covalent biotinylation of cell surface proteins was achieved by treating cells with various concentration of sulfo-NHS-LC-biotin (0.025; 0.1; 0.5; 1; 2 mM). Two milliliters of sulfo-NHS-LC-biotin solution was used to treat 1×106 cells for 1 hour in a 15 mL centrifuge tube on ice and rinsed with ice-cold DPBS twice. One milliliter of 0.5 μg/mL streptavidin-phycoerythrin (SAPE, Invitrogen) mixed with extra streptavidin protein (0, 20, 60, 80 or 90 μg/mL) in PBS was used to label 1×106 of biotinylated cells for 1hr on ice. After rinsing with ice-cold PBS twice, the fluorescence of PE-labeled cells was analyzed by flow cytometry in FL2 channel.
QuantiBRITE™ PE purchased from BD Biosciences is a phycoerythrin (PE) fluorescence quantitation kit and includes PE calibration beads of known quantity of PE conjugated per bead. Red PE fluorescence was collected in FL2 channel (585/40 nm bandpass filter) by flow cytometry. The mean FL2 fluorescence per PE was correlated from a linear regression and was used for quantitation of PE fluorophore bound to each cell. The number of available biotin groups on each cell event was calculated through PE calibration curve (Figure S1).
Activated or naïve HUVECs were trypsinized as before and rinsed twice with PBS. 100 μL of 2.5 μg/mL biotinylated human ICAM1 antibody in ice-cold PBS was added for every 1×106 HUVECs and incubated for 30 min on ice. After rinsing twice with PBS, cells were incubated in 500 μL of 0.5 μg/mL SA-PE in PBS. After PBS rinsing, the fluorescence of surface-anchored PE was analyzed through flow cytometry in FL2 channel, and a relative measure of ICAM1 per cell was determined using the PE calibration curve (Figure S1).
After coating with antibody against human ICAM1, coated MSCs were incubated with 350 μL of PE-conjugated secondary antibody (Invitrogen) against mouse for 45 min. The PE fluorescence acquired by FL2 channel of flow cytometry was compared with calibration PE curve to determine the number of ICAM1 antibody coated on a cell by the assumption of 1:1 binding ratio between anti-ICAM1 and secondary antibody.
Preparation of hICAM1-coated slides
Human recombinant ICAM1 protein (R&D systems) was reconstituted at 250 μg/mL in sterilized PBS and stored at 4°C before use. The density of hICAM1 protein was estimated by specifically fluorescent targeting on hICAM1 molecules coated on protein-reactive epoxy glass surfaces (CEL associates). Eleven concentrations of hICAM1 protein (250; 150; 100; 50; 25; 12,5; 6.25; 4; 2; 1 μg/mL and PBSA negative control) were tested in this study. Epoxide slides were placed in a covered petri dish, coated with 30μL of hICAM1 protein solution in a 1 cm2 area for 3 h, and then rinsed twice with 0.1% BSA in PBS. ICAM1-immobilized slides were loaded into a Whatman Chip Clip, blocked with 0.1% BSA for 30 min, and washed with 1X PBS twice. Each ICAM1-coated area was labeled with 50 μL of 30 μg/mL biotinylated human ICAM1 antibody for 1 h and rinsed twice with PBS. Biotinylated immobilized sites were fluorescently tagged through the incubation of 70 μL diluted SA-Cy3 (1:40 in PBS, Invitrogen) in each well for 30 min. After rinsing with PBS three times, slides were dried in a stream of air and scanned in an Affymetrix 428 Microarray Scanner at 30 dB gain in the Cy3 channel. The number of Cy3 molecules were estimated by Cy3 calibration curve (Figure S2) prepared using a Cy3 calibration slide from Full Moon Biosystems and image analysis using ImageJ software.
In vitro adhesion of anti-ICAM1 coated MSCs on ICAM-immobilized surfaces under shears
A 1 cm by 1 cm region of an epoxy slide was treated with 30 μL of human ICAM1 protein in PBS at 6.25 μg/mL for 3 h at room temperature in a sealed dish. The protein-coated slides were blocked with 1 mg/mL BSA in PBS for 30 min. 50 μL of 0.5×106 MSCs were added onto the ICAM1-coated surface for 5 or 45 min at room temperature. The adhered cell slide was loaded into a rectangular parallel flow chamber as described above. The initial count of adhered cells was acquired after the washout of cells under 2 dyne/cm2 shear for 15 seconds. Cells were sheared for 30 s at each shear stress level and counted to calculate the percentage of cell retention.
RESULTS AND DISCUSSION
Intramyocardial delivery of mMSCs in an infarct mouse model
The direct injection of MSCs into the heart tissue has been effective in improving the function of hearts following a heart attack. The effective dose of MSCs present in the heart tissue after injection is a critical limitation, and this study tests the hypothesis that the synthetic incorporation of antibodies against ICAM1 onto the peripheral membrane of the injected MSCs will increase the number of MSCs retained at the site of injection. Our strategy is to first biotinylate MSCs through incubation in 1 mM sulfo-NHS-LC-biotin (NHS-biotin), then coat the MSC surface through incubation in 25 μg/mL streptavidin, and finally use the excess biotin binding sites of streptavidin to bind a biotinylated antibody against mouse ICAM1 (100 μg/mL) (Figure 1b). Since this method targets the post-MI inflammatory environment, we induced MI in our mice with an established infarct model where the left anterior descending coronary artery is sutured. The animals are then injected with either 1.25×105 MSCs in PBS or 1.25×105 anti-ICAM1 coated MSCs in PBS. There was one injection per heart, 25 μL per injection, and all injections were into the peri-infarct heart wall, and the infarct zone was determined through visual tissue discoloration. Three days following MI and MSC injection, the mice were sacrificed, and the resident heart cells were dispersed and analyzed by flow cytometry (Figure 2a). Our utilization of GFP-labeled MSCs injected into a non-GFP host allows for simple determination of the number of injected cells remaining in the host. The number of the injected MSCs remaining in the heart on day 3 were significantly higher (p=0.02) for the MSCs coated with antibodies against ICAM1 (16 ± 3.7 cells/mg) than for the unmodified MSCs (4 ± 1.4 cells/mg, Figure 2c). This finding is qualitatively supported through the analysis of tissue sections from other animals in the study, where GFP+ cells are more frequent in peri-infarct tissue sections from the anti-ICAM1 coated MSCs than the uncoated MSCs (Figure 2b). In related studies, inflammation-adhesive cell coatings have also been designed on MSCs by incorporating functional ligands that target endothelial antigens36–38. Coatings with a CAM-specific peptide also allowed MSC to target an inflamed endothelium39, while surface incorporation of an ICAM1 antibody enhanced MSC homing to the site of inflammation by MSC adhesion on ICAM1-upregulated endothelium36. Finally, antibodies against VCAM and addressins have also been used to target inflamed tissues in a mouse model of inflammatory bowel disease37. In all, our study strongly supports, for the first time, that the ICAM1 coated MSC are retained in the infarcted heart wall more than the established treatment using unmodified MSCs.
Figure 2. In vivo Intramyocardial delivery of ICAM1 antibody-coated MSCs in infarcted mouse model.
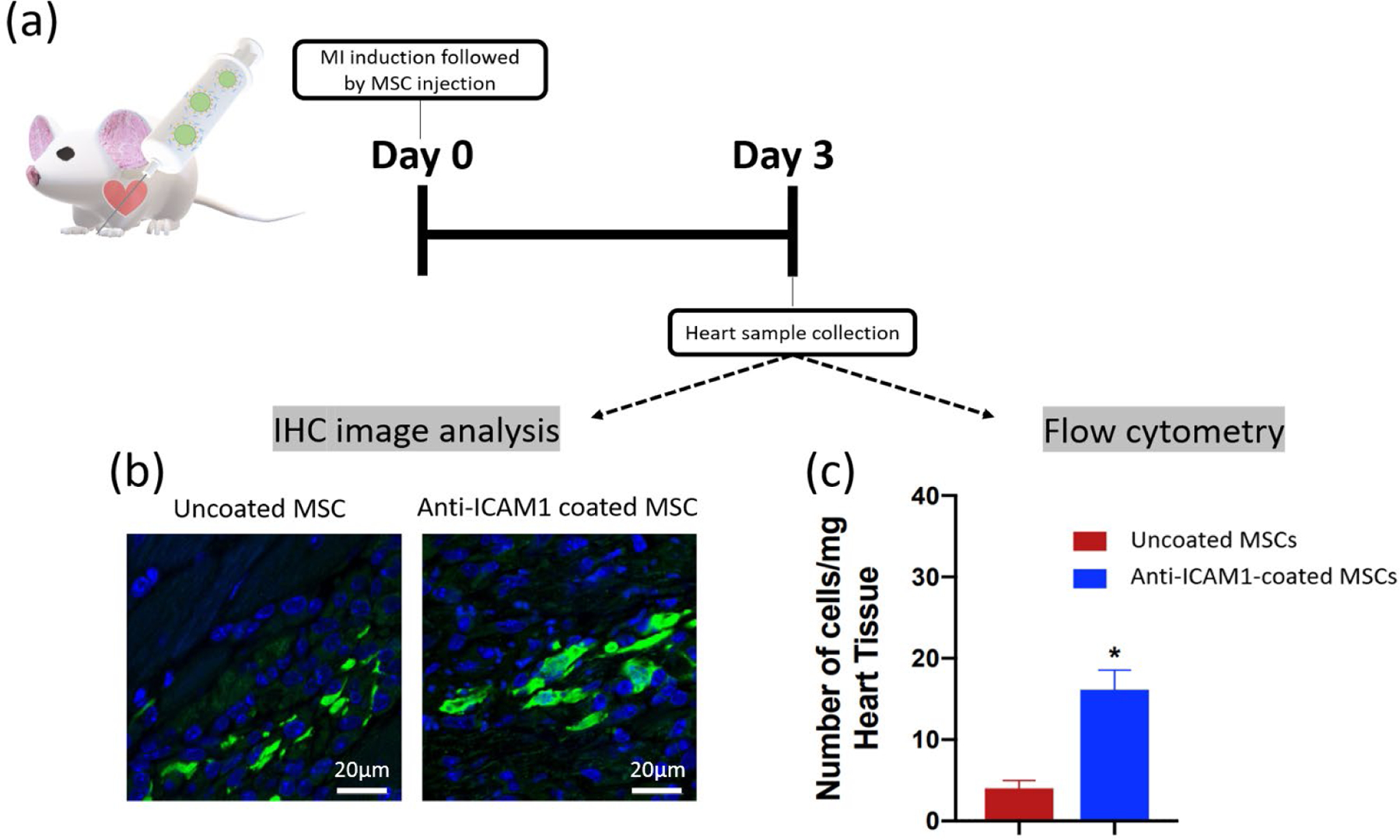
(a) Timeline of MI induction, MSC injection and sample collection. The coated or uncoated cells were directly injected into peri-infarct area 2 hour after left anterior descending artery ligation. At day 3, the heart sample was collected and digested for flow cytometry and immunohistochemistry analysis. (b) Representative immunofluorescent analysis of uncoated and anti-ICAM1 coated GFP-MSCs retention in infarcted heart tissues. The scale bar is 20 μm. (c) Quantitative retention analysis of uncoated MSCs (n=4) and anti-ICAM1-coated MSCs (n=3) through flow cytometry.
Viability and immunosuppressive properties of coated cells
Therapeutic MSCs play multiples biological roles in regenerating new cardiac tissues, regulating extracellular immune responses, secreting angiogenesis-inducing paracrine factors. Therefore, it is essential that our MSC coating strategy preserves the biological functions required for MI therapy. We coated MSCs with anti-ICAM1 through 1 mM ester biotin treatment and the viability of coated cells is preliminarily supported through MTT assays as well as MSC expansion in culture. As compared with uncoated cells, the MSC viability had a ~20% reduction in cell viability after the anti-ICAM1 coatings (Figure S3). Our previous work40 noted the potential for damage caused by surface biotinylation, and there is some alteration of proliferative function here.
Uncoated MSCs or MSCs coated with anti-ICAM1 were prepared seeded onto pre-coated tissue flasks in media. After 30 minute of cell culture, all groups exhibited similar numbers of attached MSCs and similar morphologies, and these groups were indistinguishable two days after treatment (Figure S4), suggesting the minimal interference on MSC expansion. In addition, the intensity of the GFP signal in the cells is correlated to viability and protein production, and the GFP signals were also similar across these groups.
The immunoregulatory function of MSCs is proposed as a dominant mode of cardioprotection post-MI. Near an infarct, ischemia-induced apoptotic and necrotic cardiomyocytes secrete proinflammatory cytokines which drive macrophages into a pro-inflammatory phenotype. As demonstrated in vivo and in vitro, the introduction of MSCs to proinflammatory macrophages will reduce the level of pro-inflammatory factors secreted from macrophages41, 42. Here, we specifically assay the capacity for our anti-ICAM1 coated MSCs to modulate the inflammatory cytokine secretion through an in vitro coculture assay with BMDMs (macrophages).
Primary BMDMs were directly contacted with uncoated MSCs, anti-ICAM1 coated MSCs or serum alone in LPS/IFN-γ supplemented medium for 3 h. Under LPS and IFN-γ stimulation, macrophages can be stimulated to M1 phenotypes which produce the proinflammatory TNF-α cytokine43. Either the uncoated or anti-ICAM1 coated MSCs cocultured with inflammatory macrophages showed significant decrease in TNF-α secretion profile when compared to macrophage alone (Figure S5). We also investigated the anti-inflammatory cytokine secretion following MSC-macrophage crosstalk. The level of immunosuppressive IL-10 in the conditioned medium exhibited higher concentration in both MSC cocultured groups compared with non-MSC exposed macrophages (Figure S5). Hence, we can conclude that the immunosuppression effects of MSCs are not adversely influenced by the coating which has not affected the secretory profile of MSCs.
Quantitation of surface group density on coated cell membrane
In addition to the preservation of the innate functions of MSCs, we have the flexibility to tune the biotin density on the surface of the cell and the density of antibodies against ICAM1 presented on our MSCs. The NHS-biotin molecule non-specifically tethers biotin groups to amine groups on the MSC’s peripheral membrane. The biotin labeling was determined by pairing accessible biotins with a streptavidin molecule labeled with phycoerythrin (PE), and comparing fluorescence of each cell to a calibration bead of known PE loading using flow cytometry (Figure S1). By simply controlling the concentration of the NHS-biotin in buffer, the surface biotin group is tunable up to 7000 biotin/μm2 (Figure 3a). This magnitude compares favorably to our previous reports of loading up to 10,000 molecules/μm2 using a similar NHS-biotin approach44. To confirm that there are not inaccuracies in the magnitude of fluorescence value based on energy transfer between adjacent molecules45, 46, we competed out the labeled streptavidin with an unlabeled streptavidin and found comparable biotin densities as the undiluted streptavidin-PE labeling.
Figure 3. Quantitation of surface biotin density on biotinylated MSCs.
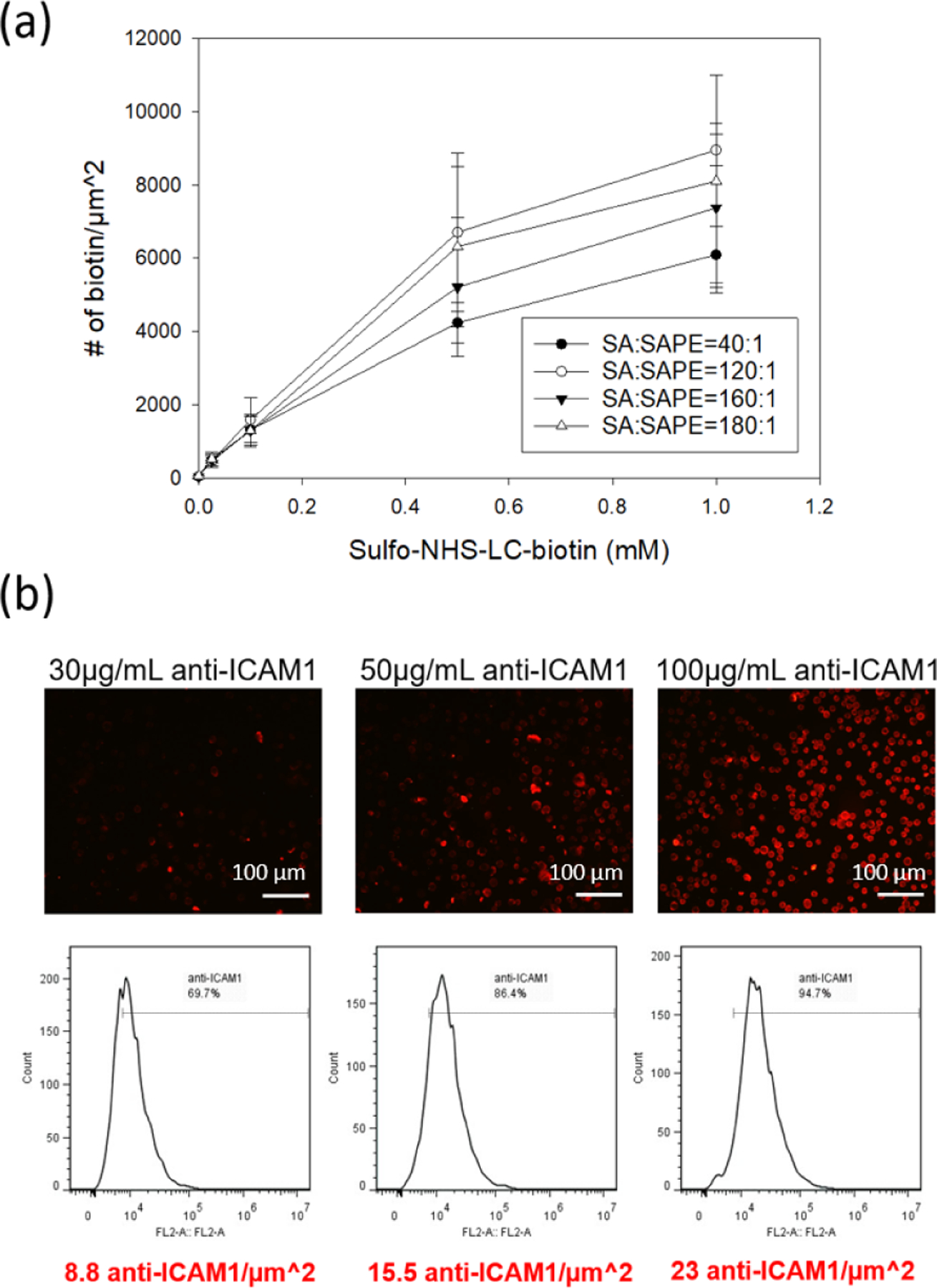
(a) Quantitation of biotin groups through competitive binding of SA/SAPE on biotinylated cells. The ester-biotin treated cells were stained with four different ratios of SA to SAPE. (b) Quantitation of ICAM1 antibodies on the MSC surface with different antibody concentrations (30, 50 and 100 μg/mL) through secondary PE labeling. The scale bar is 100 μm.
Antibody density on the MSC surface was estimated using PE-labeled secondary antibodies against mouse IgG. For an MSC treated with 1 mM NHS biotin, incubations in 30, 50, or 100 μg/mL of biotinylated antibodies against ICAM1 resulted in up to 23 antibodies/μm2 (Figure 3b). While ~1 PE is bound per secondary antibody, each primary antibody will be tagged by multiple secondary antibodies47. As a result, the actual density of anti-ICAM1 groups is likely several fold lower. The biotin density (~7,000 biotin/μm2) represents an upper limit for number of antibodies loaded onto the MSC surface. Given the larger size of the antibody (150kDa)48 over the streptavidin (53 kDa)49 and inefficiencies of sequential binding, it is reasonable there are order several orders of magnitude fewer antibodies than accessible biotin groups.
Coated cell adhesion on ICAM1-activated endothelial cells under physiological shears
Our overall MSC retention hypothesis is based on the expectation that the anti-ICAM1 coated MSCs adhere strongly to the ICAM1 presented by the inflamed endothelium near the infarct site (Figure 1b). To support these proposed mechanisms, we contrasted the force of adhesion between anti-ICAM1 coated MSCs and an activated endothelial cell with the shear stress exerted by a flowing buffer (Figure 4a). A near-confluent layer of HUVECs were grown on a microscope slide and were activated with a 5 or 20 h exposure for TNF-α, and activation was confirmed with fluorescent labeling of ICAM1 on HUVECs (Figure S6 and S7). MSCs were treated with 0.1 mM NHS-biotin, streptavidin, and 100 μg/mL biotinylated antibodies against human ICAM1. MSCs were allowed to settle on the HUVECs for 5 minutes and then subjected to wall shear stress up to 31 dyne/cm2. While greater shear stresses than 31 dyne/cm2 delaminated the HUVECs, the chosen shear range captures the physiological wall shear stress range observed in capillary beds (<1 dyne/cm2), veins (1–10 dyne/cm2), and many human arteries (10–70 dyne/cm2)26, 50–52.
Figure 4. Cell adhesion on ICAM1-expressed endothelium and cell detachment under the presence of shear flow.
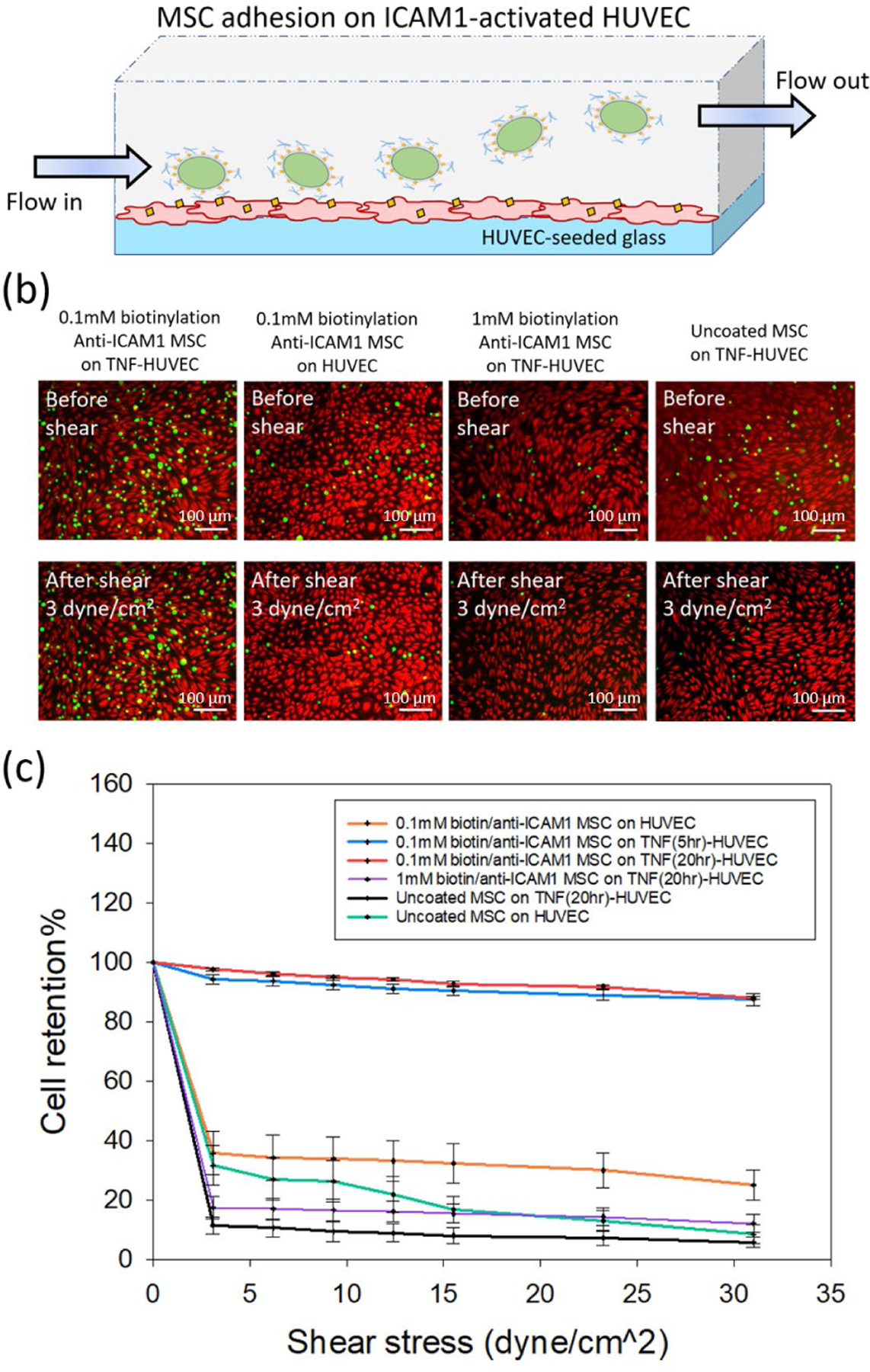
(a) Schematic diagram of experimental setup of shear-detached cells on HUVEC monolayer. (b) Fluorescence images of adhesion behavior of uncoated or anti-ICAM1 coated MSCs (GFP+) on untreated or TNFα-treated HUVECs (calcein-red staining) under shear flow. The anti-ICAM1 coated cells were treated with 0.1mM or 1mM ester-biotin, respectively. Cell adhesion behavior was observed before and after shear (3dyne/cm2). The scale bar is 100 μm. (c) Relationship between cell retention % and shear stress under different cell adhesion conditions (n= 7 for uncoated cells on activated or unactivated HUVECs; n=5 for 0.1mM biotin/anti-ICAM1 cells on all HUVEC conditions; n= 4 for 1mM biotin/anti-ICAM1 cells).
For all shear stresses studied, a greater number of the anti-ICAM1 coated MSCs were retained on the activated HUVEC cells than unmodified MSCs (Figure 4b and 4c). The retention of anti-ICAM1 coated MSCs was comparable with 5 or 20 h exposure to TNF-α. This is attributed to the surface density of ICAM1 on the activated HUVECs being in large excess (>500 ICAM1/μm2, Figure S7) compared to the surface density of the anti-ICAM1 ligands on the MSCs (<23 antibodies/μm2). In the absence of TNF-α induced ICAM1 activation, neither the anti-ICAM1 coated MSCs nor the unmodified MSCs were retained on the HUVEC surface under flow. This is expected as MSCs do not typically interact with endothelial cells through direct physical interactions. The statistically insignificant increase in mean retention of the anti-ICAM1 coated MSCs over uncoated MSCs on non-activated HUVECs is potentially attributed to the low basal expression of ICAM1 on HUVECs is <10 ICAM1/μm2, Figure S7).
Coated cell adhesion on ICAM1-localized glass under physiological shears
While specificity is not required for this application, the antibody-driven mechanism of retention is expected to be specific. These in vitro shear studies on activated HUVECs support the proposed antibody antigen mode of retention as the driver for the increased MSC retention in vivo. To further confirm the proposed mechanism for MSC retention, we evaluated cell adhesion to an ICAM1-coated surface under shearing flow (Figure 5a). The use of these purified protein surfaces also eliminates the potential for unintended interactions between mouse MSCs and human endothelial cells. Epoxy coated glass slides were incubated in human ICAM1, and the density of ICAM1 on the glass surface was determined through fluorescent labeling of ICAM1, and correlation of fluorescent intensity to a calibration slide in a microarray scanner. This approach provided surface densities of purified protein up to 5,000 ICAM1/μm2 (Figure 5b), and this range encompasses the surface density of ICAM1 on non-activated and activated HUVEC cells (Figure S7).
Figure 5. Cell adhesion on ICAM1-modifed glass and cell detachment under the presence of shear flow.
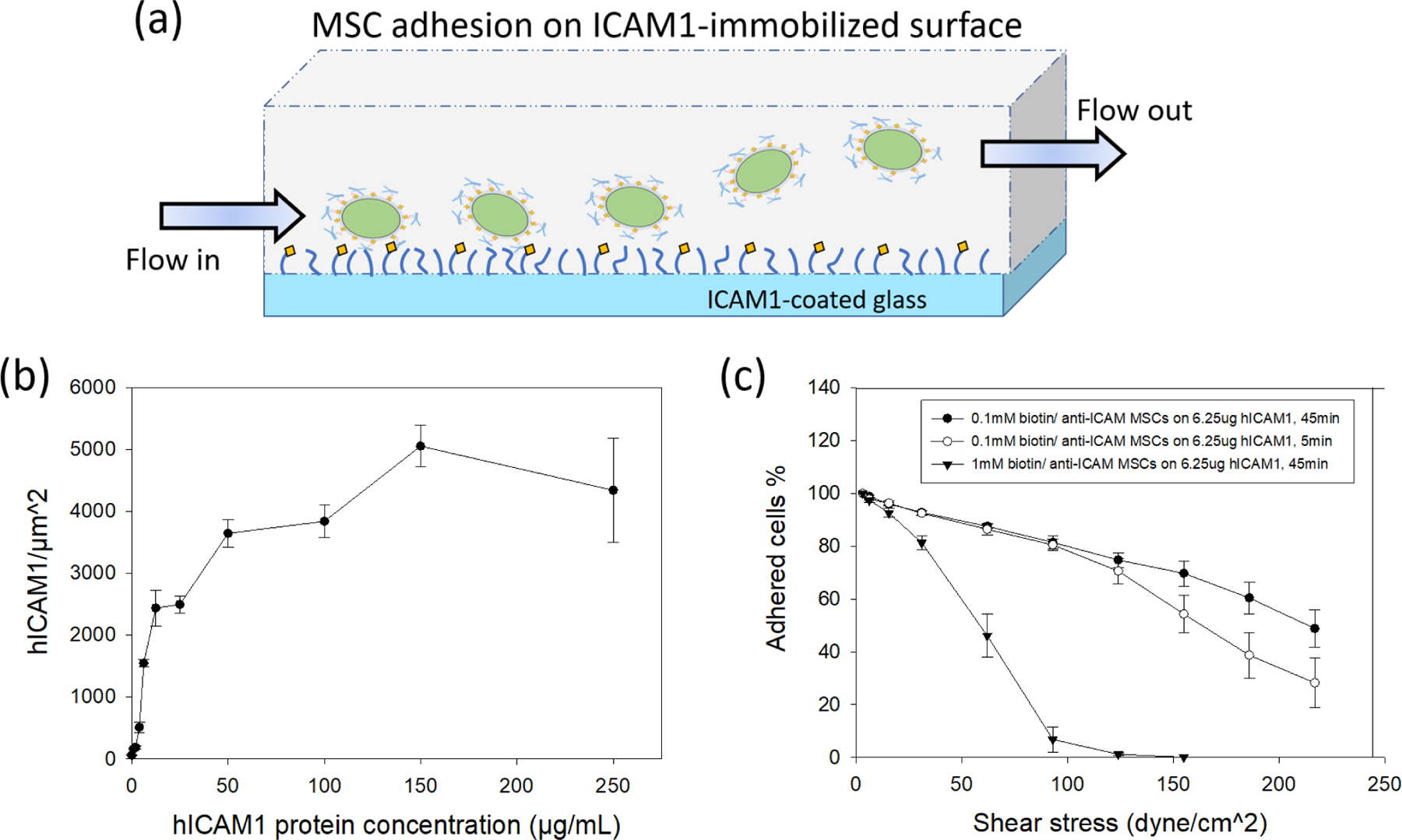
(a) Schematic diagram of experimental setup of shear-detached cells on ICAM1-modifed glass. (b) Relationship of human ICAM1 protein density and protein concentration used for epoxide glass modification. (c) Relationship between attached cells % and shear stress under different cell adhesion conditions (n=5 for 0.1mM biotin/anti-ICAM1 MSCs with 5 min; n=7 for 0.1mM biotin/anti-ICAM1 MSCs with 45 min; n=4 for 1mM biotin/anti-ICAM1 cells).
In particular, we focused on incubations of 6.25 μg ICAM1/mL to give a surface density of 2,400 ICAM1/μm2 to replicate the surface density of HUVECs activated with 20 h exposure to TNF-α (1,500 ICAM1/μm2). MSCs were added to the ICAM1 surfaces, incubated for 5 minutes at 37°C, and subjected to shearing flows. For all groups of anti-ICAM1 coated MSCs, the majority of cells remained adhered for the expected physiological range of wall shear stress (<70 dyne/cm2). Increasing the contact time between the MSC and the ICAM1 surfaces to 45 minutes did not have a significant increase in the fractional retention of cells, suggesting the adhesion is dominated by rapid binding events, including the designed antibody-antigen interactions. Importantly, the fractional retention of anti-ICAM1 coated MSCs on the purified ICAM1 surface compares favorably with the fractional retention of for anti-ICAM1 coated MSCs on the activated HUVECs. These two systems were run using the same MSC-surface contact times, antigen type, and antigen density. The quantitative agreement in MSC retention supports the antibody-antigen mode of retention in the HUVEC study, and by extension supports the role of antibody coatings in the increase in anti-ICAM1 coated cell retention in vivo. We were unable to retain any uncoated MSCs on the ICAM1-coated surface under the most minimal shear (Figure S8 and S9), and this supports our significant enhancement of retention for the coated cells in vivo and in the in vitro assay of MSC attachment to activated endothelial cells. Throughout our in vitro shear assays, MSCs that were biotinylated using 1 mM NHS-biotin were retained to a lesser degree than the MSCs that were biotinylated using 0.1 mM NHS-biotin (Figure 4c and 5c). The lower cell retention on 1mM biotin-modified cells is counter intuitive but reproducible in our MSC studies. The fact that this decrease persists in the study without the HUVEC cells (Figure 5), suggests that the harsher biotinylation conditions compromised the cell’s ability to tether. The mechanism for this loss of adhesion is still under investigation, but is likely related to a loss of cell function at higher biotinylation levels seen in our previous studies. Given the damage reported with >1 mM NHS-biotin in other cell lines40, we suspect this higher concentration is causing damage to the MSCs. It is reasonable to think that excessive conjugation of cell surface proteins will have a significant impact on the function of these proteins.
The shear assays of coated cells on purified surfaces of ICAM1 (Figure 5c) still support the antibody-antigen mechanism, but the magnitude of the effect is lower with the 1 mM NHS-biotin than for the 0.1 mM NHS biotin.
CONCLUSIONS
Direct injection of cells is commonly thought of as a successful method of directly controlling the position of cells in vivo. But on its own, direct injection is not capable of retaining the majority of these cells at the injection site across multiple clinical applications22, 53–56. While other studies have demonstrated ICAM1 based retention of cells in inflammatory bowel disease37, this study is the first direct evidence that anti-ICAM coatings increase the number MSCs retained in the heart tissue following a heart attack. We support the in vivo retention phenomenon with a mechanistic study of the adhesion of the coated MSCs when subjected to fluidic shears. The anti-ICAM coating significantly increases the number of cells retained on activated endothelial cells. To eliminate other cellular processes, we further supported this mechanism by coating microscope slides with ICAM1 protein and demonstrated the enhanced retention of the anti-ICAM1 coated MSCs over unmodified MSCs. In all, we connected the design of a cell surface modification with significantly increase in the number of MSCs remaining in a post-infarct heart by promoting strong adhesion between MSCs and ICAM1 presented by the inflamed endothelium. More broadly, this study is additional evidence that the intentional engineering of a cell’s peripheral membrane is an effective strategy for improving the localization of injected cells in the ever-expanding range of emerging cell-based therapies proposed in modern medicine.
Supplementary Material
Acknowledgment and Funding Sources
This work was partially supported by R01 HL127682 and the National Science Foundation under Award CBET-1351531. Dr. Abdel-Latif is supported by the University of Kentucky COBRE Early Career Program (P20 GM103527) and the NIH Grant R01 HL138488.
ABBREVIATIONS
- MI
myocardial infarction
- MSC
mesenchymal stem cell
- CD4
cluster of differentiation 4
- CD8
cluster of differentiation 8
- TNF-α
tumor necrosis factor alpha
- IFN-γ
Interferon gamma
- IDO
Indoleamine-pyrrole 2,3-dioxygenase
- PGE2
Prostaglandin E2
- TSG6
TNFα-stimulated gene-6
- CAM
cell adhesion molecules
- IgG
Immunoglobulin G
- ICAM1
Intercellular Adhesion Molecule 1
- VCAM
Vascular cell adhesion protein
- IBD
Inflammatory bowel disease
- PBS
Phosphate buffered saline
- GFP
green fluorescent protein
- PE
phycoerythrin
- HUVEC
Human Umbilical Vein Endothelial Cells
Footnotes
SUPPORTING INFORMATION
Quantitative surface group density calibration of PE and Cy3 fluorophore. Cell viability of ICAM-1 coated cells through MTT assay. Microscopy images of cell attachment after anti-ICAM1 coating on GFP+ MSCs. Immunosuppressive cytokine secretion from macrophage-interacted MSCs. Immunostaining of ICAM1 protein on activated HUVECs. Quantitative analysis of human ICAM1 surface density on activated HUVEC through flow cytometry. Stationery adhesion of anti-ICAM1 coated MSCs on hICAM1-modified glass with different hICAM1 density.
REFERENCES
- 1.Frangogiannis NG, The inflammatory response in myocardial injury, repair, and remodelling. Nature Reviews Cardiology 2014, 11 (5), 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prabhu SD; Frangogiannis NG, The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circulation research 2016, 119 (1), 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torre-Amione G, Immune activation in chronic heart failure. The American journal of cardiology 2005, 95 (11), 3–8. [DOI] [PubMed] [Google Scholar]
- 4.Westman PC; Lipinski MJ; Luger D; Waksman R; Bonow RO; Wu E; Epstein SE, Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. Journal of the American College of Cardiology 2016, 67 (17), 2050–2060. [DOI] [PubMed] [Google Scholar]
- 5.Sato K; Ozaki K; Oh I; Meguro A; Hatanaka K; Nagai T; Muroi K; Ozawa K, Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 2007, 109 (1), 228–234. [DOI] [PubMed] [Google Scholar]
- 6.Selmani Z; Naji A; Zidi I; Favier B; Gaiffe E; Obert L; Borg C; Saas P; Tiberghien P; Rouas‐Freiss N, Human leukocyte antigen‐G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+ CD25highFOXP3+ regulatory T cells. Stem cells 2008, 26 (1), 212–222. [DOI] [PubMed] [Google Scholar]
- 7.Aggarwal S; Pittenger MF, Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105 (4), 1815–1822. [DOI] [PubMed] [Google Scholar]
- 8.Xu G; Zhang Y; Zhang L; Ren G; Shi Y, The role of IL-6 in inhibition of lymphocyte apoptosis by mesenchymal stem cells. Biochemical and biophysical research communications 2007, 361 (3), 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J; Hematti P, Mesenchymal stem cell–educated macrophages: A novel type of alternatively activated macrophages. Experimental hematology 2009, 37 (12), 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.François M; Romieu-Mourez R; Li M; Galipeau J, Human MSC suppression correlates with cytokine induction of indoleamine 2, 3-dioxygenase and bystander M2 macrophage differentiation. Molecular Therapy 2012, 20 (1), 187–195. [DOI] [PubMed] [Google Scholar]
- 11.Melief SM; Schrama E; Brugman MH; Tiemessen MM; Hoogduijn MJ; Fibbe WE; Roelofs H, Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti‐inflammatory macrophages. Stem Cells 2013, 31 (9), 1980–1991. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y; Chen L; Scott PG; Tredget EE, Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem cells 2007, 25 (10), 2648–2659. [DOI] [PubMed] [Google Scholar]
- 13.Roura S; Bagó JR; Soler-Botija C; Pujal JM; Gálvez-Montón C; Prat-Vidal C; Llucià-Valldeperas A; Blanco J; Bayes-Genis A, Human umbilical cord blood-derived mesenchymal stem cells promote vascular growth in vivo. PloS one 2012, 7 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadivar M; Khatami S; Mortazavi Y; Shokrgozar MA; Taghikhani M; Soleimani M, In vitro cardiomyogenic potential of human umbilical vein-derived mesenchymal stem cells. Biochemical and biophysical research communications 2006, 340 (2), 639–647. [DOI] [PubMed] [Google Scholar]
- 15.Nishiyama N; Miyoshi S; Hida N; Uyama T; Okamoto K; Ikegami Y; Miyado K; Segawa K; Terai M; Sakamoto M, The significant cardiomyogenic potential of human umbilical cord blood‐derived mesenchymal stem cells in vitro. Stem cells 2007, 25 (8), 2017–2024. [DOI] [PubMed] [Google Scholar]
- 16.Sheng CC; Zhou L; Hao J, Current stem cell delivery methods for myocardial repair. BioMed research international 2013, 2013, 547902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinhoff G; Nesteruk J; Wolfien M; Grosse J; Ruch U; Vasudevan P; Mueller P, Stem cells and heart disease-Brake or accelerator? Advanced drug delivery reviews 2017, 120, 2–24. [DOI] [PubMed] [Google Scholar]
- 18.Kurtz A, Mesenchymal stem cell delivery routes and fate. International journal of stem cells 2008, 1 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbash IM; Chouraqui P; Baron J; Feinberg MS; Etzion S; Tessone A; Miller L; Guetta E; Zipori D; Kedes LH, Systemic delivery of bone marrow–derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation 2003, 108 (7), 863–868. [DOI] [PubMed] [Google Scholar]
- 20.Kraitchman DL; Tatsumi M; Gilson WD; Ishimori T; Kedziorek D; Walczak P; Segars WP; Chen HH; Fritzges D; Izbudak I, Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation 2005, 112 (10), 1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freyman T; Polin G; Osman H; Crary J; Lu M; Cheng L; Palasis M; Wilensky RL, A quantitative, randomized study evaluating three methods of mesenchymal stem cell delivery following myocardial infarction. European heart journal 2006, 27 (9), 1114–1122. [DOI] [PubMed] [Google Scholar]
- 22.Dow J; Simkhovich BZ; Kedes L; Kloner RA, Washout of transplanted cells from the heart: a potential new hurdle for cell transplantation therapy. Cardiovascular research 2005, 67 (2), 301–307. [DOI] [PubMed] [Google Scholar]
- 23.Terrovitis J; Lautamäki R; Bonios M; Fox J; Engles JM; Yu J; Leppo MK; Pomper MG; Wahl RL; Seidel J, Noninvasive quantification and optimization of acute cell retention by in vivo positron emission tomography after intramyocardial cardiac-derived stem cell delivery. Journal of the American College of Cardiology 2009, 54 (17), 1619–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quevedo HC; Hatzistergos KE; Oskouei BN; Feigenbaum GS; Rodriguez JE; Valdes D; Pattany PM; Zambrano JP; Hu Q; McNiece I, Allogeneic mesenchymal stem cells restore cardiac function in chronic ischemic cardiomyopathy via trilineage differentiating capacity. Proceedings of the National Academy of Sciences 2009, 106 (33), 14022–14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Müller-Ehmsen J; Krausgrill B; Burst V; Schenk K; Neisen UC; Fries JW; Fleischmann BK; Hescheler J; Schwinger RH, Effective engraftment but poor mid-term persistence of mononuclear and mesenchymal bone marrow cells in acute and chronic rat myocardial infarction. Journal of molecular and cellular cardiology 2006, 41 (5), 876–884. [DOI] [PubMed] [Google Scholar]
- 26.Mongrain R; Rodés-Cabau J, Role of shear stress in atherosclerosis and restenosis after coronary stent implantation. Revista espanola de cardiologia 2006, 59 (1), 1–4. [PubMed] [Google Scholar]
- 27.Toma C; Pittenger MF; Cahill KS; Byrne BJ; Kessler PD, Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation 2002, 105 (1), 93–98. [DOI] [PubMed] [Google Scholar]
- 28.Grieve SM; Bhindi R; Seow J; Doyle A; Turner AJ; Tomka J; Lay W; Gill A; Hunyor SN; Figtree GA, Microvascular obstruction by intracoronary delivery of mesenchymal stem cells and quantification of resulting myocardial infarction by cardiac magnetic resonance. Circulation: Heart Failure 2010, 3 (3), e5–e6. [DOI] [PubMed] [Google Scholar]
- 29.Lipowsky HH; Bowers DT; Banik BL; Brown JL, Mesenchymal stem cell deformability and implications for microvascular sequestration. Annals of biomedical engineering 2018, 46 (4), 640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao W; Green TI; Liang X; Delint RC; Perry G; Roberts MS; Le Vay K; Back CR; Ascione R; Wang H, Designer artificial membrane binding proteins to direct stem cells to the myocardium. Chemical science 2019, 10 (32), 7610–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frangogiannis NG, Targeting the inflammatory response in healing myocardial infarcts. Current medicinal chemistry 2006, 13 (16), 1877–1893. [DOI] [PubMed] [Google Scholar]
- 32.Muller WA, Leukocyte–endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends in immunology 2003, 24 (6), 326–333. [DOI] [PubMed] [Google Scholar]
- 33.Granger DN; Senchenkova E In Inflammation and the Microcirculation, Colloquium series on integrated systems physiology: from molecule to function, Morgan & Claypool Life Sciences: 2010; pp 1–87. [PubMed] [Google Scholar]
- 34.Gottipati A; Chelvarajan L; Peng H; Kong R; Cahall CF; Li C; Tripathi H; Al-Darraji A; Ye S; Elsawalhy E, Gelatin Based Polymer Cell Coating Improves Bone Marrow-Derived Cell Retention in the Heart after Myocardial Infarction. Stem Cell Reviews and Reports 2019, 15 (3), 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Darraji A; Haydar D; Chelvarajan L; Tripathi H; Levitan B; Gao E; Venditto VJ; Gensel JC; Feola DJ; Abdel-Latif A, Azithromycin therapy reduces cardiac inflammation and mitigates adverse cardiac remodeling after myocardial infarction: potential therapeutic targets in ischemic heart disease. PloS one 2018, 13 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko IK; Kean TJ; Dennis JE, Targeting mesenchymal stem cells to activated endothelial cells. Biomaterials 2009, 30 (22), 3702–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ko IK; Kim B-G; Awadallah A; Mikulan J; Lin P; Letterio JJ; Dennis JE, Targeting improves MSC treatment of inflammatory bowel disease. Molecular Therapy 2010, 18 (7), 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis KA; Wu P-J; Cahall CF; Li C; Gottipati A; Berron BJ, Coatings on mammalian cells: interfacing cells with their environment. Journal of biological engineering 2019, 13 (1), 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeong JH; Schmidt JJ; Kohman RE; Zill AT; DeVolder RJ; Smith CE; Lai M-H; Shkumatov A; Jensen TW; Schook LG, Leukocyte-mimicking stem cell delivery via in situ coating of cells with a bioactive hyperbranched polyglycerol. Journal of the American Chemical Society 2013, 135 (24), 8770–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu P-J; Lilly JL; Arreaza R; Berron BJ, Hydrogel patches on live cells through surface-mediated polymerization. Langmuir 2017, 33 (27), 6778–6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dayan V; Yannarelli G; Billia F; Filomeno P; Wang X-H; Davies JE; Keating A, Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic research in cardiology 2011, 106 (6), 1299–1310. [DOI] [PubMed] [Google Scholar]
- 42.Cho D-I; Kim MR; Jeong H.-y.; Jeong HC; Jeong MH; Yoon SH; Kim YS; Ahn Y, Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Experimental & molecular medicine 2014, 46 (1), e70–e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez FO; Gordon S, The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports 2014, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lilly JL; Berron BJ, The role of surface receptor density in surface-initiated polymerizations for Cancer cell isolation. Langmuir 2016, 32 (22), 5681–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhuang X; Ha T; Kim HD; Centner T; Labeit S; Chu S, Fluorescence quenching: A tool for single-molecule protein-folding study. Proceedings of the National Academy of Sciences 2000, 97 (26), 14241–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ballerstadt R; Schultz J, Competitive-binding assay method based on fluorescence quenching of ligands held in close proximity by a multivalent receptor. Analytica Chimica Acta 1997, 345 (1–3), 203–212. [Google Scholar]
- 47.Van der Loos CM; Das PK; Houthoff H, An immunoenzyme triple-staining method using both polyclonal and monoclonal antibodies from the same species. Application of combined direct, indirect, and avidin-biotin complex (ABC) technique. Journal of Histochemistry & Cytochemistry 1987, 35 (11), 1199–1204. [DOI] [PubMed] [Google Scholar]
- 48.Dancil K-PS; Greiner DP; Sailor MJ, A porous silicon optical biosensor: detection of reversible binding of IgG to a protein A-modified surface. Journal of the American Chemical Society 1999, 121 (34), 7925–7930. [Google Scholar]
- 49.Zijlstra P; Paulo PM; Orrit M, Optical detection of single non-absorbing molecules using the surface plasmon resonance of a gold nanorod. Nature nanotechnology 2012, 7 (6), 379–382. [DOI] [PubMed] [Google Scholar]
- 50.Cunningham KS; Gotlieb AI, The role of shear stress in the pathogenesis of atherosclerosis. Laboratory investigation 2005, 85 (1), 9–23. [DOI] [PubMed] [Google Scholar]
- 51.Jeong S-K; Lee J-Y; Rosenson RS, Association between ischemic stroke and vascular shear stress in the carotid artery. Journal of Clinical Neurology 2014, 10 (2), 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papaioannou TG; Stefanadis C, Vascular wall shear stress: basic principles and methods. Hellenic J Cardiol 2005, 46 (1), 9–15. [PubMed] [Google Scholar]
- 53.Baragi VM; Renkiewicz RR; Qiu L; Brammer D; Riley JM; Sigler RE; Frenkel SR; Amin A; Abramson SB; Roessler BJ, Transplantation of adenovirally transduced allogeneic chondrocytes into articular cartilage defects in vivo. Osteoarthritis and Cartilage 1997, 5 (4), 275–282. [DOI] [PubMed] [Google Scholar]
- 54.Malhi H; Gupta S, Hepatocyte transplantation: new horizons and challenges. Journal of hepato-biliary-pancreatic surgery 2001, 8 (1), 40–50. [DOI] [PubMed] [Google Scholar]
- 55.Hofmann M; Wollert KC; Meyer GP; Menke A; Arseniev L; Hertenstein B; Ganser A; Knapp WH; Drexler H, Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation 2005, 111 (17), 2198–2202. [DOI] [PubMed] [Google Scholar]
- 56.Brenner W; Aicher A; Eckey T; Massoudi S; Zuhayra M; Koehl U; Heeschen C; Kampen WU; Zeiher AM; Dimmeler S, 111In-labeled CD34+ hematopoietic progenitor cells in a rat myocardial infarction model. Journal of Nuclear Medicine 2004, 45 (3), 512–518. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.